Back to Archived Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 16
Facing Antitubercular Resistance: Identification of Potential Direct Inhibitors Targeting InhA Enzyme and Generation of 3D-pharmacophore Model by in silico Approach
Authors EL Haddoumi G ![]() , Mansouri M
, Mansouri M ![]() , Bendani H
, Bendani H ![]() , Bouricha EM
, Bouricha EM ![]() , Kandoussi I, Belyamani L, Ibrahimi A
, Kandoussi I, Belyamani L, Ibrahimi A
Received 23 October 2022
Accepted for publication 21 January 2023
Published 28 April 2023 Volume 2023:16 Pages 49—59
DOI https://doi.org/10.2147/AABC.S394535
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Alex MacKerell
Ghyzlane EL Haddoumi,1,2 Mariam Mansouri,1,2 Houda Bendani,1,2 El Mehdi Bouricha,1,2 Ilham Kandoussi,1,2 Lahcen Belyamani,2– 4 Azeddine Ibrahimi1– 3
1Biotechnology Lab (MedBiotech), Rabat Medical and Pharmacy School, University Mohammed V, Rabat, Morocco; 2Centre Mohammed VI for Research & Innovation (CM6), Rabat, Morocco; 3Mohammed VI University of Health Sciences (UM6SS), Casablanca, Morocco; 4Emergency Department, Military Hospital Mohammed V, Rabat, Morocco
Correspondence: Azeddine Ibrahimi, Mohammed VI University of Health Sciences (UM6SS), Casablanca, Morocco, Tel +212660240131, Email [email protected]
Purpose: The enoyl-acyl carrier protein reductase (InhA) is one of the important key enzymes employed in mycolic acids biosynthesis pathway and an important component of mycobacterial cell walls. This enzyme has also been identified as major target of isoniazid drug, except that isoniazid needs to be activated first by the catalase peroxidase (KatG) protein to form the isonicotinoyl-NAD (INH-NAD) adduct that inhibits the action of InhA enzyme. However, this activation becomes more difficult and unreachable with the problem of mutation-related resistance caused mainly by acquired mutations in KatG and InhA protein. Our main interest in this study is to identify direct InhA inhibitors using computer-aided drug design.
Methods: Computer-aided drug design was used to solve this problem by applying three different approaches including mutation impact modelling, virtual screening and 3D-pharmacophore search.
Results: A total of 15 mutations were collected from the literature, then a 3D model was generated for each of them and their impact was predicted. Of the 15 mutations, 10 were found to be deleterious and have a direct effect on flexibility, stability and SASA of the protein. In virtual screening, from 1,000 similar INH-NAD analogues obtained by the similarity search method, 823 compounds passed toxicity filter and drug likeness rules, which were then docked to the wild-type of InhA protein. Subsequently, 34 compounds with binding energy score better than that of INH-NAD were selected and docked against the 10 generated mutated models of InhA. Only three leads showed a lower binding affinity better than the reference. The 3D-pharmacophore model approach was used to identify the common features between those three compounds by generating a pharmacophoric map.
Conclusion: The result of this study may pave the way to develop more potent mutant-specific inhibitors to overcome this resistance.
Keywords: molecular docking, CADD, Mycobacterium tuberculosis, InhA, mutational profile
Introduction
Tuberculosis (TB) is still sounding the alarm of emergency worldwide, according to the World Health Organization’s 2022 global tuberculosis report, a total of 10.6 million people fell ill with tuberculosis in 2021 and 1.6 million people died from it (including 187,000 among HIV positive).1 TB is the 13th leading cause of death and the second leading infectious killer after COVID-19 (above HIV/AIDS) (World Health Organization, Global tuberculosis report 2021), The WHO recommended treatment for pulmonary TB (HIV-negative patients) is six months of quadruple therapy: two months of four-drug regimen (isoniazid, rifampicin, pyrazinamide, and ethambutol), followed by four months of isoniazid and rifampicin with once-daily or three times weekly dosing.2
Tuberculosis (TB) aggravates the situation with the problem of drug-resistance, multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB) and new pharmaceutical agents are urgently required to control this situation.
Using genetic methods, Jacobs and his team identified the protein product of the mycobacterial INHA gene as the putative target for isoniazid.3 InhA (Enoyl-[acyl-carrier-protein] reductase, NADH), catalyses the NADH-dependent reduction of the double bond of 2-trans-enoyl-[acyl-carrier protein], an essential step in the fatty acid elongation cycle of the FAS-II pathway.4
Since its first discovery in 1952, isoniazid (INH) has been a cornerstone of tuberculosis chemotherapy, showing a potent bactericidal activity against MTB.5,6 This drug causes acid-fastness loss by targeting InhA protein after its activation by KatG protein due to the inhibition of mycolic acid synthesis, a-alkyl-b-hydroxyl long-chain fatty acids (60–90 carbons long), a major component of the mycobacterial cell wall.7,8
Knowing that isoniazid itself does not directly inhibit the enzyme, but it must be converted first by catalase peroxidase (KatG) into the active form of the drug9,10 and then becomes covalently linked to the nicotinamide ring of the NADH and form in return INH-NAD complex, this complex inhibits the action of the enzyme and prevents the formation of the protein responsible for the formation of mycolic acids, with the problem of resistance, mutations detected in the active site of the target protein, make the activation of isoniazid more difficult and unreachable,11 the need to explore INH-NAD analogues is strongly required, which can target directly the InhA enzyme without needing to be activated first.
