Back to Journals » International Journal of Nanomedicine » Volume 16
Formulation and Characterization of Phytostanol Ester Solid Lipid Nanoparticles for the Management of Hypercholesterolemia: An ex vivo Study
Authors Shrestha SC ![]() , Ghebremeskel K
, Ghebremeskel K ![]() , White K
, White K ![]() , Minelli C
, Minelli C ![]() , Tewfik I
, Tewfik I ![]() , Thapa P, Tewfik S
, Thapa P, Tewfik S ![]()
Received 14 August 2020
Accepted for publication 4 December 2020
Published 9 March 2021 Volume 2021:16 Pages 1977—1992
DOI https://doi.org/10.2147/IJN.S276301
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Sony Chandi Shrestha,1,2 Kebreab Ghebremeskel,1 Kenneth White,1 Caterina Minelli,2 Ihab Tewfik,3 Panna Thapa,4 Sundus Tewfik5
1School of Human Sciences, London Metropolitan University, London, UK; 2Surface Technology, National Physical Laboratory, London, UK; 3Life Sciences, University of Westminster, London, UK; 4Department of Pharmacy, Kathmandu University, Dhulikhel, Nepal; 5Department of Applied Nanomolecules, Bloomsnano Limited, London, UK
Correspondence: Sony Chandi Shrestha Email [email protected]
Background: Phytostanols are naturally occurring compounds that reduce blood cholesterol levels significantly. However, their aqueous insolubility poses formulation challenges.
Aim: To formulate and characterize solid lipid nanoparticle carriers for phytostanol esters to enhance the bioavailability of phytostanols.
Methods: Phytostanol ester solid lipid nanoparticles were formulated by the microemulsion method. They were characterized for particle size distribution, polydispersity index, shape, surface charge, entrapment efficiency, stability, chemical structure, and thermal properties. The uptake of the formulation by cell lines, HepG2 and HT-29, and its effect on cell viability were evaluated.
Results: The formulation of solid lipid nanoparticles was successfully optimised by varying the type of lipids and their concentration relative to that of surfactants in the present study. The optimised formulation had an average diameter of (171 ± 9) nm, a negative surface charge of (− 23.0 ± 0.8) mV and was generally spherical in shape. We report high levels of drug entrapment at (89 ± 5)% in amorphous form, drug loading of (9.1 ± 0.5)%, nanoparticle yield of (67 ± 4)% and drug excipient compatibility. The biological safety and uptake of the formulations were demonstrated on hepatic and intestinal cell lines.
Conclusion: Phytostanol ester solid lipid nanoparticles were successfully formulated and characterized. The formulation has the potential to provide an innovative drug delivery system for phytostanols which reduce cholesterol and have a potentially ideal safety profile. This can contribute to better management of one of the main risk factors of cardiovascular diseases.
Keywords: cardiovascular diseases, cholesterol, phytostanol ester, PSE, solid lipid nanoparticles, SLNPs, hypercholesterolemia
Introduction
Cardiovascular diseases are the leading cause of mortality accounting for approximately 31% of annual global deaths (17.9 million people) in 2016.1 High total cholesterol and low-density lipoprotein cholesterol (LDL-C) are modifiable risk factors.2 LDL-C plays a major role in the progression of atherosclerosis, and an elevated level of cholesterol was responsible for circa 4.5% of total deaths (2.6 million lives) in 2004.3,4 Lowering of LDL-C levels significantly reduces cardiovascular morbidity and mortality risks.3 These risk factors are typically treated with standard hypolipidemic agents, including statins, fibrates, bile acid-binding resins and cholesterol absorption inhibitors.5 However, these agents are typically associated with various adverse drug reactions such as muscle diseases, elevated activity of liver enzymes and gastrointestinal symptoms.6–8 In addition to adverse reactions, statin intolerance remains a therapeutic challenge, together with familial disorders, treatment resistance, non-compliance and optimization of lipid-lowering therapy especially in severe hypercholesterolemia.9,10 Hence, there is a need for novel, effective and safe therapeutic compounds that lower cholesterol without the adverse effects of the current lipid-lowering agents.
Phytosterols (plant sterols) and phytostanols (plant stanols) are naturally occurring substances that are found in variable amounts in fruits, vegetables, cereals, nuts, seeds, etc.11,12 Phytosterols and phytostanols were chemically identified in the 1920s by Anderson and Shriner.13,14 Subsequently, they were shown to have cholesterol-lowering potential in experimental animals and humans in the 1950s.15–18 Phytostanols are saturated derivatives of sterols, saturated at C5-6 without double bonds.19 Stanols block both dietary and biliary intestinal cholesterol absorption by competing with cholesterol for its access to receptors and increasing its excretion. The efficacy and safety of plant stanols have been demonstrated in a multitude of well-designed and rigorously conducted clinical studies.20–23 It has been shown that a daily intake of 2–3 grams of plant stanols reduces LDL-C levels by 10–15% without side effects.19 There is also some evidence that it reduces triglycerides.24–26
However, plant stanols are insoluble in water and consequently have low bioavailability.27,28 The aqueous insolubility poses a challenging problem during the formulation of stanol products.29,30 Esterified forms of phytostanols with improved fat solubility are currently available in the market through various delivery vehicles such as margarine, yogurt, vegetable spread, cereal bar, soy products, chocolate, biscuit.23,31 These fortified foods add to the daily caloric intake requiring isocaloric diet management and are more expensive compared to ordinary foods.32,33 In order to realize the benefits of phytostanols, there is a need to devise a novel way of delivery.
Solid Lipid Nanoparticles (SLNPs), which were first reported by Speiser and co-workers34 in 1986, are colloidal delivery systems consisting of a lipid core and stabilized by an outer shell made of an amphiphilic surfactant. Their typical size may vary from 50 nm to 1 µm.35 As with other nanoparticle-based systems, they have the potential to improve hydrophobic molecule bioavailability.36,37 Furthermore, particles under 200 nm have been shown to achieve longer circulation times in blood, compared with larger particles regardless of their surface modifications,37 further improving their bioavailability.36,37 Reported manufacturing processes for phytosterol nanocarriers involve the use of organic solvents such as hexane, isopropyl alcohol, ethanol and acetone.38–40 However, there is concern that the organic solvents may interact with the target drug and cause toxicological and environmental problems.41 An aqueous-based formulation that avoids the use of organic solvents is desirable to improve safety, as well as for environmental and economic impact. Various studies demonstrate that the physical state and structure of the carrier, formulation type, and particle size of phytostanol formulation are important parameters that influence its cholesterol-lowering efficacy.42–48 Water-soluble SLNPs have the potential to be novel and effective delivery systems for phytostanols. The aim of this study was to contribute to the better management of hypercholesterolemia through the synthesis and characterization of SLNPs loaded with plant stanol ester (PSE).
Materials and Methods
Materials
Stearic acid (98%), palmitic acid (95%) and lecithin (refined solid) were purchased from Alfa Aesar (Haverhill, MA, US). Lauric acid (99.5+%) and sodium taurocholate (98%) were procured from Sigma Aldrich (St Louis, MO, US) and Acros Organics, Fisher Scientific (Hampton, NH, US), respectively. Plant stanol ester (91.2% sitostanol, 8.8% campestanol) was received as an in-kind donation from Raisio Nutrition Ltd. (Finland). Double deionized water (18 MΩ·cm) purified by a purite system was used throughout the experiments.
Anhydrous ethanol (99.9%) and n-heptane (99%) were obtained from Fisher Scientific. Potassium hydroxide pellets (≥85%) were purchased from Sigma Aldrich. Silylation reagent, Bis (trimethylsilyl) trifluoroacetamide (BSTFA) + 1% Trimethylsilyl chloride (TMCS) (99%) was procured from Supleco (Bellefonte, US). Pyridine (99+%) was bought from Acros Organics (Geel, Belgium). Sitostanol standard (>97%) was obtained from Matreya (Pennsylvania, US).
The growth medium used for cultivating HepG2 was Dulbecco’s Modified Eagle’s Medium (DMEM) (1X) + Glutamax (Gibco Life Technologies, Waltham, MA, US) mixed with 10% fetal bovine serum (FBS, PAA Laboratories GmbH, Pasching, Austria), 1% Penicillin (100 units/mL) Streptomycin (100 µg/mL) (Sigma Aldrich) and 1% Non-essential amino acids (Sigma Aldrich). DMEM with L-glutamine (VWR International, Radnor, PA, US) was used as the growth medium for HT-29 cells. The other reagents used were sterile dimethyl sulfoxide (DMSO, Fisher Scientific), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylphenyltetrazolium bromide (MTT reagent, Sigma Aldrich), Phosphate Buffered Saline (PBS, Sigma Aldrich), Trypsin-EDTA (VWR International), Trypan blue (Sigma Aldrich) and Rhodamine 123 (Sigma Aldrich).
Methods
Formulation of Solid Lipid Nanoparticles
SLNPs were formulated by using biodegradable, biocompatible lipids (stearic acid, palmitic acid, lauric acid) and surfactants (lecithin and sodium taurocholate) by an aqueous-based formulation process. PSE-free and PSE-loaded SLNPs were prepared by the microemulsion method developed and optimised by Gasco’s group (Figure 1).49 The composition of each formulation (Table 1) was varied in order to evaluate the effect of different types of lipids used, lecithin-to-sodium taurocholate ratio and stearic acid-to-lecithin ratio. The melting points of lipids were determined using a Stuart SMP10 melting point apparatus (Bibby Scientific Limited, Stone, Staffordshire, UK). The melting points of stearic acid, palmitic acid and lauric acid were ~70 °C, ~63 °C and ~45 °C, respectively, consistent with the reported values.50–52 Lipids (450 mg) were melted by heating at approximately 10 °C above their melting point. PSE (75 mg) and lecithin were added to the molten lipid phase. An aqueous solution of sodium taurocholate was heated to the same temperature. The lipid phase and aqueous phase were mixed together to yield a hot microemulsion under magnetic stirring. The hot micro-emulsion was then dispersed under magnetic stirring at 1000 rpm in cold water maintained between 2 °C and 8 °C for 30 minutes.
