Back to Journals » Journal of Inflammation Research » Volume 15
Ezh2 Inhibits Replicative Senescence of Atrial Fibroblasts Through Promotion of H3K27me3 in the Promoter Regions of CDKN2a and Timp4 Genes
Authors Li Y, Fang G, Cao W, Yuan J, Song S, Peng H, Wang Y, Wang Q
Received 16 May 2022
Accepted for publication 23 July 2022
Published 16 August 2022 Volume 2022:15 Pages 4693—4708
DOI https://doi.org/10.2147/JIR.S374951
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Adam Bachstetter
Yingze Li,* Guojian Fang,* Wei Cao,* Jiali Yuan,* Shuai Song, Hong Peng, Yuepeng Wang, Qunshan Wang
Department of Cardiology, Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine, Shanghai, 200092, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qunshan Wang; Yuepeng Wang, Department of Cardiology, Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine, Shanghai, 200092, People’s Republic of China, Email [email protected]; [email protected]
Background: In most cell types, replicative senescence (RS) is supposed to be a principle causative factor for aging. Atrial fibrosis, pathologically characterized by proliferation of atrial fibroblasts (AFs) and excessive accumulation of extracellular matrix proteins, is the most common substrate of atrial fibrillation (Afib) in the elderly. However, whether AFs’ RS develops in the aged and fibrotic left atrium (LA) and, if yes, what is the key regulator for the pathogenesis of AFs’ RS remain largely unknown.
Methods: We obtained the left atrial tissues from young (6– 8 weeks old) and aged (24 months old) C57BL/6 male mice. Screening and validation of differential genes were performed using comparative analysis of RNA-seq results. Replicative senescence was examined in primary AFs after cell passage. Further gain-of-function and loss-of-function experiments were performed to explore the regulation of the AFs’ RS progression.
Results: In the present study, we demonstrated that there was a considerable extent of AFs’ RS in the aged and fibrotic LA. Transcriptome screening showed that Ezh2 (Enhancer of zeste homolog 2) was significantly downregulated in the LA tissue of aged mice. Ezh2 is a histone methyltransferase that catalyzes H3K27me3 and mediates transcriptional silencing. We confirmed that Ezh2 was downregulated in the isolated pure senescent AFs. Knockdown of Ezh2 by siRNA or inhibition of Ezh2ʹs methyltransferase activities by GSK-126 and GSK-343 accelerated RS in the early passage of AFs, while its overexpression deaccelerated RS in the late passage of AFs. Mechanistically, Ezh2 suppressed CDKN2a (p16, p19) and Timp4 gene transcription by forming canonical H3K27me3 modifications in their promoter regions. Furthermore, the functional balance between Timp4 and MMP8 in AFs could be collapsed by changes in Ezh2 expression.
Conclusion: These results thus indicate that Ezh2 is a key regulator of AFs’ RS and this work may provide a basis for future treatments for atrial fibrosis in the elderly.
Keywords: aging, replicative senescence, atrial fibrosis, Ezh2, H3K27me3
Graphical Abstract:

Introduction
Atrial fibrosis is a key substrate for the development of atrial fibrillation (Afib) and the severity of atrial fibrosis progresses during aging.1 Epidemiologically, aging is also the most important risk factor for Afib. Afib in the elderly population is not only of high prevalence itself but also frequently associated with complications, like stroke, dementia, and cardiovascular death and thus dramatically increases social health-care burden. Although not being thoroughly clarified, development of atrial fibrosis in the aged atrium undergoes a similar but more chronic process as cardiac fibrosis post-acute myocardial infarction and hypertension.2 Atrial fibroblasts (AFs) are provoked by various stimulants like Ang-II, TGFβ, inflammatory cytokines, and ROS, which undergo differentiation into myofibroblasts, proliferate, migrate, and secret extracellular matrix (ECM) proteins. AFs play a fundamental role in maintaining the dynamic balance of ECM synthesis and degradation in the atrium by adjusting the balance between tissue inhibitors of metalloproteinases (Timp) and matrix metalloproteinases (MMP). Excessive deposition of ECM in the interstitial space between atrial cardiomyocytes gradually results in atrial fibrosis, which disturbs normal cardiac electric conduction and finally causes signal reentry and rotor leading to Afib.
Senescence is a gradually developed functional deterioration in living organism and is divided into developmentally programmed senescence, stress-induced premature senescence, proteasome-inhibited senescence, and replicative senescence (RS).3,4 RS is a state of irreversible cell growth arrest with permanent loss of proliferation potential and is now considered to be the principle driving force of organismal aging.5–7 RS cells secrete large amounts of inflammatory cytokines, chemokines, and matrix proteases, which is termed the senescence-associated secretory phenotype (SASP), formation of so-called “inflammaging” microenvironment also contributes to the process of organismal aging. Moreover, the ability of RS cells to maintain Timp-MMP balance is severely impaired.8 In some extra-cardiac organs, RS inhibits fibrosis, like satellite cells in the liver and skeletal muscle.
However, there has been few studies of the senescent process in AFs so far.9 Some fundamental points of its pathogenesis are not elucidated, and it is usually supposed that AFs’ senescence may promote the atrial fibrosis progression. For example, whether AFs in the fibrotic and aged left atrium (LA) develop similar RS as other cell-types do? If yes, what are the key pathogenetic regulator and the underlying mechanism of the RS? All these points remain largely unknown.
In this study, we aimed to research AFs’ senescence and found that there was considerable extent of AFs’ RS in the aged and fibrotic LA. We then screened from mice transcriptomics of old vs young LA tissue and found that Ezh2 (Enhancer of zeste homolog 2) was significantly downregulated. Enhancer of zeste homolog 2 (EZH2) is a histone-lysine N-methyltransferase enzyme, using its SET domain to form di- and trimethylated versions of lysine residue 27 within histone H3 (H3K27me3).10 Previous studies have shown that Ezh2 play an important role in cell differentiation with transcriptional activations in some genes or repressions in others.11,12 Recent studies illustrated Ezh2 took part in the pathogenesis of a number of age-related diseases, such as intervertebral disc degeneration, regenerative failure in pancreatic islet β cells and osteoporosis.13–15 Therefore, it is of great interest to explore whether EZH2 might be involved in the regulation of AFs’ senescence. We found that Ezh2 was down-regulated in both aged LA tissues and isolated senescent AFs. By establishing an in vitro cellular model, we discovered that Ezh2 regulates the RS phenotypes of AFs. Finally, we explored the possible mechanisms whereby Ezh2 regulates CDKN2a and Timp4 gene transcription through forming canonical H3K27me3 modifications in the corresponding promoter regions. Our study reveals that Ezh2 functions as a key regulator for AFs’ RS and this work may provide a therapeutic target against age-related atrial fibrosis and the subsequent Afib.
Materials and Methods
Animals Studies
Young (6–8 weeks old) and aged (24 months old) C57BL/6 male mice were used for the experiments. The study was approved by the ethical review committee of Xinhua Hospital at SJTUSM (approval number: XHEC-NSFC-2019-382). All procedures were conducted in compliance with both the Guide for the Animal Care and Use Committee of Xinhua Hospital and the guidance for the care and use of experimental animals published by NIH (the 8th Edition, NRC 2011).