Figure S1 shows the explanatory pathway of isoniazid activation as well as the role of the InhA protein in mycolic acid synthesis, it also shows the scope of our problem, and the need for direct inhibition of this protein.
Drug discovery is a veritably expensive and time-consuming process, and to discover a new medicine it may take several years, this task has become more accessible using computer-aided drug design (CADD) approaches like a very powerful tool to simulate molecular interactions of large systems. It provides valuable insights on experimental results and the mechanism of action, predict new molecules to synthetize, and most important it can help to make cost effective decisions before starting expensive molecule synthesis. Many compounds were discovered and optimized using computer-aided drug design methods or even gained US FDA approval.12 Among the techniques used are virtual screening analysis to discover and optimize novel potent molecules by using molecular docking,13–16 the computational prediction methods in order to predict the impact of mutation on protein properties,17 toxicity prediction has also been done on the basis of the molecule criteria which show the physicochemical properties and biological activity of the selected drugs and pharmacophore modelling for the classification of the key features of the best compounds found.18
The aim of this study is to develop direct InhA protein inhibitors based on the understanding of isoniazid mechanism using CADD like a powerful technology, for this purpose, three different approaches were used: molecular modelling including a virtual screening to determine the best leads with the lowest bindings energy against InhA protein that still have a nice binding affinity score with the mutant models generated; computational prediction method in order to predict the impact of all the mutations described in the literature on protein properties; and pharmacophore mapping to discover common features between the best ligands found.
Materials and Methods
Workflow
Figure 1 shows the workflow of the present study.
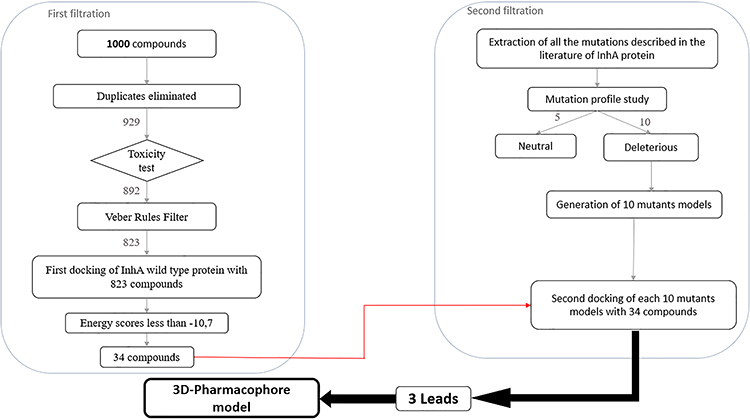 |
Figure 1 In silico study workflow. |
Dataset Generation and Preparation of Wild-type and Mutated Models of InhA Protein
The wild-type X-ray structure of InhA in complex with the isonicotinic inhibitor-ACYL-NADH (PDB ID: 1ZID) with a resolution of 2.70 Å was extracted from the Protein Data Bank,19 and the attached ligand was removed. Fifteen known InhA mutations were collected from literature. These mutations are: D148G, I16T, I21V, I47T, I95P, I194T, K8N, K165A/Q, S94A, T266A/D, V78A, and Y158A/S.4,20–25 Then, a mutant model was generated for each of them using “Rotamer” function of UCSF Chimera tool V1.16.26 Finally, the protein structures (including mutated models) were prepared using AutoDock Tools – V.1.5.7,27 by removing water molecules and adding polar hydrogens and Kollman charges then saved under pdbqt format.
Ligands Preparation and Filtration
To search for similar INH-NAD molecules, a search by similarity was carried out using PubChem, 1,000 molecules were selected with a Tanimoto threshold 90%, and then downloaded under sdf format with 2D conformation, the conversion to 3D, the elimination of duplicate and then the conversion into a pdb file format was carried out by graphical interface of OpenBabel.28
The toxicity properties of compounds were evaluated using two software products, STopTox29,30 and ProTox-II.31 In this step, 892 molecules with a toxicity class-4/5 with a predicted LD50 (mg/kg) ranged from 2,000 to 5,000 for Class V and more than 5,000 for class VI by ProTox-II and noncardiotox by STopTox were kept.
Drug likeness score predictions of the best compounds were evaluated using DruLiTo.32 The 823 compounds were selected for molecular docking preparation, all the ligands were prepared and contain atom types supported by AutoDock tools plus extra records that specify rotatable bonds and then saved in pdbqt format.
Mutation Effect Prediction
To study the impact of mutation on protein function, the site directed mutator (SDM)33 and DynaMut34 online were used to predict the mutations impact on protein stability and flexibility respectively.
