Back to Journals » OncoTargets and Therapy » Volume 12
FGFR1 Induces Acquired Resistance Against Gefitinib By Activating AKT/mTOR Pathway In NSCLC
Authors Zhang D, Han L, Du F, Liu X, Li J, Wang H, Song M, Li Z, Li G ![]()
Received 22 June 2019
Accepted for publication 31 October 2019
Published 18 November 2019 Volume 2019:12 Pages 9809—9816
DOI https://doi.org/10.2147/OTT.S220462
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianmin Xu
Dan Zhang,1,2,* Li-li Han,3,* Fen Du,4,* Xiao-meng Liu,1 Jin Li,1 Hui-hui Wang,1 Ming-hui Song,1 Zeng Li,2 Guo-yin Li1
1College of Life Science and Agronomy, Zhoukou Normal University, Zhoukou, Henan, People’s Republic of China; 2Department of Oncology, Hanzhong 3201 Hospital Affiliated to Xi’an Jiaotong University, Xi’an, Shaanxi, People’s Republic of China; 3Department of Respiratory, Zhoukou Central Hospital, Zhoukou, Henan, People’s Republic of China; 4Department of Nursing, Hanzhong Vocational Technical College, Hanzhong, Shaanxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guo-yin Li
College of Life Science and Agronomy, Zhoukou Normal University, 6 Changle Wenchang Road, Zhoukou 466000, People’s Republic of China
Email [email protected]
Objective: As an epidermal growth factor, receptor-tyrosine kinase inhibitor (EGFR-TKI), gefitinib demonstrates a good therapeutic effect in patients with EGFR-mutant non-small-cell lung cancer (NSCLC). However, an overwhelming majority of these patients inevitably develop resistance against gefitinib. Unfortunately, the mechanism underlying this phenomenon is still not fully understood. Here we aim to reveal the mechanism of gefitinib resistance in NSCLC induced by FGFR1.
Materials and methods: We used high-throughput sequencing to compare the mRNA expression profiles of PC9 and PC9-GR (gefitinib-resistant) cells. The clinical significance of fibroblast growth factor receptor 1 (FGFR1) in NSCLC was also investigated using immunohistochemistry and Kaplan-Meier survival analysis. Finally, the in vitro molecular mechanisms were analyzed using confocal laser microscopy, Western blotting, transwell assay, colony formation assay, CCK-8 assay, and apoptosis assay.
Results: We observed that FGFR1 was highly expressed in NSCLC tissues and was closely associated with poor prognosis. Cytological experiments showed that FGFR1 promoted the proliferation and migration of PC9-GR cells and mediated their resistance to gefitinib. Furthermore, studies aimed at unraveling this mechanism revealed that FGFR1 activated the AKT/mTOR signaling pathway. These findings show that the FGFR1/AKT/mTOR signaling pathway plays a vital role in acquired resistance against gefitinib in NSCLC.
Conclusion: This work provides new evidence that FGFR1 functions as a key regulator of gefitinib resistance, thereby demonstrating its potential as a novel biomarker and therapeutic target for NSCLC.
