Back to Journals » Journal of Hepatocellular Carcinoma » Volume 12
EZH2: From Oncogenic Driver to Therapeutic Target for Overcoming Drug Resistance in Hepatocellular Carcinoma
Authors Tang W ![]() , Cao J, Wang N, Tang L, Cao J
, Cao J, Wang N, Tang L, Cao J
Received 16 July 2025
Accepted for publication 23 December 2025
Published 31 December 2025 Volume 2025:12 Pages 3091—3104
DOI https://doi.org/10.2147/JHC.S554181
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Jörg Trojan
Weijing Tang,1 Jianzhong Cao,1 Nan Wang,2 Liwen Tang,2 Jianxiong Cao1,2
1School of Traditional Chinese Medicine, Hunan University of Chinese Medicine, Changsha, Hunan, People’s Republic of China; 2Department of Oncology, the First Hospital of Hunan University of Chinese Medicine, Changsha, Hunan, People’s Republic of China
Correspondence: Liwen Tang, Department of Oncology, the First Hospital of Hunan University of Chinese Medicine, Changsha, Hunan, People’s Republic of China, Email [email protected] Jianxiong Cao, School of Traditional Chinese Medicine, Hunan University of Chinese Medicine, Changsha, Hunan, People’s Republic of China, Email [email protected]
Abstract: Hepatocellular carcinoma (HCC) remains a therapeutic challenge due to the high prevalence of drug resistance. The histone methyltransferase Enhancer of Zeste Homolog 2 (EZH2), a core component of the Polycomb Repressive Complex 2, is frequently overexpressed in HCC and drives of drug resistance. This review delineates the multifaceted mechanisms by which EZH2 promotes resistance to chemotherapy, targeted therapy, and immunotherapy. We detail how EZH2 orchestrates pro-survival pathways by modulating cell cycle checkpoints, inhibiting apoptosis, enhancing DNA repair, and fostering an immunosuppressive tumor microenvironment. Furthermore, we evaluate the current landscape of EZH2 inhibitors, from clinically approved agents to novel therapeutic modalities like PROTACs, and discuss their potential to re-sensitize HCC to treatment. Finally, we outline future research directions, emphasizing combination strategies and biomarker development, to advance EZH2-targeting therapies for HCC.
Keywords: EZH2, hepatocellular carcinoma, mechanisms of drug resistance, epigenetics
Introduction
Hepatocellular carcinoma (HCC) ranks as the third leading cause of cancer-related mortality worldwide.1 Its development is predominantly associated with chronic liver diseases, including viral hepatitis, alcohol-related liver disease, non-alcoholic fatty liver disease, and cirrhosis.2 Despite progress in early detection and therapeutic interventions, the five-year survival rate for HCC patients remains dismal at below 20%,3 largely attributable to the tumor’s aggressive behavior,4 high recurrence rates,5 and a constrained arsenal of effective treatments.6
Current clinical management of HCC includes surgical resection, liver transplantation, locoregional therapies, and systemic agents such as sorafenib and lenvatinib.6 However, the efficacy of these strategies is consistently hampered by limited effectiveness, the almost inevitable development of drug resistance, and significant adverse effects. For instance, sorafenib achieves an objective response rate of only 2–3%, with most patients developing acquired resistance within 6 to 8 months of treatment initiation.7 While immune checkpoint inhibitors have emerged as a promising modality, their monotherapy response rates remain modest at 10–20%,8 highlighting the pervasive and unresolved challenge of drug resistance in HCC management. This underscores the urgent and unmet need to identify novel therapeutic targets and strategies to improve patient outcomes.
Recent research has highlighted the critical involvement of enhancer of zeste homolog 2 (EZH2) in driving drug resistance in HCC.9,10 As the catalytic core of the polycomb repressive complex 2 (PRC2), EZH2 is predominantly localized in the nucleus, where it mediates the trimethylation of histone H3 at lysine 27 (H3K27me3), leading to the epigenetic silencing of numerous tumor suppressor genes and promoting HCC cell tolerance to both therapeutic agents.9–12 For instance, a recent study revealed that EZH2 functions as a central driver in hepatocellular carcinoma, where its upregulation directly reactivates ErbB2/ErbB3 signaling, culminating in acquired resistance to the FGFR inhibitor infigratinib.9 Furthermore, EZH2 undermines immunotherapy by remodeling the tumor immune microenvironment, facilitating immune cell exclusion and evasion mechanisms.13 The promising activity of EZH2 inhibitors, such as tazemetostat, in reversing resistance and sensitizing HCC cells to treatment positions EZH2 as a compelling therapeutic target.9 This review aims to comprehensively summarize the mechanisms by which EZH2 mediates drug resistance in HCC. By synthesizing current knowledge, we seek not only to elucidate the intrinsic drivers of therapeutic failure but also to inform the rational development of effective EZH2-targeting strategies to overcome resistance and improve the prognosis for HCC patients.
EZH2
Structure and Function
Polycomb group (PcG) proteins are essential epigenetic regulators of gene silencing, functioning within multi-subunit complexes, primarily polycomb repressive complex 1 (PRC1) and PRC2. PRC2 is distinguished as the principal complex responsible for catalyzing the trimethylation of H3K27me3.14 Structurally, the canonical PRC2 complex comprises three core subunits: the catalytic methyltransferase EZH2, along with embryonic ectoderm development (EED) and suppressor of zeste 12 (SUZ12), which are essential for its stability and activity. The methyltransferase function is housed within the C-terminal SET domain of EZH2, which interacts with EED and SUZ12 to form an active site that utilizes s-adenosyl methionine (SAM) as a methyl donor. This catalytic process drives H3K27me3 deposition, leading to chromatin compaction and transcriptional repression of target genes, including key tumor suppressors.15 EED plays a critical role in allosterically stabilizing the active conformation of the complex.
