Back to Journals » Journal of Inflammation Research » Volume 17
FTO Stabilizes MIS12 to Inhibit Vascular Smooth Muscle Cell Senescence in Atherosclerotic Plaque
Authors Sun J, Wang M, Jia F, Song J, Ren J ![]() , Hu B
, Hu B
Received 30 October 2023
Accepted for publication 28 February 2024
Published 20 March 2024 Volume 2024:17 Pages 1857—1871
DOI https://doi.org/10.2147/JIR.S447379
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Adam Bachstetter
Jingzhao Sun,1 Mengqi Wang,2 Fengming Jia,2 Jiantao Song,2 Jinlin Ren,2 Bo Hu2
1Department of Emergency, Shandong Provincial Hospital, Shandong University, Jinan, Shandong, 250021, People’s Republic of China; 2Department of Emergency, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, 250021, People’s Republic of China
Correspondence: Bo Hu, Department of Emergency, Shandong Provincial Hospital Affiliated to Shandong First Medical University, No. 324, Jingwu Road, Jinan, Shandong, 250021, People’s Republic of China, Email [email protected]
Purpose: Atherosclerosis is the main cause of atherosclerotic cardiovascular disease (CVD). Here, we aimed to uncover the role and mechanisms of fat mass and obesity-associated genes (FTO) in the regulation of vascular smooth muscle cell (VSMC) senescence in atherosclerotic plaques.
Methods: ApoE−/− mice fed a high-fat diet (HFD) were used to establish an atherosclerotic animal model. Immunohistochemistry, and the staining of hematoxylin-eosin, Oil Red O, Sirius red, and Masson were performed to confirm the role of FTO in atherosclerosis in vivo. Subsequently, FTO expression in primary VSMCs is either upregulated or downregulated. Oxidized low-density lipoprotein (ox-LDL) was used to treat VSMCs, followed by EdU staining, flow cytometry, senescence-associated β-galactosidase (SA-β-gal) staining, immunofluorescence, telomere detection, RT-qPCR, and Western blotting to determine the molecular mechanisms by which FTO inhibits VSMC senescence.
Results: Decreased FTO expression was observed in progressive atherosclerotic plaques of ApoE−/− mice fed with HFD. FTO upregulation inhibits atherosclerotic lesions in mice. FTO inhibits VSMC aging in atherosclerotic plaques by helping VSMC withstand ox-LDL-induced cell cycle arrest and senescence. This process is achieved by stabilizing the MIS12 protein in VSMC through a proteasome-mediated pathway.
Conclusion: FTO inhibits VSMC senescence and subsequently slows the progression of atherosclerotic plaques by stabilizing the MIS12 protein.
Keywords: vascular smooth muscle cell, FTO, HFD, senescence, MIS12 protein
Introduction
Atherosclerosis is a chronic disease that is widely distributed worldwide and is characterized by high incidence and mortality rates. Vascular smooth muscle cells (VSMCs) are the basic components of arterial stromal cells and fibrous caps. The continuous accumulation of atherosclerotic plaques in the intima of large and medium-sized arteries leads to vascular stenosis, which restricts blood flow and causes severe tissue hypoxia.1 Senescent VSMCs accumulate during atherosclerosis and drive plaque progression.2 VSMCs from human atherosclerotic plaques show lower proliferation levels and higher p21 and p16 expression levels, as well as increased senescence associated beta-galactosidase (SA-β-gal) activity, compared to cells from healthy arterial medium.3 In atherosclerotic plaques of patients with ischemic heart disease, abdominal aortic aneurysm, and peripheral artery disease, a large number of senescent VSMCs accumulated,4 and approximately 20% of VSMCs present age-related markers in the carotid artery plaques.5 In addition, VSMCs from atherosclerotic fibrous caps had shorter telomeres than normal ones.6 Therefore, studying VSMC senescence can help to elucidate the detailed mechanisms underlying the occurrence of arterial sclerosis.
The fat mass and obesity-associated gene (FTO) was one of the earliest genes found in the human genome, and its function is associated with obesity, lipid metabolism, and energy regulation.7 Current research suggests that FTO gene polymorphisms may be involved in the pathogenesis of metabolic diseases, such as obesity, weight gain, and diabetes.8 Initially, FTO was thought to be related only to human body fat content, body mass index (BMI), and other indicators. However, recent evidence has shown that the FTO gene-encoded protein is a key regulator of metabolic processes, such as energy metabolism, appetite regulation, insulin sensitivity, glucose, and fatty acid metabolism.8–10 Additionally, studies have found associations between FTO gene variations and cognitive ability, neurodevelopment, and personality traits.11,12
Recent studies have demonstrated a relationship between FTO expression and atherosclerosis. Prospective or retrospective cohort studies have found that FTO gene polymorphisms (rs1421085, rs17817449, and rs1121980) are associated with an increased risk of atherosclerosis.13 FTO gene polymorphisms are significantly correlated with atherosclerosis-related indicators such as total cholesterol, triglycerides, and the risk of hypertension.14 Mutations or pathogenic missense mutations in the FTO gene, such as Alstrom syndrome and Bardet-Biedl syndrome.15 Although the causal relationship between FTO and atherosclerosis is not yet fully understood, an increasing number of studies have recognized that FTO may play a role in the occurrence and development of atherosclerosis.
MIS12 is a microtubule-associated protein and a constituent of the MIND complex that can affect mitosis and cell cycle via interactions with other proteins. Interestingly, Zhang et al suggested that FTO may stabilize MIS12 protein via a proteasome-mediated pathway to inhibit the senescence of human mesenchymal progenitor cells (hMPCs)16 and is involved in cell cycle progression.16,17 However, there is no evidence that MIS12 is correlated with an increased risk of atherosclerosis. We propose the use of ApoE−/− mice fed with a high-fat diet (HFD) as a model of atherosclerosis to investigate the role and cellular and molecular mechanisms by which FTO regulates atherosclerosis in VSMCs. This study aimed to deepen our understanding of atherosclerosis and to identify new drug targets and preventive strategies.