Keywords: fibroblast growth factor receptor 1, FGFR1, acquired resistance, gefitinib, non-small-cell lung cancer, NSCLC, AKT/mTOR pathway
Introduction
Lung cancer is one of the most common malignant neoplasms and is a leading cause of cancer mortality worldwide.1,2 Non-small-cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancers, and is characterized by relative insensitivity to radiation and chemotherapy.3 However, the discovery of somatic mutations in epidermal growth factor receptors (EGFRs) and the use of targeted therapy via oral tyrosine kinase inhibitors (TKIs) has improved therapeutic prospects of patients with advanced NSCLC. The incidence of EGFR mutations has been reported to be approximately 62% in Asian populations and 10–15% in North American and European populations.4 Consequently, gefitinib and erlotinib were approved as first-line treatments for advanced NSCLC associated with EGFR-activating mutations.5 Both these drugs significantly prolong the progression-free survival (PFS) of NSCLC patients harboring EGFR-activating mutations.6 Approximately 80% of NSCLC patients harboring EGFR-activating mutations respond well to EGFR-TKIs.7 Unfortunately, an overwhelming majority of these patients inevitably develop resistance to EGFR-TKIs.8
Acquired resistance plays a vital role in the failure of EGFR targeted therapy, which eventually leads to the death of NSCLC patients. Thus, researchers have extensively tried to clarify the mechanisms behind secondary resistance towards EGFR-TKIs. The T790M mutation in EGFR and amplification of the MET oncogene have been proved to be the leading reasons behind EGFR-TKI acquired resistance.9–12 In addition, hepatocyte growth factor (HGF) overexpression, EGFR amplification, epithelial-mesenchymal transition (EMT), and conversion to small-cell lung cancer have also been shown to be crucial mechanisms supporting the development of EGFR-TKI acquired resistance.13–15 However, approximately 30% of EGFR-TKI secondary resistance mechanisms remain undefined.7
Fibroblast growth factor receptor 1 (FGFR1) is a receptor tyrosine kinase that belongs to the FGFR family. It plays a pivotal role in multiple biological processes, including cell survival, migration, proliferation, and differentiation.16 Previous studies have shown that FGFR1 is overexpressed in a variety of cancers, including NSCLC, ovarian cancer, and prostate cancer.17 Silencing of FGFR1 expression or inhibiting its activity inhibits NSCLC proliferation.18,19 Although FGFR1 plays an important role in the development of resistance against EGFR-TKI in tumors, its precise role in NSCLC is currently being debated.7,20 In this study, we show that FGFR1 is upregulated in PC9-GR cells, and that it is correlated with acquired resistance against gefitinib. Furthermore, we show that overexpression of FGFR1 activates the AKT/mTOR signaling pathway, which promotes the proliferation of cancer cells.
Materials And Methods
Cell Culture
PC9 wild-type and gefitinib-resistant cells (PC9-GR) were obtained from the cell bank of the Chinese Academy of Sciences (Shanghai, China). They were cultured in Dulbecco’s Modified Eagle Medium (DMEM; GIBCO, New York, NY, USA) containing 10% fetal bovine serum (FBS; GIBCO) and maintained in an incubator with constant temperature and CO2 (Thermo Fisher Scientific, Waltham, MA, USA).
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was extracted using RNAiso Plus (#9109, TaKaRa, Kusatsu, Shiga, Japan) according to the manufacturer’s instructions. Reverse transcription for gene expression was performed using the PrimeScript™ RT Master Mix (#RR036A, TaKaRa). RT-qPCR was performed using SYBR Green dye (#RR820A, TaKaRa) according to the manufacturer’s protocol. The following paired primers were used: β-actin, forward: 5′-CGGGAAATCGTGCGTGAC-3′ and reverse: 5′-CAGGAAGGAAGGCTGGAAG-3′; and FGFR1, forward: 5′-TCAAATGCCCTTCCAGTG-3′ and reverse: 5′-CATAACGGACCTTGTAGCC-3′.
Western Blotting
Cells were lysed for 20 min in ice-cold RIPA lysis buffer supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and a cocktail of protease inhibitors. Western blotting was performed using antibodies against FGFR1 (#9740, Cell Signaling Technology), Akt (#2920, CST), phospho-Akt (#4060, CST), β-Actin (#3700, CST), mTOR (#2983, CST), and phospho-mTOR (#5536, CST). Goat anti-rabbit and goat anti-mouse immunoglobulin horseradish peroxidase-linked F(ab’)2 fragments (Millipore) were used as secondary antibodies.
Colony Formation Assay
Cells were seeded onto 6-well plates (200 cells/well) and cultured for 14 days. They were then fixed using 4% paraformaldehyde for 15 min and stained with crystal violet for 20 min, following which they were washed with ddH2O and air dried. All the above steps were performed at room temperature. Each treatment was repeated thrice and the number of clones was counted.
Transwell Migration Assay
Briefly, 1 × 104 cells suspended in serum-free medium (200 μL) were plated onto the top chamber of a transwell system (24-well insert; 8 μm; Millicell). Complete medium (500 μL) was used as a chemoattractant in the lower chamber. The cells were incubated for 24 h. Cells that did not migrate through the pores were removed using a cotton swab. Cells on the lower surface of the membrane were stained with crystal violet, air dried, and photographed.
Immunohistochemistry Assay
A standard hematoxylin and eosin staining protocol was employed for staining the NSCLC samples. Briefly, the following steps were performed: formalin fixing, paraffin embedding, dewaxing of paraffin sections, antigen repair, serum blocking, primary antibody incubation, secondary antibody incubation, coloration, counterstaining, dehydration, and blocking. Imaging was performed by Servicebio (http://www.servicebio.com).