The predominant and most characterized EZH2 variant is isoform A, an 86 kDa protein of 751 amino acids. Its modular structure features an N-terminal regulatory domain that mediates interactions with other PRC2 subunits,10 and a C-terminal catalytic domain that directly governs its methyltransferase activity.16 It is also important to note the existence of two distinct PRC2 variants, PRC2.1 and PRC2.2, which share the core EZH2-EED-SUZ12 trimer but incorporate different accessory proteins. For instance, JARID2 is exclusively associated with PRC2.2, suggesting functional specialization in their regulatory targets (Figure 1).
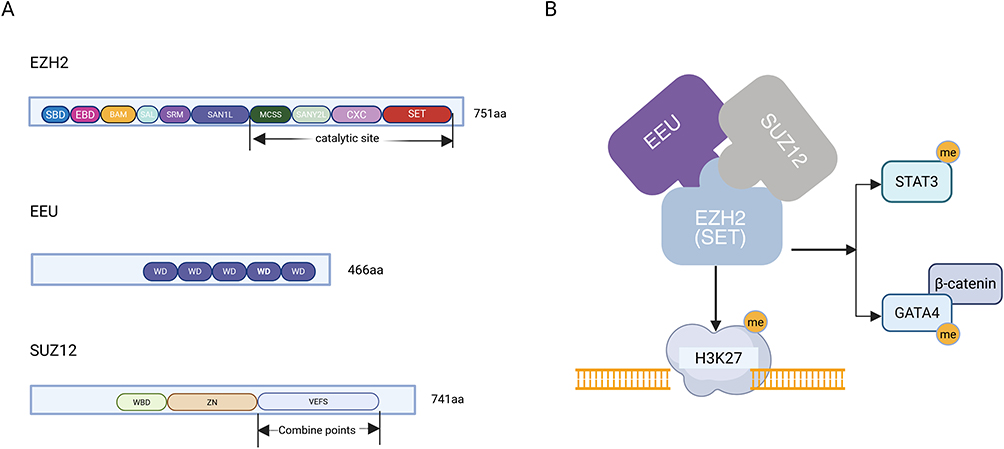 |
Figure 1 Architectural and functional overview of PRC2. (A) Schematic representation of the canonical domain organization for the core subunits EZH2 (isoform A), EED, and SUZ12, which illustrates their primary protein domains. (B) The PRC2 complex, led by EZH2’s methyltransferase activity, exerts dual regulatory functions: it silences gene transcription via H3K27me3 and directly regulates protein activity by methylating non-histone substrates. Figure was created in https://BioRender.com. Abbreviations: PRC2, Polycomb Repressive Complex 2; EZH2, enhancer of zeste homolog 2; EED, embryonic ectoderm development; SUZ12, suppressor of zeste 12; H3K27me3, histone H3K27 trimethylation. |
Beyond its canonical role in histone methylation, EZH2 exhibits multifaceted oncogenic functions. It can augment its own enzymatic activity through automethylation and directly influence oncogenic signaling by methylating non-histone proteins. A key example in HCC is the formation of a ternary complex with β-catenin and the long non-coding RNA LNC-β-CATM, where EZH2-mediated methylation of β-catenin stabilizes the protein, leading to constitutive activation of the Wnt signaling pathway and driving tumor growth.17 Furthermore, EZH2 drives tumor immune escape through its ability to epigenetically silence miRNAs and reprogram the immune microenvironment by controlling the functions of mature immune cells, including lymphocytes and myeloid cells.18 Through these diverse mechanisms, EZH2 establishes itself as a pivotal therapeutic target, with its aberrant activation being strongly correlated with tumor invasion, metastasis, and poor prognosis in HCC and other malignancies.
Expression Characteristics and Clinical Significance of EZH2 in HCC
EZH2 is consistently overexpressed in HCC tissues compared to normal liver parenchyma and is strongly associated with aggressive clinicopathological features,19 including advanced tumor grade, enhanced invasive potential, and promoted angiogenesis.20 Consequently, EZH2 overexpression has been validated as a powerful, independent prognostic biomarker, reliably predicting reduced overall survival in HCC patients.20
The oncogenic impact of EZH2 stems from its direct role in driving malignant phenotypes. It facilitates aberrant cell cycle progression and suppresses apoptosis primarily through the epigenetic silencing of critical tumor suppressor genes, such as p21 and LINC00978.21,22 Furthermore, EZH2 gene amplification or acquired somatic mutations can potentiate its activity, fostering drug resistance and metastasis by enhancing the self-renewal capacity of cancer stem cells.