Materials and Methods
Animals and Experimental Procedures
ApoE−/− mice on a C57BL6 background (all male, 8 weeks old) were obtained from Gempharmatech Co., Ltd. (Nanjing, China) and maintained under a 12-hour dark-light cycle. Animal experiments were approved by the Institutional Animal Care and Use Committee of Shandong Provincial Hospital, Shandong University and performed according to the Guidelines for the Care and Use of Laboratory Animals approved by Shandong Provincial Hospital, Shandong University. Mice in the HFD group were fed a HFD (containing 0.15% cholesterol and 21% lipid) for 16 weeks. Mice in the chow group were fed a regular diet as a control. An adeno-associated virus (AAV)9 vector carrying the FTO gene (1 × 1011 viral genomes) was injected into the mice via the tail vein once before HFD feeding.18 Mice that received AAV9 injections containing a null transgene (AAV9-null) were used as controls.
Metabolism Test
After the experiments were completed, overnight-fasted mice were anesthetized and euthanized, and blood samples were harvested for the detection of high-density lipoprotein (HDL), low-density lipoprotein cholesterol (LDL-C), total cholesterol (TC), and triglycerides (TG) using the corresponding kits (Nanjing Jiancheng Biological Engineering Research Institute, Nanjing, China).
Hematoxylin and Eosin (H&E) Staining
An H&E staining kit (C0105S, Beyotime, China) was used to stain aortic tissue sections. Briefly, the sections were stained with hematoxylin for 10 min and counterstained with eosin for 30s at room temperature. Tissue sections were imaged using a light microscope (Eclipse 80i; Nikon Corporation).
Sirius Red and Masson Staining
Aortic tissues were fixed in 4% formalin fixation, processed to dehydration and embedding, and sectioned into 5 μm thickness. Tissue sections were respectively stained with Modified Sirius Red Stain Kit (G1472, Solarbio, China) and Masson’s Trichrome Staining Kit (C0189S, Beyotime, China) according to the manufacturers’ instructions. After washing with purified water, the tissue sections were dehydrated with ethanol, cleared in xylene, and mounted.
Primary VSMC Culture
As described before,19 primary VSMCs from the mouse aorta were extracted. To obtain primary VSMCs, thoracoabdominal aortas were extracted from healthy C57BL6 mice. The collected tissues were washed with DMEM (BasalMedia, Shanghai, China) and cut into 1 mm3 pieces. The pieces were plated onto 24-well plates and digested with 1 mg/mL collagenase (Sigma-Aldrich) and 0.5 mg/mL elastase (Sigma-Aldrich) for 1.5 h at 37°C. Collected VSMCs were cultured in DMEM with 100 U/mL penicillin, streptomycin, and 10% FBS (BasalMedia) in a humidified atmosphere containing 5% CO2. Cells from passages to 3–5 were used for subsequent analyses. VSMCs were treated with 50 μg/mL oxidized low-density lipoprotein (ox-LDL; Solarbio, China) for 72 h of incubation at 37°C to induce VSMC senescence.
Cell Transfection
The siRNA sequences were as follows: FTO siRNA, 5’-GACCCCCAAAGATGATGAGTT-3’ and scrambled siRNA, 5’-GGATTATTCCCGACGAGACAA-3’. MIS12 siRNA, 5’-GACTTGCCTGCTGAGGATCTA-3’; scrambled siRNA, 5’-GGCGCACTTCCGTAAGAGTTT-3’ The siRNAs were obtained from Tsingke (Beijing, China), and the FTO overexpression plasmids and empty control pcDNA3.1 were from FulenGen (Guangzhou, China). Lipofectamine 3000 (Invitrogen) was used for transfection for 48 h following the manufacturer’s instructions.
Oil-Red-O Staining Assay
Oil Red O staining of mouse aortic tissues or cells was performed using Oil Red O staining kits (G1262 and G1261, Solarbio, China). Briefly, 6 μm tissue slices were fixed with 4% paraformaldehyde for 10 min and rinsed with distilled water. After washing with 60% isopropanol for 30s, the tissue slices were incubated with Oil Red O staining solution for 15 min and stained with hematoxylin solution for 2 min. The stained tissues were imaged using a light microscope (Eclipse 80i; Nikon Corporation).
EdU Staining
An EdU staining kit (C0071S; Beyotime) was used to detect cell proliferation. VSMCs in 6-well plates and treated with ox-LDL. The cells were then incubated with 10 μM EdU for 3 h, fixed with 4% paraformaldehyde for 15 min, and permeated with 0.3% Triton X-100 for another 15 min. After incubating the cells with the Click Reaction Mixture for 30 min at room temperature, they were stained with DAPI, scanned, and imaged under a Nikon Eclipse Ni fluorescence microscope (Nikon, Tokyo, Japan).
Cumulative Population Doubling (CPD) Level
CPD was calculated to evaluate the long-term cell growth. The following formula was used:
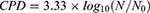
Where N is the cell number at the end of the experiment and N0 at the beginning. Growth curves were constructed by calculating CPD, which is the sum of the individual CPDs.
Cell Cycle Detection
A cell cycle detection kit (C1052, Beyotime, China) was used to detect the cell cycle. The cells were mixed with the RNase reagent at 37°C for 1 h after overnight immobilization in 70% ethanol. After staining the cells for 30 min with PI, the mixed solution was analyzed by a flow cytometer (Accuri‑C6 plus; BD Biosciences).
Cell Immunofluorescence
In 24-well plates, cells were cultured on glass coverslips (5 × 104 cells/well) for 48 h. After washing with PBS, cells were fixed for 1 h at room temperature with 4% paraformaldehyde in PBS. Next, PBS supplemented with 0.4% Triton X-100 and 3% BSA was added to the cells for 30 min, and the cells were labeled with an anti-p16 antibody (ab185620, Abcam, dilution 1:50) for 1 h at room temperature. After washing three times with PBST, the cells were incubated with goat anti-rabbit secondary antibody (ab150077, Abcam, dilution 1:200) for 1 h at room temperature and washed again before being mounted and imaged.