Calculation of RMSD of Re-docking Test
As a positive control, and to perform re-docking calculation from Protein Data Bank, the X-ray crystallographic complex protein-ligand (PDB ID: 1ZID). The re-docking test was evaluated using BALLView software.35
Docking and Scoring
After selecting the active site residues of the target protein using AutoDock Tools V.1.5.7,36 allowed the preparation of the grid box, with a spacing of 1 Å for each macromolecular protein and center coordinates fixed at X= −8.411 Å, Y=38.576 Å, Z=11.141 Å and a size of X=20 Å, Y= 20 Å, Z=20 Å. The grid settings file was saved from the grid menu output option.
In order to choose ligands with lower affinity scores, Autodock Vina was used to assess the binding affinity and bound conformation of those selected ligands. This tool is designed for protein-ligand docking, using multiple CPUs at a time, making it faster and more accurate and uses the Lamarckian genetic algorithm and semi-empirical free energy force field that generates free binding energy of 10 ligand poses after docking.37 The 3D protein-ligand interactions were visualized using PyMOL V.2.5.2.38
3D-pharmacophore Model Building
To build a pharmacophore profile of the three lead compounds, pharmacophore query methodology was used, this method is based on searching 3D distances between features like HBD/HBA hydrogen bond donors/acceptors, PI positive ionizable, ARO aromatic ring, Hyd hydrophobic centres.39 For this purpose, the Molecular Operating Environment (MOE) tool was used. An MOE database was created using the builder option, the energy was minimized and then the alignment was launched to generate the best pharmacophoric model with a pharmacophoric map with the first common features.
Results
Mutation Effect Prediction
In order to start the second molecular docking, a detailed study of the mutational profile of InhA protein was carried out.
Fifteen mutations were collected in the literature and the first filtration was carried out by PROVEAN by which it was possible to identify the neutral mutations from those deleterious (Figure 2) and also their effect on the solvent accessibility of the protein (Figure 3A). Subsequently the ten putative deleterious mutations were chosen to study their effect on the flexibility and stability of the protein using SDM and DynaMut (Figure 3B).
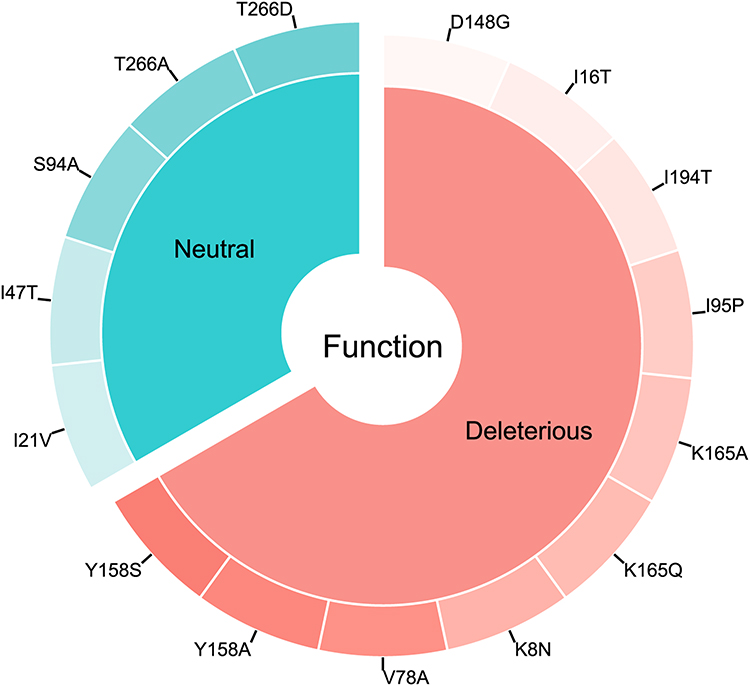 |
Figure 2 The distribution of the 15 mutations according to the function, deleterious or neutral. |
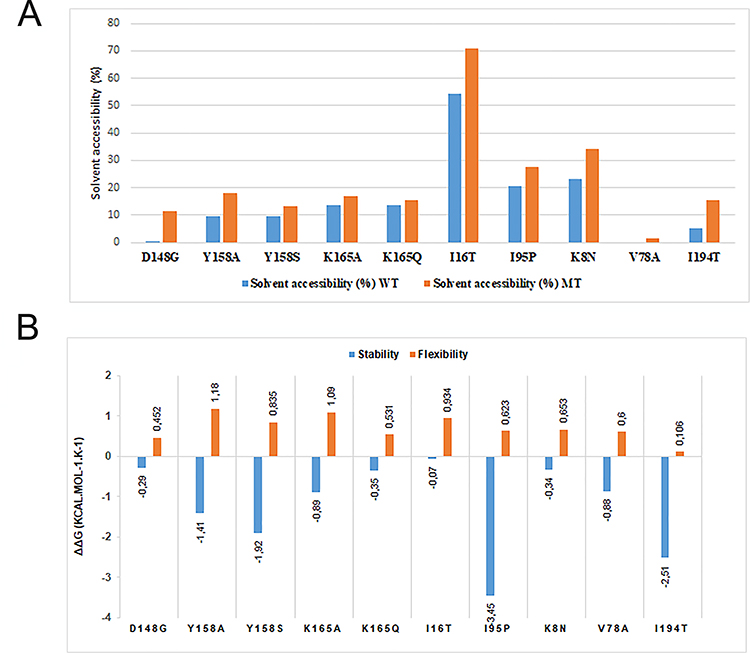 |
Figure 3 (A) The effect of the deleterious mutations on solvent accessibility surface area (SASA) between the wild-type protein (in blue) and the mutanttype protein (in orange). (B) The effect of the deleterious mutations on the flexibility (in orange) and stability (in blue) of the InhA protein. |
Re-docking Score
The re-docking test was evaluated by calculating the RMSD between the predicted pose and the co-crystallographic ligand. The score of RMSD between the two complexes show a value lower than 2 Å with a value of 0.38 Å.