The clinical utility of EZH2 is underscored by its integration into prognostic models. For instance, Zhou et al developed an EZH2-based prognostic model using a pathological score based on histopathological images.23 A score >0.4628 defined high-risk patients, who showed significantly increased mortality, indicating EZH2 as a potential target for non-invasive prognosis and precision therapy in HCC. Targeting EZH2 represents a promising avenue to overcome drug resistance, and a detailed elucidation of its molecular mechanisms, as summarized in Table 1 and Figure 2, is paramount for developing innovative diagnostic and therapeutic strategies against this lethal malignancy.
 |
Table 1 Mechanisms by Which EZH2 Regulates Drug Resistance in HCC |
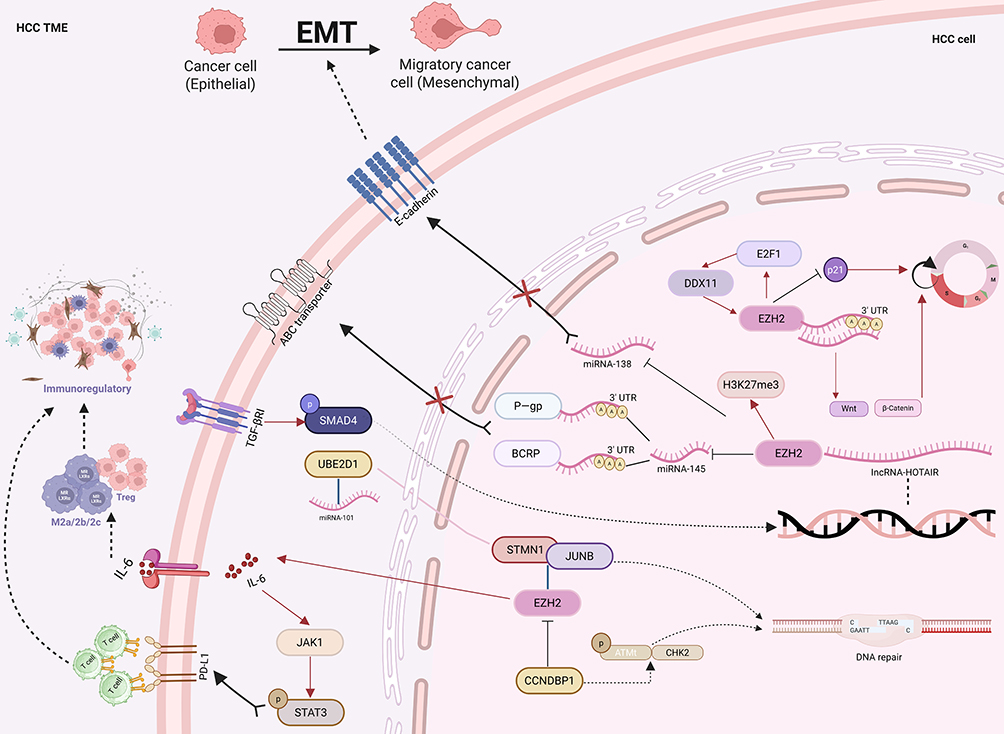 |
Figure 2 This schematic illustrates the key molecular pathways through which EZH2 confers resistance to chemotherapy, targeted therapy, and immunotherapy in HCC. The core mechanisms include: dysregulation of the cell cycle and DNA repair via the E2F1/DDX11/EZH2 axis that silences p21 to abrogate G1/S checkpoint control, alongside enhanced DNA repair through EZH2-mediated stabilization of the STMN1/JUNB complex and, conversely, CCNDBP1-suppressed activation of the miR-101/UBE2D1 and ATM/CHK2 pathways; remodeling of the immunosuppressive microenvironment via EZH2-induced IL-6 secretion that promotes expansion of Tregs and MDSCs, combined with JAK/STAT3 activation to upregulate PD-L1 and directly inhibit CD8⁺ T cell function; activation of epithelial–mesenchymal transition through EZH2-directed H3K27me3 epigenetic silencing of the tumor suppressor miR-138; and induction of drug efflux pump activity by the TGF- β1/SMAD4 axis upregulating HOTAIR, which recruits EZH2 to repress miR-145, thereby relieving its inhibition of the efflux transporters P-gp and BCRP and reducing intracellular drug accumulation. Figure was created in https://BioRender.com. Abbreviations: EZH2, enhancer of zeste homolog 2; HCC, Hepatocellular Carcinoma; Tregs, Regulatory T cells; MDSCs, Myeloid-derived suppressor cells; PD-L1, Programmed cell death 1 ligand 1; H3K27me3, histone H3K27 trimethylation; P-gp, P-Glycoprotein; BCRP, breast cancer resistance protein. |
Mechanisms of EZH2-Mediated Drug Resistance in HCC
Regulation of Cell Cycle and Apoptosis
Dysregulation of the cell cycle is a hallmark of cancer, and EZH2 is a pivotal contributor to this process in HCC, directly conferring resistance to chemotherapeutic agents like cisplatin, paclitaxel, and doxorubicin.19,31,37 As a critical negative regulator of the cell cycle, p21 inhibits cyclin-dependent kinase complexes to arrest cell cycle progression.39,40 EZH2 suppresses p21 expression via H3K27me3 modification, facilitating the G1/S phase transition and enabling continuous proliferation even under genotoxic stress.22,41 Similarly, EZH2 represses the cell cycle inhibitor miR-122, further accelerating proliferation.42 Importantly, pharmacological inhibition of EZH2 restores the expression of both p21 and miR-122, leading to suppressed proliferation, induced apoptosis, and the reversal of chemoresistance in preclinical HCC models,42 validating the therapeutic potential of this approach.