Echocardiographic Measurements of the Left Atrium (LA)
Mice were anesthetized by 2% isoflurane inhalation and were placed on a heated pad to keep 37 °C body temperature prior to echocardiography examinations (15 MHz transducer; Vivid 7, GE Healthcare). The LA dimensions, LV end-systolic and end-diastolic dimensions, ejection fraction (EF), and fractional shortening (FS) were measured from M-mode images guided by a parasternal long-axial 2-dimensional view.16 The sonographer was blinded to the information of mice.
Isolation and Culture of AFs
The primary AFs were isolated from young mice and cultured (37 °C and 5% CO2) in DMEM containing 10% fetal calf serum and 100 U/mL penicillin/streptomycin, as described previously.17 The isolated AFs were seeded at a density of 104/6-well dish on day-0, took about 4~6 h to get attached to the bottom of the cultural dish, further proliferated profoundly and reached 90~100% confluent on the next day-1 were termed and harvested as P0. On day-1, a part of cells was passaged for the first time with trypsin digestion, proliferated and became 80~100% confluent on day-4 were termed and harvested as P1. Then, a part of cells was passaged for the second time, proliferated and became 70~90% confluent on day-9 were termed and harvested as P2. Thereafter, P2, cell proliferation was totally and irreversibly ceased, but they still kept alive for further 3~4 days until day-12 when they reach the dead end of RS. If the cells at day-9 (P2) were further passaged for the third time the cells could not be attached and grow anymore.
Histology and Tissue Immunofluorescence
LA tissues from mice were fixed with 4% phosphate-buffered formalin for 24 h, paraffin embedded, serial sectioned at 5μM thick, deparaffinized, stained with Masson’s trichrome staining kit (HT15 Trichrome Staining Kit; Sigma-Aldrich), sealed and stored at 4°C. Immunofluorescence was performed on deparaffinized sections using primary antibodies: vimentin (Abcam, ab8978, 1:200), CDKN2A/p16INK4a (Abcam, ab211542, 1:200) and p21 (Abcam, ab188224, 1:200). Then, the sections were incubated with secondary antibodies Alexa Fluor 594-AffiniPure Donkey Anti-Rabbit IgG (H+L) (Yeason, 34212ES60, 1:300) or Alexa Fluor 488-AffiniPure Donkey Anti-Mouse IgG (H+L) (Yeason, 34106ES60, 1:300). DAPI was used to stain the nuclei.
Cellular Immunofluorescence
For cellular immunofluorescence analysis, primary cultured AFs were fixed in 4% paraformaldehyde for 15 min and permeabilized with 0.1% TritonX-100 in PBS for 10 min. After blocking with 5% BSA for 1 h at room temperature, the cells were incubated with mouse antibody against H3K27me3 (ABclonal, A16199, 1:200), vimentin (Abcam, ab45939, 1:200), CDKN2A/p16INK4a (Abcam, ab211542, 1:200) and p21 (Abcam, ab188224, 1:200) at 4 °C overnight, rabbit normal IgG (Abcam, ab6702) was used as negative control. The cells were then incubated with Alexa Fluor 488-AffiniPure Donkey Anti-Mouse IgG (H+L) (Yeason, 34106ES60, 1:300), Alexa Fluor 594-AffiniPure Donkey Anti-Rabbit IgG (H+L) (Yeason, 34212ES60, 1:300) and DAPI at room temperature for 1 h.
Senescence-Associated β-Galactosidase (SA-β-gal) Staining
For SA-β-gal staining, we used senescence β-gal staining kit (Cell Signaling Technology, #9860; USA) according to the manufacturer’s instructions.
Adenovirus
Mouse full-length Ezh2 (NM_007971; 2241bp) cDNA sequence was cloned into the pAdM-FH vector and packaged into adenovirus particles (Adv-Ezh2) with a titer of about 1.26 × 1010 PFU/mL under technical support from Hanbio (Shanghai, China). For infection, AFs at 60–70% confluency at day-6 were used at a viral dose based on the multiplicity of infection (MOI) of the fibroblasts (=100). Infected cells were harvested at day-9 (P2).
RNA Interference
Three small-interfering RNA (siRNA) designed and synthesized against Ezh2 are GGATAATCGAGATGATAAA (si-Ezh2-1), GCTGAAGCCTCCATGTTTA (si-Ezh2-2), and CATCGTAAGTGCAGTTATT (si-Ezh2-3; RiboBio, Guangzhou, China). When the cells seeded in 6 cm dishes reached 40~60% confluence, 100nM siRNA was transfected with Lipofectamine 3000 (Invitrogen) in accordance with the manufacturer’s protocols. The cells were harvested for evaluation of knockdown efficiencies and for other assays 72 h after transfection.
Quantitative Real-Time PCR
Total RNA was extracted from LA tissues or cultured AFs using TRIzol Reagent (Takara, Cat# 9109), according to the provided manual instructions. cDNA was synthesized from 1000 ng total RNA using the Prime-ScriptTM RT Master Mix (Takara; Cat# RR036A). SYBR qRT-PCR was performed using Takara SYBR mix (Takara, SYBR Premix Ex Taq II, #RR820A) and normalized to GAPDH expression. The sequences of the primers synthesized by Sangon Biotech (Shanghai, China) are shown in S-Table 1.
RNA Sequencing Transcriptome Analyses
Total RNAs were extracted individually from LA tissues of young (6–8 weeks old; n = 3) and aged (24 months old; n = 3) mice using TRIzol (Takara, Cat# 9109), followed by RT. cDNA sequencing was carried out on a sequencer (BGISEQ500) by Weihuan Biotech. Co., Ltd. (Shanghai, China). DEGs were identified through log2 |fold change| >0.7 as well as p-values (≤0.05), followed by GO and KEGG analysis.
Cleavage Under Targets and Tagmentation (CUT&Tag) Assay
The CUT&Tag, a novel method to profile histone modifications, can quickly obtain high resolution and low background chromatin components.18 AFs harvested from mice were prepared with Hieff NGS® G-Type In-Situ DNA Binding Profiling Library Prep Kit for Illumina® (Yeason, China) under the manufacturer’s instructions. An H3K27me3 antibody (Cell Signaling Technology, #9733) was used to pull down protein-DNA complexes and rabbit normal IgG (Abcam, ab6702) was used as negative control. The DNA fragments were extracted, amplified by PCR and sequenced using specific primers (S-Table 2).
Western Blotting (WB)
Protein samples were prepared from mice LA tissues or from cultured AFs. An equal amount of protein (10~40 μg) was resolved by SDS/PAGE and transferred to PVDF membranes. The membranes were blocked with 5% non-fat milk in TBST (Tris-HCl buffered saline supplemented with 0.05% Tween-20) for 2 h at room temperature and then incubated with primary antibodies against Ezh2 (Abclonal, A11085), H3K27me3 (CST, #9733S), Timp1 (Abcam, ab179580), Timp2 (Abcam, ab180630), Timp3 (Abcam, ab39184), Timp4 (Abclonal, A6416), MMP1 (Abcam, ab137332), MMP8 (Abcam, ab53017), MMP13 (Abcam, ab39012), p16 (Abcam, ab211542), p19 (Abcam, ab80), p21 (Abcam, ab188224), collagen-I (Abcam, ab254113), collagen-III (Servicebio, GB111323), or GAPDH (Servicebio, GB11002) at 4°C overnight. GAPDH was utilized as an internal control. The membranes were then incubated with secondary antibodies conjugated with horseradish peroxidase (Sigma-Aldrich) for 1 h at room temperature followed by triplicate washing out with TBST. The signals were revealed by enhanced chemiluminescence ECL (Thermo Scientific) on a Gel Imaging System (GE, AI600) and were quantified by densitometry software (Image J).
