Back to Journals » Journal of Inflammation Research » Volume 17
FcγRIIb Exacerbates LPS-Induced Neuroinflammation by Binding with the Bridging Protein DAP12 and Promoting the Activation of PI3K/AKT Signaling Pathway in Microglia
Authors Han Y ![]() , Wang L
, Wang L ![]() , Ye X
, Ye X ![]() , Gong X
, Gong X ![]() , Shao X
, Shao X ![]()
Received 5 July 2023
Accepted for publication 19 December 2023
Published 4 January 2024 Volume 2024:17 Pages 41—57
DOI https://doi.org/10.2147/JIR.S428093
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
YingWen Han,* Luyao Wang,* Xiaokun Ye, Xue Gong, Xiaoyi Shao
Department of Immunology, Medical School, Nantong University, Nantong, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaoyi Shao, Department of Immunology, Medical School, Nantong University, 19 Qixiu Road, Nantong, Jiangsu, 226001, People’s Republic of China, Email [email protected]
Introduction: This paper focuses on the expression and role of FcγRIIb in neuroinflammation, exploring the molecular mechanisms by which FcγRIIb interacts with the bridging protein DAP12 to regulate the PI3K-AKT signaling pathway that promote neuroinflammation and aggravate neuronal injury.
Methods: LPS-induced neuroinflammation models in vivo and in vitro were constructed to explore the role and mechanism of FcγRIIb in CNS inflammation. Subsequently, FcγRIIb was knocked down or overexpressed to observe the activation of BV2 cell and the effect on PI3K-AKT pathway. Then the PI3K-AKT pathway was blocked to observe its effect on cell activation and FcγRIIb expression. We analyzed the interaction between FcγRIIb and DAP12 by Immunoprecipitation technique. Then FcγRIIb was overexpressed while knocking down DAP12 to observe its effect on PI3K-AKT pathway. Finally, BV2 cell culture supernatant was co-cultured with neuronal cell HT22 to observe its effect on neuronal apoptosis and cell activity.
Results: In vivo and in vitro, we found that FcγRIIb expression was significantly increased and activated the PI3K-AKT pathway. Contrary to the results of overexpression of FcγRIIb, knockdown of FcγRIIb resulted in a significant low level of relevant inflammatory factors and suppressed the PI3K-AKT pathway. Furthermore, LPS stimulation induced an interaction between FcγRIIb and DAP12. Knockdown of DAP12 suppressed inflammation and activation of the PI3K-AKT pathway in BV2 cells, and meantime overexpression of FcγRIIb suppressed the level of FcγRIIb-induced AKT phosphorylation. Additionally, knockdown of FcγRIIb inhibited microglia activation, which induced neuronal apoptosis.
Discussion: Altogether, our experiments indicate that FcγRIIb interacts with DAP12 to promote microglia activation by activating the PI3K-AKT pathway while leading to neuronal apoptosis and exacerbating brain tissue injury, which may provide a new target for the treatment of inflammatory diseases in the central nervous system.
Keywords: LPS-induced neuroinflammation, FcγRIIb, DAP12, microglia, PI3K-AKT
Introduction
Neuroinflammation is defined as an inflammatory response that occurs within the central nervous system (CNS). The progression of neuroinflammation is characterized by a complex cascade of cellular and biomolecular reactions, with brain triggering a nonspecific immune response upon stimulation.1–3 Common abnormal features in the brains of patients with neuroinflammation include increased expression of pro-inflammatory factors, macrophage activation, peripheral leukocyte infiltration, and damage to neural tissue. Various glial cells are present in the human brain, among which, microglia play a key role in neuroinflammation. In the normal brain environment, microglia are branched and highly motile, and they serve to monitor fluctuations in the brain microenvironment, removing apoptotic neurons and maintaining the stability of the central nervous system.4–6 Microglia are activated and polarized into the proinflammatory phenotype or the anti-inflammatory phenotype;7–9 the proinflammatory phenotype secrete large amounts of inflammatory and neurotoxic factors, which exacerbate neuronal damage, while the anti-inflammatory phenotype secrete large amounts of anti-inflammatory and trophic factors upon activation, alleviating inflammation and maintaining the stability of the brain internal environment.10,11
FcγRIIb, a glycoprotein of approximately 40 kDa, consists of two IgG-like extracellular domains, a transmembrane region, and a cytoplasmic tail containing (immunoreceptor tyrosine-based inhibition motif), and it is the only IgG-inhibitory Fc receptor known to be found in both humans and mice. Many of the typical functions of FcγRIIb are based on signaling through the cytoplasmic region ITIM.12–15 FcγRIIb is a potent modulator in lupus nephritis, while in arthritis models, it effectively attenuates collagen-induced arthritis symptoms. Although ITIM-mediated inhibition through the cytoplasmic compartment is thought to be the main feature of FcγRIIb-mediated immune regulation, evidence also suggests that inhibition or activation of immune responses can occur independently of ITIM signaling in some specific cases. Following a review of the literature, we found reports indicating a 3.54-fold increase in FcγRIIb expression in RAW264.7 cells after lipopolysaccharide (LPS) treatment.16,17 Furthermore, there is evidence to suggest that activation of PI3K is required for FcγR to exert its function, and its activity can be monitored using p-Akt (Ser-473) antibody to determine the change in Akt activity.18,19 These reports have opened new ideas about what role FcγRIIb actually plays in a typical model of neuroinflammation. Based on the above logic, we conducted a preliminary exploration of the role of FcγRIIb in LPS-induced neuroinflammation.
DAP12, a member of the type I transmembrane bridging protein family, consists of a short extracellular structural domain, a single transmembrane structural domain, and a cytoplasmic region, (immunoreceptor tyrosine-based activation motif).20–22 DAP12 is mainly expressed in microglia in the CNS and macrophages in the periphery, and it does not possess ligand-binding capacity owing to its short extracellular structural domain, resulting in the need to form complexes with specific chaperone membrane receptors to transmit signals downstream.23–26 Several signaling pathways involved in DAP12, such as PI3K, PKC, and NF-κB, have been associated with pro-inflammatory responses, phagocytosis, and microglia migration and proliferation.27–30 Notably, the induction of pro-inflammatory cytokines and their related molecules by DAP12 in microglia may lead to a phenotypic shift to a toxic phenotype, resulting in neuronal damage.
PI3K-AKT is a highly conserved intracellular signaling pathway that is required for important activity processes in multiple classes of cells, including the synthesis of key proteins, cell proliferation and activation, neuronal activation, and morphological changes.31–34 This pathway can be activated by a variety of signals, including growth factors (GF), LPS, and insulin (INS), which produce phospholipids and contribute to protein phosphorylation.35–37 Dysregulation of the PI3K-AKT signaling pathway involves multiple pathological changes in multiple systems, such as neurological diseases. Experimental data suggest that activation of the PI3K-AKT signaling pathway in LPS-activated microglia leads to increased levels of proinflammatory factors. Conversely, other data suggest that activation of the PI3K-AKT signaling pathway helps to reduce brain damage and can inhibit inflammatory processes and apoptosis. Aberrant PI3K-AKT signaling is also involved in various diseases, including cancer, neurodegeneration, and neuroinflammatory diseases. In the brain, changes in AKT activity are critical for neuronal development and glial cell activity.38–41 In the present study, FcγRIIb promotes microglia inflammatory activation. FcγRIIb promotes microglia activation by activating the PI3K/AKT signaling pathway. FcγRIIb and DAP12 interact to promote microglia activation-mediated LPS-induced neuroinflammation. The “FcγRIIb-PI3K-AKT” positive feedback loop exacerbates neuroinflammation-mediated neuronal injury.
Materials and Methods
Construction of Animal Models
Animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Research Council, 1996, USA) and approved by Jiangsu Branch of the National Committee for the Use of Laboratory Animals for Medical Purposes, China. C57BL/6 whole male mice (6–8 weeks, 25–30 g) were used for experiments. The mice were kept in the experimental animal center for 7 days before the induction of the LPS-induced neuroinflammation model to acclimatize and reduce stress. After the mice were stabilized, LPS (Escherichia coli O111-B4; Sigma, St. Louis, MO, USA) was injected intraperitoneally into the mice for 6 h, 12 h, 1 d, 3 d, 5 d, and 7 d, while the control mice were injected with an sterile saline (n = 7 groups). After the appropriate length of stimulation, the mice were injected with anesthetic (lidocaine hydrochloride) and then sacrificed, the eyeballs were removed to obtain blood, and brain tissue was removed. The brain tissue samples were stored at –80°C and used for subsequent experiments. During the animal experiments, the pain felt by the mice was minimized as much as possible to ensure the objectivity of the experiments.