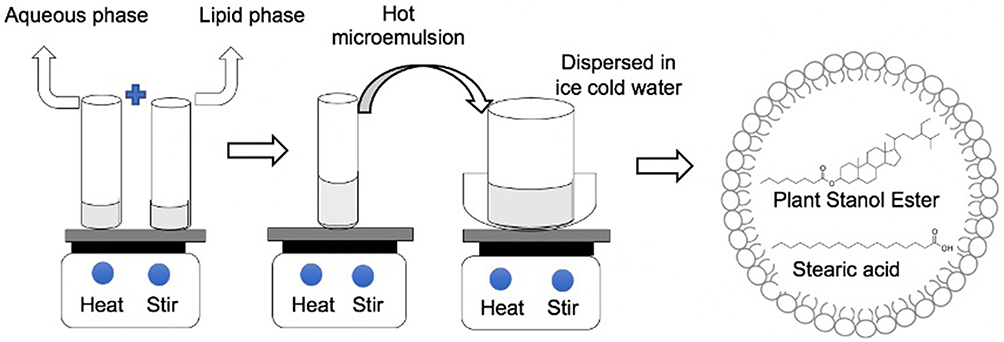 |
Figure 1 Formulation of solid lipid nanoparticles (SLNPs) by the microemulsion technique. |
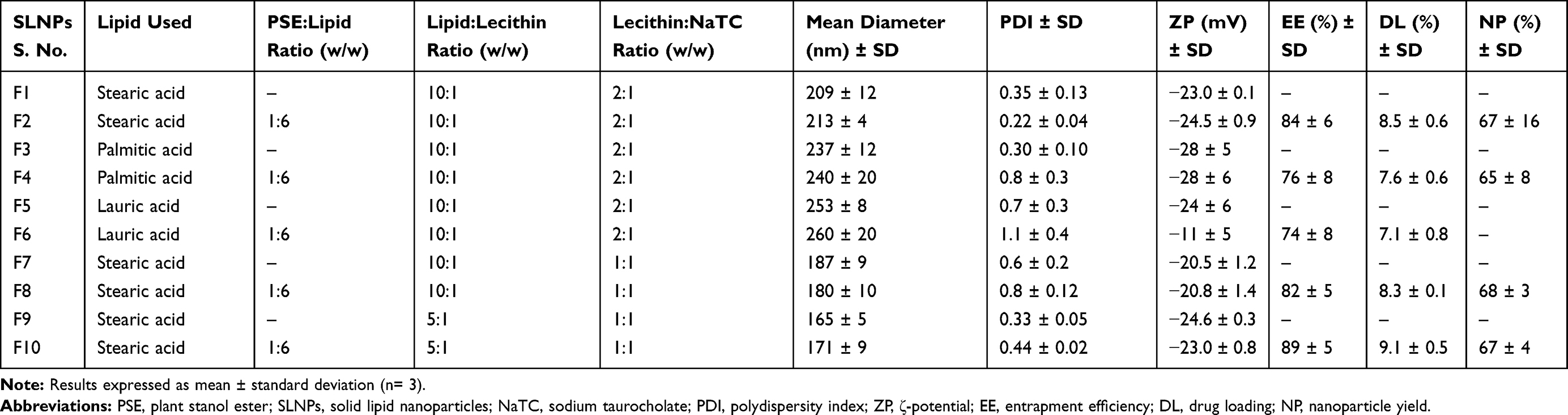 |
Table 1 Composition and Responses of PSE Free and PSE SLNPs |
Analytical Method
Quantification of phytostanols was carried out on a high-resolution gas chromatography (HRGC) MEGA 2 series (Fisons Instruments, Manchester, UK) gas-liquid chromatograph equipped with a flame ionization detector. The analytical column was an HP-5, 5% phenyl methyl siloxane stationary phase (30 m, 0.32 mm I.D., 0.25 µm). Gas chromatographic conditions were optimized as follows: detector temperature, 300 °C; injector temperature, 280 °C; column temperature 250 °C isothermal and injection volume, 1 µL. Samples were injected under a 1:50 split mode using hydrogen as carrier gas (Claind Brezza, Lake Como, Italy) maintained at 6 bars with a run time of 20 minutes. A computer chromatography data system (EZ Chrome 6.6 Chromatography Data System, Scientific Software, Inc., San Ramon, CA, US) was used to analyze the chromatograms.
Particle Size and ζ-Potential
Particle size and ζ-potential, which relates to surface charge, were measured by tunable resistive pulse sensing (TRPS). This is a type of Coulter counter method that employs a pore of tunable size immersed in a flow chamber. The qNano instrument (Izon Science Ltd., Christchurch, New Zealand) was calibrated with monodisperse, carboxylated polystyrene particles (CPC200, nominal mean diameter of 210 nm and ζ-potential of −20 mV) from Izon. The nanopore membranes (NP200, from Izon) were specified for particles with diameters ranging from 100 to 400 nm. The calibrant, as well as the samples, were dispersed in PBS and subjected to a brief vortexing for uniform mixing before measurements. The resistive current pulse signals were analyzed using the Izon Control Suite software 3.3.
Entrapment Efficiency and Drug Loading
A gas chromatography (GC) method was developed for the identification, separation, and quantification of phytostanols. Methods used by the Food and Agricultural Organization53 and Shapiro et al54 were followed to prepare phytostanol samples for GC. The sample preparation steps included saponification with ethanolic potassium hydroxide, liquid-liquid extraction with heptane, and derivatization with BSTFA + 1% TMCS and pyridine. The method was validated for linearity, selectivity, and specificity. Entrapment efficiency and drug loading were calculated by quantifying phytostanols in PSE SLNPs.
A volume of PSE SLNPs equivalent to 7.5 mg of phytostanol ester was subjected to centrifugation at 4400 g for 45 minutes. The resulting supernatant and pellet were separated. The samples were saponified, extracted with heptane and derivatized to determine the amounts of free PSE and PSE entrapped within the lipid, respectively, following the methods of Nandini et al55 and Tan et al.56 Entrapment efficiency (EE) and drug loading (DL) were calculated according to equations 1 and 2, respectively.
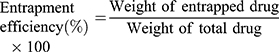
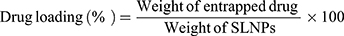
Powder Flowability and Nanoparticle Yield
PSE free and PSE SLNP samples were freeze-dried for these tests. Samples with different lipids as carriers were frozen at −86 °C (Forma Scientific, Inc., Marietta, Ohio, US) overnight. The freeze-drying process was carried out at a condenser surface temperature of (−54 ± 3) °C at a pressure of 19 Pa in a Heto PowerDry PL3000 freeze dryer (Thermo Fisher Scientific, Waltham, US).
Powder flowability was evaluated using the angle of repose method. The angle of repose is defined as the angle made by a material with respect to the horizontal when piled.57 Approximately 1 gm of lyophilised PSE SLNPs was poured through a funnel onto a flat surface. The funnel was positioned 2 cm from the horizontal surface, and the powders were allowed to flow freely. Both the diameter (d) and height (h) of the pile were measured and recorded. Equation 3 was used to calculate the angle of repose (θ).
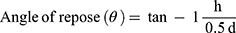
where d and h are, respectively, the diameter and height of the pile.
The lyophilized SLNPs were used for spectroscopic and thermal analysis, and stability studies. Nanoparticle yield was calculated according to Nandini et al55 as the weight of freeze-dried SLNPs in relation to the sum of the starting material (equation 4).
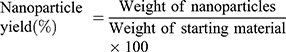
One of the ten produced formulations of SLNPs was selected based on desirable average particle diameter, ζ-potential, maximum entrapment efficiency, maximum drug loading, high nanoparticle yield and high quality of the lyophilizate. The optimized formulation was further evaluated for particle size, stability, shape and morphology, spectroscopic properties, thermal behaviour, biological safety, and cellular uptake.
Shape and Surface Morphology
Sample preparation involved depositing 10 µL of optimized PSE SLNPs on a silicon wafer substrate. After drying for 24 hours at ambient temperature, images were collected. An Asylum Research Cypher atomic force microscope (AFM, Oxford Instruments, Santa Barbara, CA, US) in tapping mode was used to record topographic and phase images of SLNPs. AFM images were obtained by measurement of the interaction forces between the silicon tip of a high resonant frequency cantilever and the sample surface. The experiments were performed in air at ambient temperature. The topographical AFM images were processed by Proprietary Scanning Probe Image Processor (SPIP) software version 6.64 (Image Metrology, Copenhagen, Denmark).
Spectroscopic Analysis
The Fourier transform infrared spectroscopy (FTIR) spectra were obtained for PSE, stearic acid, physical mixture of PSE and stearic acid, and lyophilized PSE SLNPs using a Cary 630 FTIR spectrometer (Agilent Technologies Inc, Santa Clara, CA, US). The samples were scanned at a wavenumber ranging from 400 to 4000 cm−1.