Measurement of the Relative Average Telomere Length
Based on the RT-qPCR telomere assay described previously,20 the relative average telomere length was examined. RT-qPCR was used to determine the Ct values of telomeres (T) and 36B4 (S), which served as the reference genes. Telomere length (T/S) can be assessed based on the ratio of telomere (T) repetitive copy number to single-copy internal reference gene (S), as follows: In contrast, T/S ratio is proportional to the telomere length.

Senescence-Associated β-Galactosidase (SA-β-Gal) Staining
SA-β-gal staining was performed using an SA-β-gal staining kit (C0602, Beyotime). Briefly, the cells were washed twice with PBS and fixed in fixation solution for 10 min. After washing, the cells were incubated with 1 mL SA-β-gal staining solution overnight at 37°C in a CO2-free atmosphere. The captured images were analyzed using an image analysis program (BioQuant).
Co-Immunoprecipitation (Co-IP)
Vectors expressing FLAG-LUC or FLAG-FTO (TransGen Biotech, Beijing, China) were used to transfect the VSMCs. VSMCs were lysed in NP-40 Lysis Buffer with a complete protease inhibitor cocktail (Roche). After centrifugation at 4°C, 12,000 r/min, for 30 min, the supernatant was collected and incubated with an anti-FLAG Affinity Gel (Sigma-Aldrich) at 4°C overnight. FTO-interacting protein complexes were obtained for Western blotting using competitive elution with FLAG peptides.
Telomerase Activity Assay
Extracts from cells were used to conduct the telomeric repeat amplification protocol. Telomerase activity was quantified using the Telomerase PCR ELISA kit (Roche, Mannheim, Germany) according to the manufacturer’s instructions.
RT-qPCR
The TRIzol reagent (Thermo Fisher Scientific) was used for RNA extraction. cDNA was generated using the PrimeScript RT-PCR kit (RR014, Takara, Dalian, China) and subjected to RT-qPCR analysis using TB Green® Premix Ex Taq™ II (RR820, Takara) in a CFX-Connect Real-Time PCR system (Bio-Rad). Primers used for RT-qPCR analysis were designed using the online primer-blast tool7 and are listed below:
FTO:
forward 5’-TTGGGACATCGAGACACCAG-3’,
backward 5’-GGCATTGAGGTCATCCAGCA-3’.
β-actin:
forward 5’-CACTGTCGAGTCGCGTCC-3’
backward 5’-TCATCCATGGCGAACTGGTG-3’
Western Blot
Proteins were extracted from VSMCs and aortic tissues using RIPA lysis buffer (Beyotime, China) and quantified using a BCA kit (Beyotime, China). Briefly, 20 µg of protein per sample was subjected to SDS-PAGE and electrotransferred onto a PVDF membrane (Millipore). The membrane was blocked with 5% non-fat milk, and incubated with antibodies at 4°C overnight, followed by incubation with an HRP-conjugated secondary antibody at room temperature for 1 h. Subsequently, imaging was performed using a ECL Plus (Beyotime) and the intensity of the target protein bands was determined using ImageJ software (v7.6.5, Tree Star, Inc.). The antibodies used for Western blotting were anti-FTO (27,226-1-AP; Proteintech), anti-MIS12 (ab70843; Abcam), anti-β-actin (ab8227; Abcam), anti-p53 (ab32389; Abcam), anti-p16 (10,883-1-AP; Proteintech), anti-p21 (ab109520; Abcam), anti-Tert (27,586-1-AP, Proteintech) and a secondary antibody goat anti-rabbit IgG (ab6721; Abcam).
Statistical Analysis
Experiments were performed at least in triplicate, and the data are presented as the mean ± SD. Statistical differences were analyzed using Student’s t-test or one-way analysis of variance (ANOVA), followed by Dunnett’s post-hoc test using the GraphPad Prism software (GraphPad Software, Inc.). Statistical significance was set at p < 0.05.
Result
Low-Level Expression of FTO in Progressed Atherosclerotic Plaque
From the RT-qPCR data, we observed low-level FTO expression in the aortic tissues of the model group of mice compared to that in the control group (Figure 1A). Western blot analysis showed results similar to those obtained using RT-qPCR (Figure 1B). In addition, the downregulation of FTO expression in the aortic tissues of the model group of mice was accompanied by the upregulation of the aging marker p16. These results indicate that FTO is significantly downregulated in atherosclerotic aortic tissues at both transcriptional and translational levels.
 |
Figure 1 Low-level expression of FTO in progressed atherosclerotic plaque. (A) Statistical results of FTO transcription level in mouse aortic tissues in chow and HFD groups were detected using RT-qPCR; (B) Western blot detected FTO protein level in aortic tissues of mouse in the chow and HFD groups. *p < 0.05. |
FTO Inhibits the Atherosclerotic Lesions in ApoE−/− Mice Fed with HFD
ApoE−/− mice were infected with AAV9 carrying FTO overexpression vectors (AAV9-FTO) or control vector (AAV9-null). Vascular Oil Red O staining showed that, in contrast to the control group, the model group exhibited significant atherosclerotic plaques in the aorta. However, compared to mice infected with AAV9-null, mice infected with AAV9-FTO exhibited a significant reduction in atherosclerotic plaque area in the aorta (Figure 2A). Subsequently, H&E and Oil Red O staining of the aortic slices of the mice were performed. The HFD group exhibited significant plaques, whereas AAV9-FTO reduced the atherosclerotic plaque area in the aorta (Figure 2B–D). These results indicate that upregulation of FTO expression can reduce the atherosclerotic plaque area in the aorta of mice. Compared to the chow group, the model group of mice had high blood levels of TC, TG, and LDL-C and low levels of HDL-C (Figure 2E). However, compared to AAV9-null mice, AAV9-FTO significantly reduced the levels of TC, TG, and LDL-C and increased the level of HDL-C. Overall, these findings suggest that FTO inhibits the formation and progression of atherosclerotic plaques to a certain extent.