Western Blotting
After protein extraction, the protein concentration was determined by Bio-Rad protein assay (BioRad, Hercules, CA, USA), the samples were subjected to sodium polyacrylamide gel electrophoresis (SDS-PAGE), and then the proteins were transferred to polyvinyl fluoride (PVDF) membranes. Closure was performed using 5% skim milk. The membranes were incubated with anti-FcγRIIb (anti-rabbit, 1:1000; Signalway Antibody), anti-FcγRIIb (anti-rat, 1:500; RnDSystems), anti-DAP12 (anti-rabbit, 1:1000. Abcam), iNOS (anti-rabbit, 1:1000; Abcam), GAPDH (anti-mouse, 1:5000; Proteintech), p-AKT (Ser473) (anti-rabbit, 1:1000. Cell Signaling Technology), AKT (anti-rabbit, 1:1000; Cell Signaling Technology), PI3K (anti-mouse, 1:500; Santa Cruz Biotechnology), and Bax (anti-rabbit, 1:1000; Cell Signaling Technology) overnight at 4°C, followed by incubation with the corresponding secondary antibodies for 2 h at room temperature. Finally, the protein bands were visualized by an enhanced chemiluminescence system (ECL, Pierce Company, Appleton, WI, USA).
Plasmid Transfection
The small interfering and negative control of FcγRIIb were purchased from Reebok Biologicals, and the overexpression plasmid and negative control of FcγRIIb were purchased from PPL Genetics: si-DAP12#196, 5’-GGGAUUGUUCUGGGUGAVUTT-3’; si-DAP12#104, 5’-UCCUGACUGUGGGAGGAUUTT-3’;si-DAP12#288, 5’-GGAAGGACCCGGAAACAATT-3’. Myc-RNA was transfected into BV2 cells using Lip2000 transfection reagent (Invitrogen Life Technologies, Carlsbad, CA, USA).
Mouse Brain Tissue Processing and Sectioning
The mouse cerebral cortex was removed, fixed in 4% paraformaldehyde, and then treated with 10%, 20%, and 30% sucrose solution for gradient dehydration. The tissues were subsequently sliced with a freezing microtome at a thickness of 6 μm.
Immunohistochemical Fluorescence
The tissues were pre-incubated with 1% bovine serum albumin (1% BSA) for 2 h at room temperature to block non-specific binding of the primary antibody to the tissue membrane, followed by F4/8O (anti-rat, 1:100, Abcam) and a mixture of anti-FcγRIIb (anti-rabbit, 1:100; Signalway Antibody) incubated overnight at 4°C in the refrigerator. Subsequently, the tissues were incubated with corresponding secondary antibody for 2 h at room temperature protected from light, shaken dry, and incubated for 30 min in Hoechst dye. A blocking solution (glycerol: ddH2O= 1:1) was used to block the slices before observing them under a fluorescence microscope (Leica, DM 5000B, Leica CTR 5000, Germany).
Immunocytofluorescence
BV2 cells were spread on 24-well plates and then stimulated with LPS (100 ng/mL LPS, Escherichia coli 055:B5, Sigma Aldrich, L6529). Next, 4% paraformaldehyde was used to fix the cells for 1 h at room temperature. Subsequently, to enhance the permeability of the cell membrane, 0.1% TritonX-100 was added to the 24-well plate containing BV2 cells at 25°C for 15 min before blocking non-specific binding by incubating the cells with 1% bovine serum albumin for 2 h at room temperature. After the blocking solution (1% bovine serum albumin) was discarded, a mixture of FcγRIIb and Iba-1 anti-FcγRIIb (anti-rat, 1:500; RnDSystems) was added to the 24-well plate and refrigerated overnight at 4°C. Next, the nuclei were incubated with Alexa 488 anti-rabbit IgG (H + L) (Thermo Fisher Scientific) for 2 h, before adding Hoechst dye (Thermo Fisher Scientific) to stain the nuclei. A drop of glycerol was added to the slide, which was then covered with a coverslip, and fluorescence microscopy (Leica, DM 5000B, Leica CTR 5000, Germany) was used for observation.
RNA Extraction and RT-PCR
Total RNA was extracted from the mouse cerebral cortex and LPS-stimulated BV2 cells using a TRIzol kit, while cDNA was synthesized by reverse transcription-polymerase chain reaction using a RevertAidTM RT kit (Thermo Fisher Scientific). The amplified products were subjected to quantitative analysis by q-PCR. This procedure was performed with the following primers (Sunny Shanghai) for DNA sequences: GAPDH Forward primer (5′-3′) AGGTCGGTGTGAACGGATTTG Reverse primer (5′-3′) TGTAGACCATGTAGTTGAGGTCA FcγRIIb Forward primer (5′-3′) ATGGGAATCCTGCCGTTCCTA Reverse primer (5′-3′) CCGTGAGAACACATGGACAGT TYROBP Forward primer (5′-3′) GGTGCCTTCTGTTCCTTCCTGTC Reverse primer (5′-3′) ATCAGCAGAGTCAACACCAAGTCAC TNFα Forward primer (5′-3′) GACGTGGAACTGGCAGAAGAG Reverse primer (5′-3′) TTGGTGGTTTGTGAGTGTGAG IL-6 Forward primer (5′-3′) TAGTCCTTCCTACCCCAATTTCC Reverse primer (5′-3′) TTGGTCCTTAGCCACTCCTTC IL-1β Forward primer (5′-3′) GAAATGCCACCTTTTGACAGTG Reverse primer (5′-3′) TGGATGCTCTCATCAGGACAG
Enzyme-Linked Immunosorbent Assay (ELISA)
The expression levels of TNF-α, IL-6, and IL-1β in the collected mouse sera and cell supernatants were measured using an ELISA kit (Proteintech or MULTI SCIENCES). The optical density (OD) of each well was measured at 450 nm using an enzyme marker with 630 nm as the calibration wavelength. The standard curve was generated by four-parameter fitting using ELISACalc curve making software and the corresponding concentrations were deduced from the standard curve according to the OD values of the samples. The entire operation was performed according to the manufacturer’s instructions.
Cell Culture, Cell Stimulation, and Neuronal Injury Model Construction
BV2 cells (Purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd; Item No. ZQ0397) and HT22 neurons (Purchased from Shanghai Cell Bank of Chinese Academy of Sciences, respectively) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco 8121170) with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C and under 5% CO2. LPS stimulation was used to induce BV2 cells activation (100 ng/mL LPS, Escherichia coli 055:B5, Sigma Aldrich, L6529). Construction of the neuronal injury model: The supernatant of activated BV2 cell culture (MG-CM) was collected, mixed with DMEM complete medium in a 1:1 ratio, and then added to HT22 neurons to generate the neuronal injury model.
Cell Counting Kit 8 Assay (CCK-8)
Briefly, 100 μL of cell suspension was added to each well of a 96-well plate and cultured for 24 h after LPS stimulation for 24 h. Next, 10 μL of CCK-8 reagent (MCE, China) was added to each well and incubated for 2 h protected from light. Finally, the absorbance at 450 nm was measured with an enzyme marker. The data were analyzed according to the manufacturer’s instructions.
Immunoprecipitation (IP)
Briefly, the cells were washed three times with phosphate-buffered saline (PBS), before adding 1 mL of PBS to break the cells. Next, the precipitate was centrifuged, before adding 1 mL of RIPA lysate and rotating at 4°C for 1.5 h. The supernatant was centrifuged and 50 μL was aspirated as the input group. The samples were stored at –20°C until required. After that, 25–30 μL of magnetic beads (Protein A/G PLUS-Agarose sc-2003, immunoprecipitation reagent; Santa Cruz Biotechnology) was added to the supernatant to adsorb impurities, and rotated at 4°C for 2–3 h. The samples were centrifuged to settle the beads, before adding FcγRIIb (anti-rat, 4 μL; RnDSystems) antibody to the supernatant and rotating overnight. The next day, 30 μL of magnetic beads was added and continued to rotate for 3 h. Subsequently, the precipitate was centrifuged before adding 500 μL of RIPA lysate to gently explode the beads, before centrifuging and repeating the above operation once more. Finally, 2x loading buffer (Beyotime Biotechnology) was added to the precipitate before immunoblotting.