Immunofluorescence Assay
Cells were seeded into a glass-bottom cell culture dish (NEST, 15 mm) and cultured for 12 h. They were then fixed with 4% paraformaldehyde and permeabilized with Triton X-100 (0.1%). Next, the cells were labeled overnight with primary antibodies at 4°C, following which, they were incubated with secondary antibodies for 1 h at room temperature, and then counterstained with DAPI (0.1%) for 1 min. Imaging was performed using confocal laser scanning microscopy.
RNA Sequencing
Isolation of total RNA from the samples was performed using TRIzol reagent (Invitrogen, Carlsbad, VI, USA); this RNA was reverse transcribed into cDNA. RNA sequencing was then performed by Annoroad (Beijing, China).
CCK-8 Assay
Cells were plated at a density of 2 × 103 cells/well in 96-well plates and incubated in complete medium. After incubation, viable cells were quantified using cell counting kit-8 (CCK-8; 7Sea Pharmatech, Shanghai, China). Briefly, 10 μL of CCK-8 and 100 μL of medium were added to each well. Following incubation at 37°C for 3 h, a microplate reader (iMARK™; Bio-Rad) was used to estimate the optical density at 450 nm.
Apoptosis Assay
Cells were plated at a density of 2 × 105 cells/well into 6-well plates, incubated for 24 h in complete medium, serum starved for 12 h, and then treated as indicated. Complete medium containing gefitinib and/or PD173074 (a specific inhibitor of FGFR1) was added into the wells and incubated for 24 h. Flow cytometry was used to detect the percentage of dying cells using an Annexin V-FITC/PI apoptosis detection kit (Beyotime, Shanghai, China) according to the manufacturer’s protocol. Flow cytometry was performed via a FACScan system using the CellQuest™ software.
Patients And Follow-Up
Samples from primary NSCLC patients were collected from the clinical samples bank of Hanzhong 3201 China starting in 2010. A total of 58 human NSCLC samples were obtained from Chinese patients at Hanzhong 3201 Hospital (Hanzhong, China). Target genes expression levels variation between cancerous and paracancerous tissues was examined. The clinical characteristics of patients were obtained from hospital records. All patients signed informed consent and were in accordance with the the Declaration of Helsinki. Use of clinical specimens were approved by the Ethics Committee of Zhoukou Normal University. Overall survival (OS) was calculated from the date of surgery until death, or until the date of the last follow-up visit for patients who were still alive. The authors have access to information that could identify individual participants during or after data collection. Stratified analysis of OS was performed as described previously.21
Statistical Analysis
All data were analyzed using SPSS standard version 19.0 and GraphPad Prism version 5.0. Data were obtained from 3 independent experiments and presented as mean ± standard error. Survival rate was analyzed using the Kaplan-Meier method and compared using a log rank test. P< 0.05 was considered to be statistically significant.
Results
FGFR1 Is Highly Expressed In PC9-GR Cells
The cytological function of PC9-GR cells changed significantly when compared to PC9 cells. CCK-8 and colony formation assays were used to examine cell proliferation. The results showed that the proliferation potential and clone-forming abilities of PC9-GR cells were significantly higher than those of PC9 cells (Figure 1A and D). Transwell assay was used to detect cell migration. The migration ability of PC9-GR cells was observed to be approximately 2-fold higher than that of PC9 cells (Figure 1B and C). Furthermore, we used high-throughput sequencing to identify genes that mediated NSCLC resistance to gefitinib by comparing mRNA expression profiles of PC9 and PC9-GR cells. Notably, we found 973 upregulated and 744 downregulated mRNAs in PC9-GR cells (Figure 1E). Among the upregulated mRNAs, we focused on FGFR1 and confirmed that it was upregulated in PC9-GR cells via quantitative real-time PCR (qRT-PCR), Western blotting, and cell immunofluorescence assays (Figure 1F–I). Altogether, these findings suggest that FGFR1 plays a vital role in the development of resistance towards gefitinib in NSCLC.