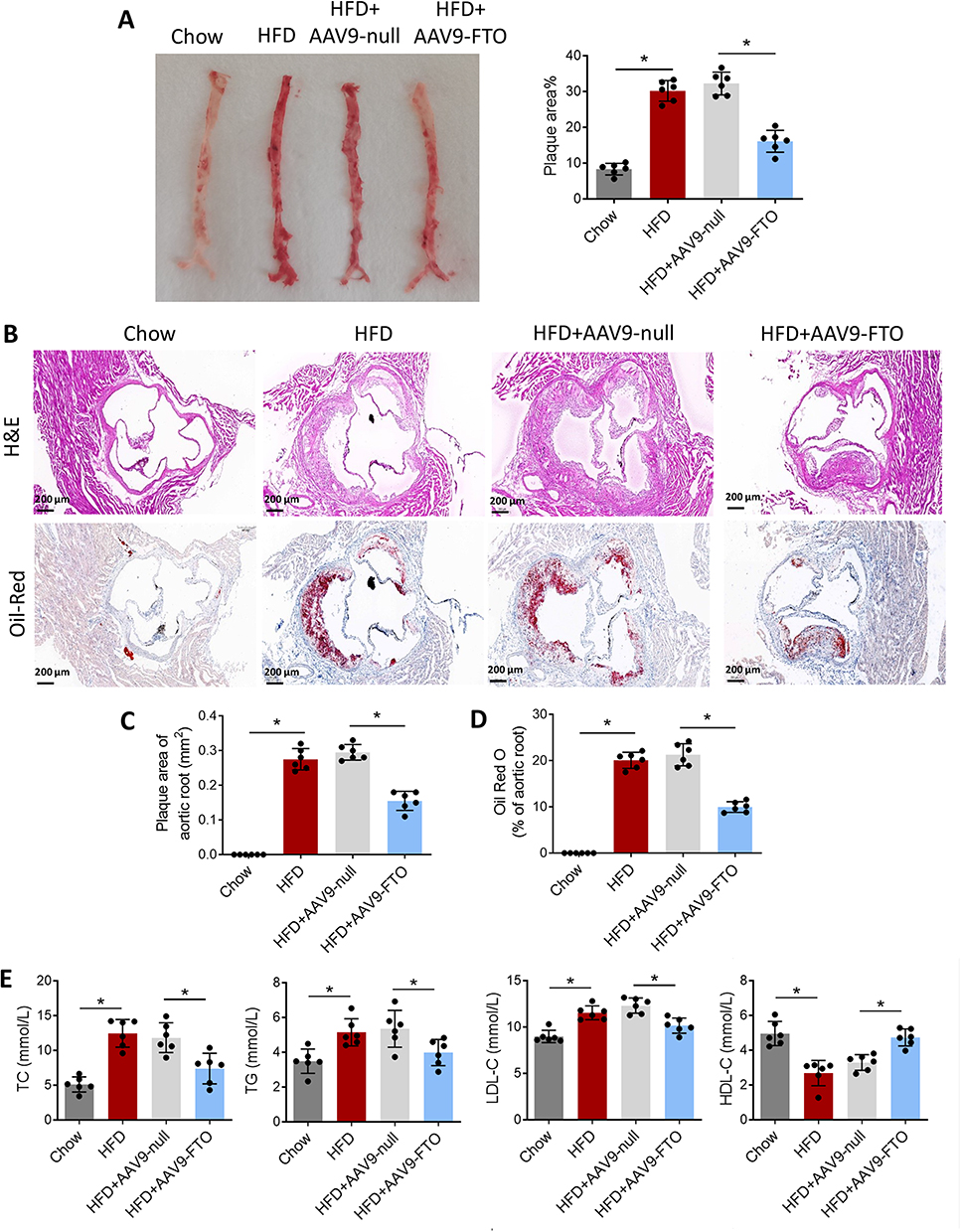 |
Figure 2 FTO inhibits the atherosclerotic lesions in ApoE−/− mice fed with HFD. (A) Oil Red O staining detected the distribution of aortic plaques in each group of mice; (B) Mouse aortic sections were stained with H&E and Oil Red O; (C) Plaque area of aortic root and (D) Oil Red O staining area of aortic root; (E) TC, TG, LDL-C, and HDL-C levels in mouse blood were detected. *p < 0.05. |
FTO Inhibits Senescence-Associated Markers in Atherosclerotic Plaque
Aortic stiffening is one of the major hallmarks of the ageing vascular in atherosclerotic animals. Here, we stained aortic tissue sections with Sirius red and Masson to evaluate age-related collagen deposition. Results presented in Figure 3A showed that, Sirius red- and Masson-positive areas were remarkably increased in HFD group as compared to the chow group. Injection of AAV9-FTO reduced Sirius red- and Masson-positive areas as compared to the injection of AAV9-null. RNA was extracted from the aortic tissue homogenate and qPCR was used to measure telomere length which is an indicator of cell senescence. Compared to the control group, the HFD group had shorter telomeres (Figure 3B). Infection with AAV9-FTO partially suppressed telomere shortening compared to AAV9-null infection. Subsequently, Western blotting was performed to detect the expression of cell senescence-associated markers. In contrast to the chow group, the aortic tissues of mice in the HFD group exhibited higher expression levels of p53, p21, and p16. AAV9-FTO infection partially suppressed the expression of these proteins compared with AAV9-null infection (Figure 3C and D). These results indicated that FTO can effectively suppress senescence-associated markers in an animal model of atherosclerosis.
 |
Figure 3 FTO inhibits senescence-associated markers in atherosclerotic plaque. (A) Sirius red and Masson staining detected the age-related collagen deposition of aortic plaques in each group of mice; (B) RNA was extracted from mouse aortic tissues and telomere length was detected via RT-qPCR; (C) Representative Western blot images of changes in the expression of cellular senescence-associated markers; (D) The relative expression of p53, p21 and p16 obtained from Western blot analysis. *p < 0.05. |
FTO Prevents VSMC Against Ox-LDL-Induced Cell Cycle Arrest
Next, primary mouse VSMCs were isolated and the FTO overexpression vector or empty vector control was transfected into the cells. RT-qPCR and Western blot analysis revealed that ox-LDL significantly downregulated FTO expression, whereas transfection with the FTO overexpression vector significantly upregulated FTO expression (Figure 4A and B). EdU staining revealed that ox-LDL significantly inhibited VSMC proliferation. Compared to transfection with the empty vector, transfection with the FTO overexpression vector partially alleviated the inhibitory effect of ox-LDL on cell proliferation (Figure 4C). Figure 4D shows that ox-LDL largely inhibited VSMC cell cycle progression, leading to cell arrest in the G0/G1 phase. Transfection with the FTO overexpression vector partially reversed cell cycle arrest compared with transfection with the empty vector. FTO can promote VSMC cycle progression, thereby preventing cell arrest in the G0/G1 phase.