Statistical Analysis
Data were analyzed in SPSS 19.0 statistical software or GraphPad Prism 8.0 and presented as mean ± SD. Two-tailed Student’s tests were used for two-group comparisons. ANOVA followed by post hoc Tukey’s test was used for multiple-group comparisons. A value of p<0.05 was regarded as statistically significant.
Results
Age-Induced Fibrosis and AFs’ RS in LA
Previous studies reported that the aged LA had two obvious remodeling in structure: enlarged cavity and fibrotic wall.19,20 We therefore first in this study confirmed these two changes in mice. On echocardiography, aged mice (24 months old) showed significantly enlarged LA as compared with young mice (6–8 weeks old; Figure 1A). As identified by Masson’s trichrome staining, the severity of LA fibrosis remarkably increased from 0.81 ± 0.08% to 7.76 ± 1.04% in the aged mice (Figure 1B). WB results showed that both collagen-I and collagen-III were upregulated to near 3 folds in the aged LA tissues. Thus, we confirmed the previous results (Figure 1C).
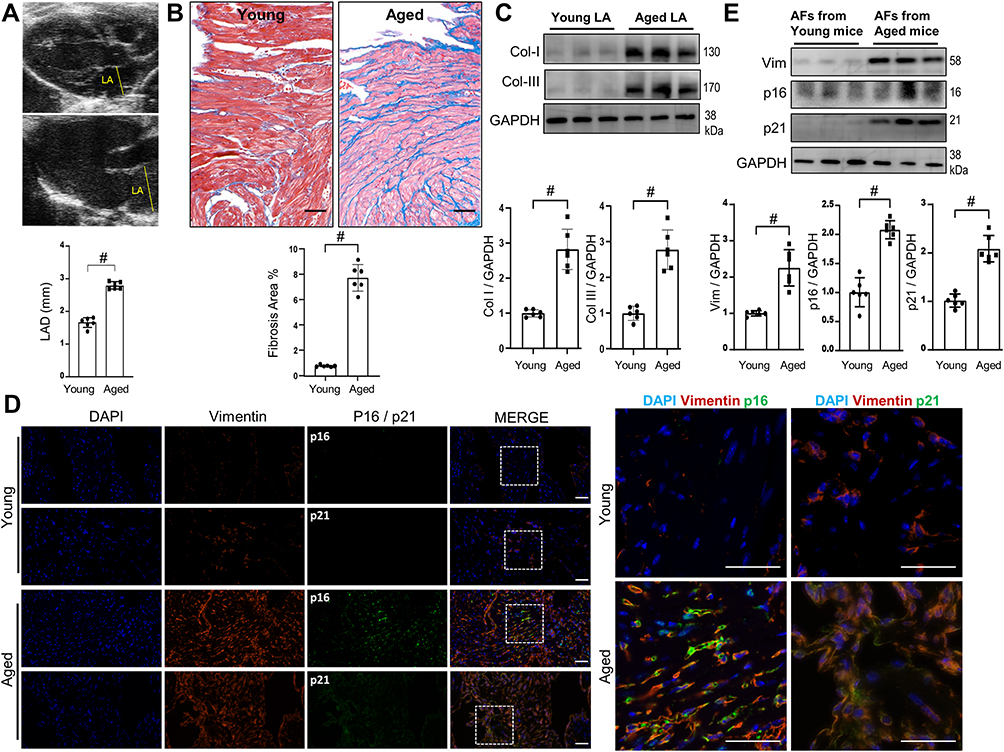 |
Figure 1 Age-related LA fibrosis in mice. Both young (6~8 weeks) and aged (24 months old) mice were received echocardiography examinations, followed by harvesting of LA tissues for Masson trichrome, WB, immunofluorescence staining or isolation of AFs. (A) Representative echocardiographic images of LA dimension and their quantitative analyses. n=6. (B) Representative images of Masson trichrome staining of LA sections and their quantitative analyses. n=6. bar=50μm. (C) Representative WB images and their densitometry analyses of collagen-I and collagen-III protein expressions in LA tissues. (D) Representative immunofluorescence images of LA sections stained with DAPI, vimentin, p16 and p21 as indicated. bar=50μm. (E) Representative WB images and their densitometry analyses of vimentin, p16 and p21 protein expressions in isolated pure AFs. n=6. Data are presented as mean ± SD. #p<0.001. |
To elucidate whether the RS of AFs develops in the aged and fibrotic LA in vivo, we checked the expressions of vimentin, p16 and p21. Their expressions identified by immunofluorescence were all intensified in the fibrotic areas in the aged LA, where vimentin was highly colocalized with either p16 or p21 (Figure 1D), indicating an accumulation of senescent AFs around the fibrotic areas. WB of the aged LA tissues also illustrated upregulations of all three proteins (Figure 1E). These results demonstrated that there is a considerable extent of AFs’ RS in the aged and fibrotic LA. AFs’ RS may contribute to the pathogenesis of age-related atrial fibrosis.
Ezh2 as a Potential Regulator in Age-Induced AFs’ RS
What are the underlying regulating molecules and mechanisms in the pathogenesis of AFs’ RS in the aged LA remain largely unknown. Here, we explored them by comparing transcriptome signatures between young and old LA tissues in mice. We obtained 1866 (945 up; 921 down) differentially expressed genes (DEGs; a log2 |fold change| >0.7; p<0.05) (Figure 2A). Heatmap illustrated the hierarchical clustering of all the DEGs between two groups using the Spearman correlation coefficient (Figure 2B). GO analysis demonstrated that the top five most regulated Biological Process were “covalent chromatin modification”, “negative regulation of cell cycle”, “aging”, “collagen metabolic process” and “replicative senescence”. “Extracellular organelle” and “collagen trimer” were ranked the top two most significantly changed Cellular Component. Top three items for Molecular Function were “extracellular matrix structural constituent”, “transcription compressor activity” and “histone binding” (Figure 2C). Aged LV tissues appeared to mainly have altered Cellular Processes and Molecular Functions related to transcription regulations and ECM proteins.
