")
Back to Journals » Journal of Pain Research » Volume 12
First evaluation of tapentadol oral solution for the treatment of moderate to severe acute pain in children aged 6 to <18
Authors Finkel JC, Goldberg J, Rosenburg R, Ariyawansa J, Sun T, Ochs-Ross R, Zannikos P, Zhang L, Etropolski M
Received 7 December 2018
Accepted for publication 27 May 2019
Published 28 June 2019 Volume 2019:12 Pages 1925—1936
DOI https://doi.org/10.2147/JPR.S197348
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Katherine Hanlon
Julia C Finkel,1 Jutta Goldberg,2 Ronald Rosenburg,2 Jay Ariyawansa,3 Tao Sun,3 Rachel Ochs-Ross,3 Peter Zannikos,3 Liping Zhang,3 Mila Etropolski3
1Children’s National Medical Center, Washington, DC, USA; 2Grünenthal GmbH, Aachen, Germany; 3Janssen Pharmaceutical, Titusville, NJ, USA
Background: This is the first clinical trial in the global pediatric clinical development program for the use of the analgesic tapentadol in children and adolescents.
Patients and methods: This multicenter, open-label clinical trial investigated pharmacokinetics, safety and tolerability, and efficacy of tapentadol and its major metabolite tapentadol-O-glucuronide after administration of a single dose of tapentadol oral solution (OS) in pediatric patients aged 6 to <18 years experiencing moderate to severe acute pain after surgery. Efficacy (change in pain intensity after tapentadol intake) was assessed in an exploratory manner using the McGrath Color Analog Scale and Faces Pain Scale-Revised. Adverse events were monitored throughout the trial.
Results: Forty-four patients who received a single dose of 1 mg/kg tapentadol OS were included in this investigation. Maximum serum concentrations of tapentadol (111 ng/mL) and tapentadol-O-glucuronide (2,400 ng/mL) observed in this trial were within the range of individual maximum concentrations observed in healthy adults administered a comparable dose (range for tapentadol 23.2–129 ng/mL, for tapentadol-O-glucuronide 1,040–4,070 ng/mL). Following tapentadol administration, pain intensity scores improved from baseline at all timepoints. Treatment-emergent adverse events, none of which were serious, were experienced by 45.5% of the patients; the most commonly reported were vomiting (29.5%) and nausea (9.1%).
Conclusions: Tapentadol OS administered as a single dose of 1 mg/kg in children aged 6 to <18 years was generally well tolerated and produced similar serum concentrations as administration of 50–100 mg tapentadol immediate-release tablets in adults. A decrease in postsurgical pain was observed using exploratory subject-reported pain assessments. Tapentadol OS may provide a new treatment option in the management of moderate to severe acute pain in children and adolescents.
Keywords: tapentadol, acute pain, children, μ-opioid, pharmacokinetics, efficacy and safety
Introduction
Acute pain in children
Acute pain is a sensory symptom of short duration typically associated with trauma.1 Research into neurologic pain transmission pathways has shown that infants and children of all ages have the capacity to perceive pain.2 Acute pain is common among infants, children, and adolescents; however, it is undertreated in both the clinical and community settings.3
Current treatment options for the management of acute pain in children include opioids, NSAIDs, and acetaminophen. Although indisputably efficacious in the treatment of postsurgical pain, use of opioids may be limited in this setting by tolerability issues such as nausea or vomiting or constipation.
Tapentadol
Tapentadol is a centrally active analgesic drug with an immediate-release (IR) tablet formulation and an oral solution (OS) approved for the treatment of moderate to severe acute pain in adults.4,5 A prolonged-release formulation is also approved for the treatment of severe chronic pain in adults.6 Tapentadol differs from other marketed analgesic opioids by virtue of a dual mechanism of action. The mechanism of µ-opioid receptor agonism is combined with a complementary analgesic mechanism (noradrenaline reuptake inhibition).7 Both contribute synergistically to a strong analgesic effect (comparable to classical strong opioid analgesics) but with a reduced µ-opioid load8 which might reduce the incidence of opioid-typical side effects.7 Tapentadol is a pure stereoisomer that acts directly on the central nervous system (CNS) and has no active analgesic metabolites.9 Tapentadol is mainly metabolized by Phase 2 glucuronidation and only a small amount is metabolized by Phase 1 oxidative pathways.9 As glucuronidation is a high-capacity/low-affinity system, clinically relevant interactions caused by Phase 2 metabolism are unlikely, as confirmed by drug–drug interaction trials in humans.4,10 In vitro tapentadol did not inhibit or induce cytochrome P450 enzymes (CYP).9 Thus, clinically relevant interactions mediated by the CYP system are unlikely to occur. As tapentadol IR was shown to be efficacious and better tolerated than pure µ-opioid analgesics (due to a reduced µ-opioid load) in adults for the treatment of acute pain,11–14 tapentadol may also offer advantages for pain management in children.