Statistical Analysis
SPSS 19.0 software was used to calculate the data related to the experiment in this thesis, and the data were expressed using the mean ± standard deviation. The analysis of differences between two groups of data was done by t-test, and all three and more groups of data were subjected to One-way ANOVA analysis was performed on all three and more groups. Statistical plots were plotted by GraphPad Prism. p<0.05 indicates a statistical difference. p<0.05 indicates a statistical difference.
Results
Upregulation of FcγRIIb Expression is Involved in Microglia Activation in a Mouse Model of CNS Inflammation
To investigate the role of FcγRIIb in LPS-induced neuroinflammation, a mouse CNS inflammation model was constructed by intraperitoneal injection of LPS (9 mg/kg) into C57BL/6 experimental mice. The ELISA results showed that the expression of pro-inflammatory factors TNF-α, IL-6, and IL-1β in mouse serum was increased sharply after LPS injection and peaked at 6 hours (Figure S1). Additionally, the results of q-PCR showed that the inflammatory factors (TNF-α, IL-6, IL-1β) in the mouse cerebral cortex peaked at 6 hours, while FcγRIIb expression peaked at 12 hours after LPS injection (Figure 1A). The protein expression level of iNOS started to increase at 6 hours and peaked at 3 days, while FcγRIIb expression started to increase after 6 hours and peaked at 12 hours (Figure 1B). The immunofluorescence results showed that FcγRIIb in the LPS-stimulated group was significantly co-localized with the microglia marker F4/80, while the expression of FcγRIIb was significantly increased in the mouse cerebral cortex in the LPS-stimulated group compared to the control group (Figure 1C). The above results suggest that the mouse CNS inflammation model was successfully constructed and that FcγRIIb was involved in microglia activation and neuroinflammatory response.
 |
Figure 1 The expression and role of FcγRIIb in a mouse model of CNS inflammation. C57BL/6 mice were injected intraperitoneally with LPS (9 mg/kg), and serum was collected at 6 h, 12 h, 1 d, 3 d, 5 d, and 7 d, while the control group (N) was injected with an equal amount of saline. (A) Q-PCR was performed to detect the mRNA expression levels of IL-6, TNF-α, IL-1β, and FcγRIIb in the mouse cerebral cortex. (B) Western blot was performed to detect the protein expression levels of FcγRIIb, iNOS, PI3K, p-AKT, AKT and DAP12 in the cerebral cortex of experimental mice. (C) Immunofluorescence staining was used to detect the expression of FcγRIIb in the cerebral cortex tissue of control and LPS mice. FcγRIIb (green), F4/80 (red), Hoechst (blue); scale bar: 100 μm (mean ± SEM represents the resultant values, n = 3, *Indicates compared to the control, #indicates compared to the control, p < 0.05). |
Upregulation of FcγRIIb Expression in an in vitro Model of LPS-Induced Neuroinflammation
To further explore the mechanism by which FcγRIIb mediates LPS-induced neuroinflammation during the activation of BV2 cells, we stimulated BV2 cells with LPS (100 ng/mL) at different time points to construct an in vitro activation model. The ELISA results showed that the expression levels of TNF-α, IL-6, and IL-1β were significantly increased in BV2 cells treated with LPS (Figure S2). Additionally, the q-PCR results showed that the expression levels of TNF-α, IL-6, IL-1β and FcγRIIb were significantly increased in BV2 cells treated with LPS (Figure 2A). The results of Western blot showed that the expression of iNOS increased and reached a peak at 12 hours, while the expression of FcγRIIb increased after 3 hours and reached a peak at 12 hours (Figure 2B). To further investigate the significance of FcγRIIb expression in BV2 cells, we observed the localization of FcγRIIb in inflammation-activated BV2 cells. The results of the immunohistochemical fluorescence assay showed that FcγRIIb in the LPS-stimulated group was significantly co-localized with the BV2 cell marker F4/80, while the expression of FcγRIIb in BV2 cells was found to be significantly increased in the LPS-stimulated group compared to the N group (Figure 2C). The above experimental results indicated that the in vitro activation model of BV2 cells was successfully constructed and that FcγRIIb was involved in the process of microglial inflammatory activation.
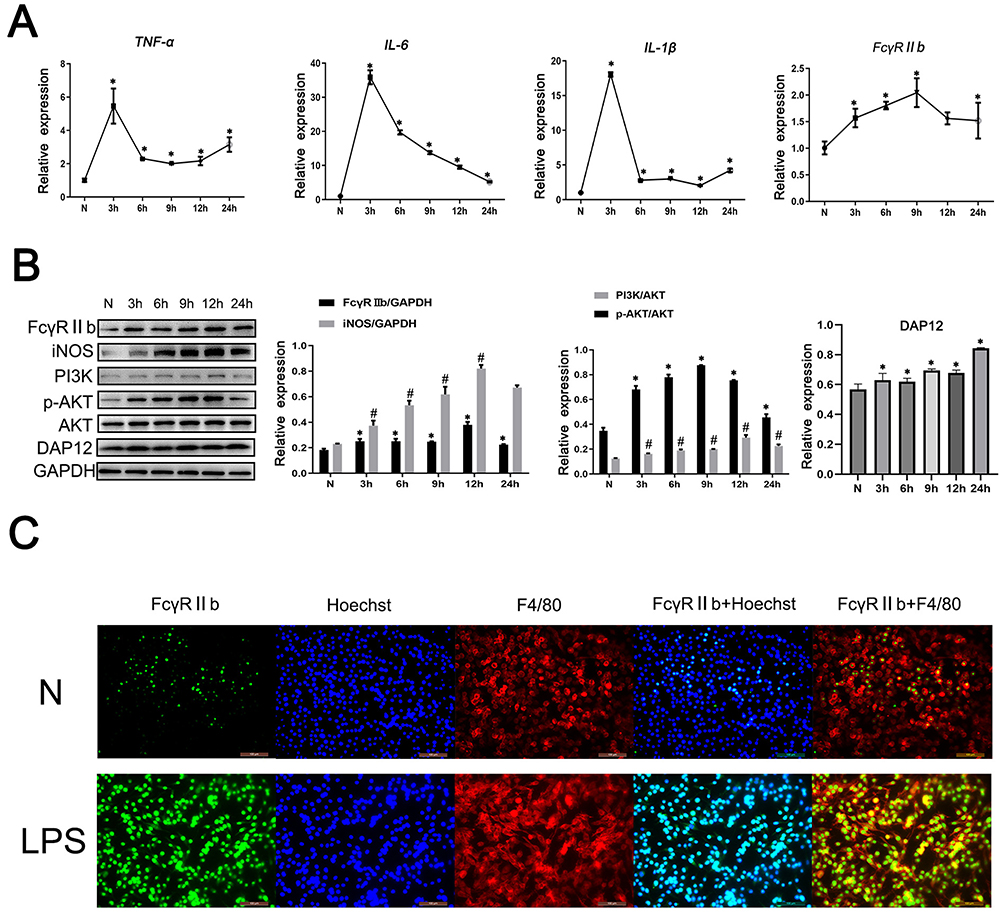 |
Figure 2 The expression and role of FcγRIIb in an in vitro model of LPS-induced neuroinflammation. BV2 cells were stimulated with LPS (100 ng/mL) for 0, 3, 6, 9, 12, and 24 h, and then the cells and the cell culture supernatants were collected. (A) Q-PCR was performed to detect the mRNA expression levels of TNF-α, IL-6, IL-1β, and FcγRIIb in BV2 cells. (B) Western blot was used to detect the protein expression of FcγRIIb, iNOS, PI3K, p-AKT, AKT and DAP12 in BV2 cells. (C) Immunofluorescence staining was employed to detect the expression and localization of FcγRIIb in the N group and LPS-stimulated group of BV2 cells. FcγRIIb (green), F4/80 (red), Hoechst (blue); scale bar: 100 μm (mean ± SEM represents the resultant values, n = 3, *Indicates compared to the control, #indicates compared to the control, p < 0.05). |
FcγRIIb Promotes Inflammatory Activation of Microglia
The above results showed that FcγRIIb expression showed an upregulation trend in inflammation-activated BV2 cells. To confirm the mechanism of action of FcγRIIb in the activation of BV2 cells, we knocked down FcγRIIb in BV2 cells and observed whether there was an effect on the inflammatory activation of BV2 cells. We transfected plasmids expressing an siRNA targeting FcγRIIb into BV2 cells. The Western blot results showed that expression of FcγRIIb and iNOS was reduced in BV2 cells (Figure 3A). The q-PCR results also showed that the expression of FcγRIIb and inflammatory factors (TNF-α, IL-6, and IL-1β) was decreased in siRNA-treated BV2 cells (Figure 3B and D). Meanwhile, the results of ELISA showed a downregulated expression trend for inflammatory factors (TNF-α and IL-6) in the cell culture supernatants of siRNA-treated BV2 cells (Figure 3C). The q-PCR assay results were consistent with the ELISA results, in that expression knocking-down of FcγRIIb in BV2 cells significantly inhibited the expression of inflammatory factors in LPS stimulated group, and the expression levels of TNF-α, IL-6, and IL-1β showed a decreasing trend.