Thermal Analysis
Differential scanning calorimetry (DSC) was performed using model Q2000 equipped with an automated computer-controlled refrigerated cooling system (RSC-90) and TZero pans (TA Instruments, New Castle, DE, US). The weights of the sample were measured using a calibrated Sartorius CC6 balance. A predetermined weight of the sample was placed in TZero alodine-coated aluminum DSC pans and then hermetically sealed with a TZero encapsulation press (TA Instruments, New Castle, DE, US). An empty aluminium pan was used as the reference. Phase transition temperatures and the melting point of the samples were measured under a 50 mL per minute dry ultra-high purity nitrogen gas (99.9998% purity) purge. PSE, stearic acid, physical mixture of PSE and stearic acid, lyophilized PSE free and PSE SLNPs were heated from 0 °C to 330 °C at the rate of 10°C per minute. Triplicate melting scans were carried out to ensure melting point temperature reproducibility.
The heat stability of PSE, stearic acid, lyophilized PSE free and PSE SLNPs was determined using a Q5000 thermogravimetric analysis (TGA) instrument (TA Instruments, New Castle, US). Samples were heated in a 100 µL platinum crucible from 25 °C to 600 °C at a heating rate of 10 °C per minute under a constant nitrogen flow of 25 mL per minute. The thermal decomposition profile of the samples was obtained. The weight loss data for each sample were acquired in triplicate. The DSC and TGA thermograms were analyzed using a TA universal analysis software package.
Stability Studies
The size distribution of PSE SLNPs was measured in triplicate by particle tracking analysis (PTA) for the assessment of stability. The measurements were taken on day 1, day 7, day 15 and at monthly intervals up to 6 months. The average particle diameter was monitored at two different storage conditions, ie fridge and room temperature protected from light. An LM10 Nanosight instrument (Malvern Panalytical, Malvern, UK) equipped with a 638 nm laser was used for the measurement of particle size distribution. The lyophilizate was reconstituted in purified water in the amount of the original volume. Samples were diluted in purified water and injected into the sample chamber. Three videos per sample of 60 seconds duration were recorded. Data acquisition and processing were performed using NTA 2.3 analytical software.
Cellular Studies
Human hepatocellular carcinoma, HepG2 and colon adenocarcinoma, HT-29 cells were obtained from the European Collection of Authenticated Cell Cultures (ECACC). HepG2, derived from a fifteen-year-old Caucasian male, express 3-hydroxy-3-methylglutaryl-CoA reductase and hepatic triglyceride lipase activities.58 HT-29 was isolated from a primary tumour of a 44 years old Caucasian female.59 Both the adherent cell lines were cultured in a complete growth medium in a humidified incubator (Heraeus Hera cell incubator, Thermo Electron Corporation, Waltham, MA, US) of 5% CO2 in the air at 37 °C. For routine maintenance, the cell medium was replaced every 48 hours. Cells were sub-cultured when 80% confluent by trypsinization with a 0.05% trypsin and 0.02% ethylenediaminetetraacetic acid (EDTA) solution. All cell culture procedures were undertaken in a BioMAT 2 microbiological safety cabinet (Thermo Electron Corporation, Waltham, MA, US) using an aseptic technique to ensure sterility.
Cytotoxicity Assay
Cells were seeded in a 96-well plate, at a density of 3×104 (HepG2) and of 4×104 (HT-29) cells per well in 100 µL cell culture medium. Cells were allowed to equilibrate at 37 °C overnight to promote adhesion. At time zero, PSE free and PSE SLNPs (0.5 mg mL−1) were added to each replicate well and incubated for 24 hours. 20 µL of 3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyl-tetrazolium bromide (MTT) (5 mg mL−1 in PBS) reagent was added to each well and incubated at 37 °C for 4 hours. The intracellular formazan crystals were solubilized with dimethyl sulfoxide (200 µL per well). The plates were shaken for 15 minutes in R100 Rotatest Shaker (Luckham Ltd., Burgess hill, Sussex, England) in dark, and absorbance values were measured using a FLUOstar Omega microplate reader (BMG Labtech GmbH, Ortenberg, Germany). Cell survival was expressed as a % absorbance at 570 nm compared with control, ie untreated cells. HepG2 and HT-29 treated with Triton X-100 were used as the negative control, as the detergent action disrupts the cells. An average of three separate experiments with three replicates in each has been taken into consideration to determine HepG2 and HT-29 survival after exposure to PSE free and PSE SLNPs.
The cell survival of the samples was calculated using equation 5.
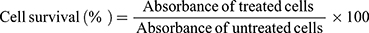
Cellular Uptake
To evaluate whether the SLNPs could efficiently deliver the drug into the cells, Rhodamine 123 (R123) was encapsulated into the SLNPs (F10) instead of PSE, as a fluorescent probe. The concentration of the dye with respect to the lipid was 0.5%. The SLNPs so obtained were sterilized by filtration through 0.2 µm filter units. Particle Tracking Analysis (PTA) was used for the particle sizing of PSE SLNPs (F10) and R123 SLNPs.
HepG2 and HT-29 cells were cultured and seeded at a density of 5.8 × 105 and 6 × 105 cells per mL, respectively, in a 12-well plate (COSTAR) for incubation overnight. At time zero, cells were treated with R123 SLNPs (100 μg per mL medium) and incubated for 2 hours at 37 °C. HepG2 and HT-29 cells incubated with the medium without the SLNPs served as controls. The cells were also incubated with fluorescent SLNPs at 4 °C to investigate the role of endocytosis in SLNPs uptake. After incubation, cell media was removed, and samples were washed with sterile PBS two times. Cells were examined using an inverted fluorescence microscope (FM) (Olympus IX81, Olympus Corporation, Tokyo, Japan) in order to visualize the fluorescence of the cargo fluorescent dye, Rhodamine 123. After capturing images, uptake of R123 was assessed by flow cytometry. 300 µL of pre-warmed trypsin/EDTA was added to each well to detach the adherent cells and incubated for about 5 minutes. 700 µL of media was added to each well to stop trypsinization. Cell solutions were centrifuged (Eppendorf centrifuge 5702, Hamburg, Germany) at 125 g for 5 minutes. The cell pellets were washed with ice-cold PBS twice and re-suspended in cold PBS. At least ten thousand cells of each sample were analyzed using a Guava Easycyte 8HT flow cytometer (FCM) (EMD Millipore Corporation, Billerica, MA, US). Guava Express FCS4 express flow cytometry was used for data analysis and for making overlay histograms.
Statistical Analysis
The data were presented as mean ± standard deviation (SD). Quantitative data were analyzed by a one-way analysis of variance (ANOVA) followed by Fisher’s least significant difference test when significance was indicated. p < 0.05 was considered statistically significant. Statistical Package for the Social Sciences (SPSS, version 25) was used to analyze the data.
Results
Particle Size and ζ-Potential
PSE-free and PSE-loaded SLNPs employing three different lipids (stearic acid, palmitic acid, lauric acid) and varying concentration of surfactants were produced. The particles appear spherical (see Figure S1 of the supplementary information) with an average particle diameter of PSE SLNPs in the range of (171 ± 9) nm to (260 ± 20) nm (Table 1). The type of lipid used to synthesize the SLNPs had a significant impact on the resulting size of the particles. PSE SLNPs with stearic acid produced significantly smaller nanoparticles than PSE SLNPs with palmitic acid. SLNPs with lauric acid as the lipid produced significantly larger nanoparticles compared to SLNPs with stearic acid. Representative number-based size distributions of the optimized formula of SLNPs, F9 and F10 are shown in Figure 2. The D10, D50, D90 (particle size corresponding to 10%, 50% and 90% of the cumulative undersize particle size distribution, respectively) for F10 were (138 ± 8) nm, (162 ± 8) nm, and (210 ± 8) nm, respectively, generating the lowest mean particle size of (171 ± 9) nm. Increasing the concentration of lecithin and sodium taurocholate in SLNPs had a significant effect on decreasing their mean diameter. F6 had the highest PDI of 1.1 ± 0.4 whereas F2 had the lowest PDI of 0.22 ± 0.04 (Table 1). The type of lipid used had a significant effect on the PDI, with stearic acid producing the least polydisperse PSE SLNPs.
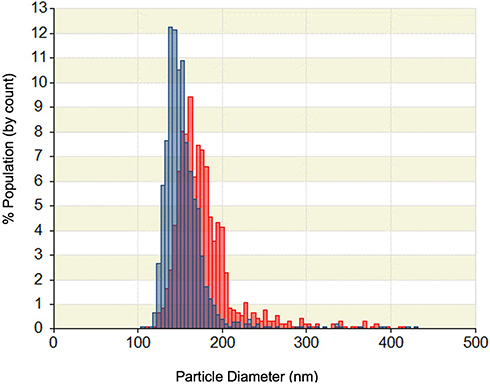 |
Figure 2 Representative number-based particle size distribution of F9 (blue) and F10 (red) solid lipid nanoparticles as measured by tunable resistive pulse sensing. |
A stability study was conducted for the optimized formula of PSE SLNPs (F10) by measuring their average size by PTA over a period of 6 months. We evaluated two different storage conditions and the results are shown in Figure S2 of the supplementary information. The mean diameter of F10 stored at 4 °C was found to increase by 7.6% from (184 ± 6) nm after 6 months. On the other hand, when stored at room temperature, the same formulation increased 20% in size over the same period.