Pediatric trials are challenging; they potentially need to include a wide age, body weight, and developmental range from (preterm) neonates to <18-year-old patients. Age-appropriate medication formulations need to be developed, and trial designs and assessments appropriate for different age groups need to be chosen. A comprehensive review of these challenges was recently published.15 Regulatory frameworks by both the European Medicines Agency and the Food and Drug Administration (FDA) are now in place for guidance with the development of pediatric medications.16–19 Tapentadol is the first analgesic to undergo a formal pediatric clinical trial program. This program includes several clinical investigations starting with single-dose pharmacokinetic (PK) trials to derive appropriate tapentadol dose regimens for the different pediatric age groups. The current article provides the first PK data for tapentadol and its analgesically inactive major metabolite tapentadol-O-glucuronide after administration of a single dose of tapentadol OS 1 mg/kg in children and adolescents aged 6 to <18 years after scheduled surgical procedures that routinely produce moderate to severe acute postsurgical pain. Although analgesically inactive, the O-glucuronide metabolite is the major tapentadol metabolite with concentrations in adults considerably higher than the parent compound. Concentrations of the metabolite were measured in this pediatric trial to provide a comparison with adult data. Safety, tolerability, and efficacy of tapentadol OS were additionally assessed.
Patients and methods
Recruitment
This multicenter, open-label, single-dose trial was conducted from 12 October 2011 to 23 March 2013 at 32 trial centers in Canada, Spain, and the US. The trial was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with Good Clinical Practices and applicable regulatory requirements. The trial protocol and amendments were reviewed by independent ethics committees/institutional review boards in the participating countries (Supplementary material). Informed consent was obtained from all parents of the participants included in the trial; assent forms were signed by those patients able to understand the risks and benefits of participation (as per local regulations). The trial is registered with ClinicalTrials.gov (identifier NCT01134536).
Recruited for this trial were male and female surgical patients aged 6 to <18 years with a maximum body weight of 85 kg and body mass index (BMI) <95th percentile for children, based on Center for Disease Control growth charts.20 Pediatric surgery patients were separated into 2 groups (Group 1: 12 to <18 years old, Group 2: 6 to <12 years old) in accordance with a Written Request by the FDA. Patients undergoing surgical procedures with long and difficult recovery periods or possible heavy blood loss were excluded from this trial. Consistent with those exclusion criteria, patients were excluded if the qualifying surgery involved a large body cavity, upper or lower airway (excluding tonsillectomies), or the brain and required prolonged ventilation or intensive care.
Patients were to be treated with tapentadol after scheduled surgical procedures that routinely produce moderate to severe acute postsurgical pain. To be eligible for the trial, patients had to complete scheduled surgery per investigator’s judgment and have a postsurgical pain intensity score ≥4 on the McGrath Color Analog Scale (CAS21) or in the investigator’s clinical judgment (ie, investigator judgment relying on standard of care rather than the McGrath CAS) have a pain level that required opioid treatment. The latter led to the inclusion of subjects with a CAS score of 0 as the investigator was expecting these subjects to have significant pain resulting from the surgery and found it unethical to wait for the pain to increase before administration of tapentadol.
Dose selection
Tapentadol OS in two concentrations (20 mg/mL and 4 mg/mL) was developed for pediatric trials to allow flexible dosing in all age groups. The formulation was first used in adults, and bioequivalence was demonstrated between tapentadol OS and tapentadol IR tablets (data on file). The dose levels used in the current trial were 1 mg/kg tapentadol OS for patients with a body weight <75 kg and 75 mg for patients with a body weight of 75–85 kg. These doses were selected based on efficacy and safety experiences in adult trials and on trial simulation using a population PK model. The safety and efficacy of tapentadol IR tablets of 50–100 mg administered 4–6 times daily in adults were demonstrated during a completed Phase 3 program in acute pain. As the underlying pain mechanisms and the exposure–effect curve of tapentadol were expected to be similar in adults and children >2 years of age, the serum exposure levels achieved in adults were taken as the target exposure range in this pediatric trial. A nonlinear mixed effects modeling population PK model and a 2-compartment model described the serum PK of tapentadol after oral administration of IR formulations.22 Evaluations demonstrated that oral clearance and central volume of distribution of tapentadol increased with an increase in body weight. Simulations showed that 0.7 mg/kg and 1.4 mg/kg doses of tapentadol in children were expected to produce exposures similar to those after administration of 50 mg and 100 mg tapentadol IR in adults. Therefore, a 1 mg/kg tapentadol OS dose for patients up to 75 kg body weight and 75 mg for patients with a body weight between 75 and 85 kg were considered appropriate, allowing characterization of the exposure and disposition of the drug without compromising the safety of the pediatric patients.
Trial design
The trial included a screening phase (≤30 days, including pre- and postsurgical evaluations, surgical procedure, and postsurgical recovery period), a treatment phase (Day 1) with a single oral dose of tapentadol OS 1 mg/kg with PK sampling, and an end-of-trial phase (Day 2) with end of trial/early withdrawal (discontinuation) assessments. The patients were confined to the trial center (during surgery, postsurgical phase, entire 15-hr post dose evaluation phase, and end-of-trial phase) as per standard of care.
Pain intensity was measured using 2 scales. The McGrath CAS pain intensity rating scale has been established as a valid self-reported measure of pain intensity in children aged from 6 to 16 years.21 The CAS is a visual analog scale from 0=no pain (light pink color) to 10=worst imaginable pain (deep red hue). The Faces Pain Scale - Revised (FPS-R) scale has been established as a valid self-reported measure of pain intensity in children aged 4–15 years.23,24 The FPS-R shows six faces with scores of 0, 2, 4, 6, 8, and 10 where 0=no pain and 10=very much pain. The McGrath CAS and FPS-R were shown to have strong convergent validity, discriminative validity, and responsivity in patients 4–17 years of age.25 Across ages, different pain assessment scales are used considering the developmental status of the child. The pediatric development program for tapentadol included an efficacy trial in children aged from 2 to less than 18 years requiring the use of different pain scales in the same efficacy trial. Therefore, bridging pain intensity measurements from different scales was needed. The use of the two validated scales in the current trial provided information on the similarity of pain intensity scores rated by children aged from 6 to <12 years on 2 different rating scales at similar time points.