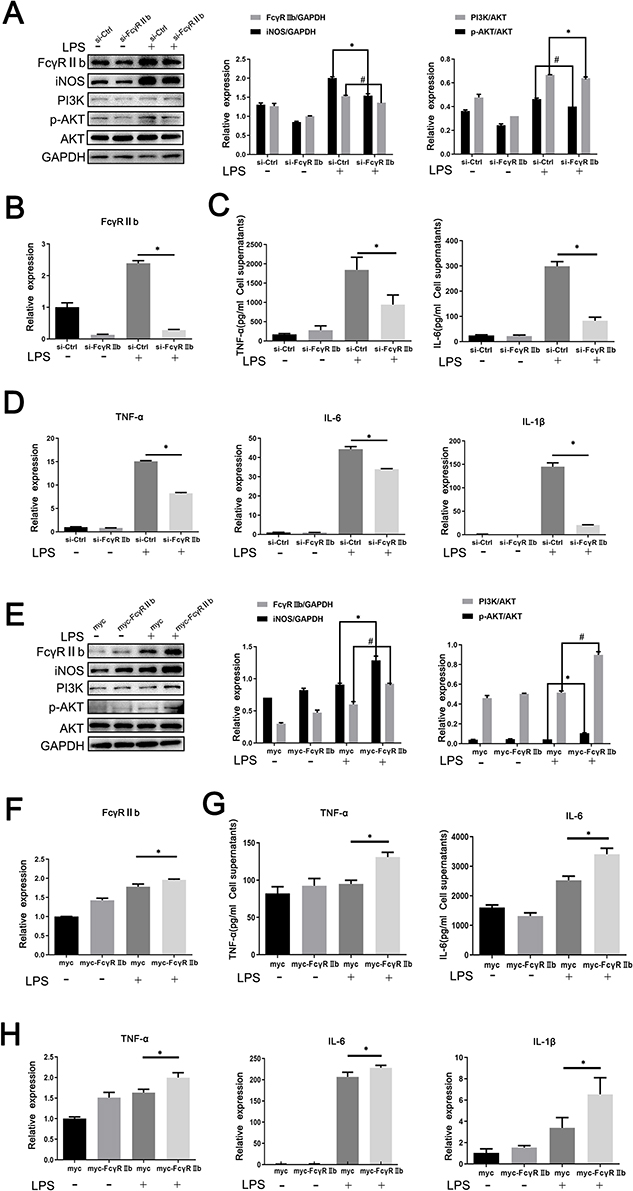 |
Figure 3 Effect of inhibition or overexpression of FcγRIIb on inflammatory activation in microglia. FcγRIIb interference knocking-down or overexpressing plasmids were transfected into BV2 cells for 48 h and then stimulated with LPS for 12 h. (A) Western blot detection of FcγRIIb, iNOS, PI3K, p-AKT and AKT protein expression levels after interference with BV2 cells. (B) Q-PCR assay of the mRNA expression levels of the inflammatory factors FcγRIIb in BV2 cells after interference with FcγRIIb. (C) ELISA the assay of the expression levels of TNF-α and IL-6 in the cell supernatants collected after interference with BV2 cells. (D) Q-PCR assay of the mRNA expression levels of the inflammatory factors TNF-α, IL-6, and IL-1β in BV2 cells after interference with FcγRIIb. (E) Western blot assay of the protein expression levels of FcγRIIb, iNOS, PI3K, p-AKT and AKT after overexpression with BV2 cells. (F) Q-PCR assay of the mRNA expression levels of FcγRIIb after overexpression with BV2 cells. (G) ELISA assay of the expression levels of inflammatory factors TNF-α and IL-6 in BV2 cells.(H) Q-PCR assay of the mRNA expression levels of TNF-α, IL-6, and IL-1β after overexpression with BV2 cells. (mean ± SEM represents the result values, n = 3, *Indicates compared to the si-Ctrl group, or myc+LPS group, #Indicates compared to the si-Ctrl+LPS group, or myc+LPS group, p < 0.05). |
Meanwhile, we transfected FcγRIIb overexpressing plasmid into BV2 cells and then stimulated the cells with LPS (100 ng/mL) for 12 hours. Western blot detection showed that in LPS-stimulated group, the expression levels of iNOS and FcγRIIb were significantly increased in the overexpression group compared to the no-load group (Figure 3E). Moreover, q-PCR results demonstrated that the overexpression group showed increased expression of FcγRIIb, TNF-α, IL-6, and IL-1β in BV2 cells (Figure 3F and H), while ELISA results showed that the expression levels of TNF-α, and IL-6 were significantly upregulated in LPS-stimulated BV2 cells (Figure 3G). The above experimental results suggest that the changes in FcγRIIb expression are positively correlated with microglia activation and that FcγRIIb can promote the inflammatory activation of microglia, thus mediating LPS-induced neuroinflammation.
PI3K-AKT Pathway is Involved in the Development of LPS-Induced Neuroinflammation Mediated by Activated Microglia
Many recent studies have found that the PI3K-AKT signaling pathway is involved in various diseases, including neurodegenerative and neuroinflammatory diseases. In BV2 cells treated with LPS, the level of p-AKT in the cerebral cortex of CNS inflammation model mice gradually increased after LPS intraperitoneal injection, reaching the highest level at 12 h, while PI3K expression reached the highest level at 7d (Figure 1C). The results of the cellular immunofluorescence staining assay showed that the expression of p-AKT was increased in the stimulated group compared to the control group (Figure 4A), which is consistent with the results of the Western blot assay. Meanwhile, in vitro stimulation of BV2 cells with LPS for 0, 3, 6, 9, 12, and 24 h showed that the expression of p-AKT increased in BV2 cells and reached the highest level at 9 h, while the expression of PI3K reached the highest level at 12 h (Figure 2C). The results of cell immunofluorescence staining also verified that the p-AKT expression in the stimulated group was much higher than that in the control group (Figure 4B). The above experimental data can be used to tentatively verify that the PI3K-AKT signaling pathway is involved in the inflammatory activation of microglia. To further validate the role of the PI3K-AKT pathway in microglia inflammatory activation, we blocked the PI3K-AKT pathway using the pathway blocker GDC0941,the experimental results showed that the expression of iNOS and p-AKT was significantly decreased in BV2 cells treated with LPS (Figure 4C). The results of ELISA and q-PCR assays were consistent with these findings, in that the expression of pro-inflammatory factors was relatively reduced after inhibition of PI3K-AKT pathway activity (Figure 4D and E). The above experimental results confirm that PI3K-AKT pathway activation is involved in the development of LPS-induced neuroinflammation mediated by activated microglia.
 |
Figure 4 PI3K/AKT signaling pathway is involved in microglia-mediated LPS-induced neuroinflammation. (A) Cellular immunofluorescence staining of the expression and localization of p-AKT in control and LPS-stimulated groups in a mouse CNS inflammation model. p-AKT (green), Iba-1 (red), and Hoechst (blue); scale bar: 100 μm. (B) Cellular immunofluorescence staining of the expression and localization of p-AKT in control and LPS-stimulated groups in an in vitro model. p-AKT (green), Iba-1 (red), and Hoechst (blue); scale bar: 100 μm. (C) BV2 cells were pretreated with PI3K-AKT inhibitor GDC0941 for 1 h and then stimulated with LPS (100 ng/mL) for 9 h. Western blotting was performed to detect the protein expression levels of p-AKT, AKT, iNOS and FcγRIIb in BV2 cells. (D) Q-PCR detection of the mRNA expression levels of TNF-α, IL-6 and IL-1βin BV2 cells. (E) ELISA was performed to detect changes in expression levels of TNF-α and IL-6 in the supernatants of treated BV2 cells. (mean ± SEM represents the resultant values, n = 3, *Indicates compared to the control, or N+DMSO+LPS group, #Indicates compared to the control, or N+DMSO+LPS group, p < 0.05). |
FcγRIIb Promotes Microglia Activation Through PI3K-AKT Pathway Activation
The combined experimental data show that the change in FcγRIIb expression was closely associated with PI3K-AKT pathway activation during the inflammatory activation of BV2 cells. Previous works in the literature have reported that FcγRIIb can contribute to disease onset and progression through the activation of the PI3K-AKT signaling pathway during immune regulation. To investigate the mechanism by which FcγRIIb promotes inflammatory activation in BV2 cells, we transfected si-FcγRIIb and si-Ctrl into BV2 cells and observed changes in PI3K-AKT signaling pathway activity in BV2 cells in the LPS-stimulated group after interference with FcγRIIb expression. The results showed that the protein-level of p-AKT and PI3K were significantly reduced after knockdown of FcγRIIb in BV2 cells (Figure 3A). Conversely, the protein expression of p-AKT and PI3K was significantly increased by transfection of the overexpression plasmid (myc-FcγRIIb) compared to the control group (Figure 3E). These results suggest that FcγRIIb mediates microglia inflammatory activation through the positive regulation of PI3K-AKT pathway activity.