The regulatory network extends to other tumor-suppressive miRNAs, which are master regulators of cell cycle, apoptosis, and chemoresistance.43,44 For instance, miR-98 directly targets the 3’-untranslated region (3’-UTR) of EZH2 mRNA, repressing its expression and subsequently inhibiting the Wnt/β-catenin signaling pathway, leading to G1/S phase arrest and suppression of proliferation,24 thereby overcoming sorafenib resistance.45 Conversely, loss of miR-98 leads to EZH2 accumulation, Wnt pathway activation, and uncontrolled proliferation.24 EZH2 also functions downstream of such regulatory miRNAs; it drives hepatocarcinogenesis and chemoresistance by epigenetically upregulating SETDB1 through H3K27me3, which in turn silences miR-381 and activates AKT signaling pathway.26
In addition to cell cycle dysregulation, the suppression of apoptosis is a critical mechanism of drug resistance. EZH2 is targeted by several apoptosis-inducing miRNAs. miR-1297, for example, exerts dual anti-tumor effects by directly targeting EZH2: its ectopic expression suppresses proliferation and sensitizes cells to apoptosis,46 while its-mediated downregulation of EZH2 attenuates cisplatin resistance.47,48
The central tumor suppressor phosphatase and tensin homolog (PTEN), a negative regulator of the PI3K/AKT pathway, is a frequent target of EZH2-mediated silencing, and its loss is a well-established driver of resistance to doxorubicin, oxaliplatin, and 5-fluorouracil (5-Fu).49,50 In HCC, EZH2 is recruited by the long non-coding RNA CASC11 to repress PTEN transcription through H3K27me3 deposition, effectively blocking apoptosis.27 Furthermore, the lncRNA SNHG9 recruits EZH2 to silence PTEN, thereby enhancing the self-renewal and tumorigenicity of liver cancer stem cells.28 These findings illustrate a consistent theme where EZH2, often in concert with various non-coding RNAs, disrupts both cell cycle checkpoints and apoptotic pathways to foster a resistant and aggressive tumor phenotype.
DNA Repair and Genomic Stability
The efficacy of DNA-damaging chemotherapeutic agents, which exert their anticancer effects by inducing DNA double-strand breaks (DSB), is limited by enhanced DNA repair capacity of cancer cells.51 The DNA damage response (DDR) acts as a guardian of genomic integrity, coordinating DNA repair and cell cycle checkpoints. However, DDR dysregulation presents a “double-edged sword”: while its failure promote tumorigenesis, its hyperactivation in established tumors confers resistance to genotoxic therapies like cisplatin (cDDP) and 5-FU.52 Tumor cells achieve this by aberrantly activating DSB repair pathways, such as non-homologous end joining (NHEJ) and homologous recombination (HR). This rationale underpins the strategy of pharmacologically targeting DDR components to subvert tumor cell survival, thereby converting genotoxic stress into a lethal vulnerability and overcoming chemoresistance.
A critical epigenetic regulator of this process is EZH2, which governs cellular responses to DNA damage by controlling the expression of repair genes, thereby directly influencing sensitivity to radiotherapy and DNA-targeting chemotherapy. In HCC, EZH2 is a central element of the miR-101 regulatory network, where it synergizes with STMN1 and JUNB to promote hepatocarcinogenesis and resistance.29–31 Moreover, miR-101/UBE2D1 axis has been shown to determines HCC cell responsiveness to cDDP and 5-FU by modulating DDR pathways through EZH2.34
Further illustrating the complex role of the DDR, the typically tumor-suppressive protein Cyclin D1 binding protein 1 (CCNDBP1) is paradoxically overexpressed and exhibits oncogenic activity by augmenting DNA repair capacity.35 CCNDBP1 mediates radio- and chemotherapy resistance through a coordinated mechanism involving the transcriptional repression of EZH2 and concomitant hyperactivation of the ATM/CHK2 signaling pathway, evidenced by significantly elevated levels of phosphorylated ATM (p-ATM) and CHK2 (p-CHK2).53 Validation in CCNDBP 1-knockout murine models confirms this pathway’s role and highlights the therapeutic potential of targeting EZH2 to counteract this adaptive resistance mechanism in DNA damage-based therapies.53
Tumor Microenvironment Regulation
The immunosuppressive tumor microenvironment (TME) is a well-established extrinsic driver of immune escape in HCC, critically limiting the efficacy of immunotherapies.32,33,54 Multi-omics analyses reveal that this hostile niche orchestrates immune evasion through two interconnected mechanisms: the specific overexpression of immune checkpoint molecules, including programmed cell death 1 ligand 1 (PD-L1) and programmed cell death 1 (PD-1),55,56 and substantial enrichment of immunosuppressive cellular populations. These alterations collectively induce a state of functional immune paralysis, characterized by impaired MHC-I antigen presentation and the exhaustion of CD8⁺ T cells,57 often marked by TIM-3 expression.58 The self-reinforcing immunosuppressive network correlates strongly with poor clinical outcomes, as reflected in the low objective response rates (<20%) to immune checkpoint blockade,8 thereby establishing the TME as a fundamental therapeutic target for overcoming immunotherapy resistance in HCC.
A pivotal cytokine mediating this immunosuppressive reprogramming is IL-6. Acting as a core inflammatory signal, IL-6 reshapes the tumor immune microenvironment (TIME) to foster immune escape and broad therapeutic resistance.59 Sustained IL-6 secretion from both cancer cells and cancer-associated fibroblasts (CAFs) activates the JAK/STAT3 signaling pathway. This activation induces PD-L1 upregulation, directly inhibits CD8⁺ T cell function, and promotes the expansion of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), thereby establishing a potent immunosuppressive circuit.36 Concurrently, IL-6 drives M2-type tumor-associated macrophage (TAM) polarization via STAT3, stimulating the secretion of VEGF and TGF-β to promote angiogenesis and matrix fibrosis, which further aggravates HCC invasion and resistance.36 The clinical relevance of this axis is underscored by the positive correlation between IL-6 levels and resistance to sorafenib and regorafenib,60 suggesting that combining IL-6 inhibitors with existing therapies could surmount current therapeutic limitations.