 |
Figure 4 FTO prevents VSMC against ox-LDL-induced cell cycle arrest. (A) RT-qPCR detected the difference in transcription levels of FTO among groups; (B) Western blot detected the difference in FTO protein expression levels among cell groups; (C) EdU staining detected cell proliferation in each group; (D) Flow cytometry measures cell cycles for each group.*p < 0.05. |
FTO Prevents VSMC Against Ox-LDL-Induced Senescence
Furthermore, we calculated the CPD of VSMCs. ox-LDL significantly inhibited CPD in VSMCs, whereas transfection with the FTO overexpression vector significantly increased CPD compared to transfection with the empty vector (Figure 5A). RT-qPCR analysis of telomere length revealed that ox-LDL significantly shortened telomeres, whereas transfection with the FTO overexpression vector suppressed telomere shortening, compared to transfection with the empty vector (Figure 5B). As shown in Figure 5C, ox-LDL treatment increased the percentage of SA-β-gal-positive cells in VSMCs, whereas transfection with the FTO overexpression vector reduced the percentage of SA-β-gal-positive cells, in contrast to transfection with the empty vector. Furthermore, immunofluorescence results showed that ox-LDL increased the percentage of p16-positive cells in VSMCs, whereas transfection with the FTO overexpression vector reduced the percentage of p16-positive cells compared to transfection with the empty vector (Figure 5D). Western blot analysis also indicated that ox-LDL significantly upregulated the protein expression of p53, p21, and p16, while transfection with the FTO overexpression vector partially suppressed the expression of these proteins compared to transfection with the empty vector (Figure 5E). Transfection with the FTO overexpression vector significantly increased Tert expression and the telomerase activity in ox-LDL-treated VSMCs (Figure 5F–G). These results indicate that FTO inhibits ox-LDL-induced VSMC senescence.
 |
Figure 5 FTO prevents VSMC against ox-LDL-induced senescence. (A) Comparison of cumulative cell population multiplication in each group; (B) Telomere length was detected by RT-qPCR; (C) SA-β-gal staining of each group; (D) The proportion of p16-positive cells was detected by immunofluorescence; (E) The expression levels of p53, p21 and p16 in cells were detected by Western blot; (F) The expression levels of Tert was detected by Western-blot; (G) Telomerase activity was analyzed by the corresponding ELISA kit. *p < 0.05. |
FTO Stabilizes MIS12 Protein in VSMCs Through the Proteasome-Mediated Pathway
FTO inhibits cellular aging by stabilizing MIS12 expression.16 We hypothesized that FTO inhibits VSMC aging by regulating the stability of MIS12 protein. To verify this hypothesis, we first used Co-IP experiments to confirm the interaction between FTO and MIS12 in VSMCs (Figure 6A). Western blotting results, as shown in Figure 6B, showed that FTO overexpression led to the upregulation of MIS12, while FTO knockdown resulted in the downregulation of MIS12. The stability of the MIS12 protein was examined by adding the protein synthesis inhibitor CHX. The degradation rate of MIS12 protein in the siNC group was slow, whereas in the FTO knockdown cells, the degradation rate of MIS12 protein was accelerated (Figure 6C).
 |
Figure 6 FTO stabilizes MIS12 protein in VSMCs through the proteasome-mediated pathway. (A) Co-IP experiment was carried out to verify the interaction between FTO and MIS12 in VSMCs; (B) FTO and MIS12 protein expression was detected by Western blot; (C) The stability of MIS12 protein in FTO knockdown cells was detected by Western blot; (D) The effects of CHX and MG132 on the expression of MIS12 protein were detected by Western blot; (E) A mechanism diagram of FTO regulation on MIS12 protein during VSMC aging. *p < 0.05. |
Furthermore, CHX and the proteasome inhibitor MG132 were added simultaneously, and it was found that CHX downregulated the expression of MIS12 in FTO knockdown cells, while MG132 reversed the downregulation of MIS12 induced by CHX (Figure 6D). Based on the above experimental results, we proposed a mechanism by which FTO regulates MIS12 to exert its anti-aging effect, as shown in Figure 6E. FTO can stabilize the expression of MIS12 protein, avoid its degradation via the proteasome pathway, promote MIS12 to inhibit VSMC aging, and ultimately inhibit the formation and progression of atherosclerotic plaques.
FTO Prevents VSMC Senescence via Stabilizing MIS12
To further verify the above conclusion, the FTO overexpression vector and siMIS12 were co-transfected into VSMCs and treated with ox-LDL. As shown in Figure 7A, FTO significantly increased CPD, whereas siMIS12 inhibited CPD. RT-qPCR data showed that FTO alleviated telomere shortening, while siMIS12 could reverse the effect of FTO on telomere length (Figure 7B). FTO inhibited the percentage of SA-β-gal- and p16-positive VSMCs (Figure 7C and D). siMIS12 reversed the effect of FTO on the percentage of SA-β-gal- and p16-positive cells. FTO downregulated the protein expression of p53, p21, and p16, whereas siMIS12 reversed the regulatory effect of FTO on the aforementioned proteins (Figure 7E). The above experimental results validate the hypothesis that FTO inhibits VSMC aging by stabilizing MIS12.