The screening phase consisted of informed consent, assent, medical history, physical examination, 12-lead electrocardiogram (ECG), clinical laboratory tests, vital signs, and review of pain intensity scales. Baseline assessments included vital signs, alertness (ability to drink), and pain intensity measurements (McGrath CAS for all patients, FPS-R for Group 2) after surgery, followed by administration of tapentadol OS 1 mg/kg. During the postsurgical phase, vital signs of the patients were assessed, pain intensity measurements were taken at 15 and 30 mins, 1, 2, 4, 6, 11, and 15 hrs post-dose, and PK sampling was performed (one sample for each time range) between 15 mins and 1 hr, 1 and 4 hrs, 4 and 11 hrs, and 11 and 15 hrs post-dose. Serum samples were analyzed for tapentadol and tapentadol-O-glucuronide concentrations using validated liquid chromatography–tandem mass spectrometry (LC-MS/MS) bioanalytical method. The quantification range was 0.2–200 ng/mL for tapentadol and 10.0–10,000 ng/mL for tapentadol-O-glucuronide. End-of-trial assessments included physical exam, clinical laboratory tests, 12-lead ECG, vital signs, and pain intensity measurements.
Patients experiencing persistent pain during the treatment phase were allowed to take supplemental analgesic medication, including oral acetaminophen (paracetamol) every 4–6 hrs, ibuprofen or naproxen according to approved posology beginning 1 hr after tapentadol administration, or morphine according to posology commonly used beginning 2 hrs after tapentadol administration. Intravenous diphenhydramine could be administered every 6 hrs as needed for treatment of itching. Ondansetron and naloxone were allowed to treat nausea and clinically significant respiratory depression, respectively.
Neuroleptics, monoamine oxidase inhibitors, selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, tricyclic antidepressants, triptans, anticonvulsants, antiparkinsonian drugs, and dextromethorphan (within 2 days of tapentadol administration), rifampin, or any drugs that impair metabolism of serotonin were prohibited within 24 hrs of tapentadol administration and during the trial. Sedatives were not allowed unless used during surgery. Concomitant use of other CNS depressants was restricted and if used required close observation. Administration of analgesic medications, including narcotic analgesia, prior to the start of tapentadol, was to be avoided. An interval of 2 hrs was encouraged between the last narcotic dose and administration of tapentadol OS.
Statistical analysis and plan
Sample size estimation
The sample size was chosen to limit the exposure in pediatric patients while providing sufficient data to characterize primary PK parameters in the studied age range with a population PK modeling approach.
The trial was powered to target a 95% confidence interval within 71% and 140% of the point estimate for the geometric mean estimates of clearance and volume of distribution for tapentadol in each age group in accordance with Wang and colleagues.26 An approximately equal distribution of male and female subjects was pursued to ensure representation across the range of ages.
Statistical analysis
PK data (concentration-time data of tapentadol and tapentadol-O-glucuronide) were summarized using descriptive methods. If factors influencing serum concentrations (eg, vomiting) were present, the responsible pharmacokineticist was to decide whether to include or exclude the specific sample or patient. All patients receiving tapentadol OS were included in safety and efficacy analyses (safety set). Safety and tolerability were assessed by physical examination, ECG, clinical laboratory tests, vital signs, and the occurrence of treatment-emergent adverse events (TEAEs). Adverse events were encoded using the Medical Dictionary for Regulatory Activities (MedDRA), version 16.0. As the trial was powered for PK measurements, efficacy assessments were exploratory. Pain intensity data collected for both CAS and FPS-R were summarized descriptively; pain intensity assessments at early withdrawal were included in the end-of-trial results. The number of intakes of supplemental analgesic medication by a patient and the time between tapentadol OS administration and first intake of supplemental analgesic medication were summarized by type of medication.
Results
Trial population
A total of 44 patients (20 males and 24 females) received tapentadol and were included in the safety set. Of these, 6 patients were discontinued for protocol-defined discontinuation criteria, most commonly due to vomiting within the first 3 hrs following tapentadol administration (n=5). Patients were white (84.1%), black or African American (6.8%), Pacific Islander (2.3%), and other (6.8%; Table 1).