Crucially, EZH2 sits upstream of this process, directly elevating IL-6 production and secretion to promote MDSC recruitment and impair CD8⁺ T cell activity.36,61 The finding that the efficacy of anti-PD-1/PD-L1 therapy is markedly enhanced following EZH2 inhibition,62 positioning EZH2-targeting agents as potent modulators of the immunosuppressive TME.
Regulation of EMT and Metastasis-Related Genes
Epithelial-mesenchymal transition (EMT) is a fundamental molecular program that drives tumor progression by conferring cancer cells with enhanced invasiveness, anti-apoptotic capacity, and therapeutic resistance through extensive epigenetic and signaling network reprogramming Gene expression profiles associated with EMT are strongly correlated with drug resistance in various cancers,63 including HCC.64 EZH2 directly orchestrates EMT by coordinating the expression of key transcription factors such as Snail, Slug, and Twist. This is achieved through a dual mechanism: the suppression of epithelial markers like E-cadherin and the concurrent activation of mesenchymal effector proteins such as Vimentin. This EZH2-driven reprogramming enhances metastatic potential and fosters resistance to therapy.
A specific example is the miR-138/EZH2 axis. Zeng et al identified that reduced miR-138 expression promotes cisplatin resistance by targeting EZH2.37 MiR-138 mimics lower EZH2 protein levels and reverse cisplatin-induced EMT phenotypes, whereas EZH2 overexpression counteracts chemosensitizing effect. This delineates a critical feedback loop where the miR-138/EZH2 axis modulates the EMT program to drive the resistance cycle.
The connection between EMT and resistance is exemplified by BIRC5 (Survivin), an EMT-linked apoptosis suppressor that is elevated in HCC and correlates with poor clinical outcomes.65 EZH2 has been identified as a key gene associated with BIRC5 expression. Prognostic models incorporating EZH2 not only accurately predict patient survival but also help stratify potential responders to immune checkpoint blockade through immunophenotypic and tumor immune dysfunction and exclusion (TIDE) analyses.64 Collectively, these findings establish EZH2’s utility in prognostic assessment, providing a valuable framework for precision oncology in HCC management.
Regulation of Drug Efflux Pump
Multidrug resistance (MDR), a phenomenon where cancer cells develop resistance to a wide spectrum of chemotherapeutic agents, poses a major clinical challenge. In HCC, MDR is mechanistically driven by the aberrant activation of the TGF-β1 signaling pathway and the overexpression of ATP-binding cassette (ABC) transporter proteins.25,66 Among these transporters, P-glycoprotein (P-gp/ABCB1) and the breast cancer resistance protein (BCRP/ABCG2) are the most critical determinants of chemotherapy-induced MDR. They mediate resistance by actively effluxing drugs, thereby reducing intracellular concentrations. Specifically, P-gp facilitates the export of doxorubicin, paclitaxel, and 5-FU,67–69 while BCRP predominantly mediates resistance to sorafenib and lenvatinib.25,70
A key molecular axis linking TGF-β1 to MDR involves the long non-coding RNA HOTAIR. Previous study has shown that TGF-β1 induces HOTAIR upregulation in Smad4-dependent manner.25 HOTAIR silencing enhances the expression of miR-145, which directly binds to the 3’-UTRs of ABCB1 (P-gp) and ABCG2 (BCRP) mRNAs, suppressing their translation and counteracting drug efflux capability.
EZH2 plays a central and dual role in this regulatory cascade. First, EZH2 is required for maintaining miR-145 repression, as EZH2 knockdown reduces miR-145 levels, suggesting EZH2 regulates miR-145 stability or transcription via H3K27me3-mediated silencing. Second, HOTAIR recruits the EZH2-containing PRC2 complex to synergistically inhibit miR-145 expression. This coordinated action relieves miR-145’s suppression of the efflux pumps, thereby promoting the MDR phenotype in HCC.
Strategies to Overcome EZH2-Mediated Drug Resistance in HCC
The emergence of acquired drug resistance poses a critical barrier to the success of chemotherapy, targeted therapy, and immunotherapy in HCC. Current therapeutic strategies prioritize targeting EZH2 directly or modulating its regulatory network, which includes PRC2 complex components and DNA damage repair pathways; the objective is to overcome resistance through multi-target synergistic approaches. Therefore, combination therapies integrating EZH2 inhibitors with immune checkpoint inhibitors now represent a promising therapeutic paradigm for HCC management.
Application of EZH2 Inhibitors
EZH2 represents a compelling therapeutic target for HCC, with significant progress achieved over the past decade. Current inhibitors fall into three mechanistic categories: direct suppression of EZH2 methyltransferase activity, disruption of EZH2-PRC2 complex interactions, and enhancement of EZH2 ubiquitination-mediated degradation (Table 2 and Figure 3).