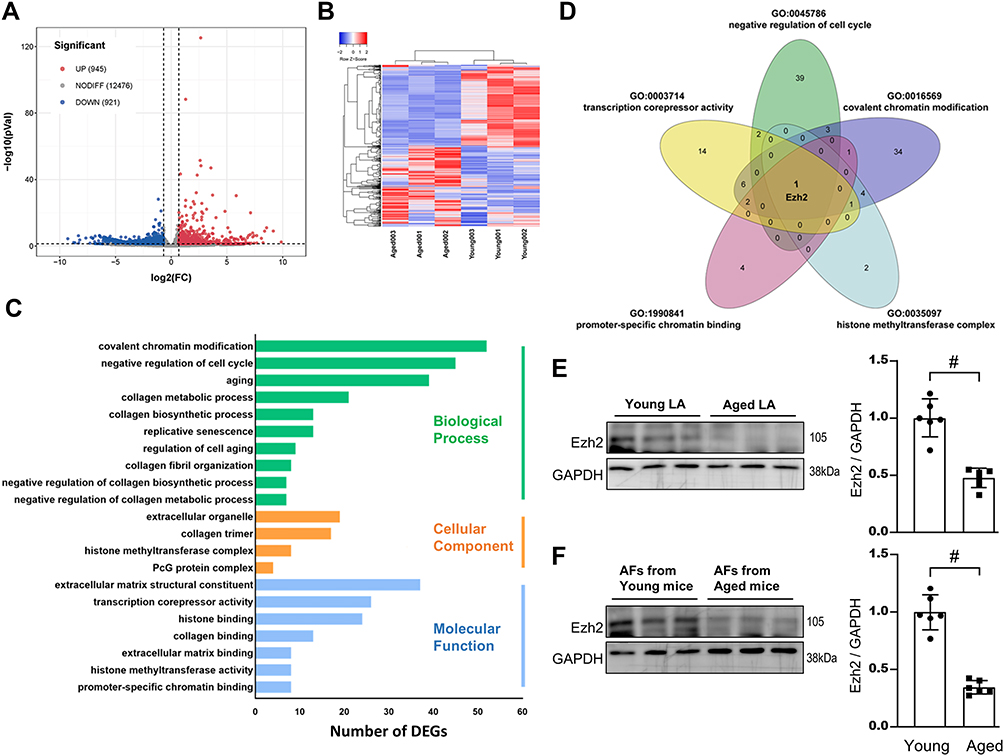 |
Figure 2 Transcriptome analyses reveals Ezh2 is a potential regulator of age-related LA fibrosis. Total RNA were extracted from mice LA tissues and reversely transcribed to cDNA for RNA-Seq. (A) Volcano plots revealed that 945 of DEGs were up-regulated (log2 fold change >0.7; p<0.05; red dots) and 921 down-regulated (log2 fold change<0.7; p<0.05; blue dots) in aged vs young LA. (B) Heatmap summaries. (C) GO term enrichment analyses. The vertical coordinates are the terms (the biological process in green; the cellular component in Orange; the molecular function in blue) and the horizontal coordinates are the numbers of DEGs. (D) Venn diagram showing the overlap of GO terms as indicated, and Ezh2 gene was chosen. (E and F) Down-regulated level of Ezh2 was validated by WB of either mice LA tissues (E) or isolated pure AFs (F). Representative WB images of Ezh2 and their densitometry analyses. n=6 mice. Data are presented as mean ± SD. #p<0.001. |
By further GO (functional processes) overlapped analysis, we found that Ezh2 (the enhancer of zeste homolog 2), and a core component of Polycomb repressive complex 2 (PRC2), was significantly downregulated in the aged LA (0.56-fold; Figure 2D). As a histone methyltransferase, Ezh2 catalyzes the addition of methyl groups to histone H3 at lysine 27 (H3K27me3) by using the cofactor S-adenosyl-L-methionine and ultimately results in transcriptional repression.
Validation WB results showed that Ezh2 was indeed downregulated in either aged LV tissues (Figure 2E) or in isolated pure senescent AFs (Figure 2F).
Downregulation of Ezh2 in a RS Model of AFs
We thus hypothesized that Ezh2 regulates the AFs’ RS progression. Based on our experience, the primary culture of isolated AFs from young mice usually undergoes a rapid RS and after reaching a maximal two passages cell proliferation ceases completely. Next, we designed an AFs’ in vitro model to study Ezh2ʹs regulatory functions on their RS progression (see detailed descriptions in Materials and Methods; Figure 3A).
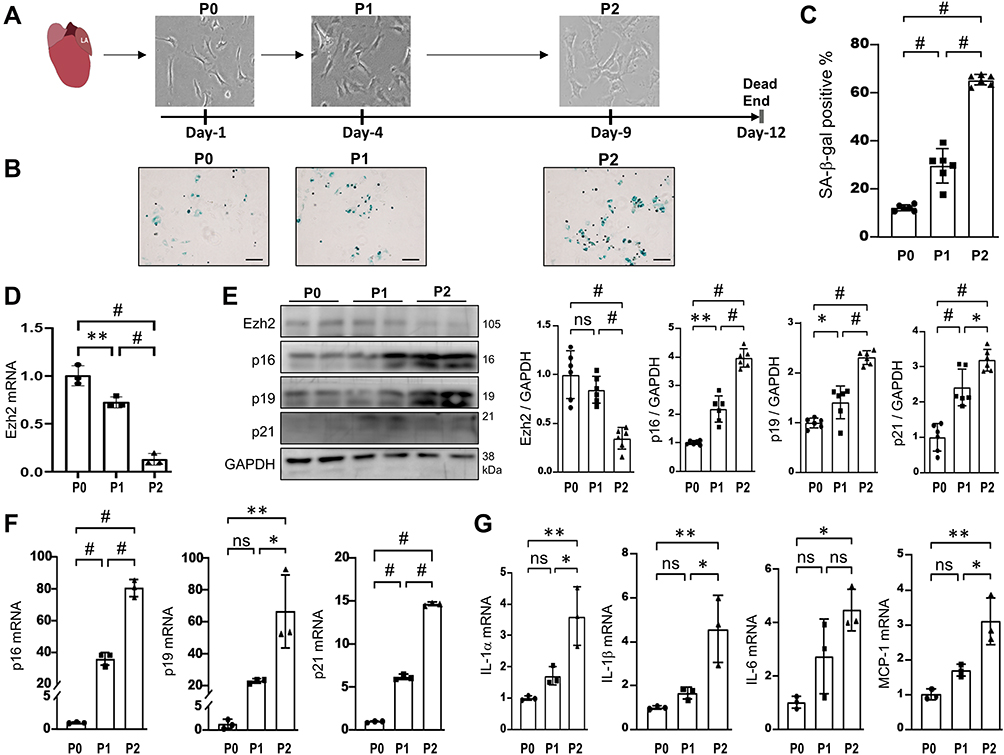 |
Figure 3 Ezh2 is down-regulated in a RS model of AFs. (A) Schematic diagram of our RS model of AFs isolated from young mice and classified as P0, P1 and P2. bar=20μm. P0, P1 and P2 cells were harvested for multiple analyses in B~G. (B and C) Representative images of SA-β-gal staining and their quantitative analyses (n=6 mice in each group). (D) Transcriptions of Ezh2 was assessed by qRT-PCR (n=3 in each group). (E) Representative WB images of Ezh2 and CDKIs markers as indicated, as well as their densitometry analyses (n=6). (F and G) Transcriptions of CDKIs (F) and SASP genes (IL-1α, IL-1β, IL-6, MCP-1; (G) were evaluated by qRT-PCR (n=3 in each group). Data are presented as mean ± SD. *p<0.05, **p<0.01, #p<0.001. Abbreviation: ns, no significance. |
We found that the activity of SA-β-gal got stepwise increases from P0 to P1 and from P1 to P2 (Figure 3B and C), indicating this cellular model is reliable. Compared with P0, Ezh2ʹs transcription and translation were gradually downregulated in P1 and P2 cells (Figure 3D and E). For CDKIs (p16, p19, p21), stepwise upregulations of both transcriptions and translations were observed in P1 and P2 cells (Figure 3E and F). Of the three CDKIs, we further confirmed p16 and p21ʹs upregulations by immunofluorescence (S-Figure 1). In addition, four SASP genes (IL-1α, IL-1β, IL-6, MCP-1) were found to be transcriptionally activated in P2 cells (Figure 3G). On the other hand, previous study reported that typical AF’s RS has classic features, ie an increased expression of vimentin and the well-organized structure of the vimentin filaments.21 We therefore further performed immunofluorescence of vimentin and the results were in line with our expectations (S-Figure 2). These results indicate that Ezh2ʹs downregulation is implicated in AFs’ RS.