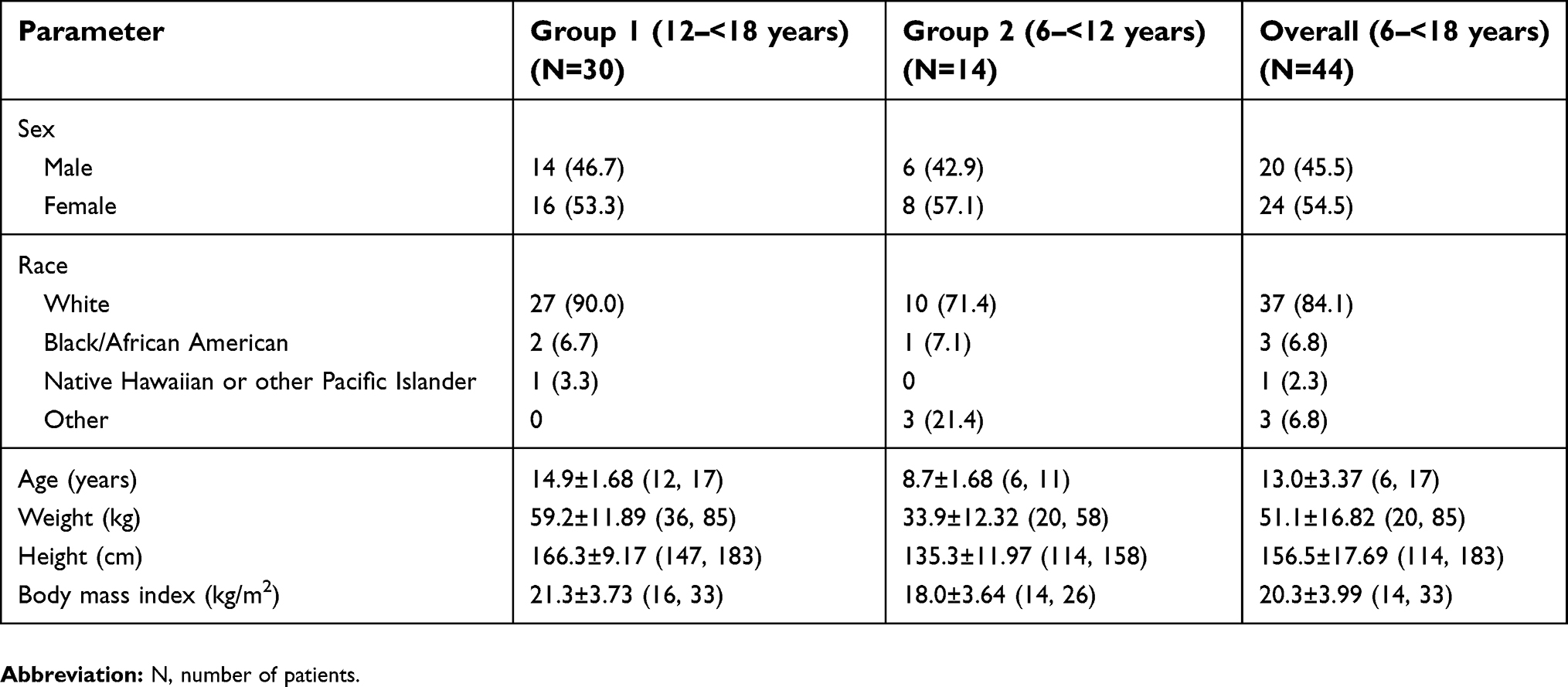 |
Table 1 Demographic and baseline characteristics |
Pharmacokinetics
PK data were available from 44 patients. The concentrations of tapentadol and tapentadol-O-glucuronide in serum are summarized in Table 2. Data included in this descriptive analysis included valid concentrations from all patients who received tapentadol. Over the first 12 hrs following drug administration, mean concentrations ranged from 6.97 to 59.2 ng/mL and from 167 to 1,250 ng/mL for tapentadol and tapentadol-O-glucuronide, respectively, for those intervals which included data for >1 patient (Table 2). Individual serum concentrations vs time are shown as a semi-log scatter plot in Figure 1 with different symbols used for children in the age groups of 12 to <18 and 6 to <12 years of age. It is apparent from this figure that similar profiles were observed for both age groups, with slightly lower concentrations in the younger children.
 |
Table 2 Serum concentrations of tapentadol and tapentadol-O-glucuronide following single-dose administration of tapentadol to pediatric patients |
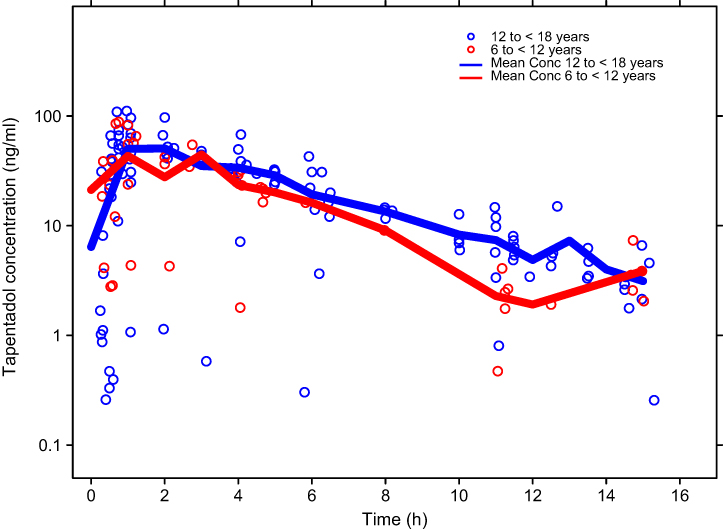 |
Figure 1 Individual serum concentrations of tapentadol in pediatric patients in the age groups from 12 to <18 years (N=30) and from 6 to <12 years (N=14). |
The highest serum tapentadol concentration observed in any of the pediatric patients was 111 ng/mL. This is within the range of individual maximum concentrations (23.2–129 ng/mL) observed in healthy adult subjects who were administered 80 mg of tapentadol (which approximates to the 1 mg/kg dosing regimen used in the current pediatric trial).10 Similarly, the highest tapentadol-O-glucuronide concentration of 2,400 ng/mL in the serum of a pediatric patient in the present trial was within the range of maximum concentrations (1,040–4,070 ng/mL) reported in adults who received 80 mg of tapentadol.10
A population PK model was developed to describe tapentadol PK in children. This model includes the PK data collected in this trial as well as the PK data collected in a second single-dose pediatric trial in children aged from 2 to <18 years of age. The other PK trial as well as the population PK model will be published in the thematic tapentadol series in this journal.