 |
Table 2 Mechanism and Clinical Trials of Drugs Against EZH2 |
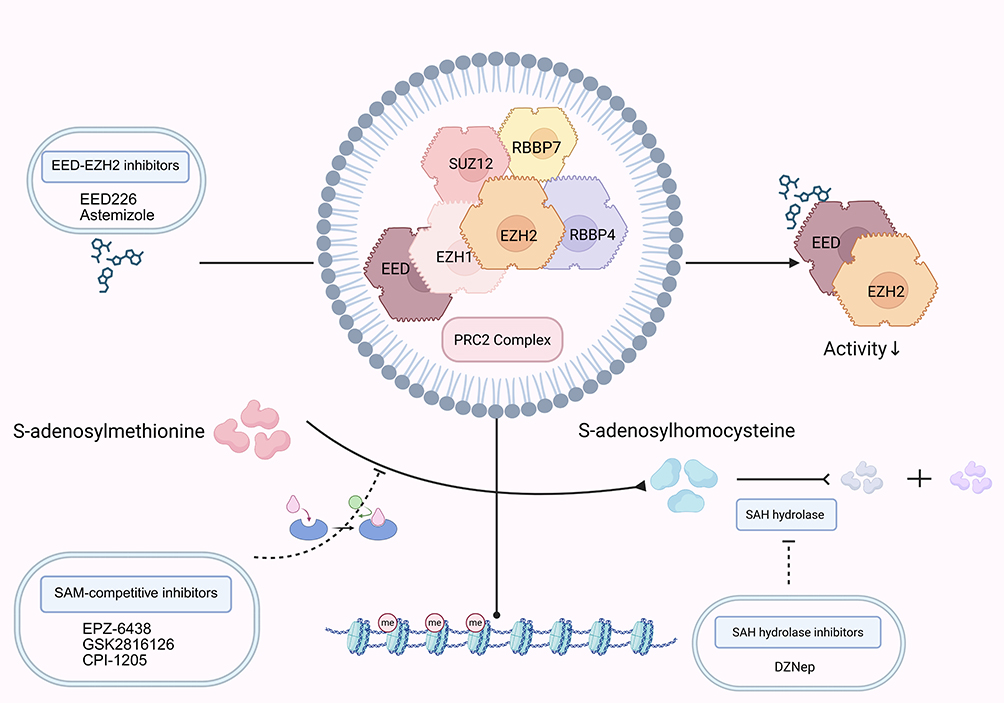 |
Figure 3 This schematic summarizes the principal approaches for targeting EZH2, categorized by their mechanisms of action: direct catalytic blockade using SAM-competitive inhibitors that occupy the substrate-binding site; targeted protein degradation via PROTACs that induce E3 ligase-mediated ubiquitination and proteasomal breakdown; and allosteric inhibition through disruption of protein–protein interactions within the PRC2, leading to its disassembly. The downward arrow indicates decreased activity. Figure was created in https://BioRender.com. Abbreviations: EZH2, enhancer of zeste homolog 2; SAM, S-adenosyl methionine; PROTAC, Proteolysis–targeting chimera; PRC2, Polycomb repressive complex 2. |
Tazemetostat (EPZ-6438) remains the most clinically advanced EZH2 inhibitor, having received Food and Drug Administration approval in 2020 for advanced epithelioid sarcoma and follicular lymphoma.71 This SAM-competitive inhibitor binds EZH2’s SET domain, abolishing aberrant H3K27 methylation.71 Preclinical study demonstrates that tazemetostat potentiates chimeric antigen receptor T-cell immunotherapy (CAR-T) efficacy by upregulating adhesion molecules and inflammatory mediators across various solid and hematologic malignancies.72 EZH1/EZH2 dual-target inhibitor valemetostat similarly synergizes with CAR-T therapy to enhance treatment responses. Moreover, computational and in vitro study established tazemetostat’s triple inhibition of ABCB1, ABCC1, and ABCG2 efflux transporters as an effective multidrug resistance reversal strategy.73
Targeting PRC2 subunit interactions provides an alternative inhibition strategy.74 Next-generation PRC2 inhibitors compromise complex assembly through selective binding to EZH2’s SET domain or disruption of SUZ12-EED protein-protein interfaces.74 The protein-protein interaction inhibitor LG1980 exerts anti-tumor effects by binding EED, triggering EZH2 degradation and PRC2 complex destabilization while disrupting the non-canonical EZH2-STAT3 pathway in chemotherapy-resistant prostate cancer.75 Investigations revealed that co-administration of EED226 with SAM-competitive inhibitors synergistically reduces H3K27me3 levels and suppresses proliferation in EZH2-mutant resistant cell lines, unveiling new avenues for overcoming therapeutic resistance.76
Collectively, EZH2 inhibitor research has generated preclinical and clinical advancements. Nevertheless, challenges persist—the valemetostat, despite regulatory approval for adult T-cell leukemia/lymphoma, presents CYP450-mediated metabolic complications that may compromise treatment efficacy, underscoring the need for pharmacokinetic refinement and safety optimization.77 These developments signal a paradigm shift from monotherapy to combination regimens in EZH2-targeted approaches, though metabolic toxicity remains a pivotal challenge requiring resolution.
Multi-Target Combination Therapy Strategy
Research demonstrates that nearly 80% of HCC cases display aberrant expression of fibroblast growth factor (FGF) and its receptors (FGFRs).78 FGFR4 exhibits specific overexpression in HCC tissue,79 and its inhibition triggers compensatory EZH2 upregulation, exacerbating tumor invasiveness and conferring resistance to both chemotherapeutic agents and immunotherapies.38 Confirmatory study established that this dual-target approach synergistically induces apoptosis and overcomes drug resistance, unmasking compensatory FGFR4/EZH2 signaling activation as a key resistance mechanism while validating the synergistic anti-tumor efficacy of combinatorial inhibition.38
Another investigations revealed that IGF1R inhibitors effectively reverse EZH2-mediated sorafenib resistance, proposing coordinated targeting of the EZH2/miRNA/IGF1R axis as a novel strategy to disrupt chemoresistance.80 Furthermore, BRD4, a BET family protein that promotes HCC proliferation and suppresses apoptosis through interactions with transcriptional regulators and epigenetic modifiers, shows remarkable synergy with EZH2 inhibition. Combined treatment with BRD4 inhibitor ZBC260 and EZH2 inhibitor CPI-169 synergistically augmented HCC cell apoptosis,81 highlighting this combination’s potential to circumvent therapeutic resistance.