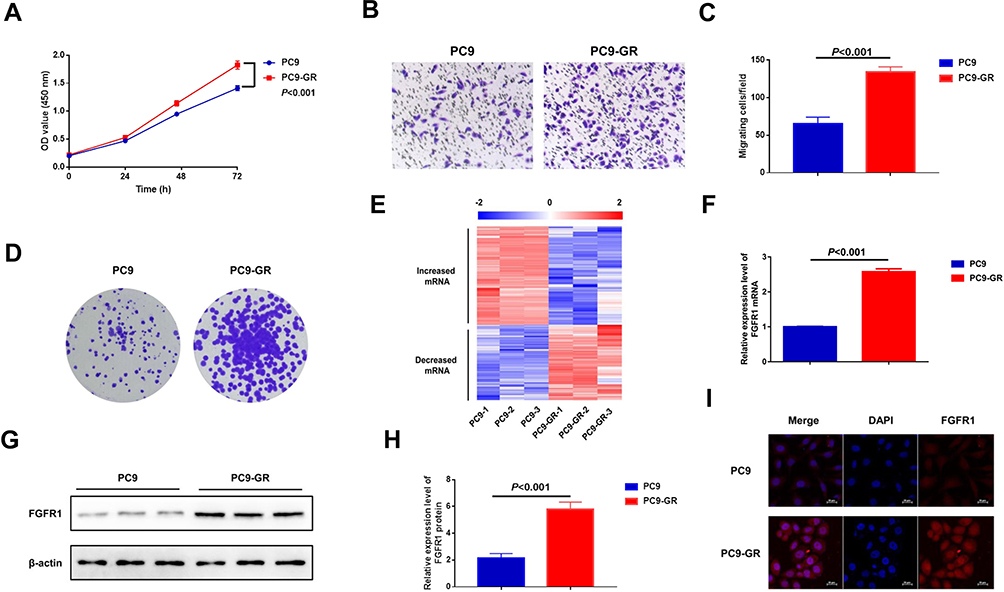 |
Figure 1 FGFR1 is highly expressed in PC9-GR cells. (A) CCK-8 assay of normally cultured cells. (B) Transwell migration assay of PC9 and PC9-GR cells. (C) Quantitative analysis of cell migration in (B) (mean ± SEM, n = 3). (D) Colony formation assay of PC9 and PC9-GR cells. (E) A heatmap depicting the high-throughput sequencing analysis of altered mRNA expression profiles in PC9 and PC9-GR cells. (F) Relative mRNA levels of FGFR1 in PC9 and PC9-GR cells. (G) Western blot analysis of PC9 and PC9-GR cells. (H) Quantitative analysis of FGFR1 expression levels in (G) (mean ± SEM, n = 3). (I) Immunofluorescence microscopy analysis. |
FGFR1 Is Highly Expressed In NSCLC And Results In Poor Outcomes In NSCLC Patients
To determine its clinical significance, we first examined FGFR1 expression levels in the NSCLC samples comprised of cancerous tissues and para-carcinoma tissues using immunohistochemistry and Western blot assays. The results showed that compared to paracarcinoma tissues, FGFR1 was expressed at higher levels in cancerous tissues (Figure 2A–D). We further examined if FGFR1 expression is correlated with the clinical outcomes of NSCLC after lung cancer resection. Kaplan-Meier’s analysis of the NSCLC patients revealed that high FGFR1 expression in NSCLC cancerous tissues was significantly correlated with a reduction in overall survival (Figure 2E), thus confirming its crucial role in the pathogenesis and prognosis of NSCLC .
 |
Figure 2 FGFR1 is highly expressed in NSCLC and predicts poor outcomes in NSCLC patients. (A) Representative immunohistochemical staining using cancerous and paracarcinoma tissues. (B) Western blot analysis of FGFR1 in representative NSCLC samples. (C) Relative protein levels of FGFR1 in cancerous and paracarcinoma tissues. Each data point represents an individual NSCLC sample. (D) Expression ratio of FGFR1 in cancerous and para-carcinoma tissues. (E) Patients were grouped according to FGFR1 expression in the carcinomas and enrolled in follow-up investigations. The percentage of surviving patients was plotted. |
FGFR1 Promotes The Proliferation And Migration Of PC9-GR Cells And Inhibits Their Apoptosis
In order to confirm if FGFR1 plays a key role in PC9-GR cell resistance to gefitinib, we performed apoptosis assays. PC9-GR cells were treated with gefitinib (100 nM) and/or PD173074 (20 nM) for 24 h. The results showed that PD173074 significantly enhanced the sensitivity of PC9-GR cells to gefitinib (Figure 3A and B). Subsequently, we used small interfering RNA (siRNA) to knockdown FGFR1 in PC9-GR cells, and then incubated them in complete medium containing 100 nM gefitinib for 24 h. The results showed that the tolerance of PC9-GR cells towards gefitinib decreased significantly after FGFR1 knockdown (Figure 3C and D). Furthermore, to investigate if FGFR1 was associated with the proliferative and migratory potential of NSCLC cells in vitro, we performed transwell, colony formation, and CCK-8 assays. The results showed that the proliferation, migration, and cloning abilities of PC9-GR cells decreased significantly after FGFR1 knockdown or inhibition (Figure 3E–K). Thus, these results indicate that FGFR1 plays a vital role in the proliferation, migration, and apoptosis of NSCLC cells.