Pain intensity
Mean pain intensity at baseline in the older age group was 3.9 (SD 2.1) on the CAS; the efficacy of tapentadol increased in the first 2 hrs of treatment and then plateaued around a score of 3 (Figure 2A). Pain intensity assessments for the younger group included both CAS and FPS-R scores. Mean baseline pain intensity was similar to the older age group (CAS 3.8 [SD 2.1]) but with greater improvements from baseline during treatment. The improvement was similar for both pain measurement tools (Figure 2B). “No pain” was reported by 8 of 14 patients and 9 of 13 patients at Day 1, 2 hrs and Day 1, 6 hrs post-dose, respectively, when using the FPS-R. At the end of the trial, 13 of 14 patients had scores ranging from 0 to 2 on the FPS-R compared to 7 patients at baseline with similar scores.
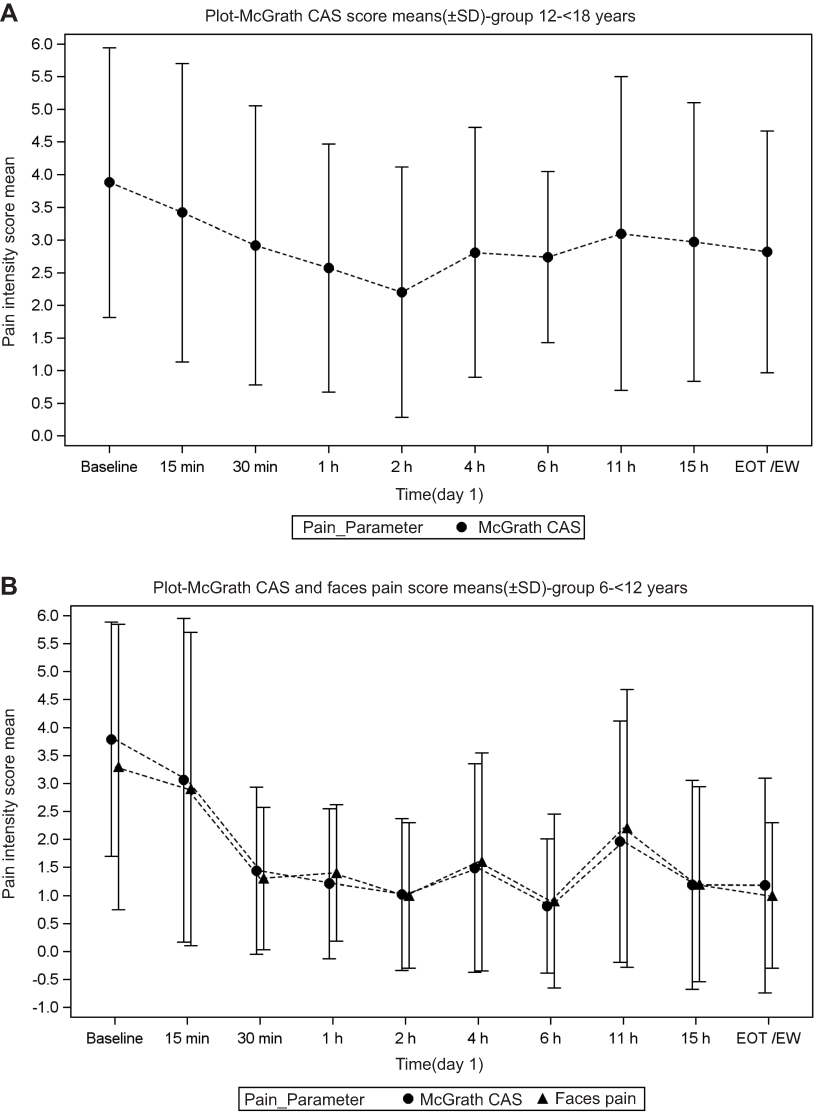 |
Figure 2 Mean pain intensity over time in (A) patients aged from 12 to <18 years and (B) patients aged from 6 to <12 years. Abbreviations: CAS, Color Analog Scale; EOT, end of trial; EW, early withdrawal. |
Supplemental analgesic medications
Of the 44 patients enrolled in the trial, 32 (72.7%) received supplemental analgesic medications during the 15-hr post-dose evaluation period. Ibuprofen or naproxen was the most frequently (45.5%) administered analgesic medication followed by opioids (36.4%), opioid in addition to acetaminophen (paracetamol) or ibuprofen or naproxen (29.5%), and acetaminophen (paracetamol) (27.3%). Half of the patients (50%) received supplemental analgesic medications for persistent pain at least twice during the trial. The median time to first intake of any supplemental analgesic medication after tapentadol administration was approximately 5 hrs (range: 0.6–21.3 hrs). The median time to first intake after tapentadol administration for ibuprofen or naproxen was 6.3 hrs, for opioids 4.1 hrs, and for acetaminophen (paracetamol) 4.3 hrs.