Knockdown of Ezh2 Accelerates RS in Early Passage of AFs
As described above, Ezh2 were down-regulated in P2 cells, and we next examined whether knockdown of Ezh2 from P1 could accelerate RS? P1 cells on day-1 were transfected with three kinds of siRNAs against Ezh2 and on day-4 (P1) the most effective si-Ezh2-2 was selected out by WB (Figure 4A and B). si-Ezh2-2 was shown to decrease Ezh2ʹs transcription to 16.80% of the scramble siRNA’s (Figure 4C). Thereafter, all experiments were performed with si-Ezh2-2. Ezh2 silence increased SA-β-gal activity (Figure 4D), up-regulated CDKIs at both mRNA and protein levels (Figure 4E and F), and activated SASP genes transcription (Figure 4G). Consistently, Ezh2 silence increased immunofluorescence of vimentin (S-Figure 3A). In other words, these data indicated that knockdown of Ezh2 could induce RS phenotypes in the early passage of AFs.
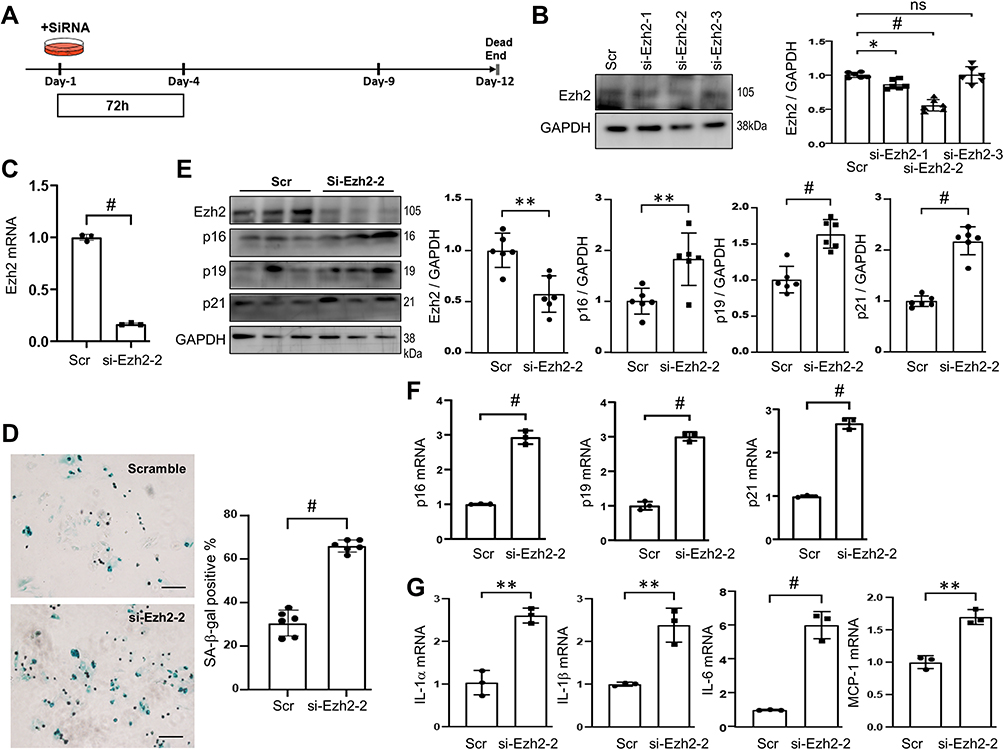 |
Figure 4 Knockdown of Ezh2 induces RS in the early passage of AFs. (A) Schematic diagram of Ezh2 silencing in AFs. Isolated, plated and stably attached P1 AFs on Day-1 were transfected with scramble (Scr) or Ezh2 siRNAs (100nM) for 72h and harvested on Day-4 for analyses. (B) Representative WB images showing the efficiency of Ezh2 knocking-down (left), as well as its densitometry analyses (right; n=6). Si-Ezh2-2 was chosen to be used in the thereafter experiments. (C) Transcriptions of Ezh2 after transfection of si-Ezh2-2 was assessed by qRT-PCR (n=3 in each group). (D) Representative images of SA-β-gal staining. SA-β-gal activity was quantitated and expressed as percent of the positive cells (n=6). bar=100μm. (E) Representative WB images of Ezh2 and CDKIs, as well as their densitometry analyses (n=6). (F and G) Transcriptions of CDKIs (F) and SASP genes (G) were evaluated by qRT-PCR (n=3). Data are presented as mean ± SD. *p<0.05, **p<0.01, #p<0.001. Abbreviation: ns, no significance. |
Overexpression of Ezh2 Deaccelerates RS in Late Passage of AFs
Next, whether the overexpression of Ezh2 by Adv-Ezh2 could retard the RS progression? In Adv-Ezh2 infected AFs, Ezh2 were upregulated for 2.88 ± 1.04 folds as identified by qRT-PCR and for 2.60 ± 0.49 folds as shown by WB (Figure 5A–C). Ezh2 overexpression decreased SA-β-gal activity (Figure 5D), the mRNA and protein levels of CDKIs (Figure 5C and E) and repressed transcriptions of the SASP genes (Figure 5F). Also, Ezh2 overexpression decreased immunofluorescence of vimentin (S-Figure 3B). These data suggest that Ezh2 upregulation in late passage AFs retarded senescence phenotypes development.
 |
Figure 5 Overexpression of Ezh2 alleviates RS in the late passage of AFs. (A) Schematic diagram of Ezh2 overexpression in P2 AFs. AFs on Day-6 were transduced with Adv-Ezh2 and harvested 72h later for analyses. (B) Transcriptions of Ezh2 was assessed by qRT-PCR (n=3 in each group). (C) Representative WB images of Ezh2 and CDKIs, as well as their densitometry quantitative analyses (n=6). (D) Representative images of SA-β-gal staining, SA-β-gal activity was quantified and expressed as percentage of the positive cells (n=6). bar=100μm. (E and F) Transcriptions of CDKIs (E) and SASP genes (F) were evaluated by qRT-PCR (n=3). Data are presented as mean ± SD. *p<0.05, **p<0.01, #p<0.001. |
Associations of Ezh2 Expressions or Its Methyltransferase Activities with H3K27me3 Levels
Whether Ezh2 expression level is closely related to the functional levels of H3K27me3 in AFs? As described above, Ezh2 expression was down-regulated naturally in P2 cells, was silenced by si-Ezh2-2 in P1 cells, and was up-regulated in P2 cells by infection of Adv-Ezh2. Consistently, we found that the levels of H3K27me3 were changed in similar trends as Ezh2 did under three situations (Figure 6A–E). In detail, P2 cells with down-regulated Ezh2 was associated with a decreased level of H3K27me3 in WB (Figure 6A). In P1 cells with silenced Ezh2 had decreased level of H3K27me3 in both WB and the immunofluorescence (Figure 6B and C). Meanwhile, in P2 cells with overexpressed Ezh2 had increased level of H3K27me3 in both WB and the immunofluorescence (Figure 6D and E). These results indicate that H3K27me3 levels in AFs were completely and proportionally determined by Ezh2 expression levels.