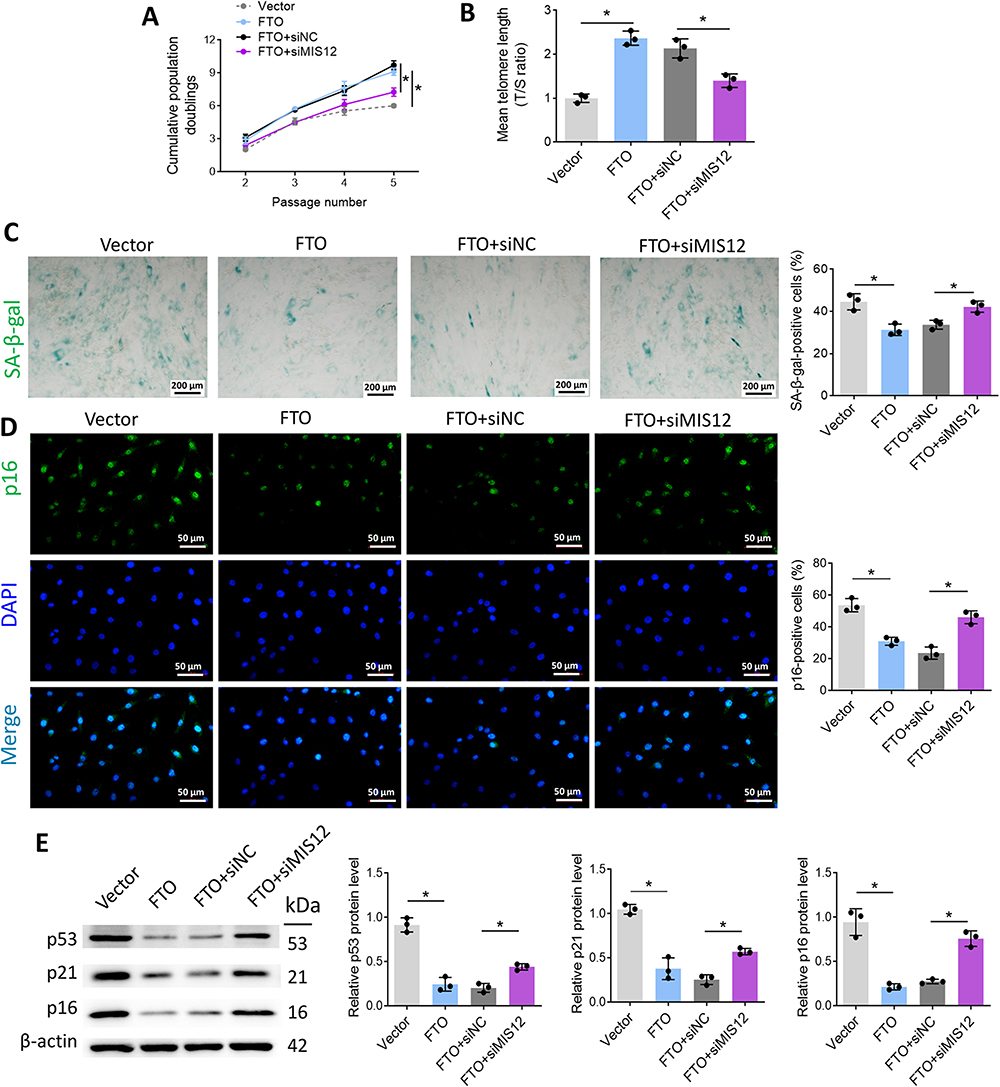 |
Figure 7 FTO prevents VSMC senescence via stabilizing MIS12. (A) Comparison of cumulative cell population multiplication after ox-LDL treatment; (B) Telomere length was measured by RT-qPCR; (C) SA-β-gal staining of VSMCs in each group; (D) The percentage of p16-positive cells was detected by immunofluorescence; (E) p53, p21, and p16 protein expression in cells was measured by Western blot. *p < 0.05. |
Discussion
In the current study, FTO expression was investigated in atherosclerosis, and its effects and related mechanisms in VSMC senescence were investigated. We found that FTO was weakly expressed in the aortic tissues of atherosclerotic mice. FTO inhibits plaque formation during atherosclerosis in mice. Furthermore, FTO inhibited VSMC senescence, as evidenced by the recovery of CPD values, alleviation of telomere shortening, reduction in the percentage of SA-β-gal and p16-positive cells, and downregulation of p53, p21, and p16 proteins. FTO ameliorated aortic stiffening of atherosclerotic mice as Sirius red- and Masson-positive areas were remarkably reduced. Additionally, we found that FTO inhibited MIS12 degradation in VSMCs via the proteasome pathway. By stabilizing MIS12 protein, FTO can inhibit VSMC senescence and slow the progression of atherosclerotic plaques.
Lipid metabolism disorder is one of the major underlying cause of atherosclerosis. Lipid mediates oxidative DNA damage and telomere dysfunction which induces VSMC senescence and promotes plaque progression.21 The cholesterol excess in VSMCs is likely the result of inadequate cholesterol efflux in advanced plaques.22,23 In the present study, high expression of FTO relieved the lipid metabolism disorder in atherosclerotic mice, indicating FTO is more possibly involved in the regulation of late stage of atherogenesis.
VSMCs play pivotal roles in plaque initiation and the progression of atherosclerosis and are thought to favor plaque stabilization.24,25 The absence of VSMCs leads to thinning of the fibrous cap and the formation of a necrotic core of plaques, which leads to the formation of fragile plaques. It is widely accepted that many aspects of the VSMC processes are involved in the pathogenesis of atherosclerosis, including cell proliferation, migration, cell death, phenotypic switching, and senescence.25 Recently, VSMC senescence has been recognized as a significant process in the induction of plaque progression and plaque instability.26 Thus, the inhibition of VSMC senescence has important clinical value for the prevention and treatment of atherosclerosis.21,27,28 FTO gene variants were previously reported to be associated with atherosclerosis and obesity-related metabolic diseases,29,30 and Zhang et al recognized the role of FTO in hMPC senescence.16 In the current study, we demonstrated that FTO is beneficial for controlling high-fat and ox-LDL-induced VSMC senescence.