Safety
Less than half of the patients enrolled in the trial (45.5% [20/44]) experienced at least one TEAE (Table 3). Twelve of 30 patients (40%) in Group 1 and 8 of 14 patients (57.1%) in Group 2 experienced a TEAE. The most common TEAEs reported by >1 patient were vomiting (29.5%) and nausea (9.1%). Vomiting occurred more frequently in Group 2 (50%) than in Group 1 (20%). All TEAEs were considered mild or moderate in severity by the investigator. Serious adverse events did not occur. TEAEs considered at least possibly related to tapentadol administration by the investigator included vomiting (20.5% of all patients), nausea (9.1%), hypoesthesia oral, pruritus, swelling face, dizziness, headache, and hypoxia (each in 2.3% of the patients). All other TEAEs reported during the trial were considered unrelated to tapentadol administration. There were no clinically relevant changes in hematology, chemistry, and urinalysis group mean values for Group 1 or Group 2. No clinically relevant individual subject changes in laboratory analytes from baseline to end of trial were observed. No abnormalities in laboratory values were reported as TEAEs. One patient in Group 2 reported a TEAE of oxygen saturation decreased (87% saturation) on Day 1, which resolved on the same day and was considered not related to trial medication by the investigator.
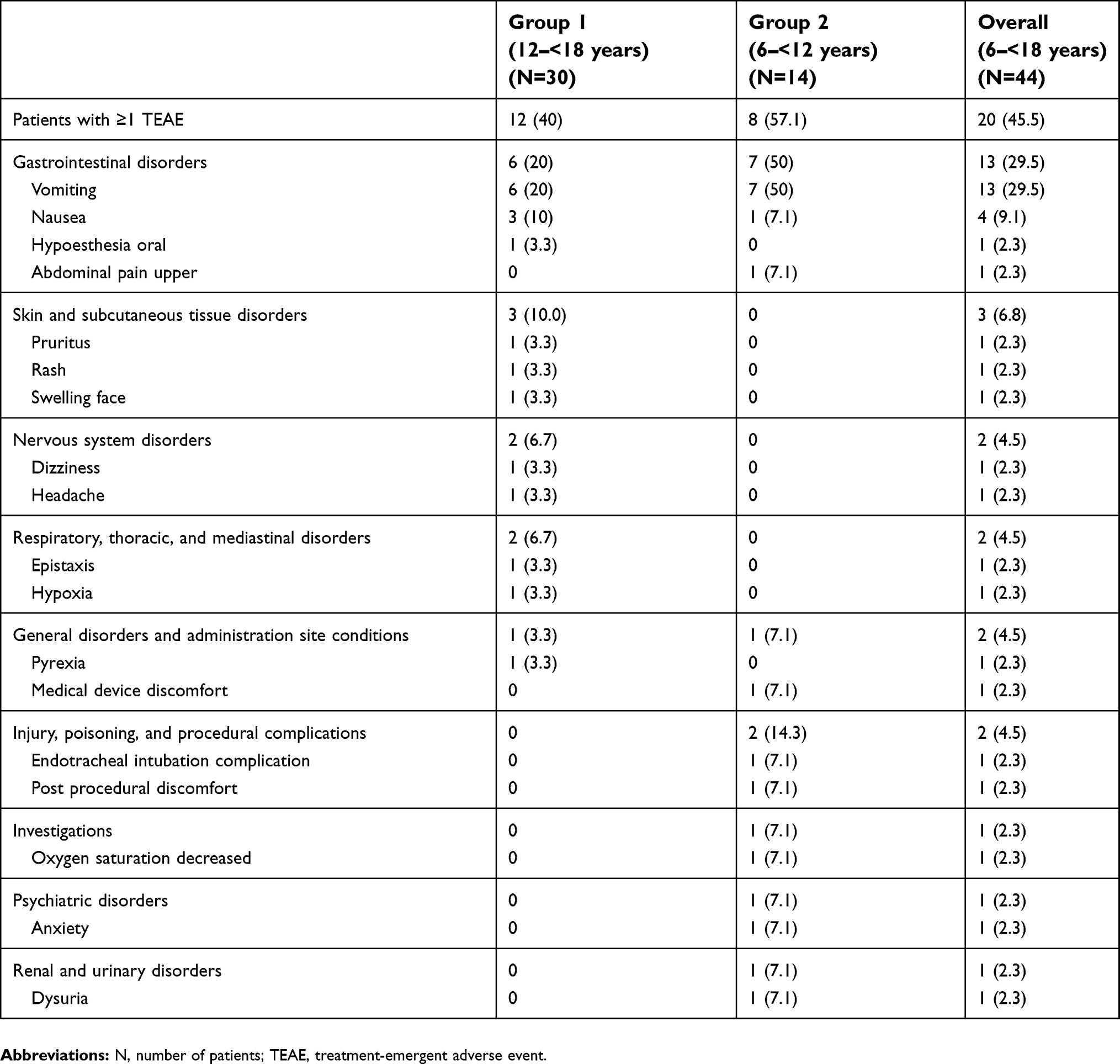 |
Table 3 Treatment-emergent adverse events by body system/organ class and preferred term |
Discussion
This first-in-children open-label clinical trial is part of a global pediatric clinical development program for the use of tapentadol in children and adolescents. The PK results indicate that the 1 mg/kg tapentadol OS dose in children aged from 6 to <18 years produced concentrations of tapentadol in serum that were within the range observed after administration of a 50–100 mg therapeutic dose of tapentadol in adults. In the pediatric patients enrolled in the present trial, the mean serum tapentadol concentrations were 59.2 ng/mL, 51.5 ng/mL, and 47.1 ng/mL during the following time periods, respectively: 45 mins to <1 hr post dose, 1 to <1.5 hrs post dose, and 2 to <3 hrs post dose. By comparison, serum tapentadol concentrations in adults with acute pain following bunionectomy who received a single dose of tapentadol IR averaged from 39.8 ng/mL to 62.8 ng/mL, when adjusted to a dose of 75 mg, at 1 hr and 3 hrs post-dose (data on file). All serum concentration data from this trial were included in further analyses to model the appropriate tapentadol OS dose for children which was then investigated in a double-blind, randomized, placebo-controlled efficacy and safety trial (see reference 15 for a trial overview). Manuscripts describing population PK modeling and the confirmatory efficacy trial are in preparation and are intended for publication in this journal as part of the thematic series ‘Tapentadol for moderate to severe acute pain in children and adolescents’.