ζ-potential values of SLNPs ranged from (−11 ± 5) mV to (−28 ± 6) mV (Table 1). Lauric acid produced SLNPs with a lower ζ-potential than stearic acid and palmitic acid. However, there were no differences in ζ-potential between drug-free and corresponding drug-loaded SLNPs with stearic acid and palmitic acid as the lipid, which appear consistent with the drug being encapsulated inside the particle carrier.
Entrapment Efficiency and Drug Loading
The gas chromatograms in Figure 3 indicate that the campestanol and sitostanol components (peaks 1 and 2, respectively) could be separated under the GC conditions. The retention time of the two compounds were 10.2 and 12.9 minutes, respectively, and the peaks are observed in both the sample containing the free phytostanol components and that containing the phytostanol loaded SLNPs. A calibration curve was plotted in the concentration range from 0.2 µg/µL to 5.5 µg/µL with five concentration points. The response was linear with a correlation coefficient (R2) of 0.995 (see Figure S3 of the supplementary information). The maximum entrapment efficiency and drug loading were found to be (89 ± 5)% and (9.1 ± 0.5)%, respectively, in F10, whereas F6 had the least amounts of drug entrapped at (74 ± 8)% and loading of (7.1 ± 0.8)% (Table 1). The lipid matrix used had a significant impact on the ability of SLNPs to entrap and load PSE. F2, which contains stearic acid, had a higher amount of PSE entrapped than F4 containing palmitic acid and F6 containing lauric acid. Likewise, SLNPs with stearic acid had higher drug loading than palmitic acid. Compared to F2, the effect of adding surfactants in F8 and F10 was not found to entrap and load significantly higher amounts of the drug. There were no differences between palmitic acid (F4) and lauric acid (F6) based SLNPs with regard to entrapment efficiency.
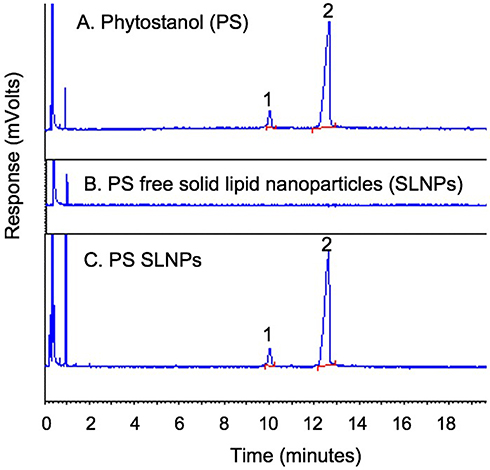 |
Figure 3 Gas chromatogram of phytostanol (A), phytostanol free SLNPs (B), phytostanol loaded SLNPs (C). |
Powder Flowability and Nanoparticle Yield
The angle of repose of PSE SLNPs formulated with stearic acid were 34 ± 2° (F2), 31 ± 1° (F8) and 33 ± 1° (F10). The angle of repose of PSE SLNPs with palmitic acid as the lipid matrix was 35 ± 3° (F4). However, SLNPs with lauric acid (F6) as the lipid yielded a sticky lump of powder. The nanoparticle yield ranged from (65 ± 8)% to (68 ± 3)% (Table 1). The nanoparticle yield of F10 was (67 ± 4)%.
F10 combined many positive critical quality attributes such as desirable average particle diameter, ζ-potential, maximum entrapment efficiency, maximum drug loading, high nanoparticle yield and high quality of the lyophilizate. Hence, it was chosen as the optimized formulation for stability assessment, shape and morphology studies, spectroscopic analysis, thermal analysis, and in vitro studies.
Spectroscopic Analysis
The FTIR spectra of PSE, stearic acid, the physical mixture of the drug with the lipid, and PSE SLNPs (F10) are presented in Figure 4 and Table 2. FTIR spectra of PSE revealed characteristic peaks at 2925 cm−1 and 2851 cm−1, both attributed to the -C-H groups.60 The two sharp bands at 1737 cm−1 and 1179 cm−1 represent absorption of the carbonyl group from the ester component.60,61 The spectra for stearic acid showed absorption bands with characteristic peaks at 2914 cm−1, 2847 cm−1 and 1700 cm−1, corresponding to –C-H, -C-H and C=0 groups, respectively.60 The prominent peaks of the drug and stearic acid were all present in the physical mixture. The FTIR spectrum of PSE SLNPs shows approximately all absorbance of the lipid matrix. However, the peaks corresponding to the carbonyl group of the plant stanol ester were absent. There were no major shifts of peak or new bands in the lyophilized PSE-loaded SLNPs.
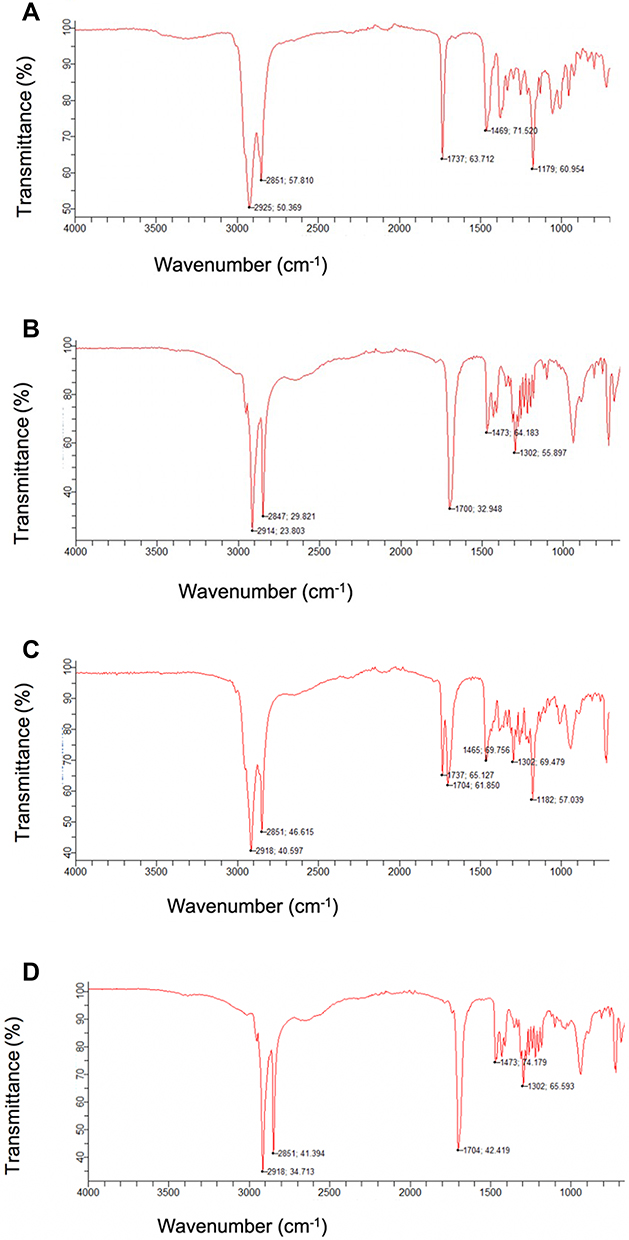 |
Figure 4 Fourier-transform infrared spectroscopy (FTIR) spectra for plant stanol ester (A), stearic acid (B), plant stanol ester + stearic acid (C) and plant stanol ester solid lipid nanoparticles (D). |
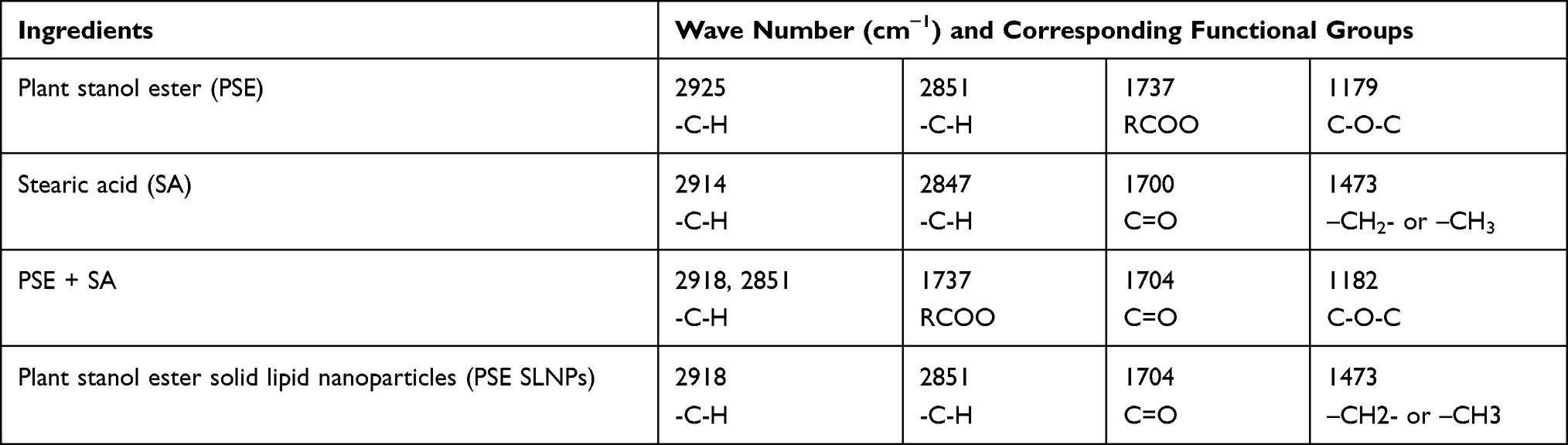 |
Table 2 FTIR Interpretation of Plant Stanol Ester, Stearic Acid, Physical Mixture and PSE SLNPs |
Thermal Analysis
Figure 5A shows the DSC curves of PSE, stearic acid, lyophilized PSE free and PSE SLNPs. PSE exhibits a melting range between 25.7 °C and 41.1 °C with the melting peak at 34.5 °C. Stearic acid melts between 62.0 °C and 81.5 °C with the melting point at 71.5 °C. There was no significant shift in the position of the endothermic peak in the melting range of stearic acid in the optimized PSE SLNPs. After formulation into SLNPs, the peak of PSE disappeared, and the only peak observed was of the lipid, stearic acid.