 |
Figure 6 Changes in H3K27me3 Levels in our RS models (A), after Knockdown of Ezh2 (B and C), and Overexpression of Ezh2 (D and E). (A) Representative WB images of H3K27me3 using SDS-protein samples of above Figure 3 and its densitometry analyses (n=6). (B) Representative WB images of H3K27me3 using SDS-protein samples of above Figure 4 and its densitometry analyses (n=6). (C) Representative immunofluorescence images of H3K27me3 (green) expression in cells manipulated as above Figure 4. bar=100μm. (D) Representative WB images of H3K27me3 using SDS-protein samples of above Figure 5 and its densitometry analyses (n=6). (E) Representative immunofluorescence images of H3K27me3 (green) expression in cells manipulated as above Figure 5. bar=100μm. Data are presented as mean ± SD. #p<0.001. Abbreviation: ns, no significance. |
Furthermore, previous studies have reported that Ezh2 inhibitors, GSK-126 and GSK-343, suppressed Ezh2 methyltransferase activity and thus decrease H3K27me3 levels without interference on Ezh2 expression.22 Therefore, we further checked H3K27me3 levels after treatment of P1 cells with either GSK-126 or GSK-343 (Figure 7A). As expected, the cells treated had no effect on Ezh2 expression, while H3K27me3 expression was significantly reduced as identified by both WB (Figure 7B) and immunofluorescence (Figure 7C). These results further indicate that H3K27me3 levels in AFs were closely related to Ezh2ʹs methyltransferase activities.
 |
Figure 7 Inhibition of Ezh2ʹs methyltransferase activities promotes RS in the early passage of AFs. (A) Schematic diagram of Ezh2 inhibition in P1 AFs. AFs on Day-1 were treated with GSK-126 (1μM, 72h) or GSK-343 (1μM, 72h) and harvested on Day-4 for the following analyses. (B) Representative WB images of Ezh2, H3K27me3 and CDKIs, as well as their densitometry quantitative analyses (n=6). (C) Representative immunofluorescence images of H3K27me3 expression (green). bar=100μm. (D) Representative images of SA-β-gal staining and SA-β-gal activity was quantitated and expressed as a percentage of SA-β-gal-positive cells (n=6). bar=100μm. (E and F) Transcriptions of CDKIs (E) and SASP genes (F) were evaluated by qRT-PCR (n=3). Data are presented as mean ± SD. *p<0.05, **p<0.01, #p<0.001. Abbreviation: ns, no significance. |
In addition, Ezh2 inhibition had similar effects as above si-Ezh2-2 on SA-β-gal activities (Figure 7D), the mRNA and protein levels of CDKIs (Figure 7E) and activated transcription of the SASP genes (Figure 7F). Taken together, these results show that Ezh2 induces RS phenotype mainly through its methyltransferase activity.
H3K27me3 is Enriched in Both CDKN2A and Timp4 Gene Promoter Regions
To further study the underlying mechanism by which H3K27me3 marks regulates RS phenotypes, we performed CUT&Tag assay for the detection of H3K27me3 deposition in AFs at the genome-wide level, given its higher sensitivity as compared with ChIP-seq. First, we analyzed the distribution of H3K27me3-binding peaks within the genome that was classified into functional categories analysis, the results showed that 34.25% genomic regions with specific enrichment of H3K27me3 locate in the promoter regions in AFs (Figure 8A), indicating that H3K27me3 is certainly involved in the regulation of transcriptional process.
 |
Figure 8 H3K27me3 is enriched in CDKN2A and Timp4 gene promoters. AFs in P0 were harvested for CUT&Tag assay, and AFs from P0, P1 and P2 were harvested for qRT-PCR and WB analyses. (A) Pie chart illustrating the distribution of H3K27me3 depositing sites relative to RefSeq functional categories. (B) Heatmap representing normalized H3K27me3 reads across promoters (±3 kb of TSS; Up) and averaged profiles (Bottom). TES; transcriptional end site. (C) CUT&Tag tracks show H3K27me3 potential localizations on the CDKN2a and Timp4 genes. Areas shadowed in red indicate the regions chosen for PCR validations. (D) Schematic diagram (left) showing the relative positions of the primers on the promoters of the CDKN2a (p16 and p19) and Timp4 genes. Arrows indicate the transcription start site (TSS) and direction and short black lines beneath indicate the locations for PCR. PCR bands shown on agarose gels demonstrate that H3K27me3 specifically binds to the promoter regions of CDKN2a (p16 and p19) and Timp4 genes. (E) Transcriptions of Timp1, Timp2, Timp3 and Timp4 genes were evaluated by qRT-PCR (n=3). (F) WB for analyzing Timp1~4 and three collagenase-type MMPs (n=6). Data are presented as mean ± SD. *p<0.05, **p<0.01, #p<0.001. Abbreviation: ns, no significance. |
To further confirm the distribution pattern of the peaks, we analyzed the histone mark reads distribution across transcriptional start site (TSS; ±3.0 Kb) genome wide and found that H3K27me3 enrichment peaked at TSS regions (Figure 8B).
As CDKIs’ significant roles in the progression of RS phenotype and Timps’ function in atrial fibrosis,23 we next analyzed the enrichment of H3K27me3 at the CDKN1a, CDKN2a and Timp1~4 genes’ promoters. As a result, the enrichments were found in CDKN2a and Timp4 genes (Figure 8C), but not in CDKN1a gene and Timp1~3 genes (S-Figure 4). We further designed specific primers pairs for the binding sites of Ezh2 on the promoter regions of these three genes and performed PCR using the CUT&Tag products as templates to testify whether we can obtain the anticipated size of DNA fragments. The results specifically verified our anticipations (Figure 8D). These results indicate not only that our CUT&Tag assay results own high reliability but also that the underlying mechanism by which H3K27me3 marks regulates RS phenotypes is specifically related to the regulation of CDKN2a (p16 and p19) and Timp4ʹs transcriptions.
We further assessed the changes in four Timps and three collagenase-typed MMPs in our RS model of AFs. Of the four Timps, only was Timp4 significantly upregulated in both mRNA and protein levels (Figure 8E and F). Of the three collagenases, only was MMP8 significantly down-regulated (Figure 8F). Surprisingly, these results indicate an imbalance of Timp4-MMP8 in senescent AFs. This imbalance may contribute to fibrotic accumulation of ECM, ie decreased proteolysis and increased collagen deposition.
Either Knockdown or Inhibition of Ezh2 Causes Imbalance of Timp4-MMP8
Whether the imbalance of Timp4-MMP8 is resulted from Ezh2? As shown in S-Figure 5, either knockdown of Ezh2 by Si-Ezh2-2 or inhibition of Ezh2 by GSK-126 or GSK-343 exacerbated the Timp4-MMP8 imbalance in P1 cells. Meanwhile the overexpression of Ezh2 by Adv-Ezh2 partially alleviated the Timp4-MMP8 imbalance in P2 cells (S-Figure 5). The results suggest that either knockdown or inhibition of Ezh2 exacerbates the Timp4-MMP8 imbalance in P1 cells.
Discussion
In the present study, we demonstrated for the first time that there is a huge extent of AFs’ RS in the aged and fibrotic LA and clarified the role and mechanism of Ezh2 in AFs’ RS through the following findings: 1) Ezh2 expression decreased during RS process both in vitro and in vivo; 2) knockdown of Ezh2 expression or inhibition of Ezh2 methyltransferase activity in early passage AFs induced RS phenotype; 3) Overexpression of Ezh2 in late passage AFs alleviated the senescent phenotype; 4) repressive H3K27me3 marks were enriched at both Cdkn2a and Timp4 gene promoter regions.