Meanwhile, we pretreated BV2 cells with a GDC0941 pathway inhibitor, before stimulating with LPS for 9 h to analyze the effect of blocking the PI3K-AKT pathway on FcγRIIb protein expression in activated BV2 cells. The results revealed that FcγRIIb expression was significantly downregulated in BV2 cells of the LPS-stimulated group after inhibition of the PI3K-AKT signaling pathway (Figure 4C). Therefore, we conclude that PI3K-AKT pathway activity has a positive regulatory effect on the expression of FcγRIIb in inflammation-activated BV2 cells, which can form a positive feedback loop in BV2 cells to mediate the development of LPS-induced neuroinflammation.
FcγRIIb and DAP12 Interact to Promote Microglia Activation-Mediated LPS-Induced Neuroinflammation
The above experimental results suggest that FcγRIIb plays a role in LPS-induced neuroinflammation through the PI3K-AKT signaling pathway. We first confirmed the expression of DAP12 in the cerebral cortex of CNS inflammation model mice and in LPS-activated BV2 cells by Western blot. The results showed that DAP12 was significantly increased in the cerebral cortex of LPS intraperitoneally injected mice. Moreover, the expression of DAP12 was significantly increased in the cerebral cortex of LPS-injected mice (Figure 1C), while the expression in LPS-stimulated BV2 cells also showed an increasing trend (Figure 2C). The above results suggest that DAP12 plays an important role in the inflammatory activation process of BV2 cells. Indeed, numerous studies have reported an irreplaceable role of DAP12 in the development of neuroinflammatory diseases.27 Therefore, we speculated that FcγRIIb has a specific connection with DAP12. To verify this idea, we performed immunoprecipitation experiments on proteins extracted from BV2 cells treated with LPS for 9 h. The experimental results showed that FcγRIIb in LPS-treated BV2 cells interacted with DAP12 (Figure 5A). Taken together, these results suggest that FcγRIIb and DAP12 interact in inflammation-activated microglia.
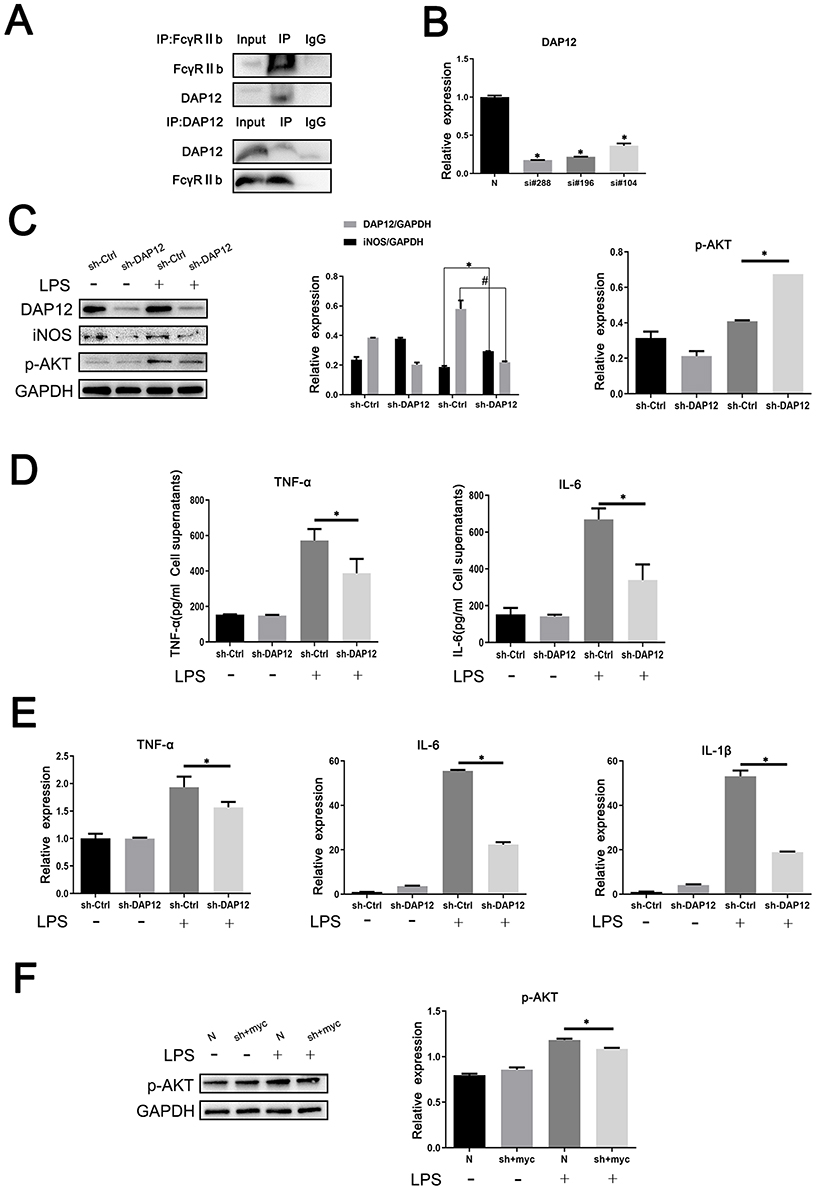 |
Figure 5 FcγRIIb and DAP12 interact to promote microglia activation-mediated LPS-induced neuroinflammation. (A) Analysis of the interaction between FcγRIIb and DAP12 by immunoprecipitation assay. (B) Si-RNA at different loci of DAP12 (si-DAP12#288, si-DAP12#196, and si-DAP12#104) were transfected into BV2 cells, before treating with LPS for 9 h. Q-PCR was performed to detect the transfection efficiency of knocking down si-RNA at different loci of DAP12. (C) Western blot detection of the changes in p-AKT, DAP12 and iNOS protein expression levels in BV2 cells after knockdown of DAP12. (D) ELISA assay of the expression levels of TNF-α and IL-6 in the supernatants of cells collected by centrifugation. (E) Q-PCR assay of the changes in NF-α, IL-6, and IL-1β expression in BV2 cells. (F) BV2 cells were transfected and then stimulated with LPS. Western blot detected the change in p-AKT protein expression in BV2 cells after overexpressed FcγRIIb while interfering with DAP12 (mean ± SEM represents the result value, n = 3, *Compared to the control, or si-Ctrl+LPS group, or N+LPS group, or si-DAP12+myc-FcγRIIb+LPS group, #Indicates compared to the sh-Ctrl+LPS group, p < 0.05). |
Although we know that DAP12 interacts with FcγRIIb, we speculate that it is the bridging protein DAP12 that binds to FcγRIIb before inflammation activation of BV2 cells. To test our conjecture, we investigated the association between DAP12 and microglial inflammatory activation by interfering with DAP12 expression in BV2 cells. To this end, we used the cell transfection technique to transfect BV2 cells with siRNA targeting different DAP12 loci (si-DAP12#288, si-DAP12#196, and si-DAP12#104) before treating them with LPS for 9 h. The q-PCR results showed that transfection with si-DAP12#288 had the strongest disrupting effect on DAP12 (Figure 5B). After knockdown of DAP12, the Western blot results showed that the expression levels of DAP12 and iNOS proteins were reduced in the LPS-stimulated group (Figure 5C). The ELISA results were consistent with those of Western blot, and showed that interference with DAP12 expression in BV2 cells inhibited the expression of TNF-α and IL-6 (Figure 5D). The q-PCR assay results also clearly showed that knockdown of DAP12 decreased the expression levels of TNF-α, IL-6, and IL-1β in the stimulated group compared to the control group (Figure 5E). These results indicate that the expression of DAP12 was positively correlated with the degree of inflammatory activation in BV2 cells. Additionally, we found that the phosphorylation level of AKT in the PI3K-AKT signaling pathway was altered after knocking down DAP12; as shown by the Western blot assay, the phosphorylation level of AKT was reduced in the stimulated group compared to the null group (Figure 5C).