Emerging evidence particularly underscores the therapeutic value of combining EZH2 inhibitors with immune checkpoint blockade in HCC treatment. Epigenetic modulation via EZH2 inhibition reduces H3K27me3 levels at CD274 and IRF1 promoters, thereby elevating PD-L1 expression.13 A team from Beijing University elucidated that EZH2 inhibition stabilizes PD-L1 through USP22-mediated deubiquitylation, potentiating tumor responsiveness to anti-PD-1 therapy.82 This mechanistic insight supports clinical exploration of EZH2 inhibitor-immunotherapy combinations. Supporting this approach, Palomba et al documented enhanced anti-tumor responses and prolonged progression-free survival in diffuse large B-cell lymphoma patients receiving EZH2 inhibitor/anti-PD-L1 combination therapy.83
Collectively, these studies delineate multi-target combinatorial approaches as effective countermeasures against EZH2-mediated drug resistance in HCC. Future investigations should prioritize identifying optimal therapeutic partners for EZH2 inhibitors to refine resistance management paradigms.
Targeted Protein Degradation Strategy: PROTACs
Currently, the clinical translation of EZH2 inhibitors still faces multiple obstacles.84 The pronounced heterogeneity of HCC underlies marked variability in patient responses to EZH2 inhibitors, with some patients experiencing limited long-term benefit due to acquired tolerance or severe adverse effects such as myelosuppression. Drug resistance arises not only through compensatory activation of pro-survival pathways-including PI3K/AKT,17 Wnt/β-catenin,13 and ATM/CHK2 signaling following EZH2 inhibition-but also involves PRC2-independent EZH2 functions.64 Capitalizing on EZH2’s non-canonical roles in chromatin dynamics and transcriptional control, recent advances have pioneered targeted protein degradation approaches.
Proteolysis-targeting chimera (PROTAC) technology employs heterobifunctional molecules to recruit E3 ubiquitin ligases for targeted protein degradation via the ubiquitin-proteasome system. PROTACa comprises three structural components: a target protein-binding warhead, an E3 ligase-recruiting moiety, and a flexible linker. Unlike conventional inhibitors requiring active-site engagement, PROTACs catalytically degrade entire proteins regardless of mutation status or overexpression levels. For EZH2 inactivation, conjugation of EPZ6438 with CRBN ligand thalidomide yields EZH2 PROTAC 1, which abolishes EZH2-driven oncogenicity. Development of VHL ligand-conjugated EPZ6438 disrupts EZH2’s non-enzymatic functions.85 Notably, PROTACs utilizing VHL or CRBN ligases bind both EZH2’s WD40 domain and E3 ligases,86,87 inducing polyubiquitination and subsequent proteasomal destruction. This mechanism circumvents resistance from EZH2 catalytic site mutations (Y641, A687) and overexpression-driven oncogenicity observed in lymphomas and solid tumors.87 Preclinical study demonstrates PROTAC-mediated EZH2 degradation reduces H3K27me3 levels by >80% within 24 hours, compared to 50% inhibition with EPZ6438 alone.88 Clinical-stage candidates like ARV-110 and ARV-471 validate PROTAC’s translational potential, with Phase I trials showing tumor regression in treatment-resistant cancers.89
Challenges and Future Perspectives
In conclusion, this review has systematically delineated the pivotal role of the epigenetic regulator EZH2 as a core epigenetic regulator of multidrug resistance in HCC. EZH2 orchestrates resistance to chemotherapy, targeted therapy, and immunotherapy in HCC through multifaceted mechanisms, including H3K27me3-dependent gene silencing, activation of key oncogenic pathways, and remodeling of the immune microenvironment.77
EZH2 exerts multidimensional control over the tumor immune microenvironment. Particularly noteworthy is its “bidirectional regulation” of PD-L1 expression: on one hand, EZH2 stimulates IL-6 production, which stabilizes PD-L1 protein expression while directly driving the expansion of MDSCs and Tregs, thereby shaping an immunosuppressive microenvironment;36 on the other hand, its canonical catalytic function can suppress PD-L1 transcription by elevating H3K27me3 levels on the promoters of CD274 and interferon regulatory factor 1.13 Consequently, for HCC characterized as an “immunologically cold” tumor, future EZH2-targeting therapeutic strategies must simultaneously account for its dual—and potentially opposing—impacts on both innate and adaptive immunity.
Despite this compelling rationale for targeting EZH2, contemporary clinical efforts face three principal challenges: (1) compensatory activation of alternative signaling pathways that drive adaptive resistance;17 (2) target site-specific drug escape mechanisms arising from structural peculiarities of the PRC2 complex;90 (3) the persistent ineffectiveness of conventional catalytic inhibitors against EZH2’s non-canonical, scaffolding functions in chromatin remodeling.91 This limitation directly explains the clinical failure of specific catalytic inhibitors such as EPZ-6438 in certain HCC subtypes: while effectively suppressing EZH2’s methyltransferase activity, they fail to disassemble the PRC2 complex, allowing EZH2 to maintain its scaffolding function. Through recruitment of DNA methyltransferases and other protein partners, it continues to sustain a repressive chromatin state, thereby propelling tumor progression.91
Looking forward, overcoming these obstacles requires innovative approaches. While multi-target combination therapies have demonstrated synergistic potential, critical hurdles remain. Overcoming resistance dynamics driven by the profound molecular heterogeneity of HCC, refining sequential dosing protocols, and establishing optimal therapeutic ratios are urgent unmet needs. The advent of PROTACs, which achieves sustained tumor control through continuous EZH2 degradation and can synergize with immunotherapy, represents a groundbreaking strategy. However, several limitations must be resolved: the restricted diversity of exploitable E3 ligases necessitates the development of PROTACs with enhanced selectivity; structural refinements are required to minimize off-target toxicity risks; and advanced smart delivery systems are needed to achieve spatially controlled degradation. Crucially, deciphering the complex interdependencies between PROTAC pharmacokinetics and the ensuing epigenetic reprogramming will be vital to establishing novel, durable paradigms for overcoming therapeutic resistance in HCC.