 |
Figure 5 (A) Differential scanning calorimetry (DSC) thermograms of plant stanol ester (PSE), stearic acid, PSE + stearic acid, lyophilized PSE-free solid lipid nanoparticles (SLNPs) and PSE SLNPs. (B) Thermogravimetric Analysis (TGA) curve of PSE, stearic acid, lyophilized PSE-free SLNPs and PSE. SLNPs. |
The thermal degradation profiles of PSE, SA, PSE free and PSE SLNPs are depicted in Figure 5B. PSE presented a thermal event at 306.6 °C, which was followed by 97.2% degradation when heated to 420 °C. The TGA curve of stearic acid showed thermal decomposition of approximately 95% when heated from 190.4 °C to 288 °C followed by elimination of carbonaceous material. PSE SLNPs showed thermal decomposition at temperatures above 130 °C. All the tested samples degraded to a minimal amount of residue when heated to 600 °C.
Cytotoxicity Assay
The results of the MTT assay are shown in Figure 6. When HepG2 cells were incubated with PSE free and PSE SLNPs for 24 hours, cell survival was (99 ± 4)% and (98 ±8) % compared with untreated controls, respectively. Likewise, the percentage survival of HT-29 after exposure to PSE free and PSE SLNPs overnight were (97 ± 2)% and (97 ± 6)%, respectively. There were no significant differences in cell survival between untreated cells and cells incubated with drug-free or drug-loaded SLNPs.
 |
Figure 6 HepG2 and HT-29 survival after exposure to plant stanol ester (PSE) free and PSE solid lipid nanoparticles (SLNPs) overnight, *p > 0.05, **p < 0.05. |
Cellular Uptake
The PTA mean diameters of PSE SLNPs (F10) and R123 SLNPs were (183 ± 5) nm and (187 ± 4) nm, respectively. A strong green fluorescence signal was observed with liver and intestinal cells treated with Rhodamine 123 SLNPs at 37° C (see Figure S4 of the supplementary information). The untreated cells and the cells incubated with Rhodamine 123 free SLNPs did not exhibit fluorescence. The cells treated with free dye and incubated at 4° C with R123 SLNPs exhibited very negligible fluorescence. A significant shift in mean fluorescence intensity was observed in HepG2 and HT-29 treated with R123 SLNPs incubated at 37 °C, using flow cytometry (Figure 7).
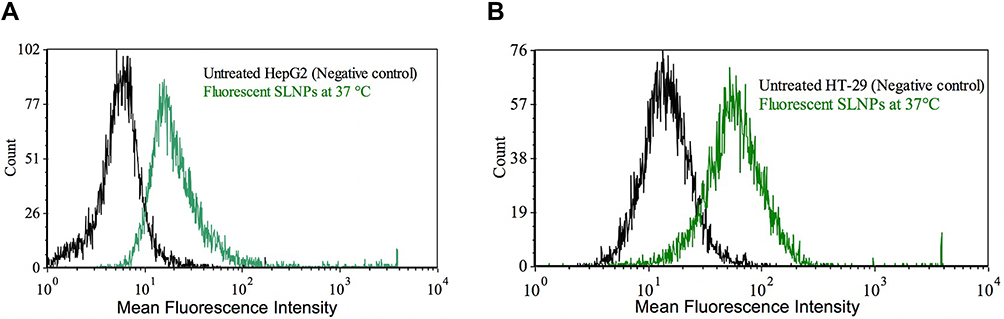 |
Figure 7 Flow cytometry data of HepG2 (A) and HT-29 (B) showing the mean fluorescence intensity of untreated cells (black) and the uptake of fluorescent solid lipid nanoparticles (SLNPs) by cells at 37 °C (green). |
Discussion
SLNPs represent an advancement of drug delivery systems such as emulsions, liposomes, microparticles and polymer-based nanoparticles.62 The rationale for choosing SLNPs as a vehicle for delivery of plant esters, such as their small size, stability, high drug entrapment, safety and effectiveness as drug carriers, was demonstrated. Hence, PSE SLNPs are potentially suitable for oral administration to lower LDL-C and manage hypercholesterolaemia. The intended delivery form of PSE SLNPs is as a dry powder packaged in sachets which can be sprinkled on food, as and when necessary, giving patients the flexibility to consume it with any meal during the day. The use of PSE SLNPs sachets is appealing and convenient since it is easier to incorporate these into a lipid-lowering regimen compared with dietary adjustments necessary to include food products.
SLNPs were manufactured by a simple and solvent-free microemulsion method. This technique is highly attractive for scale-up to industrial production due to the green chemistry and the low mechanical energy input, through magnetic stirring, needed to form nano-sized SLNPs. In contrast, the homogenization methods used for SLNP formulation are energy-intensive and may cause thermodynamic and mechanic stress to the product.41,63 Furthermore, commonly used solvent emulsification-evaporation, emulsification solvent diffusion and solvent-injection methods employ organic solvents.64 Solvent removal may be incomplete and costly, the residues may cause adverse effects and interaction with the drug may lead to stability issues.41 Thus, the aqueous-based microemulsion technique employed is favourable compared with solvent-based methods taking into account toxicological, environmental and regulatory concerns.
The SLNPs formulated had a favourable mean diameter (<260 nm) within the range of diameters of most clinically approved nanoparticles, from 50 to 300 nm.65 However, nanoparticles of 200 nm diameter or larger tend to activate the complement system and are removed from the circulation faster compared to nanoparticles less than 200 nm.66 Nano-sized PSE SLNPs (F10) with an average diameter (171 ± 9) nm may have a greater ability to compete with cholesterol for access to its receptors, and to be incorporated into mixed micelles, and therefore have greater cholesterol-lowering efficacy due to the increment in surface area.
The most widely used nanocarriers for delivering drugs to combat atherosclerosis have been lipid-based.67 Stearic acid (C18), palmitic acid (C16) and lauric acid (C12) were chosen based on their “generally regarded as safe” (GRAS) categorization by the Food and Drug Administration,68 high melting point and their increasing use as drug carriers.49 SLNPs formulated with the longest chain lipid, stearic acid, had the lowest average diameter, were less polydisperse, were stable, and entrapped the highest amounts of PSE. On the other hand, SLNPs fabricated with the shortest chain fatty acid, lauric acid, had the largest diameter, were highly disperse, the least stable and incorporated the least amounts of the drug. The observed inverse relation of fatty acid carbon chain length to the average diameter of SLNPs agrees with the findings of Holt et al.69 Particle samples with PDI values below 0.3 are considered to have a narrow size distribution, with PDI below 0.1 being referred to as monodispersed.70 According to this, most SLNPs synthesized in this work have a broad size distribution. Tails of the distribution at higher size values for SLNPs may be due to some level of aggregation. A key benefit of stearic acid is that, unlike palmitic acid and lauric acid, it does not appear to increase LDL-C in vivo.71,72 In fact, it is converted to a monounsaturated fatty acid, oleic acid which lowers LDL-C and triglyceride levels.73–75 Hence, the formulation combines the natural cholesterol-lowering properties of PSE and stearic acid.76 Stearic acid was also the best for the incorporation of lipophilic PSE as it had higher entrapments, 82–89% and drug loading, 8.3–9.1% compared with palmitic acid (76% and 7.6%, respectively) and lauric acid (74% and 7.1%, respectively). SLNPs have been found to entrap lipophilic and hydrophilic drugs to varying degrees. Entrapment efficiencies around 50–70% for carbamazepine,77 90% for atorvastatin,78 95% for simvastatin79 and 50% for rosuvastatin80 have been reported. The high molecular weight, melting point, and hydrophobicity due to stearic acid being a long-chain fatty acid may account for a higher incorporation of the drug in the lipid matrix during formulation, compared with the shorter fatty acids. The amount of drug within the lipid matrix co-relates with the functionality and ultimately the bioavailability from the drug delivery system.56 The high values of EE and DL demonstrate the efficiency and cost-effectiveness of this method for the preparation of PSE SLNPs.
The SLNPs were prepared using two physiological surfactants, lecithin and sodium taurocholate to stabilize the formulations. These are preferable to surfactants commonly used for SLNP fabrication such as butanol and iso-propanol with respect to safety and regulatory aspects.81,83 A 2:1 ratio of lecithin to sodium taurocholate produced SLNPs with a mean diameter (213 ± 4) nm, whereas an equal ratio produced smaller nanoparticles. This is likely due to the fact that the increased concentration of surfactant reduces the interfacial tension between the lipid matrix and dispersion medium. Increasing the amounts of surfactants did not significantly increase the amount of PSE entrapped within the stearic acid matrix. This indicates that the optimized PSE SLNPs (F10) had an optimum level of surfactants in the external phase. The concentration of lecithin and sodium taurocholate were adequate to stabilize and cover the surface of the nanoformulations effectively.