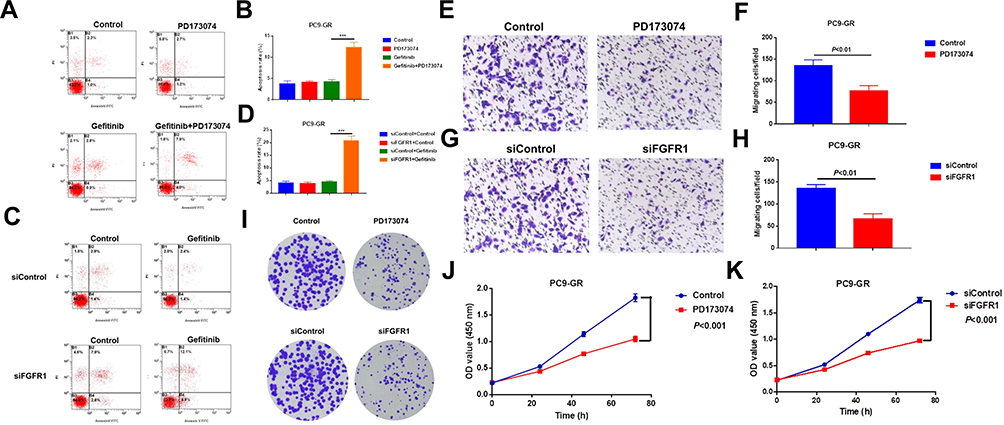 |
Figure 3 FGFR1 promotes the proliferation and migration of PC9-GR cells and inhibits their apoptosis. (A) Cells were treated with gefitinib (100 nM) and/or PD173074 (20 nM) and then subjected to Annexin V/PI staining and flow cytometry analysis for apoptosis. (B) Quantitative analysis of apoptosis rate in (A) (mean ± SEM, n = 3). (C) Cells were transfected with control or FGFR1-targeting siRNA, treated with or without gefitinib (100 nM) for 24 h, and then subjected to Annexin V/PI staining and flow cytometry analysis for detecting apoptosis. (D) Quantitative analysis of apoptosis rate in (C) (mean ± SEM, n = 3). (E) Transwell migration assay of cells treated with PD173074 (20 nM). (F) Quantitative analysis of cell migration in (E) (mean ± SEM, n = 3). (G) Transwell migration assay of cells subjected to FGFR1 knockdown. (H) Quantitative analysis of cell migration in (G) (mean ± SEM, n = 3). (I) Colony formation assay of cells treated with PD173074 or transfected with siFGFR1. (J) CCK-8 assay of cells treated with PD173074 (10 nM). (K) CCK-8 assay of cells subjected to FGFR1 knockdown. *** P < 0.001. |
FGFR1 Facilitates Gefitinib Resistance In NSCLCs By Activating The AKT/Mtor Signaling Pathway
The AKT/mTOR signaling pathway plays an important role in the drug resistance mechanism in tumors. Western blotting and immunofluorescence results showed that AKT and mTOR phosphorylation levels increased significantly in PC9-GR cells (Figure 4A and B and Supporting Information Figure S1). To determine if FGFR1 mediated gefitinib resistance in NSCLC by activating the AKT/mTOR signaling pathway, we knocked down FGFR1 expression in PC9-GR cells using 2 individual siRNAs that targeted different sequences of the transcript (Figure 4C and D). Western blotting analysis revealed that FGFR1 silencing remarkably repressed AKT and mTOR phosphorylation levels (Figure 4E). Then we overexpressed FGFR1 in PC9 and H1299 cells,Western blotting showed that over-expression of FGFR1 could promote phosphorylation of AKT and mTOR (Figure S2). PC9-GR cells were also treated with PD173074 for 24 h and AKT and mTOR phosphorylation levels were again detected using Western blotting. The results showed that PD173074 significantly inhibited AKT and mTOR phosphorylation (Figure 4F). Subsequently, immunohistochemistry was used to detect AKT and mTOR phosphorylation levels in clinical samples of lung cancer. The results showed that the phosphorylation levels of AKT and mTOR were highly expressed in cancerous tissues as compared to paracarcinoma tissues and that they were also positively correlated with FGFR1 expression (Figure 4G–I). However, exposure to rapamycin (10 nM) reduced the proliferation and clonal formation of PC9-GR cells (Figure 4J and K). Then we overexpressed FGFR1 in PC9 cells and exposed to rapamycin (10 nM). CCK-8 assay showed that rapamycin could counteract the proliferation induced by FGFR1 (Supporting Information Figure S3).