FTO has been proven to be a potential therapeutic target for various diseases.31 For example, FTO was observed to be low in animal models of heart failure, and restoration of its expression contributed to cardiac contraction in heart failure.32 Inhibition of FTO via siRNA interference or by using a specific inhibitor inhibits the activation of macrophages and reduces the inflammatory response during endotoxic shock.33 FTO expression was also reported to be essential for the proliferation of pancreatic cancer cells.34 Moreover, FTO is associated with the growth and death of various human cancers, such as breast cancer,35 pancreatic cancer,36 colorectal cancer,37 and melanoma.38 Here, we uncovered, for the first time, the effects of FTO on inhibiting atherosclerotic plaque progression by helping VSMC to withstand senescence.
As part of the MIS12/MIND type complex, MIS12 is involved in the attachment of mitotic spindle microtubules to the kinetochore and kinetochore assembly, which is essential for proper kinetochore microtubule attachment.39 In addition to its essential role of MIS12 in regulating the kinetochore function, MIS12 has recently been reported to play a regulatory role in cell senescence. Wu et al showed that METTL3 stabilizes MIS12 expression to control hMSC senescence.40 Zhang et al showed that FTO stabilizes MIS12 via a proteasome-mediated pathway to inhibit hMSC senescence.16 Our results are consistent with those of previous studies that showed that FTO promotes MIS12 stability via a proteasome-mediated pathway to inhibit VSMC senescence. This study explored the mechanisms by which FTO regulates MIS12 expression in VSMC, thereby alleviating atherosclerosis.
As an increasing understanding of the mechanisms of FTO, over ten FTO inhibitors have been discovered in vitro and in vivo to have therapeutic effects on inhibiting tumors, including Rhein, MO-I-500, MA & MA2, and R-2HG.41 Most of them exhibits their therapeutic effects by inhibiting the enzymatic activity of FTO. For instance, Rhein binds to the catalytic domain of FTO demethylase and blocks its enzymatic activity.42 R-2HG, a small molecule compound, inhibits tumor growth by competitively inhibiting the enzymatic activity of FTO.43 However, at present, the therapeutic effects of FTO inhibitors are limited to tumor researches. The uses of them in treating atherosclerosis should be evaluated in the future.
Conclusion
In conclusion, we propose for the first time that FTO inhibits VSMC senescence and thereby slows the progression of atherosclerotic plaques by stabilizing MIS12 protein. We found that FTO inhibited the degradation of the MIS12 protein through the proteasome pathway. This finding provides a theoretical basis for designing targeted drugs for the treatment of atherosclerosis involving FTO or MIS12.
Data Sharing Statement
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
Animal experiments were approved by the Institutional Animal Care and Use Committee of Shandong Provincial Hospital, Shandong University and performed according to the Guidelines for the Care and Use of Laboratory Animals approved by Shandong Provincial Hospital, Shandong University.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the TCM Science and Technology Project of the Shandong Province (Z-2023001).
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. doi:10.1038/nature01323
2. Grootaert MOJ, Moulis M, Roth L, et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res. 2018;114(4):622–634. doi:10.1093/cvr/cvy007
3. Yin H, Pickering JG. Cellular senescence and vascular disease: novel routes to better understanding and therapy. Can J Cardiol. 2016;32(5):612–623. doi:10.1016/j.cjca.2016.02.051
4. Cafueri G, Parodi F, Pistorio A, et al. Endothelial and smooth muscle cells from abdominal aortic aneurysm have increased oxidative stress and telomere attrition. PLoS One. 2012;7(4):e35312. doi:10.1371/journal.pone.0035312
5. Matthews C, Gorenne I, Scott S, et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. 2006;99(2):156–164. doi:10.1161/01.RES.0000233315.38086.bc
6. Salpea KD, Humphries SE. Telomere length in atherosclerosis and diabetes. Atherosclerosis. 2010;209(1):35–38. doi:10.1016/j.atherosclerosis.2009.12.021
7. Loos RJ, Yeo GS. The bigger picture of FTO: the first GWAS-identified obesity gene. Nat Rev Endocrinol. 2014;10(1):51–61. doi:10.1038/nrendo.2013.227
8. Yang Z, Yu GL, Zhu X, Peng TH, Lv YC. Critical roles of FTO-mediated mRNA m6A demethylation in regulating adipogenesis and lipid metabolism: implications in lipid metabolic disorders. Genes Dis. 2022;9(1):51–61. doi:10.1016/j.gendis.2021.01.005
9. Claussnitzer M, Dankel SN, Kim KH, et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N Engl J Med. 2015;373(10):895–907. doi:10.1056/NEJMoa1502214
10. Ruan DY, Li T, Wang YN, et al. FTO downregulation mediated by hypoxia facilitates colorectal cancer metastasis. Oncogene. 2021;40(33):5168–5181. doi:10.1038/s41388-021-01916-0
11. Flores-Dorantes MT, Diaz-Lopez YE, Gutierrez-Aguilar R. Environment and gene association with obesity and their impact on neurodegenerative and neurodevelopmental diseases. Front Neurosci. 2020;14:863. doi:10.3389/fnins.2020.00863
12. Vasan SK, Fall T, Neville MJ, et al. Associations of variants in FTO and Near MC4R with obesity traits in south asian Indians. Obesity. 2012;20(11):2268–2277. doi:10.1038/oby.2012.64
13. Wing MR, Ziegler J, Langefeld CD, et al. Analysis of FTO gene variants with measures of obesity and glucose homeostasis in the IRAS Family Study. Hum Genet. 2009;125(5–6):615–626. doi:10.1007/s00439-009-0656-3