The ζ-potential values indicate that the formulations are electrostatically stable, ie approximately −25 mV.72,82 The surface chemistry critically affects the way nanoparticles interact with each other and with their surrounding environment.83 The consistently negative ζ-potential values explain the stability by maintaining electrostatic repulsion among the particles. Stearic acid has a pKa value of 4.7550 and in the conditions that the SLNPs have been synthesized (pH approximately 5.8), it is negatively charged. A net negative charge of lecithin84 and the anionic nature of sodium taurocholate85 could have further contributed to the negative surface charge and electrostatic stabilisation of the SLNPs. The surface charge values of PSE free and PSE SLNPs with stearic acid and palmitic acid as the lipid matrix are comparable. This seems to indicate that all the nanoformulations, apart from F6, have the drug entrapped within the lipid and not deposited on the surface.
Aqueous dispersions of PSE SLNPs were freeze-dried to preserve the nano-size range, particle properties and improve the stability of the SLNPs. An angle of repose less than 30° indicates excellent flowability and angles between 31° and 35° denote good flowability.55 Hence, lyophilisation was found to be successful in preparing free-flowing discrete SLNPs formulated with stearic acid (F2, F8, F10) and palmitic acid (F4). The lyophilized SLNPs showed 0.5% moisture loss below 100 °C, indicating that effective lyophilization of the SLNPs is possible, since moisture content was less than the 5% acceptable in the pharmaceutical industry.86 The free-flowing dry powder formed by lyophilization of the optimized PSE SLNPs presents exciting opportunities for handling, reconstitution, and compressibility of the powder into other oral dosage forms.
TGA demonstrates that the optimized PSE SLNPs are thermally stable until approximately 130 °C. The physical state of plant stanols has implications for their efficacy.43 The microcrystalline structure of sitostanol was postulated to impair its dissolution in the small intestine46 and crystalline phytosterols were not absorbed efficiently in the intestine resulting in low bioaccessibility.39 The disappearance of the melting endotherm in DSC and absorption bands in FTIR, after entrapment in the lipid matrix, indicates the amorphous state of the drug in the optimized SLNPs. The amorphous form of PSE can be expected to have better drug dispersion and enhanced drug-matrix association suitable for controlled release compared to crystalline forms.86 The presence of surfactants might have prevented the crystallization of PSE dispersed within the lipid.
A prolonged shelf life of the formulation is indicated by the lack of change of the particle size of lyophilized PSE SLNPs when stored at 4 °C over a period of 6 months. The formulations were less stable at room temperature, at which a gradual increase in particle size was observed starting at 90 days, presumably due to the increased kinetic energy of SLNPs at higher temperatures.87
The in vitro assessment of optimized PSE SLNPs was conducted on HT-29 and HepG2 cell culture models of intestinal and hepatic cells, respectively, since phytostanols lower LDL-C by inhibiting intestinal and hepatic cholesterol absorption.88–90 According to the International Standards Organization (ISO) 10993–5, a reduction of cell viability by more than 30% is considered cytotoxic.91 The concentration of phytostanol used in MTT assay (65 μM) was much higher than normal serum physiological concentration (0.6 μM) and after dietary supplementation (1.2 μM) on cultured human blood cells.92 Therefore, the MTT assay indicates that the optimized PSE SLNPs are not cytotoxic,93 and are likely not toxic in vivo.
Rhodamine 123 was chosen as a fluorescent probe since it is lipophilic and has a molecular weight (380.8 g/mol)94 similar to stanols (sitostanol: 416.7 g/mol, campestanol: 402.6 g/mol).95,96 The entrapment of the dye did not have any significant influence on the characteristics of the SLNPs since the average diameter of R123 SLNPs was comparable to PSE SLNPs. A strong green fluorescence signal of R123 SLNPs within the hepatic and intestinal cells visualised by fluorescence microscopy and significant shift in mean fluorescence intensity by flow cytometry compared to untreated cells demonstrated efficient uptake of fluorescent SLNPs. There was no significant uptake of R123 SLNPs when incubated with cells at 4 °C, suggesting a mechanism of uptake involving endocytosis. Negligible fluorescence of cells treated with R123 free SLNPs, free dye, indicates that Rhodamine 123, by itself, was not internalized. This demonstrated the suitability of SLNPs as novel and effective drug carriers for therapeutic applications. SLNPs with a mean diameter of 80–300 nm have been reported to be taken up by different cell lines through various endocytic pathways.97 Martins and colleagues reported similar findings for Rhodamine 123-loaded SLNPs having a mean size 135–175 nm and −20 mV surface charge.98 Likewise, Rhodamine 123-loaded SLNPs (average diameter 224 nm) were taken up by multidrug-resistant breast cancer cells by endocytosis.99 The nano-scaled size, generally spherical shape and negative surface charge might have contributed to the internalization of SLNPs.100
The methodology of SLNP formulation described may also have application for other poorly soluble active compounds for drug delivery applications.
Conclusion
A novel aqueous-based formulation incorporating plant stanol ester within SLNPs as the carrier has been successfully devised, characterized, and optimized. The method is simple, energy-efficient and has excellent scale-up potential. The synthesis is based on green technology since organic solvents were not used and little waste has been generated. The optimal nano-sized formulation of PSE SLNPs has a spherical shape and a negative surface charge that stabilizes them. Approximately 89% of the drug was entrapped within the carrier in an amorphous form with drug excipient compatibility and the formulation has been found to be stable for at least six (6) months. The optimized PSE SLNPs are not toxic to hepatic and intestinal cell lines and are rapidly taken up by the cells within two (2) hours.
The formulation has the potential to be used as a therapeutic dietary supplement of cholesterol-lowering drugs leading to the administration of lower doses of drugs and reducing the incidence of adverse drug reactions. The formulation may have the potential to avoid the need for drug therapy altogether. This work offers an alternative approach in the clinical management of patients with hypercholesterolemia that could contribute to better therapeutic outcomes of cardiovascular diseases.
Future Work
The absorption of SLNPs across the intestinal barrier can be studied in vitro using cell culture models of intestinal epithelial cells in a trans-well arrangement to monitor movement from apical to basal compartments through the epithelial cell barrier. The efficacy of PSE SLNPs as a cholesterol-lowering agent in vivo needs to be demonstrated in animal models, coupled with an assessment of the uptake and pharmacokinetics of the SLNPs in the circulation. Further in vitro work could focus on the potential interaction of the SNLPs with the low density-lipoprotein receptor and LDL-C, to establish how cholesterol metabolism is affected.
Acknowledgments
This research project was enabled by the Lord Limerick Fund of the London Metropolitan University. The work at the National Physical Laboratory was funded by the UK Department of Business, Energy, and Industrial Strategy through the National Measurement Strategy (NMS) projects. The authors thank Natalie Belsey, Charles Clifford and Zeinab Al-Rekabi for support with the AFM measurements and Sam Gnaniah for support in the thermogravimetric analysis.
Disclosure
Sundus Tewfik is an employee of Bloomsnano Limited, London, UK. The authors report no other potential conflicts of interest in this work.
References
1. World Health Organization. Cardiovascular diseases. 2019. Available from: https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds).
2. Nelson RH. Hyperlipidemia as a risk factor for cardiovascular disease. Prim Care. 2013;40(1):195–211. doi:10.1016/j.pop.2012.11.003
3. Soran H, Dent R, Durrington P. Evidence-based goals in LDL-C reduction. Clin Res Cardiol. 2017;106(4):237–248. doi:10.1007/s00392-016-1069-7
4. World Health Organization. Global health risks: mortality and burden of disease attributable to selected major risks. 2009; pp. 52. Available from: https://www.who.int/healthinfo/global_burden_disease/GlobalHealthRisks_report_full.pdf.
5. Karr S. Epidemiology and management of hyperlipidemia. Am J Manag Care. 2017;23(9):S139–S148.
6. Hovingh GK, Gandra SR, McKendrick J, et al. Identification and management of patients with statin-associated symptoms in clinical practice: a clinician survey. Atherosclerosis. 2016;245:111–117. doi:10.1016/j.atherosclerosis.2015.12.015
7. Rosenson RS, Gandra SR, McKendrick J, et al. Identification and management of statin-associated symptoms in clinical practice: extension of a clinician survey to 12 further countries. Cardiovasc Drugs Ther. 2017;31(2):187–195. doi:10.1007/s10557-017-6727-0
8. Sánchez MA, Conde AC, Fort AC, et al. Challenges in oral lipid-lowering therapy: position document of the Spanish society of cardiology. Rev Esp Cardiol. 2016;69(11):1083–1087. doi:10.1016/j.rec.2016.05.033
9. Zimmerman MP. How do PCSK9 inhibitors stack up to statins for low-density lipoprotein cholesterol control? Am Health Drug Benefits. 2015;8(8):436–442.