To further illustrate the interaction between FcγRIIb and DAP12, we overexpressed FcγRIIb while interfering with DAP12 to observe the effect on inflammatory activation in BV2 cells. The results of Western blot showed that p-AKT was significantly reduced in the stimulated group compared to the no-load group in BV2 cells after mixing (Figure 5F). Taken together, the results suggest that FcγRIIb contributes to microglial inflammatory activation by interacting with the bridging protein DAP12 and relying on DAP12 to deliver activation signals, which in turn activates the PI3K-AKT signaling pathway.
“FcγRIIb-PI3K-AKT” Loop Regulates Neuronal Damage Mediated by Microglia Activation-Induced Neuroinflammation
Numerous studies have shown that there are two types of cells in the CNS: neurons and glial cells. Neuroglia are the supporting cells of neurons. When the human brain is damaged, glial cells in the brain are activated to produce large amounts of inflammatory factors that cause neuronal apoptosis; thus, neuroinflammation is closely related to neuronal damage.42 To investigate the relationship between the “FcγRIIb-PI3K-AKT” and neuronal apoptosis, we constructed a neuronal injury model by first treating BV2 cells according to experimental requirements, collecting cell culture supernatants, and then mixing the collected supernatants with DMEM medium at a ratio of 1:1. Finally, we mixed the collected supernatant with DMEM medium as conditioned medium to culture HT22 neurons for 24 h. The results of Western blot assay showed that the expression of apoptosis-related protein Bax was significantly lower in the si-FcγRIIb+LPS group compared to the control group (si-Ctrl+LPS), and when interfering with FcγRIIb and blocking the PI3K-AKT pathway (si-FcγRIIb+GDC+LPS group), the decrease in Bax expression in neurons was more obvious (Figure 6A). Moreover, the cell viability of the si-FcγRIIb+LPS, GDC+LPS, and si-FcγRIIb+GDC+LPS groups increased, with that of the si-FcγRIIb+GDC+LPS group recovering most significantly (Figure 6B). The above results showed that the positive feedback loop of “FcγRIIb-PI3K-AKT” promoted the inflammatory activation of microglia, and that interfering with FcγRIIb and blocking the PI3K-AKT pathway could reduce the neuronal damage mediated by LPS-induced neuroinflammation.
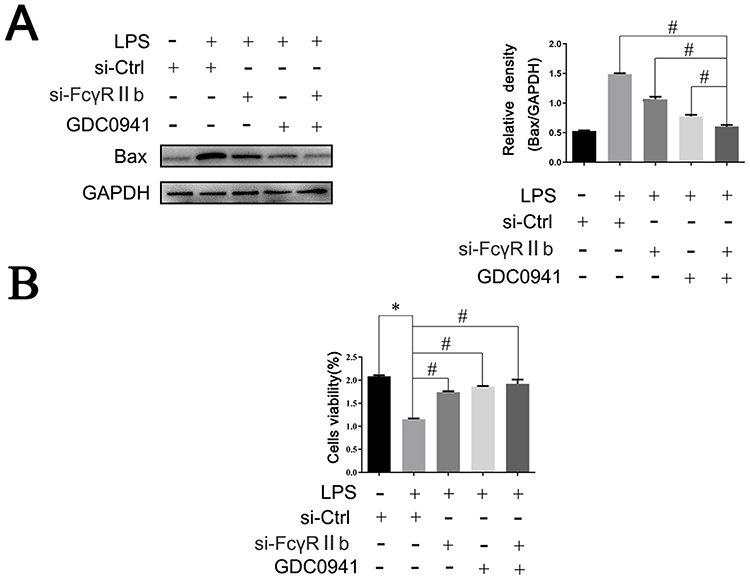 |
Figure 6 The “FcγRIIb-PI3K-AKT” positive feedback loop exacerbates neuroinflammation-mediated neuronal injury. BV2 cells were transfected with si-Ctrl or si-FcγRIIb, pretreated with GDC0941 inhibitor, transfected with si-FcγRIIb, treated with pathway inhibitor GDC0941, and then stimulated with LPS. Subsequently, the conditioned medium was prepared with DMEM complete medium and used to culture HT22 neurons for 24 h. (A) Western blot to detect the expression level of apoptosis-related protein Bax. (n = 3, # represents p < 0.05 compared to the si-Ctrl+LPS group, si-FcγRIIb+LPS group, and GDC+LPS group). (B) CCK-8 assay of HT22 cell viability (n = 3, *Compared to the si-Ctrl group, #Compared to the si-Ctrl+LPS group, p < 0.05). |
Discussion
As human life expectancy increases, neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and multiple sclerosis (MS), pose an increasing threat to human health.42–45 With the increasing mortality rate, the concern for neurodegenerative diseases is growing annually, which has resulted in in-depth exploration. Numerous studies have shown an inextricable relationship between neuroinflammation and neurodegenerative diseases. Innate immune cells, microglia, and astrocytes are important players in neuroinflammation and neurodegenerative diseases. Neuroinflammation is generally an inflammatory response in the CNS caused by various pathological injuries. The onset and progression of neuroinflammation is characterized by a significant increase in the release of pro-inflammatory factors, the release of which leads to loss of synaptic function, neuronal apoptosis, and inhibition of nerve growth.
The CNS contains neurons and glial cells. In contrast to neurons, glial cells are functionally diverse and diverse in type, including astrocytes, oligodendrocytes, and microglia, all of which perform different functions and regulate different mechanisms in the brain.46,47 Studies have shown that microglia have two distinct phenotypes (neurotoxic and neuroprotective) that keep the brain immune system in dynamic balance. Moreover, microglial phenotypic changes and neuroprotective and neurotoxic functions play different roles depending on the stage of development of neurodegenerative diseases and their severity. Thus, a simple phenotypic classification does not completely separate the various functional areas of microglia. Indeed, the complexity of this process may be the main reason why anti-inflammatory drug trials have not shown significant therapeutic effects.48,49
FcγRIIb is available for experimental exploration as an inflammatory factor. All indications suggest that FcγRIIb plays different roles and functions in neuroinflammation. In the present study, after exploring the role of FcγRIIb in LPS-induced neuroinflammation, we found that in CNS inflammation, FcγRIIb binds to the bridging protein DAP12 after LPS stimulation and forms a complex in microglia, thereby activating the PI3K-AKT signaling pathway, which ultimately leads to neuronal apoptosis and brain injury.
We constructed in vivo and in vitro models of LPS-induced neuroinflammation, and we demonstrated that FcγRIIb expression was significantly upregulated in microglia activated by inflammation both in vivo and in vitro through several experiments. Additionally, as the PI3K-AKT signaling pathway plays a non-replaceable role in microglia, we investigated and explored this pathway, with the results showing that FcγRIIb promotes microglia activation by activating the PI3K-AKT signaling pathway, while PI3K-AKT pathway activity can also inversely affect the expression of FcγRIIb. FcγRIIb forms a positive feedback loop, which promotes the development of LPS-induced neuroinflammation, and the “FcγRIIb-PI3K-AKT” loop exacerbates neuronal damage mediated by LPS-induced neuroinflammation. We now know from the available experimental data that FcγRIIb does not play an inhibitory role in LPS-induced neuroinflammation, but rather promotes the development of inflammatory activation. Therefore, we hypothesize that FcγRIIb does not play an inhibitory role because it does not stimulate the unique ITIM structure but rather binds to the bridging protein DAP12 to promote the activation of downstream pathways. DAP12 has been reported to act as a co-stimulatory membrane protein upon LPS stimulation, thereby promoting PI3K-AKT signaling pathway activation, which may be important as a mechanism for the pathogenesis of LPS-induced neuroinflammation. Due to the above speculation, we first confirmed that FcγRIIb interacts with DAP12 by immunoprecipitation assay. Therefore, we next knocked down the bridging protein DAP12, and indirectly verified that the combination of FcγRIIb and DAP12 could promote the activation of microglia. The experimental data showed that AKT phosphorylation was substantially reduced or disappeared after interfering with DAP12. Moreover, by interfering with DAP12 and overexpressing FcγRIIb, we demonstrated that the reduction in AKT phosphorylation was greatly reduced or disappeared. Although the above results widely support our conjecture, given the complexity of the molecular structure, the mechanism of action is unclear and more in-depth exploration is needed to verify our hypothesis.