Docking and Scoring
To filter out the best compounds from molecular docking, 823 compounds were docked with the wild-type InhA protein, and only 34 compounds were chosen with a binding energy to InhA protein less than −10.7 kcal/mol, since the reference ligands INH-NADH binds to InhA protein with an affinity of −10.7 kcal/mol, and NADH alone binds with an affinity of −7.5 kcal/mol. The 34 compounds selected from the first docking that showed better affinity than the reference were docked again separately with the 10 mutated model generated.
Three ligands were preferred over all compounds with their lowest binding affinity score toward the wild-type InhA protein and the 10 mutant models. Comparative docking results are depicted in Figure 4.
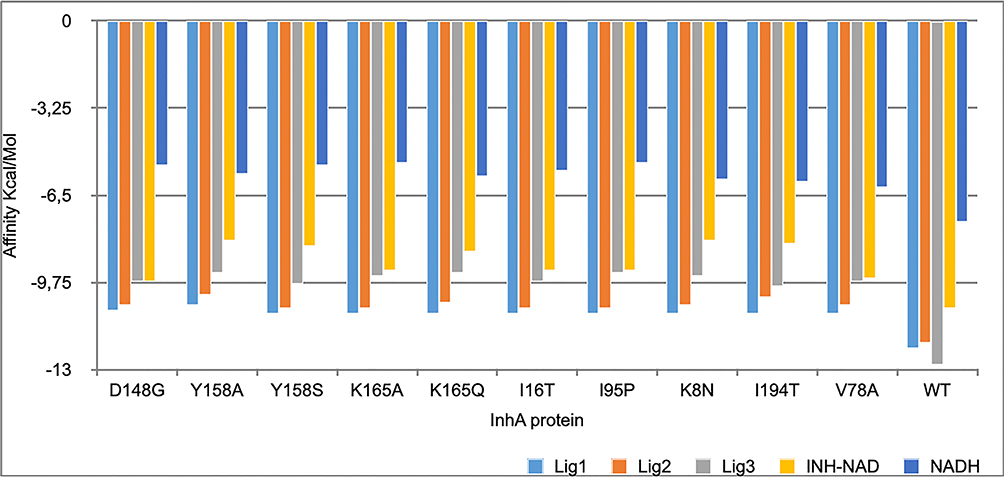 |
Figure 4 The three best leads with lowest affinity binding to the wild-type protein and the the mutant models. |
Table 1 shows the reference ligand INH-NAD, NADH and the three molecule candidates after the second docking against the mutant models with lowest binding affinity than INH-NAD ligand with their essential information: molecular formula, binding affinity energy, chemical structure, PubChem CID, molecular weight, toxicity test, and finally predicted fragment contribution in toxicity test.
 |
Table 1 The essential information about the reference ligand INH-NAD, natural ligand NADH and the Three best ligands including molecular formula, binding affinity score, hydrogen bond CID in PubChem, molecular weight, and toxicity test |
Figure 5 illustrates the 3D protein-ligand interactions visualized using PyMol. Figure 5A and B shows the interaction between the reference ligand INH-NAD and NADH alone against the InhA protein respectively. While Figure 5C–E depicts interactions between ligands (CID: 25176455, 91754233 and 101256471) and InhA protein accordingly.