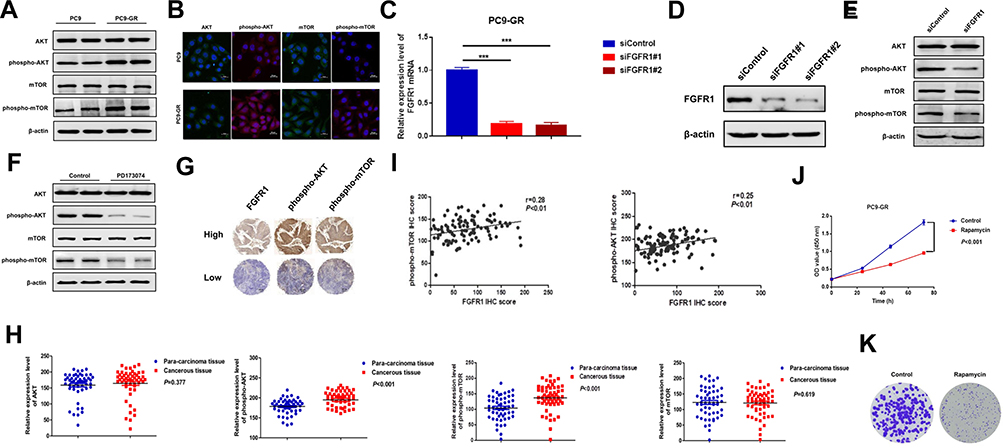 |
Figure 4 FGFR1 facilitates gefitinib resistance in NSCLCs by activating the AKT/mTOR signaling pathway. (A) Western blot analysis of PC9 and PC9-GR cells. (B) Immunofluorescence microscopy analysis of PC9 and PC9-GR cells. (C) RT-qPCR analysis of FGFR1 knockdown efficiency in PC9-GR cells. (D) Western blot analysis of FGFR1 knockdown efficiency in PC9-GR cells. (E) Western blot analysis of PC9-GR cells transfected with siFGFR1. (F) PC9-GR cells were treated with PD173074 (20 nM) for 24 h and then subjected to Western blot analysis. (G) Immunohistochemical staining for indicated proteins using tissue microarray of clinical NSCLC samples. (H) Relative protein levels of target genes in NSCLC samples. Each data point represents an individual NSCLC sample. (I) Statistical analysis of the correlations of the indicated molecules based on immunohistochemical staining scores. (J) CCK-8 assay of cells treated with rapamycin (10 nM). (K) Colony formation assay of cells treated with rapamycin (10 nM). ***P < 0.001. |
Discussion
Lung cancer is the most commonly diagnosed cancer and the leading cause of cancer-associated death worldwide.22 It is gratifying to see that the morbidity and mortality rates of lung cancer in the American population have been declining in recent years.23 Early diagnosis and innovations in the treatment of lung cancer have contributed towards improving the survival rate of patients. Currently, patients with EGFR-mutant NSCLC are administered a standard treatment regimen of EGFR-TKIs as they demonstrate better response rates and PFS as compared to chemotherapy.24,25 Unfortunately, almost all EGFR-mutant NSCLC patients who received first-generation EGFR-TKI therapy developed secondary drug resistance with a mean PFS range of 9.2–13.1 months.25 However, the mechanism underlying the development of secondary drug resistance against gefitinib is still not completely clear even though EGFR-TKIs are used commonly in clinical practice.