After Ezh2 was selected from our RNA-seq results, we found that it has a brother Ezh1, which puzzled us for a time whether we needed to simultaneously research them both in this study. We further found they are quite different in expressions and functions. For expressions, Ezh1 is ubiquitously expressed and thus is probably irrelevant to the senescence, whereas Ezh2 is expressed in the proliferating cells.24 Based on our results, we postulated that Ezh2 is expressed in proliferative AFs within non-regenerative adult organs, like LA. We verified its expression in both aged LA tissues and isolated aged AFs. Compared with aged AFs, Ezh2 is expressed abundantly in young AFs. For functions, both Ezh1 and Ezh2 are core components of PRC2 and can separately assemble other components to form PRC2-Ezh1 and PRC2-Ezh2 complexes. Margueron et al reported that PRC2-Ezh2 catalyzes H3K27me2/3 and its knockdown reduces global H3K27me2/3 levels, while PRC2-Ezh1 performs this function weakly.24 Therefore, although Ezh1 and Ezh2 share some of the downstream target genes, PRC2-Ezh1 exerts its inhibitory effect directly through other signal pathways independently of H3K27me3, while PRC2-Ezh2 exerts its inhibitory effect dependently on H3K27me2/3. Therefore, only Ezh2 was finally chosen in this study.
So far, most studies have focused on cardiac fibrosis of the ventricles, in which cardiac fibroblasts rather than AFs are used since access to the former is easy from a bulk volume of the ventricular walls. Even so, that the effects of cardiac fibroblasts’ senescence on the cardiac fibrosis are promotional or inhibitory are still inconclusive.25 Meyer et al found that Trp53−/–Cdkn2a−/− mice displayed aggravated cardiac fibrosis after transverse aortic constriction compared with wild-type controls.26 In contrast, Zhu et al reported that p53−/− mice showed less accumulated senescent cardiac fibroblasts and less infiltrated macrophages with less released levels of MMPs, but more collagen deposition after myocardial infarction.27 How do we explain these seemingly contradictory observations? Actually, the senescence has different types and in each type it has different development phases.28 For premature senescence of the cardiac fibroblast, pro-mitogenic stimuli trigger cell proliferation before inducing irreversible cell-cycle arrest and in most cases that is a short-term process but an important characteristic. Also, that is considered to be a repairing ability of the body to minimize the extent of cardiac fibrosis.29 However, for RS, the proliferation ability of cells gradually declined during the long term until reaching a stable and irreversible cell cycle arrest.30
So far, there have been few studies for AFs’ RS in which there has been no report about Ezh2ʹs role. Therefore, we adopted an in vitro AFs’ RS model to simulate the long-term aging process of AFs’ RS in vivo and found that Ezh2 was consistently down-regulated in aged atrial tissue and in isolated pure senescent AFs as well as in in vitro model. We conclude that Ezh2 is a key regulator of RS, as shown in Graphical Abstract.
Mechanistically, we found that Ezh2-mediated H3K27me3 modification in the promoter regions of CDKN2a and Timp4 genes potentially regulate the development of RS. MMPs and Timps are classically key regulators of ECM turnover during the development of atrial fibrosis.31 The ability to restore levels of collagen turnover to those observed in younger individuals may serve as a clear path toward ameliorating age-related fibrotic disease.32 In reality, however, the complicated functions of Timps and MMPs leave us with a lack of understanding of the tight balance between the two groups.33 On the one hand, as the endogenous inhibitor of MMPs, each Timp can inhibit multiple MMPs in vitro, little is known about the specific regulation process in vivo.34 On the other hand, both Timps and MMPs have functions beyond regulating ECM homeostasis.34,35 These underlie some intertwined and complicated regulatory networks that render them both to be difficult targets for therapeutic intervention. Timp4, also known as cardiac-type inhibitor of MMPs, appears to be the least studied among the four members, and therefore its role in regulating ECM proteolysis is still not fully elucidated.34 A handful of studies have suggested that the imbalance between Timp4 and MMPs occurred during the long-term process of remodeling post-myocardial infarction,36 heart failure,37 and myocardial ischemia-reperfusion injury.38,39 All of these studies showed that Timp4 were downregulated, whereas MMPs were upregulated in diseased hearts over days to months.38 The differences between our results and previous reports may be due to different cell type and experimental conditions.34 In other words, the regulatory role of Timp4 in aging process remains to be further elucidated.
Our study has two major limitations. First, we did not explore the regulatory role of Ezh2 in the process of atrial cardiomyocytes’ senescence. Second, our study was mainly performed in an in vitro model. Next in vivo study is already scheduled to establish Ezh2 conditional knockout mice. However, these limitations should not lower our expectation that restoring the levels of H3K27me3, even partially, in senescent AFs will be potentially beneficial for the treatment of age-related atrial fibrosis.
Conclusion
As shown in Graphical Abstract, this study identified that Ezh2 play a key inhibitory role in RS development by enhancing H3K27me3 levels in the promoter regions of CDKN2a and Timp4 genes in atrial fibroblasts. These findings provide reference for research on age-related atrial fibrosis.
Abbreviations
Afib, atrial fibrillation; AFs, atrial fibroblasts; CDKI, Cyclin-dependent kinase inhibitor; CUT&Tag, Cleavage Under Targets and Tagmentation Assay; DEGs, differentially expressed genes; ECM, Extracellular matrix; EF, Ejection fraction; Ezh2, Enhancer of zeste homolog 2; FS, Fractional shortening; H3K27me3, trimethylated versions of lysine residue 27 within histone H3; IL-1α, Interleukin-1α; IL-1β, Interleukin-1β; IL-6, Interleukin-6; LA, Left atrium; LAD, Left atrial dimension; MCP-1, Monocyte chemotactic protein-1; MMP, Matrix metalloproteinase; qRT-PCR, Quantitative Real-Time PCR; RNA-seq, RNA Sequencing; ROS, Reactive oxygen species; RS, Replicative senescence; SASP, Senescence-associated secretory phenotype; SA-β-gal, Senescence-associated beta-galactosidase; siRNA, Small-interfering RNA; TGF-β, transforming growth factor-β; Timp, tissue inhibitors of metalloproteinase; WB, Western Blotting.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the major research program of NSFC, Grant/Award Number: 91949128; National Natural Science Foundation of China, Grant/Award Numbers: 81873485, 81974041; Zhongnanshan Medical Foundation of Guangdong Province (ZNSA-2020017).
Disclosure
The authors declare that there is no conflict of interests.
References
1. Akoum N, Mahnkopf C, Kholmovski EG, Brachmann J, Marrouche NF. Age and sex differences in atrial fibrosis among patients with atrial fibrillation. EP Eur. 2018;20(7):1086–1092. doi:10.1093/europace/eux260
2. Dzeshka MS, Lip GYH, Snezhitskiy V, Shantsila E. Cardiac fibrosis in patients with atrial fibrillation. J Am Coll Cardiol. 2015;66(8):943–959. doi:10.1016/j.jacc.2015.06.1313
3. Gorgoulis V, Adams PD, Alimonti A, et al. Cellular senescence: defining a path forward. Cell. 2019;179(4):813–827. doi:10.1016/j.cell.2019.10.005
4. Xu J, Zhou L, Liu Y. Cellular senescence in kidney fibrosis: pathologic significance and therapeutic strategies. Front Pharmacol. 2020;11:601325. doi:10.3389/fphar.2020.601325
5. Filippi-Chiela EC, Silva MMB, Thomé MP, Lenz G. Single-cell analysis challenges the connection between autophagy and senescence induced by DNA damage. Autophagy. 2015;11(7):1099–1113. doi:10.1080/15548627.2015.1009795
6. Hou Z, Guo K, Sun X, et al. TRIB2 functions as novel oncogene in colorectal cancer by blocking cellular senescence through AP4/p21 signaling. Mol Cancer. 2018;17(1):172. doi:10.1186/s12943-018-0922-x
7. Shin EY, Park JH, You ST, et al. Integrin-mediated adhesions in regulation of cellular senescence. Sci Adv. 2020;6(19):eaay3909. doi:10.1126/sciadv.aay3909