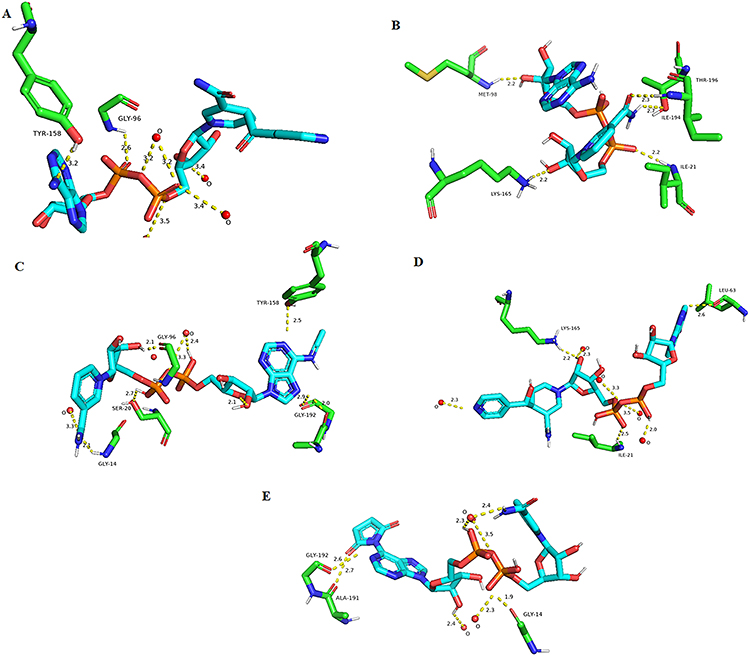 |
Figure 5 Visualization of 3D interactions against InhA protein. (A) Interaction between the reference ligand Inh-NAD and the inhA protein. (B) Interactions between NADH and InhA protein. (C) Interaction between ligand (CID:25176455) and inhA protein. (D) Interaction between ligand (CID:91754233) and InhA protein. (E) Interaction between ligand (CID:101256471) and InhA protein. |
3D-pharmacophore Modelling
Using the Molecular Operating Environment (MOE) tool, a pharmacophoric map of the best three compounds was generated.
Common features with inter-features distances of these compounds are shown in Figure S2.
Figure 6 shows a simplified view of the pharmacophoric map with the aligned ligands (Figure 6A) and without ligands (Figure 6B). The 3D-pharmacophoric model consists of a four hydrophobic (Hyd) center localized in the middle of the structure connected on both side to two Acc and ML features with a distance of 8.71Å between F11 and F1 and 5.35 Å between F2 and F8. The structure is also characterized by an aromatic centre (Aro) F6 leading to another aromatic/acceptor/metal ligator F16 (ML/Aro/Acc). The aromatic regions play an important role in binding to the enzyme and the HBA/HBD group help the ligand to bind tightly to the target.
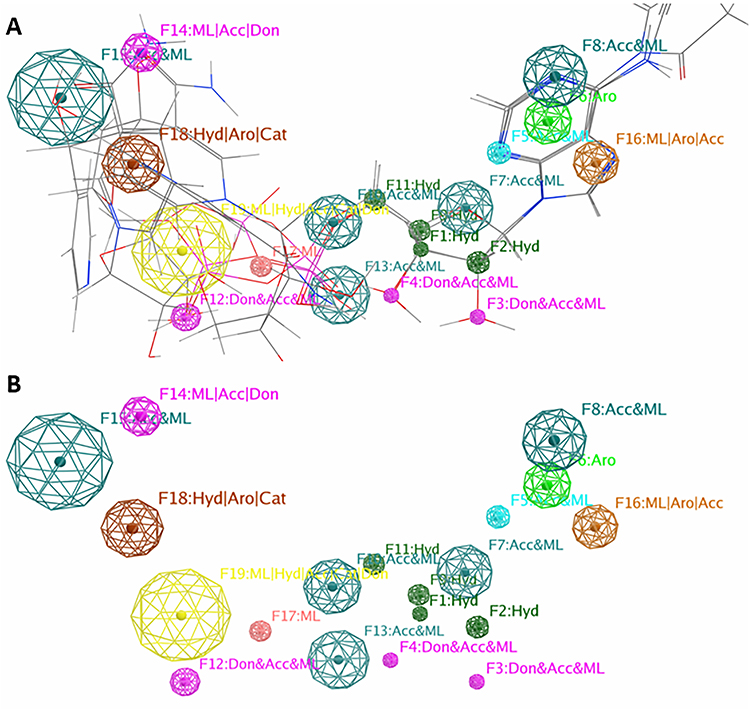 |
Figure 6 The pharmacophore model generated after the flexible alignment of the best three compounds. (A) shows the different features explored in the model structure with the aligned ligands. (B) shows a simplified view of the model without ligands. |
Discussion
In this study, computer prediction methods have been adopted to explore new inhibitors of Mycobacterium tuberculosis directly targeting the InhA protein in order to overcome the problem of resistance to treatments, linked to mutations in this bacterium. First, a prediction of the impact of different single mutations on the properties of the InhA protein was studied in order to reveal the deleterious mutations, then, a collection of 1,000 inhibitors similar to the INH-NAD complex and a study of toxicity were carried out. The interaction of the 823 nontoxic ligands with the wild-type InhA target was analyzed by docking in order to select those which present a better affinity than the INH-NAD complex and which are of the order of 34, including the interaction with the models of the InhA protein mutated by deleterious mutations was studied by a second docking. This second docking highlighted three compounds with a better affinity than that of the INH-NAD complex which were used for the generation of the pharmacophore model.