To investigate the regulatory mechanism of FcγRIIb in LPS-induced neuroinflammation in an in vitro model of LPS-induced neuroinflammation, the validation results showed that LPS stimulation activated microglia, increased FcγRIIb expression, increased release of related inflammatory factors, and resulted in neuronal damage. In an in vivo model of LPS-induced neuroinflammation, FcγRIIb expression was significantly increased in the brain tissue of mice that had undergone intraperitoneal injection of LPS. The Gene Expression Omnibus (GEO) is an international gene expression repository and a free public database, with datasets relating to the cerebellum, cortex, spinal cord, and hippocampus.50,51 Pathway analysis showed that FcγRIIb is enriched in the PI3K-AKT signaling pathway, which is essential for neuronal development and glial cell activity in the brain.52,53 Aberrant transduction of the PI3K-AKT signaling pathway can be involved in various diseases, including cancer, neurodegeneration, and neuroinflammatory diseases. Therefore, we speculate that FcγRIIb plays a unique role in CNS inflammation and that FcγRIIb can trigger LPS-induced neuroinflammation by regulating the PI3K-AKT pathway and activating microglia.
To investigate how FcγRIIb regulates the PI3K-AKT pathway and activates microglia to trigger LPS-induced neuroinflammation, we inhibited AKT phosphorylation in microglia using a PI3K inhibitor and showed that the release of inflammatory factors was reduced. Simultaneously, we found that microglia treated with LPS activated AKT phosphorylation, which confirmed the involvement of the PI3K-AKT pathway in microglia activation.54–56
As FcγRIIb is a transmembrane protein, we speculate that it triggers the lower-level signaling pathway by interacting with other receptors or proteins. Therefore, the analysis of the protein network interactions results suggested that FcγRIIb binds to another transmembrane protein DAP12 to form a complex and thus activate intracellular signaling, causing neural damage. To verify this speculation, DAP12 was confirmed to interact with FcγRIIb by immunoprecipitation assay. To further test our hypothesis, we found that AKT phosphorylation was significantly inhibited after disturbing DAP12 in activated microglia, while overexpression of FcγRIIb and DAP12 disruption in activated microglia showed a similar downregulation of AKT phosphorylation. Based on our results, we suggest that FcγRIIb binds to the transmembrane protein DAP12 to form a complex that activates intracellular signaling and causes neural damage. Many typical functions of FcγRIIb have been achieved by signaling through cytoplasmic ITIM, thus playing an inhibitory role in immunomodulatory responses. Our experimental data suggest that the ITIM structural domain is not activated in LPS-stimulated microglia and that FcγRIIb binds to the receptor adapter DAP12 in the cytoplasmic membrane to form a complex, thereby activating the PI3K-AKT pathway and causing brain injury.
It is worth noting that our experimental results only demonstrate that FcγRIIb binds to the bridging protein DAP12 to form a complex in LPS-treated microglia, which activates the PI3K-AKT pathway and causes neuronal damage, but many of the specific mechanisms involved are still unclear and need to be further explored.
In conclusion, We found that FcγRIIb expression was upregulated in microglia activated by inflammation in vitro and in vivo. FcγRIIb activates the PI3K-AKT signaling pathway by interacting with DAP12 and is involved in microglia inflammatory activation, and the PI3K-AKT pathway activity also affects FcγRIIb expression. The “FcγRIIb-PI3K-AKT” loop exacerbates neuronal damage mediated by neuroinflammation (Figure 7).
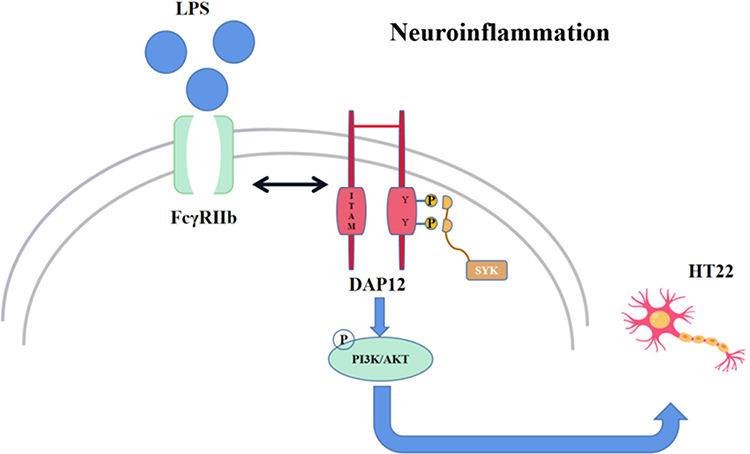 |
Figure 7 After microglia activation by LPS, FcγRIIb forms a dimer with the bridging protein DAP12, which activates the downstream PI3K/AKT signaling pathway, thereby exacerbating inflammation and causing neuronal damage. |
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
All animal experiments were approved by the Animal Care and Use Committee of Nantong University and the Jiangsu Province Animal Care Ethics Committee.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by Postgraduate Research and Practice Innovation Program in Jiangsu Province (grant No.KYCX21 3099).
Disclosure
Prof. Dr. Xiaoyi Shao reports grants from Jiangsu Education Department, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Borst K, Dumas AA, Prinz M. Microglia: immune and non-immune functions. Immunity. 2021;54(10):2194–2208. doi:10.1016/j.immuni.2021.09.014
2. Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011;22(4):189–195. doi:10.1016/j.cytogfr.2011.10.001
3. Cruceriu D, Baldasici O, Balacescu O, Berindan-Neagoe I. The dual role of tumor necrosis factor-alpha (TNF-α) in breast cancer: molecular insights and therapeutic approaches. Cell Oncol. 2020;43(1):1–18. doi:10.1007/s13402-019-00489-1
4. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Ann Rev Immunol. 2017;35(1):441. doi:10.1146/annurev-immunol-051116-052358
5. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21(10):1359–1369. doi:10.1038/s41593-018-0242-x
6. Masuda T, Sankowski R, Staszewski O, Prinz M. Microglia heterogeneity in the single-cell era. Cell Rep. 2020;30(5):1271–1281. doi:10.1016/j.celrep.2020.01.010
7. Toscano R, Millan-Linares MC, Lemus-Conejo A, Claro C, Sanchez-Margalet V, Montserrat-de la paz S. Postprandial triglyceride-rich lipoproteins promote M1/M2 microglia polarization in a fatty-acid-dependent manner. J Nutr Biochem. 2020;75:108248. doi:10.1016/j.jnutbio.2019.108248
8. Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol. 2012;32(6):463–488. doi:10.1615/CritRevImmunol.v32.i6.10
9. Xuan W, Qu Q, Zheng B, Xiong S, Fan GH. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J Leukoc Biol. 2015;97(1):61–69. doi:10.1189/jlb.1A0314-170R
10. Candelario-Jalil E, Dijkhuizen RM, Magnus TJS. Neuroinflammation, stroke, blood-brain barrier dysfunction, and imaging modalities. Stroke. 2022;53(5):1473–1486. doi:10.1161/STROKEAHA.122.036946
11. Jurcau A, Simion A. Neuroinflammation in cerebral ischemia and ischemia/reperfusion injuries: from pathophysiology to therapeutic strategies. Int J Mol Sci. 2021;23(1):14. doi:10.3390/ijms23010014
12. Varin A, Gordon S. Alternative activation of macrophages: immune function and cellular biology. Immunobiology. 2009;214(7):630. doi:10.1016/j.imbio.2008.11.009
13. Park HJ, Kim HJ, Ra J, et al. Melatonin inhibits lipopolysaccharide-induced CC chemokine subfamily gene expression in human peripheral blood mononuclear cells in a microarray analysis. J Pineal Res. 2007;43(2):121–129. doi:10.1111/j.1600-079X.2007.00452.x
14. Takai T. Fc receptors and their role in immune regulation and autoimmunity. J Clin Immunol. 2005;25(1):1–18. doi:10.1007/s10875-005-0353-8
15. Nathan C. Points of control in inflammation. Nature. 2002;420(6917):846–852. doi:10.1038/nature01320
16. Takai T. Role of Fcγ receptors in immune regulation and diseases. Jap J Clin Immunol. 2005;28(5):318–326. doi:10.2177/jsci.28.318
17. Tarasenko T, Dean JA, Bolland S. FcgammaRIIB as a modulator of autoimmune disease susceptibility. Autoimmunity. 2007;6:40.