10. Cimminiello C, Zambon A, Polo FH. Hypercholesterolemia and cardiovascular risk: advantages and limitations of current treatment options. G Ital Cardiol. 2016;17(4):6S–13. doi:10.1714/2254.24276
11. Piironen V, Toivo J, Puupponen-Pimia R, Lampi AM. Plant sterols in vegetables, fruits and berries. J Sci Food Agric. 2003;83:330–337. doi:10.1002/jsfa.1316
12. Gylling H, Plat J, Turley S, et al. Plant sterols and plant stanols in the management of dyslipidaemia and prevention of cardiovascular disease. Atherosclerosis. 2014;232(2):346–360. doi:10.1016/j.atherosclerosis.2013.11.043
13. Anderson RJ, Shriner RL. Reduction products of plant sterols. J Biol Chem. 1927;71:401–406. doi:10.1016/S0021-9258(18)84424-6
14. Bouic PJ. The role of phytosterols and phytosterolins in immune modulation: a review of the past 10 years. Curr Opin Clin Nutr Metab Care. 2001;4(6):471–475. doi:10.1097/00075197-200111000-00001
15. Peterson DW. Effect of soybean sterols in the diet on plasma and liver cholesterol in chicks. Proc Soc Exp Biol Med. 1951;78(1):143–147. doi:10.3181/00379727-78-19002
16. Peterson DW, Nichols CW, Shneour EA. Some relationships among dietary sterols, plasma and liver cholesterol levels, and atherosclerosis in chicks. J Nutr. 1952;47:57–65. doi:10.1093/jn/47.1.57
17. Pollak OJ. Successive prevention of experimental hypercholesteremia and cholesterol atherosclerosis in the rabbit. Circulation. 1953;7:696–701. doi:10.1161/01.CIR.7.5.696
18. Pollak OJ. Reduction of blood cholesterol in man. Circulation. 1953;7(5):702–706. doi:10.1161/01.CIR.7.5.702
19. Nguyen TT. The cholesterol-lowering action of plant stanol esters. J Nutr. 1999;129(12):2109–2112. doi:10.1093/jn/129.12.2109
20. Rosin S, Ojansivu I, Kopu A, Keto-Tokoi M, Gylling H. Optimal use of plant stanol ester in the management of hypercholesterolemia. Cholesterol. 2015;2015:706970. doi:10.1155/2015/706970
21. Plat J, Mackay D, Baumgartner S, Clifton PM, Gylling H, Jones PJH. Progress and prospective of plant sterol and plant stanol research: report of the Maastricht meeting. Atherosclerosis. 2012;225(2):521–533. doi:10.1016/j.atherosclerosis.2012.09.018
22. Katan MB, Grundy SM, Jones P, Law M, Miettinen T, Paoletti R; Stresa Workshop Participants. Efficacy and safety of plant stanols and sterols in the management of blood cholesterol levels. Mayo Clin Proc. 2009;78(8):965–978. doi:10.1016/S0025-6196(11)63144-3
23. Abumweis SS, Barake R, Jones PJ. Plant sterols/stanols as cholesterol lowering agents: a meta-analysis of randomized controlled trials. Food Nutr Res. 2008;52:1811. doi:10.3402/fnr.v52i0.1811
24. Demonty I, Ras RT, van der Knaap HC, et al. The effect of plant sterols on serum triglyceride concentrations is dependent on baseline concentrations: a pooled analysis of 12 randomised controlled trials. Eur J Nutr. 2013;52(1):153–160. doi:10.1007/s00394-011-0297-x
25. Naumann E, Plat J, Kesler AD, Mensink RP. The baseline serum lipoprotein profile is related to plant stanol induced changes in serum lipoprotein cholesterol and triacylglycerol concentrations. J Am Coll Nutr. 2008;27:117–126. doi:10.1080/07315724.2008.10719683
26. Plat J, Brufau G, Linga-Thie GM, Dasselaar M, Mensink RP. A plant stanol yogurt drink alone or combined with a low-dose statin lowers serum triacylglycerol and non-HDL cholesterol in metabolic syndrome patients. J Nutr. 2009;139:1143–1149. doi:10.3945/jn.108.103481
27. Cantrill R, Kawamura Y. Phytosterols, phytostanols and their esters, chemical and technical assessment.
28. Köhler J, Teupser D, Elsässer A, Weingärtner O. Plant sterol enriched functional food and atherosclerosis. Br J Pharmacol. 2017;174(11):1281–1289. doi:10.1111/bph.13764
29. Ubeyitogullaria A, Ciftci ON. Phytosterol nanoparticles with reduced crystallinity generated using nanoporous starch aerogels. RSC Adv. 2016;6(116):108319–108327. doi:10.1039/C6RA20675A
30. Plat J, Baumgartner S, Vanmierlo T, et al. Plant-based sterols and stanols in health & disease: “Consequences of human development in a plant-based environment?”. Prog Lipid Res. 2019;74:87–102. doi:10.1016/j.plipres.2019.02.003
31. Trautwein EA, Vermeer MA, Hiemstra H, Ras RT. LDL-cholesterol lowering of plant sterols and stanols-which factors influence their efficacy? Nutrients. 2018;10(9):1262. doi:10.3390/nu10091262
32. British Dietetic Association. Food fact sheet, stanols and sterols. 2015. Available from: https://www.bda.uk.com/foodfacts/PlantStanolsAndSterols.pdf.
33. Law M. Plant sterol and stanol margarines and health. BMJ. 2000;320:861–864. doi:10.1136/bmj.320.7238.861
34. Speiser P Lipid nano pellets as drug carriers for oral administration. European Patent EP0167825A2. 1986 Jan 15.
35. Duan Y, Dhar A, Patel C, et al. A brief review on solid lipid nanoparticles: part and parcel of contemporary drug delivery systems. RSC Adv. 2020;10:26777–26791. doi:10.1039/D0RA03491F
36. Galvin P, Thompson D, Ryan KB, et al. Nanoparticle-based drug delivery: case studies for cancer and cardiovascular applications. Cell Mol Life Sci. 2012;69(3):389–404. doi:10.1007/s00018-011-0856-6
37. Ventola CL. The nanomedicine revolution: part 1: emerging concepts. Pharmacol Therapeut. 2012;37(9):512–525.
38. Leong W, Lai OM, Long K, Man YBC, Misran M, Tan C. Preparation and characterization of water-soluble phytosterol nanodispersions. Food Chem. 2011;129(1):77–83. doi:10.1016/j.foodchem.2011.04.027
39. Rossi L, Hoorn JWMS, Melnikov SM, Velikov KP. Colloidal phytosterols: synthesis, characterization and bioaccessibility. Soft Matter. 2010;6:928–936. doi:10.1039/B911371A
40. Türk M, Lietzow R. Stabilized nanoparticles of phytosterol by rapid expansion from supercritical solution into aqueous solution. AAPS PharmSciTech. 2004;5(4):36–45. doi:10.1208/pt050456
41. Battaglia L, Ugazio E. Lipid nano- and microparticles: an overview of patent-related research. J Nanomater. 2019;2834941:1–22. doi:10.1155/2019/2834941
42. Christiansen LI, Lähteenmäki PLA, Mannelin MR, Seppänen-Laakso TE, Hiltunen RVK, Yliruusi JK. Cholesterol-lowering effect of spreads enriched with microcrystalline plant sterols in hypercholesterolemic subjects. Eur J Nutr. 2001;40:66–73. doi:10.1007/s003940170017
43. Denke MA. Lack of efficacy of low-dose sitostanol therapy as an adjunct to a cholesterol-lowering diet in men with moderate hypercholesterolemia. Am J Clin Nutr. 1995;61(2):392–396. doi:10.1093/ajcn/61.2.392
44. Miettinen TA, Vanhanen H. Dietary sitostanol related to absorption, synthesis and serum level of cholesterol in different apolipoprotein E phenotypes. Atherosclerosis. 1994;105(12):217–226. doi:10.1016/0021-9150(94)90052-3
45. Ostlund RE. Sitostanol formulation to reduce cholesterol absorption and method for preparing and use of same. United States patent US 5,932,562. 1999.
46. Ostlund RE, Spilburg CA, Stenson WF. Sitostanol administered in lecithin micelles potently reduces cholesterol absorption in humans. Am J Clin Nutr. 1999;70(5):826–831. doi:10.1093/ajcn/70.5.826
47. Pouteau EB, Monnard IE, Piguet-Welsch C, Groux MJ, Sagalowicz L, Berger A. Non-esterified plant sterols solubilized in low fat milks inhibit cholesterol absorption–a stable isotope double-blind crossover study. Eur J Nutr. 2003;42(3):154–164. doi:10.1007/s00394-003-0406-6
48. Trautwein EA, Koppenol WP, de Jong A, et al. Plant sterols lower LDL-cholesterol and triglycerides in dyslipidemic individuals with or at risk of developing type 2 diabetes; a randomized, double-blind, placebo-controlled study. Nutr Diabetes. 2018;8(1):30. doi:10.1038/s41387-018-0039-8
49. Gasco MR. Method for producing solid lipid microspheres having a narrow size distribution. United States patent 5250236. 1993.
50. National Center for Bioechnology Information. Stearic acid, PubChem CID=5281. 2004 Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Stearic-acid.
51. National Center for Biotechnology Information. Palmitic acid, PubChem CID=985. 2004. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/985.
52. National Center for Biotechnology Information. Lauric acid, PubChem CID=3893. 2004. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/3893.
53. Food and Agriculture Organization. Phytosterols, Phytostanols and their esters. Compendium of Food Additive Specifications. 2008;65–70.
54. Shapiro KB, Li L, Secor CA, Sugano M. Analytical method for routine quantitative analysis of plant stanols in stanol ester spread. J Oleo Sci. 2001;50(3):153–158. doi:10.5650/jos.50.153
55. Nandini PT, Doijad RC, Shivakumar HN, Dandagi PM. Formulation and evaluation of gemcitabine-loaded solid lipid nanoparticles. Drug Deliv. 2015;22(5):647–651. doi:10.3109/10717544.2013.860502
56. Tan CSW, Billa N, Roberts CJ, Scurr DJ. Properties of an oral nanoformulation of a molecularly dispersed amphotericin B comprising a composite matrix of Theobroma oil and bee’s wax. Nanomaterials. 2014;4:905–916. doi:10.3390/nano4040905
57. USP 29–NF 24. The United States pharmacopeia: the National Formulary: USP 29 NF 24. Rockville, MD: Powder flow; 2006:1174.