After having extracted all the mutations of the InhA protein described in the literature, 10 mutations among 15 were identified as deleterious. Analysis of the effect of each deleterious mutation on the protein’s flexibility, stability, and surface area solvent accessibility (SASA) revealed that all the mutations described as deleterious D148G, I16T, I21V, I47T, I95P, I194T, K8N, K165A/Q, S94A, T266A/D, V78A and Y158A/S would have a direct effect on function by altering the various properties studied, thus modifying the folding of the protein, the protein-protein or protein-ligand interaction, confirmed by several studies.17,40,41 This is confirmed by our results, which show a direct relationship between SASA and stability, especially in these mutations I95P, I16T, D148G and I194T, this relationship was also mentioned in a study that stability is the fundamental property improving biomolecular function, and regulation.42
Several studies have indicated that many diseases, including drug resistance-related mutations, are located in solvent-accessible sites, suggesting that analysis of these mutations could also shed light on the mechanisms underlying the diseases.43,44
Apparently, one can clearly see that the change in amino family makes the protein more accessible to solvent, as shown by the I16T and I194T mutations, a switch from isoleucine to tyrosine, a nonpolar aliphatic amino acid to an amino acid polar and uncharged; a clear change of family, polarity and charge loss causes an increase in solvent accessibility with a difference of +16.7% (from 54.3% to 71.0%) and 10.3% more (from 5.3% to 15.6%), respectively. I194T also causes a decrease in stability at −2.51 kcal/mol.
Regarding the I95P mutation, there was no change in the family of amino acids, although our work shows that this mutation would cause a decrease in stability at −3.45 kcal/mol.K and that it would make the protein more accessible to solvent with a difference of 7.2% (from 20.4% to 27.6%) as clearly shown in (Figure 3A). This is confirmed by a study by Kumar and Sobhia as a mutation influencing the binding energy by decreasing it by −2.01 kcal/mol and believed to disrupt the INH-NADH bond due to the change in the nature of the interacting residues, and strongly reducing the direct and water-mediated interactions of the H bonds between NADH and binding site residues allowing conformational flexibility to NADH, particularly at the pyrophosphate region, and causing a weakening of its binding to the dinucleotide binding site.45
For the D148G mutation, a clear difference was noticed in SASA between the wild-type and the mutant-type protein, a change from aspartic acid to glycine amino acid; a polar and negatively charged amino acid to a polar and uncharged amino acid, a change of family and a loss of charge made the protein more accessible to the solvent from 0.1% to 11.5% with a difference of 11.4%. This mutation is described in a study by Hartkoorn et al as indeed responsible for pyridomycin and isoniazid resistance.22
Changes in amino acid size, amino acid charge and hydrogen bond number and changes in SASA have been shown to cause the loss of stability as well as aberrant folding and aggregation of the protein.42
To explore effective inhibitors to each of the deleterious mutations studied that would prove to be involved in resistance to treatment, we analyzed the interaction of the 34 compounds by docking for the models of these mutations, which showed a better affinity than the INH-NAD complex to wild-type InhA.
Three leads showed a lower binding affinity better than the reference ligand INH-NAD using AutoDock Vina with the following CID 101256471, 91754233, and 25176455, the first one (C25H30N7O16P2+) binds with a binding affinity of −12.2 kcal/mol with the wild-type InhA protein with a number of eight hydrogen bonds; one bond of 2.7 Å with the ALA-191, five bonds with three water molecules with a distance ranged from 2.3 Å to 3.5 Å respectively, and two bonds of 1.9 Å and 2.6 Å with GLY (14–192) respectively, the second compound (C27H34N8O15P2) binds with the wild-type protein with an energy of −12.0 kcal/mol, and a number of eight hydrogen bonds; four hydrogen bonds with four water molecules with a distance ranged from 2 Å to 3.5 Å, and three bonds of 2.5 Å, 2.6 Å and 2.2 Å with ILE-21, LEU-63 and LYS-165, respectively. The last compound (C23H28N7O14P2+) binds with the wild-type protein with an energy of and −13 kcal/mol and a number 10 hydrogen bond; with three water with molecules with four hydrogen bonds ranged from 2.1 Å to 3.3 Å, two bonds of 2.0 Å and 2.9 Å with GLY-192 and two bonds of 2.1 Å and 2.1 Å with GLY (14–96), respectively, one bond of 2.5 Å with TYR-158 and one bond of 2.7 Å with SER-20 as demonstrated in Figures 4 and 5.
Based on the structure of these three compounds, the pharmacophoric map showed that the hydrophobic Hyd groups and the aromatic Aro regions play a vital role in their binding to the hA protein. HBA groups enhance the interaction between ligands and the active site of the target46,47 and should be considered as crucial structural features.48 We hope that our contribution can offer a suitable pharmacophore for further studies.
Conclusion
The number of people dying yearly because of tuberculosis is growing rapidly due to the emergence of multidrug-resistant tuberculosis, extensively drug-resistant tuberculosis and totally drug-resistant tuberculosis, which makes the discovery of a new drug an emergency to confront this infectious disease. InhA protein is one of the most effective targets for eradicating M. tuberculosis as long as this protein is a key player in the biosynthesis of mycolic acid cell wall.
In this sense we were able to propose in silico, through this study, three new direct inhibitors of InhA, for both for wild-type and resistance causing mutant-type, nontoxic showing better results than INH-NAD adduct, from which a pharmacophore map was generated to fdetermine common features between the best ligands found. This model can open up a new template for further studies to discover novel powerful anti-tuberculosis drugs.