8. Biernacka A, Frangogiannis NG. Aging and cardiac fibrosis. Aging Dis. 2011;2(2):158–173.
9. Mehdizadeh M, Aguilar M, Thorin E, Ferbeyre G, Nattel S. The role of cellular senescence in cardiac disease: basic biology and clinical relevance. Nat Rev Cardiol. 2022;19(4):250–264. doi:10.1038/s41569-021-00624-2
10. Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128(4):735–745. doi:10.1016/j.cell.2007.02.009
11. Kim J, Lee Y, Lu X, et al. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 2018;25(10):2808–2820.e4. doi:10.1016/j.celrep.2018.11.035
12. Jiang Y, Xiang C, Zhong F, et al. Histone H3K27 methyltransferase EZH2 and demethylase JMJD3 regulate hepatic stellate cells activation and liver fibrosis. Theranostics. 2021;11(1):361–378. doi:10.7150/thno.46360
13. Liu C, Liu L, Yang M, et al. A positive feedback loop between EZH2 and NOX4 regulates nucleus pulposus cell senescence in age-related intervertebral disc degeneration. Cell Div. 2020;15:2. doi:10.1186/s13008-020-0060-x
14. Chen H, Gu X, Su IH, et al. Polycomb protein Ezh2 regulates pancreatic β-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009;23(8):975–985. doi:10.1101/gad.1742509
15. Yang R, Chen J, Zhang J, et al. 1,25-Dihydroxyvitamin D protects against age-related osteoporosis by a novel VDR-Ezh2-p16 signal axis. Aging Cell. 2019;19:e13095. doi:10.1111/acel.13095
16. Wang Q, Yu Y, Zhang P, et al. The crucial role of activin A/ALK4 pathway in the pathogenesis of Ang-II-induced atrial fibrosis and vulnerability to atrial fibrillation. Basic Res Cardiol. 2017;112(4):47. doi:10.1007/s00395-017-0634-1
17. Cao W, Song S, Fang G, Li Y, Wang Y, Wang QS. Cadherin-11 deficiency attenuates Ang-II-induced atrial fibrosis and susceptibility to atrial fibrillation. J Inflamm Res. 2021;14:2897–2911. doi:10.2147/JIR.S306073
18. Kaya-Okur HS, Wu SJ, Codomo CA, et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 2019;10(1):1930. doi:10.1038/s41467-019-09982-5
19. Laredo M, Waldmann V, Khairy P, Nattel S. Age as a critical determinant of atrial fibrillation: a two-sided relationship. Can J Cardiol. 2018;34(11):1396–1406. doi:10.1016/j.cjca.2018.08.007
20. Gramley F, Lorenzen J, Knackstedt C, et al. Age-related atrial fibrosis. AGE. 2009;31(1):27–38. doi:10.1007/s11357-008-9077-9
21. Litwiniec A, Grzanka A, Helmin-Basa A, Gackowska L, Grzanka D. Features of senescence and cell death induced by doxorubicin in A549 cells: organization and level of selected cytoskeletal proteins. J Cancer Res Clin Oncol. 2010;136(5):717–736. doi:10.1007/s00432-009-0711-4
22. Asangani IA, Ateeq B, Cao Q, et al. Characterization of the EZH2-MMSET histone methyltransferase regulatory axis in cancer. Mol Cell. 2013;21:80–93.
23. Safwan-Zaiter H, Wagner N, Michiels JF, Wagner KD. Dynamic spatiotemporal expression pattern of the senescence-associated factor p16Ink4a in development and aging. Cells. 2022;11(3):541. doi:10.3390/cells11030541
24. Margueron R, Li G, Sarma K, et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell. 2008;32(4):503–518. doi:10.1016/j.molcel.2008.11.004
25. Meng X, Wang H, Song X, Clifton AC, Xiao J. The potential role of senescence in limiting fibrosis caused by aging. J Cell Physiol. 2020;235(5):4046–4059. doi:10.1002/jcp.29313
26. Meyer K, Hodwin B, Ramanujam D, Engelhardt S, Sarikas A. Essential role for premature senescence of myofibroblasts in myocardial fibrosis. J Am Coll Cardiol. 2016;67(17):2018–2028. doi:10.1016/j.jacc.2016.02.047
27. Zhu F, Li Y, Zhang J, et al. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS One. 2013;8(9):13.
28. von Kobbe C. Cellular senescence: a view throughout organismal life. Cell Mol Life Sci. 2018;75(19):3553–3567. doi:10.1007/s00018-018-2879-8
29. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15(7):482–496. doi:10.1038/nrm3823
30. Campisi J, d’Adda Di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740. doi:10.1038/nrm2233
31. Koskivirta I, Kassiri Z, Rahkonen O, et al. Mice with tissue inhibitor of metalloproteinases 4 (Timp4) deletion succumb to induced myocardial infarction but not to cardiac pressure overload. J Biol Chem. 2010;285(32):24487–24493. doi:10.1074/jbc.M110.136820
32. Podolsky MJ, Yang CD, Valenzuela CL, et al. Age-dependent regulation of cell-mediated collagen turnover. JCI Insight. 2020;5(10):e137519. doi:10.1172/jci.insight.137519
33. Vanhoutte D, Heymans S. TIMPs and cardiac remodeling: “Embracing the MMP-independent-side of the family”. J Mol Cell Cardiol. 2010;48(3):445–453. doi:10.1016/j.yjmcc.2009.09.013
34. Arpino V, Brock M, Gill SE. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015;44–46:247–254. doi:10.1016/j.matbio.2015.03.005
35. Takawale A, Sakamuri SSVP, Kassiri Z. Extracellular matrix communication and turnover in cardiac physiology and pathology. In: Terjung R, editor. Comprehensive Physiology. John Wiley & Sons, Inc.; 2015:687–719. doi:10.1002/cphy.c140045
36. Zavadzkas JA, Stroud RE, Bouges S, et al. Targeted overexpression of tissue inhibitor of matrix metalloproteinase-4 modifies post-myocardial infarction remodeling in mice. Circ Res. 2014;114(9):1435–1445. doi:10.1161/CIRCRESAHA.114.303634
37. Chaturvedi P, Tyagi SC. Epigenetic silencing of TIMP4 in heart failure. J Cell Mol Med. 2016;20(11):2089–2101. doi:10.1111/jcmm.12901
38. Schulze CJ, Wang W, Suarez-Pinzon WL, Sawicka J, Sawicki G, Schulz R. Imbalance between tissue inhibitor of metalloproteinase-4 and matrix metalloproteinases during acute myocardial ischemia-reperfusion injury. Circulation. 2003;107(19):2487–2492. doi:10.1161/01.CIR.0000065603.09430.58
39. Takawale A, Fan D, Basu R, et al. Myocardial recovery from ischemia-reperfusion is compromised in the absence of tissue inhibitor of metalloproteinase 4. Circ Heart Fail. 2014;7(4):652–662. doi:10.1161/CIRCHEARTFAILURE.114.001113
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.