In this study, we used PC9 and PC9-GR cells as models to screen genes associated with gefitinib acquired resistance via high-throughput sequencing. The results showed that FGFR1 was highly expressed in PC9-GR cells (Figure 1E). Previously, Terai et al had reported that activation of the FGF2-FGFR1 autocrine pathway led to gefitinib acquired resistance in NSCLC.7 Ware et al also confirmed the FGF2-FGFR1 autocrine growth loop to be an important mechanism that led to gefitinib resistance in cells.20 Notably, our study indicated that FGFR1 not only played a vital role in the proliferation, migration, and clonal formation of PC9-GR cells, but also mediated gefitinib resistance in PC9-GR cells (Figure 3).
Previous studies have shown that overactivation of the AKT signaling pathway was a key factor underlying cell tolerance towards gefitinib.26,27 We predicted that FGFR1 also mediated the development of gefitinib resistance in NSCLC by activating the AKT signaling pathway. To demonstrate our hypothesis, we knocked-down FGFR1 or inhibited its activity using siRNA or PD173074 in PC9-GR cells. The results showed a significant decrease in AKT and mTOR phosphorylation levels (Figure 4). Afterwards, we overexpressed FGFR1 in PC9 and H1299 cells, confirming that FGFR1 could promote the phosphorylation of AKT and mTOR (Supporting Information Fig. S2). Notably, Chen et al reported that the FGFR1/PI3K/AKT signaling pathway is the target of the cancer drug, fumagillin, that is known to mediate antiangiogenic effects.28 Zhang et al revealed that activation of the FGFR1 and TLR4 pathways by a specific signaling agonist increased Akt phosphorylation.29 We found that compared to the paracarcinoma tissues, FGFR1 was highly expressed in cancer tissues and was positively correlated with AKT and mTOR phosphorylation levels (Figure 4G–I). Combining the existing literature and our results, we propose that the FGFR1/AKT/mTOR signaling pathway plays a vital role in acquired resistance against gefitinib in NSCLC.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81903031), Zhoukou Normal University Guiding Project (ZKNUC2018011), the Key Scientific Research Projects of Higher Education Institutions of Henan Province (No. 18A180036) and the Science and Technology Development Plan of Henan Province (No. 182106000047).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi:10.3322/caac.v61:2
2. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
3. Reck M, Heigener DF, Mok T, Soria JC, Rabe KF. Management of non-small-cell lung cancer: recent developments. Lancet. 2013;382(9893):709–719. doi:10.1016/S0140-6736(13)61502-0
4. Kate S, Chougule A, Joshi A, et al. Outcome of uncommon EGFR mutation positive newly diagnosed advanced non-small cell lung cancer patients: a single center retrospective analysis. Lung Cancer (Auckl). 2019;10:1–10. doi:10.2147/LCTT.S181406
5. Lin YT, Chen JS, Liao WY, et al. Clinical outcomes and secondary epidermal growth factor receptor (EGFR) T790M mutation among first-line gefitinib, erlotinib and afatinib-treated non-small cell lung cancer patients with activating EGFR mutations. Int J Cancer. 2018;144:2887–2896.