Ethical Approval
Ethical approval for this study was not obtained as it is a pure computational study and did not involve human data and thus not require institutional review board approval.
Acknowledgments
This work was carried out under national funding from the Moroccan Ministry of Higher Education and Scientific Research (COVID-19 program) to A.I. This work was also supported by a grant from the Moroccan Institute of Cancer Research and the PPR-1 program to A.I. and Biocodex grant to AI.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Global Tuberculosis Report 2022. Available from: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022.
2. Rakotomanana F, Dreyfus A, Raberahona M, et al. 248 - Prévalence de la tuberculose et infection VIH en milieu carcéral, Antananarivo Madagascar. Rev DÉpidémiologie Santé Publique. 2022;70:S194–S195. doi:10.1016/j.respe.2022.06.180
3. Banerjee A, Dubnau E, Quemard A, et al. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263(5144):227–230. doi:10.1126/science.8284673
4. Quémard A, Sacchettini JC, Dessen A, et al. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry. 1995;34(26):8235–8241. doi:10.1021/bi00026a004
5. Fox HH. The chemical approach to the control of tuberculosis. Science. 1952;116(3006):129–134. doi:10.1126/science.116.3006.129
6. Bernstein J, Lott WA, Steinberg BA, Yale HL. Chemotherapy of Experimental Tuberculosis. Am Rev Tuberc. 1952;65(4):357–364. doi:10.1164/art.1952.65.4.357
7. Takayama K, Wang L, David HL. Effect of isoniazid on the in vivo mycolic acid synthesis, cell growth, and viability of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1972;2(1):29–35. doi:10.1128/AAC.2.1.29
8. Winder FG, Collins PBY. Inhibition by Isoniazid of Synthesis of Mycolic Acids in Mycobacterium tuberculosis. Microbiology. 1970;63(1):41–48. doi:10.1099/00221287-63-1-41
9. Nguyen M, Quémard A, Broussy S, Bernadou J, Meunier B. Mn(III) pyrophosphate as an efficient tool for studying the mode of action of isoniazid on the InhA protein of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2002;46(7):2137–2144. doi:10.1128/AAC.46.7.2137-2144.2002
10. Oliveira JS, Pereira JH, Canduri F, et al. Crystallographic and pre-steady-state kinetics studies on binding of NADH to wild-type and isoniazid-resistant enoyl-ACP(CoA) reductase enzymes from Mycobacterium tuberculosis. J Mol Biol. 2006;359(3):646–666. doi:10.1016/j.jmb.2006.03.055
11. Rozwarski DA, Grant GA, Barton DH, Jacobs WR, Sacchettini JC. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science. 1998;279(5347):98–102. doi:10.1126/science.279.5347.98
12. Liao C, Sitzmann M, Pugliese A, Nicklaus MC. Software and resources for computational medicinal chemistry. Future Med Chem. 2011;3(8):1057–1085. doi:10.4155/fmc.11.63
13. Wang HW, Noland C, Siridechadilok B, et al. Structural insights into RNA processing by the human RISC-loading complex. Nat Struct Mol Biol. 2009;16(11):1148–1153. doi:10.1038/nsmb.1673
14. Kapale SS, Mali SN, Chaudhari HK. Molecular modelling studies for 4-oxo-1,4-dihydroquinoline-3-carboxamide derivatives as anticancer agents. Med Drug Discov. 2019;2:100008. doi:10.1016/j.medidd.2019.100008
15. Mali SN, Pandey A, Bhandare RR, Shaik AB. Identification of hydantoin based Decaprenylphosphoryl-β-D-Ribose Oxidase (DprE1) inhibitors as antimycobacterial agents using computational tools. Sci Rep. 2022;12(1):16368. doi:10.1038/s41598-022-20325-1
16. Mali SN, Pandey A, Thorat BR, Lai CH. Multiple 3D- and 2D-quantitative structure–activity relationship models (QSAR), theoretical study and molecular modeling to identify structural requirements of imidazopyridine analogues as anti-infective agents against tuberculosis. Struct Chem. 2022;33(3):679–694. doi:10.1007/s11224-022-01879-2
17. Ode H, Matsuyama S, Hata M, et al. Computational characterization of structural role of the non-active site mutation M36I of human immunodeficiency virus type 1 protease. J Mol Biol. 2007;370(3):598–607. doi:10.1016/j.jmb.2007.04.081
18. Atefeh Haji Agha B, Afshin Z. Search for the pharmacophore of histone deacetylase inhibitors using pharmacophore query and docking study. Iranian J Pharmaceutical Res. 2014;1165–1172.