14. Jalili V, Mokhtari Z, Rastgoo S, et al. The association between FTO rs9939609 polymorphism and serum lipid profile in adult women. Diabetol Metab Syndr. 2021;13(1):138. doi:10.1186/s13098-021-00754-0
15. Littleton SH, Berkowitz RI, Grant SFA. Genetic Determinants of Childhood Obesity. Mol Diagn Ther. 2020;24(6):653–663. doi:10.1007/s40291-020-00496-1
16. Zhang S, Wu Z, Shi Y, et al. FTO stabilizes MIS12 and counteracts senescence. Protein Cell. 2022;13(12):954–960. doi:10.1007/s13238-022-00914-6
17. Hirayama M, Wei FY, Chujo T, et al. FTO demethylates cyclin D1 mRNA and controls cell-cycle progression. Cell Rep. 2020;31(1):107464. doi:10.1016/j.celrep.2020.03.028
18. Keeter WC, Carter NM, Nadler JL, Galkina EV. The AAV-PCSK9 murine model of atherosclerosis and metabolic dysfunction. European Heart Journal Open. 2022;2(3):oeac028. doi:10.1093/ehjopen/oeac028
19. Komaravolu RK, Waltmann MD, Konaniah E, Jaeschke A, Hui DY. ApoER2 (Apolipoprotein E Receptor-2) deficiency accelerates smooth muscle cell senescence via cytokinesis impairment and promotes fibrotic neointima after vascular injury. Arterioscler Thromb Vasc Biol. 2019;39(10):2132–2144. doi:10.1161/ATVBAHA.119.313194
20. Kawano Y, Kim HT, Matsuoka K, et al. Low telomerase activity in CD4+ regulatory T cells in patients with severe chronic GVHD after hematopoietic stem cell transplantation. Blood. 2011;118(18):5021–5030. doi:10.1182/blood-2011-06-362137
21. Grootaert MOJ, Finigan A, Figg NL, Uryga AK, Bennett MR. SIRT6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ Res. 2021;128(4):474–491. doi:10.1161/CIRCRESAHA.120.318353
22. Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129(15):1551–1559. doi:10.1161/CIRCULATIONAHA.113.005015
23. Wang Y, Dubland JA, Allahverdian S, et al. Smooth muscle cells contribute the majority of foam cells in ApoE (Apolipoprotein E)-deficient mouse atherosclerosis. Arterioscler Thromb Vasc Biol. 2019;39(5):876–887. doi:10.1161/ATVBAHA.119.312434
24. Allahverdian S, Chaabane C, Boukais K, Francis GA, Bochaton-Piallat ML. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res. 2018;114(4):540–550. doi:10.1093/cvr/cvy022
25. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118(4):692–702. doi:10.1161/CIRCRESAHA.115.306361
26. Wang J, Uryga AK, Reinhold J, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. 2015;132(20):1909–1919. doi:10.1161/CIRCULATIONAHA.115.016457
27. Xu F, Zhong JY, Lin X, et al. Melatonin alleviates vascular calcification and ageing through exosomal miR-204/miR-211 cluster in a paracrine manner. J Pineal Res. 2020;68(3):e12631. doi:10.1111/jpi.12631
28. Harman JL, Jorgensen HF. The role of smooth muscle cells in plaque stability: therapeutic targeting potential. Br J Pharmacol. 2019;176(19):3741–3753. doi:10.1111/bph.14779
29. Wing MR, Ziegler JM, Langefeld CD, et al. Analysis of FTO gene variants with obesity and glucose homeostasis measures in the multiethnic insulin resistance atherosclerosis study cohort. Int J Obes Lond. 2011;35(9):1173–1182. doi:10.1038/ijo.2010.244
30. Prakash J, Srivastava N, Awasthi S, et al. Association of FTO rs17817449 SNP with obesity and associated physiological parameters in a north Indian population. Ann Hum Biol. 2011;38(6):760–763. doi:10.3109/03014460.2011.614278
31. Lan N, Lu Y, Zhang Y, et al. FTO - A common genetic basis for obesity and cancer. Front Genet. 2020;11:559138. doi:10.3389/fgene.2020.559138
32. Mathiyalagan P, Adamiak M, Mayourian J, et al. FTO-dependent n(6)-methyladenosine regulates cardiac function during remodeling and repair. Circulation. 2019;139(4):518–532. doi:10.1161/CIRCULATIONAHA.118.033794
33. Luo J, Wang F, Sun F, et al. Targeted Inhibition of FTO demethylase protects mice against LPS-induced septic shock by suppressing NLRP3 inflammasome. Front Immunol. 2021;12:663295. doi:10.3389/fimmu.2021.663295
34. Tang X, Liu S, Chen D, Zhao Z, Zhou J. The role of the fat mass and obesity-associated protein in the proliferation of pancreatic cancer cells. Oncol Lett. 2019;17(2):2473–2478. doi:10.3892/ol.2018.9873
35. Akbari ME, Gholamalizadeh M, Doaei S, Mirsafa F. FTO gene affects obesity and breast cancer through similar mechanisms: a new insight into the molecular therapeutic targets. Nutr Cancer. 2018;70(1):30–36. doi:10.1080/01635581.2018.1397709
36. Tan Z, Shi S, Xu J. RNA N6-methyladenosine demethylase FTO promotes pancreatic cancer progression by inducing the autocrine activity of PDGFC in an m(6)A-YTHDF2-dependent manner. Oncogene. 2022;41(20):2860–2872. doi:10.1038/s41388-022-02306-w
37. Gholamalizadeh M, Tabrizi R, Bourbour F, et al. Are the FTO Gene Polymorphisms Associated with Colorectal Cancer? A Meta-analysis. J Gastrointest Cancer. 2021;52(3):846–853. doi:10.1007/s12029-021-00651-9
38. Yang S, Wei J, Cui YH, Park G, Shah P. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commu. 2019;10(1):2782.
39. Shrestha RL, Draviam VM. Lateral to end-on conversion of chromosome-microtubule attachment requires kinesins CENP-E and MCAK. Curr Biol. 2013;23(16):1514–1526. doi:10.1016/j.cub.2013.06.040
40. Wu Z, Shi Y, Lu M, et al. METTL3 counteracts premature aging via m6A-dependent stabilization of MIS12 mRNA. Nucleic Acids Res. 2020;48(19):11083–11096. doi:10.1093/nar/gkaa816
41. Han Z, Niu T, Chang J, et al. Crystal structure of the FTO protein reveals basis for its substrate specificity. Nature. 2010;464(7292):1205–1209. doi:10.1038/nature08921
42. Chen B, Ye F, Yu L, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. 2012;134(43):17963–17971.
43. Su R, Dong L, Li C, et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell. 2018;172(1–2):90–105.e123. doi:10.1016/j.cell.2017.11.031
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.