6. Meng S, Wang G, Lu Y, Fan Z. Functional cooperation between HIF-1alpha and c-Jun in mediating primary and acquired resistance to gefitinib in NSCLC cells with activating mutation of EGFR. Lung Cancer. 2018;121:82–90. doi:10.1016/j.lungcan.2018.04.024
7. Terai H, Soejima K, Yasuda H, et al. Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanism of acquired resistance to gefitinib in NSCLC. Mol Cancer Res. 2013;11(7):759–767. doi:10.1158/1541-7786.MCR-12-0652
8. Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28(2):357–360. doi:10.1200/JCO.2009.24.7049
9. Wang W, Wang H, Lu P, et al. Crizotinib with or without an EGFR-TKI in treating EGFR-mutant NSCLC patients with acquired MET amplification after failure of EGFR-TKI therapy: a multicenter retrospective study. J Transl Med. 2019;17(1):52. doi:10.1186/s12967-019-1803-9
10. Song Z, Huang S, Yu H, et al. Synthesis and biological evaluation of morpholine-substituted diphenylpyrimidine derivatives (Mor-DPPYs) as potent EGFR T790M inhibitors with improved activity toward the gefitinib-resistant non-small cell lung cancers (NSCLC). Eur J Med Chem. 2017;133:329–339. doi:10.1016/j.ejmech.2017.03.083
11. Ito T, Kumagai Y, Itano K, et al. Mathematical analysis of gefitinib resistance of lung adenocarcinoma caused by MET amplification. Biochem Biophys Res Commun. 2019;511:544–550. doi:10.1016/j.bbrc.2019.02.086
12. Nanjo S, Arai S, Wang W, et al. MET copy number gain is associated with gefitinib resistance in leptomeningeal carcinomatosis of EGFR-mutant lung cancer. Mol Cancer Ther. 2017;16(3):506–515. doi:10.1158/1535-7163.MCT-16-0522
13. Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi:10.1126/scitranslmed.3002003
14. Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68(22):9479–9487. doi:10.1158/0008-5472.CAN-08-1643
15. Thomson S, Petti F, Sujka-Kwok I, Epstein D, Haley JD. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin Exp Metastasis. 2008;25(8):843–854. doi:10.1007/s10585-008-9200-4
16. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–129. doi:10.1038/nrc2780
17. Tan Q, Wang Z, Wang Q, et al. A novel FGFR1-binding peptide exhibits anti-tumor effect on lung cancer by inhibiting proliferation and angiogenesis. Int J Biol Sci. 2018;14(10):1389–1398. doi:10.7150/ijbs.24739
18. Lemjabbar-Alaoui H, Hassan OU, Yang YW, Buchanan P. Lung cancer: biology and treatment options. Biochim Biophys Acta. 2015;1856(2):189–210. doi:10.1016/j.bbcan.2015.08.002
19. Zhang L, Yu H, Badzio A, et al. Fibroblast growth factor receptor 1 and related ligands in small-cell lung cancer. J Thorac Oncol. 2015;10(7):1083–1090. doi:10.1097/JTO.0000000000000562
20. Ware KE, Hinz TK, Kleczko E, et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis. 2013;2:e39. doi:10.1038/oncsis.2013.4
21. Li GY, Wang W, Sun JY, et al. Long non-coding RNAs AC026904.1 and UCA1: a “one-two punch” for TGF-beta-induced SNAI2 activation and epithelial-mesenchymal transition in breast cancer. Theranostics. 2018;8(10):2846–2861. doi:10.7150/thno.23463
22. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.v68.6
23. Smith RA, Andrews KS, Brooks D, et al. Cancer screening in the United States, 2018: a review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J Clin. 2018;68(4):297–316. doi:10.3322/caac.21446
24. El Kadi N, Wang L, Davis A, Korkaya H. The EGFR T790M mutation is acquired through AICDA-mediated deamination of 5-Methylcytosine following TKI treatment in lung cancer. Cancer Res. 2018;78(24):6728–6735. doi:10.1158/0008-5472.CAN-17-3370
25. Cortot AB, Janne PA. Molecular mechanisms of resistance in epidermal growth factor receptor-mutant lung adenocarcinomas. Eur Respir Rev. 2014;23(133):356–366. doi:10.1183/09059180.00004614
26. Liu X, Lu X, Zhen F, et al. LINC00665 induces acquired resistance to gefitinib through recruiting EZH2 and activating PI3K/AKT pathway in NSCLC. Mol Ther Nucleic Acids. 2019;16:155–161. doi:10.1016/j.omtn.2019.02.010
27. Yang J, Qin G, Luo M, et al. Reciprocal positive regulation between Cx26 and PI3K/Akt pathway confers acquired gefitinib resistance in NSCLC cells via GJIC-independent induction of EMT. Cell Death Dis. 2015;6:e1829. doi:10.1038/cddis.2015.197
28. Chen GJ, Weylie B, Hu C, Zhu J, Forough R. FGFR1/PI3K/AKT signaling pathway is a novel target for antiangiogenic effects of the cancer drug fumagillin (TNP-470). J Cell Biochem. 2007;101(6):1492–1504. doi:10.1002/(ISSN)1097-4644
29. Zhang R, Dong Y, Sun M, et al. Tumor-associated inflammatory microenvironment in non-small cell lung cancer: correlation with FGFR1 and TLR4 expression via PI3K/Akt pathway. J Cancer. 2019;10(4):1004–1012. doi:10.7150/jca.26277
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.