Inclusion into the trial required either a postsurgical pain intensity score ≥4 on the CAS or the clinical judgment of the investigator that the pain level experienced by the patient required opioid treatment. As the mean pain intensity score at baseline was 3.8, the investigator’s judgment was relied upon in a number of cases. Tapentadol administration led to an improvement in pain intensity scores over time and gradual decreases in the mean pain intensity scores from baseline during the first 4 hrs after administration (median time to first intake of rescue medication was approximately 5 hrs). This was shown with both pain rating scales used in the trial for the age group of patients from 6 to <12 years. Assessments showed a more pronounced improvement in the younger age group.
Tapentadol was well tolerated as evidenced by less than half (45.5%) of patients experiencing TEAEs in this trial, none of which were serious; the most commonly reported were vomiting (29.5%) and nausea (9.1%). These results are consistent with the safety profile of tapentadol in adults. Vomiting occurred more frequently in Group 2 (50%) than in Group 1 (20%). It should, however, be noted that the use of supplemental analgesics might have biased the safety and tolerability findings. Pain intensity scores decreased after the administration of tapentadol as anticipated; moreover, the use of supplemental analgesics on average started approximately 5 hrs after tapentadol administration consistent with a decrease in plasma concentrations of tapentadol. This indicates a possible dosing regimen of every 4–6 hrs in children. These data suggest that tapentadol is also effective in the treatment of moderate to severe acute pain in the pediatric population.
Trial limitations
The primary objective of the trial was to assess the PK profile of tapentadol and its metabolite. Other analyses (eg, those of efficacy measurements) were only exploratory. The inherent limitations of a single-dose trial, without comparator or control and allowance of concomitant analgesic medications do not permit firm conclusions from the pain intensity measurements assessed in the pediatric population in this trial.
Conclusions
Results from this trial showed that tapentadol OS 1 mg/kg in children aged from 6 to <18 years provided serum concentrations comparable to therapeutic levels in adults and may provide notable pain relief with a good safety and tolerability profile. Tapentadol may provide a new pain management option for children and adolescents for moderate to severe acute pain with a good safety and tolerability profile.
Abbreviation list
BMI, body mass index; CAS, Color Analog Scale; CNS, central nervous system; CYP, cytochrome P450 enzymes; ECG, electrocardiogram; FDA, Food and Drug Administration; FPS-R, Faces Pain Scale - Revised; IR, immediate-release; OS, oral solution; PK, pharmacokinetic; TEAE, treatment-emergent adverse event.
Data sharing statement
The authors will share upon request the final clinical trial report. They will also consider on a case-by-case basis requests for access to other documents and/or data including individual patient data. There is no limitation regarding document/data availability.
Acknowledgments
The authors thank all patients, parents/legal guardians, investigators, and trial site teams involved in this investigation. The trial was funded by Janssen Research & Development, LLC and Grünenthal GmbH. Writing assistance was provided by Tim Plunkett and was paid for by Grünenthal GmbH.
Disclosure
Jutta Goldberg is an employee of Grünenthal GmbH. Ronald Rosenburg was an employee of Grünenthal GmbH at the time the trial was conducted. Jay Ariyawansa, Tao Sun, Rachel Ochs-Ross, Peter Zannikos, Liping Zhang, and Mila Etropolski are employees of Janssen Pharmaceutical. Julia C Finkel reports grants from Janssen, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Cohen LL, Lemanek K, Blount RL, et al. Evidence-based assessment of pediatric pain. J Pediatr Psychol. 2008;33(9):939–955; discussion 956–957. doi:10.1093/jpepsy/jsm103
2. Fitzgerald M. The development of nociceptive circuits. Nat Rev Neurosci. 2005;6(7):507–520. doi:10.1038/nrn1701
3. Stevens BJ, Zempsky WT. Prevalence and distribution of pain in children. In: McGrath PJ, Stevens BJ, Walker SM, Zempsky WT, editors. Oxford Textbook of Paediatric Pain. Oxford: Oxford Universtiy Press; 2014:12–19.
4. Electronic Medicines Compendium. Palexia 50 mg film-coated tablets. Available from: https://www.medicines.org.uk/emc/medicine/28375.
5. Electronic Medicines Compendium. Palexia oral solution 20 mg/ml. Available from: https://www.medicines.org.uk/emc/product/5346/smpc.
6. Electronic Medicines Compendium. Palexia SR prolonged release tablets. Available from: https://www.medicines.org.uk/emc/product/5158/smpc.