19. Burley SK, Berman HM, Kleywegt GJ, Markley JL, Nakamura H, Velankar S. Protein Data Bank (PDB): the Single Global Macromolecular Structure Archive. In: Wlodawer A, Dauter Z, Jaskolski M editors. Protein Crystallography: Methods and Protocols. Methods in Molecular Biology. Springer; 2017:627–641. doi:10.1007/978-1-4939-7000-1_26
20. Basso LA, Zheng R, Musser JM, Jacobs WR, Blanchard JS. Mechanisms of isoniazid resistance in Mycobacterium tuberculosis: enzymatic characterization of enoyl reductase mutants identified in isoniazid-resistant clinical isolates. J Infect Dis. 1998;178(3):769–775. doi:10.1086/515362
21. Vilchèze C, Wang F, Arai M, et al. Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat Med. 2006;12(9):1027–1029. doi:10.1038/nm1466
22. Hartkoorn RC, Sala C, Neres J, et al. Towards a new tuberculosis drug: pyridomycin - nature’s isoniazid. EMBO Mol Med. 2012;4(10):1032–1042. doi:10.1002/emmm.201201689
23. Dessen A, Quémard A, Blanchard JS, Jacobs WR, Sacchettini JC. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science. 1995;267(5204):1638–1641. doi:10.1126/science.7886450
24. Parikh S, Moynihan DP, Xiao G, Tonge PJ. Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. Biochemistry. 1999;38(41):13623–13634. doi:10.1021/bi990529c
25. Molle V, Gulten G, Vilchèze C. Phosphorylation of InhA inhibits mycolic acid biosynthesis and growth of Mycobacterium tuberculosis. Mol Microbiol. 2010;78:6. doi:10.1111/j.1365-2958.2010.07446.x
26. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi:10.1002/jcc.20084
27. Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17(1):57–61.
28. O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminformatics. 2011;3:33. doi:10.1186/1758-2946-3-33
29. Borba JVB, Alves VM, Braga RC, et al. STopTox: an in Silico Alternative to Animal Testing for Acute Systemic and Topical Toxicity. Environ Health Perspect. 2022. doi:10.1289/EHP9341
30. Raschi E, Ceccarini L, De Ponti F, Recanatini M. hERG-related drug toxicity and models for predicting hERG liability and QT prolongation. Expert Opin Drug Metab Toxicol. 2009;5(9):1005–1021. doi:10.1517/17425250903055070
31. Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(W1):W257–W263. doi:10.1093/nar/gky318
32. Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J Med Chem. 2002;45(12):2615–2623. doi:10.1021/jm020017n
33. Worth CL, Preissner R, Blundell TL. SDM—a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011;39:W215–W222. doi:10.1093/nar/gkr363
34. Rodrigues CH, Pires DE, Ascher DB. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018;46(W1):W350–W355. doi:10.1093/nar/gky300
35. Moll A, Hildebrandt A, Lenhof HP, Kohlbacher O. BALLView: an object-oriented molecular visualization and modeling framework. J Comput Aided Mol Des. 2005;19(11):791–800. doi:10.1007/s10822-005-9027-x
36. Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi:10.1002/jcc.21256
37. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
38. Yuan S, Chan HCS, Hu Z. Using PyMOL as a platform for computational drug design. WIREs Comput Mol Sci. 2017;7(2):e1298. doi:10.1002/wcms.1298
39. Pickett SD, Jonathan S. Diversity Profiling and Design Using 3D Pharmacophores: Pharmacophore-Derived Queries (PDQ). ACS Publications; 2013; doi:10.1021/ci960039g
40. Lorch M, Mason JM, Sessions RB, Clarke AR. Effects of mutations on the thermodynamics of a protein folding reaction: implications for the mechanism of formation of the intermediate and transition states. Biochemistry. 2000;39(12):3480–3485. doi:10.1021/bi9923510
41. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):e118. doi:10.1093/nar/gkr407
42. Chen J, Shen B. Computational analysis of amino acid mutation: a proteome wide perspective. Curr Proteomics. 2014;6(4):228–234.
43. Steward RE, MacArthur MW, Laskowski RA, Thornton JM. Molecular basis of inherited diseases: a structural perspective. Trends Genet TIG. 2003;19(9):505–513. doi:10.1016/S0168-9525(03)00195-1
44. George DCP, Chakraborty C, Haneef SAS, Nagasundaram N, Chen L, Zhu H. Evolution- and structure-based computational strategy reveals the impact of deleterious missense mutations on MODY 2 (maturity-onset diabetes of the young, type 2). Theranostics. 2014;4(4):366–385. doi:10.7150/thno.7473
45. Kumar V, Sobhia ME. Molecular Dynamics Assisted Mechanistic Study of Isoniazid-Resistance against Mycobacterium tuberculosis InhA. PLoS One. 2015;10(12):e0144635. doi:10.1371/journal.pone.0144635
46. Yao X, Guo S, Wu W, et al. Q63, a novel DENV2 RdRp non-nucleoside inhibitor, inhibited DENV2 replication and infection. J Pharmacol Sci. 2018;138(4):247–256. doi:10.1016/j.jphs.2018.06.012
47. Yin W, Mao C, Luan X, et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020;368(6498):1499–1504. doi:10.1126/science.abc1560
48. Macchiagodena M, Pagliai M, Procacci P. Inhibition of the Main Protease 3CL-pro of the Coronavirus Disease 19 via Structure-Based Ligand Design and Molecular Modeling. arXiv preprint arXiv. 2020. doi:10.48550/arXiv.2002.09937
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.