18. Wang F, Li C, Shao J, Ma J. Sevoflurane induces inflammation of microglia in hippocampus of neonatal rats by inhibiting Wnt/β-Catenin/CaMKIV pathway. J Pharmacol Sci. 2021;146(2):105–115. doi:10.1016/j.jphs.2021.02.004
19. Marshall JG, Booth JW, Stambolic V, et al. Restricted accumulation of phosphatidylinositol 3-kinase products in a plasmalemmal subdomain during Fc gamma receptor-mediated phagocytosis. J Cell Biol. 2001;153(7):1369–1380. doi:10.1083/jcb.153.7.1369
20. Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal. 2010;3(122):ra38. doi:10.1126/scisignal.2000500
21. Turnbull IR, Gilfillan S, Cella M, et al. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol. 2006;177(6):3520–3524. doi:10.4049/jimmunol.177.6.3520
22. Korekane H, Matsumoto A, Ota F, et al. Involvement of ST6Gal I in the biosynthesis of a unique human colon cancer biomarker candidate, alpha2,6-sialylated blood group type 2H (ST2H) antigen. J Biochem. 2010;148(3):359–370. doi:10.1093/jb/mvq077
23. Zou W, Reeve JL, Liu Y, Teitelbaum SL, Ross FP. DAP12 couples c-Fms activation to the osteoclast cytoskeleton by recruitment of Syk. Mol Cell. 2008;31(3):422–431. doi:10.1016/j.molcel.2008.06.023
24. Nakano-Yokomizo T, Tahara-Hanaoka S, Nakahashi-Oda C, et al. The immunoreceptor adapter protein DAP12 suppresses B lymphocyte-driven adaptive immune responses. J Exp Med. 2011;208(8):1661–1671. doi:10.1084/jem.20101623
25. Humphrey MB, Daws MR, Spusta SC, et al. TREM2, a DAP12-associated receptor, regulates osteoclast differentiation and function. J Bone Miner Res. 2006;21(2):237–245. doi:10.1359/JBMR.051016
26. Paloneva J, Mandelin J, Kiialainen A, et al. DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J Exp Med. 2003;198(4):669–675. doi:10.1084/jem.20030027
27. Konishi H, Kiyama H. Microglial TREM2/DAP12 signaling: a double-edged sword in neural diseases. Front Cell Neurosci. 2018;12:206. doi:10.3389/fncel.2018.00206
28. Bouchon A, Hernández-Munain C, Cella M, Colonna M. A Dap12-mediated pathway regulates expression of Cc chemokine receptor 7 and maturation of human dendritic cells. J Exp Med. 2001;194(8):1111–1122. doi:10.1084/jem.194.8.1111
29. Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol. 2006;177(4):2051–2055. doi:10.4049/jimmunol.177.4.2051
30. Guan Z, Kuhn JA, Wang X, et al. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci. 2016;19(1):94–101. doi:10.1038/nn.4189
31. Ersahin T, Tuncbag N, Cetin-Atalay R. The PI3K/AKT/mTOR interactive pathway. Mol Biosyst. 2015;11(7):1946–1954. doi:10.1039/C5MB00101C
32. Xu F, Na L, Li Y, Chen L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020;10(1):54. doi:10.1186/s13578-020-00416-0
33. Wang J, Hu K, Cai X, et al. Targeting PI3K/AKT signaling for treatment of idiopathic pulmonary fibrosis. Acta Pharm Sin B. 2022;12(1):18–32. doi:10.1016/j.apsb.2021.07.023
34. Jain S, Dash P, Minz AP, et al. Lipopolysaccharide (LPS) enhances prostate cancer metastasis potentially through NF-κB activation and recurrent dexamethasone administration fails to suppress it in vivo. Prostate. 2019;79(2):168–182. doi:10.1002/pros.23722
35. Caja L, Dituri F, Mancarella S, et al. TGF-β and the tissue microenvironment: relevance in fibrosis and cancer. Int J Mol Sci. 2018;19(5):1294. doi:10.3390/ijms19051294
36. Chiang KW. Performance enhancement of INS/GNSS/Refreshed-SLAM Integration for acceptable lane-level navigation accuracy. IEEE Trans Veh Technol. 2020;99:1.
37. Ediriweera MK, Tennekoon KH, Samarakoon SR. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: biological and therapeutic significance. Semi Cancer Biol. 2019;59:147–160. doi:10.1016/j.semcancer.2019.05.012
38. Wolin EM. PI3K/Akt/mTOR pathway inhibitors in the therapy of pancreatic neuroendocrine tumors. Cancer Lett. 2013;335(1):1–8. doi:10.1016/j.canlet.2013.02.016
39. Xie Y, Shi X, Sheng K, et al. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol Med Rep. 2019;19(2):783–791. doi:10.3892/mmr.2018.9713
40. Fattahi S, Amjadi-Moheb F, Tabaripour R, Ashrafi GH, Akhavan-Niaki H. PI3K/AKT/mTOR signaling in gastric cancer: epigenetics and beyond. Life Sci. 2020;262:118513. doi:10.1016/j.lfs.2020.118513
41. Lyketsos CG, Carrillo MC, Ryan JM, et al. Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimers Dement. 2011;7(5):532–539. doi:10.1016/j.jalz.2011.05.2410
42. Bruce-Keller AJ. Microglial-neuronal interactions in synaptic damage and recovery. J Neurosci Res. 1999;58(1):191–201. doi:10.1002/(SICI)1097-4547(19991001)58:1<191::AID-JNR17>3.0.CO;2-E
43. Doshi A, Chataway J. Multiple sclerosis, a treatable disease. Clin Med. 2017;17(6):530–536. doi:10.7861/clinmedicine.17-6-530
44. Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368–376. doi:10.1136/jnnp.2007.131045
45. Guidolin D, Tortorella C, Marcoli M, Maura G, Agnati LF. Neuroglobin, a factor playing for nerve cell survival. Int J Mol Sci. 2016;17(11):1817. doi:10.3390/ijms17111817
46. Goedert M, Spillantini MG, Davies SW. Filamentous nerve cell inclusions in neurodegenerative diseases. Curr Opin Neurobiol. 1998;8(5):619–632. doi:10.1016/S0959-4388(98)80090-1
47. Chen Z, Zhou J, He Q, Feng S, Wa Q, Chen X. Can anti-inflammatory drugs used in plastic surgery procedures increase the risk of hematoma? Aesthetic Plast Surg. 2023;47(2):862–871. doi:10.1007/s00266-022-03194-x
48. Cheng B, Chen HC, Chou IW, Tang TW, Hsieh PC. Harnessing the early post-injury inflammatory responses for cardiac regeneration. J Biomed Sci. 2017;24(1):7. doi:10.1186/s12929-017-0315-2
49. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41(Database issue):D991–D995. doi:10.1093/nar/gks1193
50. Li Y, Gu J, Xu F, Zhu Q, Ge D, Lu C. Transcriptomic and functional network features of lung squamous cell carcinoma through integrative analysis of GEO and TCGA data. Sci Rep. 2018;8(1):15834. doi:10.1038/s41598-018-34160-w
51. Guo A, Wang W, Shi H, Wang J, Liu T. Identification of hub genes and pathways in a rat model of renal ischemia-reperfusion injury using bioinformatics analysis of the Gene Expression Omnibus (GEO) dataset and integration of gene expression profiles. Med Sci Monit. 2019;25:8403–8411. doi:10.12659/MSM.920364
52. Norden DM, Trojanowski PJ, Villanueva E, Navarro E, Godbout JP. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia. 2016;64(2):300–316. doi:10.1002/glia.22930
53. Butturini E, Boriero D, Carcereri de Prati A, Mariotto S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Arch Biochem Biophys. 2019;669:22–30. doi:10.1016/j.abb.2019.05.011
54. Mishra MK, Basu A. Minocycline neuroprotects, reduces microglial activation, inhibits caspase 3 induction, and viral replication following Japanese encephalitis. J Neurochem. 2008;105(5):1582–1595. doi:10.1111/j.1471-4159.2008.05238.x
55. Hiramatsu G, Matsuda K, Uta D, Mihara K, Kume T. Panaxytriol inhibits lipopolysaccharide-induced microglia activation in brain inflammation in vivo. Biol Pharm Bull. 2021;44(7):1024–1028. doi:10.1248/bpb.b21-00288
56. Niemi K, Teirilä L, Lappalainen J, et al. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol. 2011;186(11):6119–6128. doi:10.4049/jimmunol.1002843
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.