7. Tzschentke TM, Christoph T, Kögel BY. The mu-opioid receptor agonist/noradrenaline reuptake inhibition (MOR-NRI) concept in analgesia: the case of tapentadol. CNS Drugs. 2014;28:319–329. doi:10.1007/s40263-014-0151-9
8. Raffa RB, Elling C, Tzschentke TM. Does ‘strong analgesic’ equal ‘strong opioid’? Tapentadol and the concept of ‘µ-load’. Adv Ther. 2018;35(10):1471–1484. doi:10.1007/s12325-018-0778-x
9. Tzschentke TM, De Vry J, Terlinden R, et al. Tapentadol hydrochloride. Drugs Future. 2006;31(12):1053–1061. doi:10.1358/dof.2006.031.12.1047744
10. Smit JW, Oh C, Rengelshausen J, et al. Effects of acetaminophen, naproxen, and acetylsalicylic acid on tapentadol pharmacokinetics: results of two randomized, open-label, crossover, drug-drug interaction studies. Pharmacotherapy. 2010;30(1):25–34. doi:10.1592/phco.30.1.25
11. Hartrick C, Van Hove I, Stegmann JU, Oh C, Upmalis D. Efficacy and tolerability of tapentadol immediate release and oxycodone HCL immediate release in patients awaiting primary joint replacement surgery for end-stage joint disease: a 10-day, phase III, randomized, double-blind, active- and placebo-controlled study. Clin Ther. 2009;31:260–271. doi:10.1016/j.clinthera.2009.02.009
12. Daniels SE, Upmalis D, Okamoto A, Lange C, Häeussler J. A randomized, double-blind, phase III study comparing multiple doses of tapentadol IR, oxycodone IR, and placebo for postoperative (bunionectomy) pain. Curr Med Res Opin. 2009;25(3):765–776. doi:10.1185/03007990902728183
13. Daniels SE, Casson E, Stegmann JU, et al. A randomized, double-blind, placebo-controlled phase 3 study of the relative efficacy and tolerability of tapentadol IR and oxycodone IR for acute pain. Curr Med Res Opin. 2009;25:1551–1561. doi:10.1185/03007990902952825
14. Viscusi E, Allard R, Sohns M, Eerdekens M. Tapentadol immediate release for moderate to severe acute post-surgery pain. J Opioid Manage. 2019;15(1):51–67. doi:10.5055/jom.2019.0486
15. Eerdekens M, Beuter C, Lefeber C, van den Anker J. The challenge of developing pain medications for children: therapeutic needs and future perspectives. J Pain Res. 2019;12:1649–1664.
16. European Medicines Agency. Clinical investigation of medicinal products in the paediatric population (CPMP/ICH/2711/99: ICH E11. January 2001). Available from: https://www.ema.europa.eu/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-30.pdf.
17. European Medicines Agency. ICH E11(R1) guideline on clinical investigation of medicinal products in the pediatric population. EMA/CPMP/ICH/2711/1999. September 1, 2017. Available from: https://www.ema.europa.eu/documents/scientific-guideline/ich-e11r1-guideline-clinical-investigation-medicinal-products-pediatric-population-revision-1_en.pdf.
18. Food and Drug Administration. Guidance for Industry. E11 Clinical investigation of medicinal products in the pediatric population. December 2000. Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073143.pdf.
19. Food and Drug Administration. Guidance for Industry. E11(R1) Addendum: clinical investigation of medicinal products in the pediatric population. April 2018. Available from: https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm530012.pdf.
20. National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion. Published May 30, 2000; modified Oct 16, 2000. Available from: http://www.cdc.gov/growthcharts.
21. McGrath PA, Seifert CE, Speechley KN, Booth JC, Stitt L, Gibson MC. A new analogue scale for assessing children’s pain: an initial validation study. Pain. 1996;64(3):435–443.
22. Xu XS, Smit JW, Lin R, Stuyckens K, Terlinden R, Nandy P. Population pharmacokinetics of tapentadol immediate release (IR) in healthy subjects and patients with moderate or severe pain. Clin Pharmacokinet. 2010;49(10):671–682. doi:10.2165/11535390-000000000-00000
23. Hicks CL, von Baeyer CL, Spafford PA, van Korlaar I, Goodenough B. The Faces Pain Scale–Revised: toward a common metric in pediatric pain measurement. Pain. 2001;93(2):173–183.
24. Miró J, Huguet A. Evaluation of reliability, validity, and preference for a pediatric pain intensity scale: the Catalan version of the faces pain scale – revised. Pain. 2004;111(1–2):59–64. doi:10.1016/j.pain.2004.05.023
25. Tsze DS, von Baeyer CL, Bulloch B, Dayan PS. Validation of self-report pain scales in children. Pediatrics. 2013;132(4):e971–e979. doi:10.1542/peds.2013-1509
26. Wang Y, Jadhav PR, Lala M, Gobburu JV. Clarification on precision criteria to derive sample size when designing pediatric pharmacokinetic studies. J Clin Pharmacol. 2012;52(10):1601–1606. doi:10.1177/0091270011422812
Supplementary material
List of institutional review boards/independent ethics committees
For all sites that enrolled subjects
Canada
Hamilton Integrated Research Ethics Board (HIREB), Hamilton, ON
CHUS-Centre Hospitalier Universitaire de Sherbrooke, Sherbrooke, QC
Spain
Comité Ético de Investigación Clínica, Hospital Clinico San Carlos, Madrid
Hospital Gregorio Marañón, Oficina Técnica CEIC, Madrid
Comité Ético de Investigación Clínica, Hospital Clinico Universitario de Valladolid, Valladolid
Hospital 12 de Octubre, Unidad Administrativa CEIC, Madrid
USA
Western Institutional Review Board, Olympia, WA
Sterling Institutional Review Board, Atlanta, GA
Carolina Health System IRB, Charlotte, NC
Duke University Medical Center Institutional Review Board, Durham, NC
Biomedical Research Alliance of New York, Lake Success, NY
University of Arkansas for Medical Service Institutional Review Board, Little Rock, AR
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.