58. American Type Culture Collection. Hep G2 [HEPG2] (ATCC® HB? 8065TM). 2018. Available from: http://www.lgcstandards-atcc.org/products/all/HB-8065.aspx?geo_country=gb.
59. American Type Culture Collection. HT-29 (ATCC® HTB-38TM). 2018. Available from: http://www.lgcstandards-atcc.org/en/Products/All/HTB-38.aspx.
60. Parker FS. Applications of Infrared Spectroscopy in Biochemistry, Biology, and Medicine. New York: Plenum Press; 1971:102–106.
61. Molina-Gutiérrez M, Hakalin NLS, Rodríguez-Sanchez L, Prieto A, Martínez MJ. Green synthesis of β-sitostanol esters catalyzed by the versatile lipase/sterol esterase from Ophiostoma piceae. Food Chem. 2017;221:1458–1465. doi:10.1016/j.foodchem.2016.11.005
62. Muller R, Mader K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery - a review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–177. doi:10.1016/s0939-6411(00)00087-4
63. Ekambaram P, Sathali AH, Priyanka K. Solid lipid nanoparticles: a review. Sci Rev Chem Commun. 2012;2(1):80–102.
64. Verma S, Makkar D. Solid lipid nanoparticles: a comprehensive review. J Chem Pharm Res. 2016;8(8):102–114.
65. Kraft JC, Freeling JP, Ziyao W, Ho RJY. Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J Pharm Sci. 2014;103(1):29–52. doi:10.1002/jps.23773
66. Prokop A, Davidson JM. Nanovehicular intracellular delivery systems. J Pharm Sci. 2008;97(9):3518–3590. doi:10.1002/jps.21270
67. Schiener M, Hossann M, Viola JR, et al. Nanomedicine-based strategies for treatment of atherosclerosis. Trends Mol Med. 2014;20(5):271–281. doi:10.1016/j.molmed.2013.12.001
68. U.S. Food and Drug Administration. CFR - code of federal regulations title 21 food and drugs. 2019. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=184.1505.
69. Holt T, Shah R, Eldridge D, Palombo E, Harding I. Manufacturing solid lipid nanoparticle (SLN) dispersions using a microwave assisted microemulsion approach. In: Torres T, editor. Microemulsions: Systems, Properties & Applications. New York: Nova Science Publishers Inc; 2016.
70. Ibrahim WM, AlOmrani AH, Yassin AE. Novel sulpiride-loaded solid lipid nanoparticles with enhanced intestinal permeability. Int J Nanomedicine. 2014;9:129–144. doi:10.2147/IJN.S54413
71. Mensink RP, Zock PL, Kester AD, Katan MB. Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: a meta-analysis of 60 controlled trials. Am J Clin Nutr. 2003;77(5):1146–1155. doi:10.1093/ajcn/77.5.1146
72. Sacks FM, Lichtenstein AH, Wu JH, et al. Dietary fats and cardiovascular disease: a presidential advisory from the American Heart Association. Circulation. 2017;136(3):e1–e23. doi:10.1161/CIR.0000000000000510
73. Bruce JS, Salter AM. Metabolic fate of oleic acid, palmitic acid and stearic acid in cultured hamster hepatocytes. Biochem J. 1996;316(Pt 3):847–852. doi:10.1042/bj3160847
74. Allman-Farinelli MA, Gomes K, Favaloro EJ, Petocz P. A diet rich in high-oleic-acid sunflower oil favorably alters low-density lipoprotein cholesterol, triglycerides, and factor VII coagulant activity. J Am Diet Assoc. 2005;105(7):1071–1079. doi:10.1016/j.jada.2005.04.008
75. DiNicolantonio JJ, O’Keefe JH. Effects of dietary fats on blood lipids: a review of direct comparison trials. Open Heart. 2018;5:e000871. doi:10.1136/openhrt-2018-000871
76. Carr TP. Compound and method for enhancing the cholesterol lowering property of plant sterol and stanol esters. United States patent 0085451. 2005 Mar 17.
77. Nair R, Kumar AC, Priya VK, Yadav CM, Raju PY. Formulation and evaluation of chitosan solid lipid nanoparticles of carbamazepine. Lipids Health Dis. 2012;11:72. doi:10.1186/1476-511X-11-72
78. Panakanti PK, Panakanti G, Reddy S, Somagoni J, Rao YM. Atorvastatin loaded solid lipid nanoparticles: formulation, optimisation, and in - vitro characterisation. IOSR J Pharm. 2012;2(5):23–32.
79. Raju KK, Sudhakar B, Murthy KVR. Factorial design studies and biopharmaceutical evaluation of simvastatin loaded solid lipid nanoparticles for improving the oral bioavailability. ISRN Nanotechnol. 2014;951016:1–8. doi:10.1155/2014/951016
80. Shah P, Shah S, Dhoranwala KA. Formulation optimization of rosuvastatin calcium-loaded solid lipid nanoparticles by 32 Full-factorial design. NanoWorld J. 2016.
81. Shah R, Eldridge D, Palombo E, Harding I. Optimisation and stability assessment of solid lipid nanoparticles using particle size and zeta potential. J Phys Sci. 2014;25(1):59–75.
82. Kalaycioglue GD, Aydogan N. Preparation and investigation of solid lipid nanoparticles for drug delivery. Colloid Surf A. 2016;510:77–86. doi:10.1016/j.colsurfa.2016.06.034
83. Biswas AK, Islam MR, Choudhury ZS. Mostafa A and Kadir MF. Nanotechnology based approaches in cancer therapeutics. Adv Nat Sci-Nanosci. 2014;5:4.
84. Xu Q, Nakajima M, Liu Z, Shiina T. Soybean-based surfactants and their applications. In: Soybean- Applications and Technology. London: InTech; 2011:344.
85. Fillery-Travis AJ, Foster LH, Robins MM. Stability of emulsions stabilised by two physiological surfactants: L-a-phosphatidylcholine and sodium taurocholate. Biophys Chem. 1995;54:253–260. doi:10.1016/0301-4622(94)00136-8
86. Omwoyo WN, Ogutu B, Oloo F, et al. Preparation, characterization, and optimization of primaquine-loaded solid lipid nanoparticles. Int J Nanomedicine. 2014;9:3865–3874. doi:10.2147/IJN.S62630
87. Freitas C, Müller RH. Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLN™) dispersions. Int J Pharm. 1998;168(2):221–229. doi:10.1016/S0378-5173(98)00092-1
88. Ho SS, Pal S. Margarine phytosterols decrease the secretion of atherogenic lipoproteins from HepG2 liver and Caco2 intestinal cells. Atherosclerosis. 2005;182:29–36. doi:10.1016/j.atherosclerosis.2005.01.031
89. Kim YS, Li XF, Kang KH, Ryu BM, Kim SK. Stigmasterol isolated from marine microalgae Navicula incerta induces apoptosis in human hepatoma HepG2 cells. BMB Rep. 2014;47(8):433–438. doi:10.5483/BMBRep.2014.47.8.153
90. Yi J, Knudsen TA, Nielsen AL, et al. Inhibition of cholesterol transport in an intestine cell model by pine-derived phytosterols. Chem Phys Lipids. 2016;200:62–73. doi:10.1016/j.chemphyslip.2016.06.008
91. Shah RM, Rajasekaran D, Ludford-Menting M, Eldridge DS, Palombo EA, Harding IH. Transport of stearic acid-based solid lipid nanoparticles (SLNs) into human epithelial cells. Colloids Surf B Biointerfaces. 2016;140:204–212. doi:10.1016/j.colsurfb.2015.12.029
92. Brüll F, Mensink RP, Van Den Hurk K, Duijvestijn A, Plat J. TLR2 activation is essential to induce a Th1 shift in human peripheral blood mononuclear cells by plant stanols and plant sterols. J Biol Chem. 2010;285(5):2951–2958. doi:10.1074/jbc.M109.036343
93. Li W, Zhou J, Xu Y. Study of the in vitro cytotoxicity testing of medical devices. Biomed Rep. 2015;3(5):617–620. doi:10.3892/br.2015.481
94. National Center for Biotechnology Information. Rhodamine 123, PubChem CID=65218. 2019. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Rhodamine-123.
95. National Center for Biotechnology Information. Campestanol, PubChem CID: 119394. 2019. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Campestanol.
96. National Center for Biotechnology Information. Sitostanol, PubChem CID: 15559396. 2019. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Fucostanol.
97. Canton I, Battaglia G. Endocytosis at the nanoscale. Chem Soc Rev. 2012;41:2718–2739.
98. Martins S, Costa-Lima S, Carneiro T, Cordeiro-Da-Silva A, Souto EB, Ferreira DC. Solid lipid nanoparticles as intracellular drug transporters: an investigation of the uptake mechanism and pathway. Int J Pharm. 2012;430(1–2):216–227. doi:10.1016/j.ijpharm.2012.03.032
99. Xu W, Bae EJ, Lee MK. Enhanced anticancer activity and intracellular uptake of paclitaxel-containing solid lipid nanoparticles in multidrug-resistant breast cancer cells. Int J Nanomedicine. 2018;13:7549–7563. doi:10.2147/IJN.S182621
100. Verma A, Stellacci F. Effect of surface properties on nanoparticle-cell interactions. Small. 2010;6(1):12–21. doi:10.1002/smll.200901158
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.