Therefore, future work must focus on expanding the repertoire of EZH2-targeting degraders, minimizing off-target toxicity through structural refinement, and developing smart delivery systems. Furthermore, the implementation of multidimensional biomarker platforms—integrating genomic, epigenomic, and tumor microenvironmental data—will be paramount for precisely identifying patient populations most likely to benefit from EZH2-targeted interventions. Ultimately, a deep understanding of EZH2’s multifaceted roles in HCC resistance, coupled with advances in epigenetic drug discovery and patient stratification, paves the way for developing novel and durable therapeutic paradigms to improve outcomes for HCC patients.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Project of Hunan Provincial Natural Science Foundation (NO. 2023JJ60043) and Hunan Provincial Clinical Oncology Research Center of Traditional Chinese Medicine (NO. 2021SK4023).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–263. doi:10.3322/caac.21834
2. Marengo A, Rosso C, Bugianesi E. Liver cancer: connections with obesity, fatty liver, and cirrhosis. Annu Rev Med. 2016;67(1):103–117. doi:10.1146/annurev-med-090514-013832
3. Hassanipour S, Vali M, Gaffari-Fam S, et al. The survival rate of hepatocellular carcinoma in Asian countries: a systematic review and meta-analysis. EXCLI J. 2020;19:108–130. doi:10.17179/excli2019-1842
4. Luo J, Gong L, Yang Y, et al. Enhanced mitophagy driven by ADAR1-GLI1 editing supports the self-renewal of cancer stem cells in HCC. Hepatology. 2024;79(1):61–78. doi:10.1097/HEP.0000000000000299
5. Yang M, Song X, Zhang F, et al. Spatial proteomic landscape of primary and relapsed hepatocellular carcinoma reveals immune escape characteristics in early relapse. Hepatology. 2024;81(5):1452–1467. doi:10.1097/HEP.0000000000000979
6. Chick RC, Ruff SM, Pawlik TM. Neoadjuvant systemic therapy for hepatocellular carcinoma. Front Immunol. 2024;15:1355812. doi:10.3389/fimmu.2024.1355812
7. Tang W, Chen Z, Zhang W, et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther. 2020;5(1):87. doi:10.1038/s41392-020-0187-x
8. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, Phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–2502. doi:10.1016/S0140-6736(17)31046-2
9. Lai Y, Han X, Xie B, et al. EZH2 suppresses ferroptosis in hepatocellular carcinoma and reduces sorafenib sensitivity through epigenetic regulation of TFR2. Cancer Sci. 2024;115(7):2220–2234. doi:10.1111/cas.16186
10. Prawira A, Le TBU, Ho RZW, et al. Upregulation of the ErbB family by EZH2 in hepatocellular carcinoma confers resistance to FGFR inhibitor. J Cancer Res Clin Oncol. 2021;147(10):2955–2968. doi:10.1007/s00432-021-03703-6
11. Wang K, Jiang X, Jiang Y, et al. EZH2-H3K27me3-mediated silencing of mir-139-5p inhibits cellular senescence in hepatocellular carcinoma by activating TOP2A. J Exp Clin Cancer Res. 2023;42(1):320. doi:10.1186/s13046-023-02855-2
12. Li Q, Li B, Dong C, et al. 20(S)-ginsenoside Rh2 suppresses proliferation and migration of hepatocellular carcinoma cells by targeting EZH2 to regulate CDKN2A-2B gene cluster transcription. Eur J Pharmacol. 2017;815:173–180. doi:10.1016/j.ejphar.2017.09.023
13. Xiao G, Jin -L-L, Liu C-Q, et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J Immunother Cancer. 2019;7(1):300. doi:10.1186/s40425-019-0784-9
14. Pasini D, Di Croce L. Emerging roles for polycomb proteins in cancer. Curr Opin Genet Dev. 2016;36:50–58. doi:10.1016/j.gde.2016.03.013
15. Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647(1–2):21–29. doi:10.1016/j.mrfmmm.2008.07.010
16. Justin N, Zhang Y, Tarricone C, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun. 2016;7(1):11316. doi:10.1038/ncomms11316
17. Zhu P, Wang Y, Huang G, et al. lnc-β-Catm elicits EZH2-dependent β-catenin stabilization and sustains liver CSC self-renewal. Nat Struct Mol Biol. 2016;23(7):631–639. doi:10.1038/nsmb.3235
18. Nutt SL, Keenan C, Chopin M, Allan RS. EZH2 function in immune cell development. Biol Chem. 2020;401(8):933–943. doi:10.1515/hsz-2019-0436
19. Zhao H, Liu H, Kang W, et al. Analysis on EZH2: mechanism identification of related CeRNA and its immunoassay in hepatocellular carcinoma. BMC Med Genomics. 2023;16(1):201. doi:10.1186/s12920-023-01594-9
20. Wang B, Liu Y, Liao Z, Wu H, Zhang B, Zhang L. EZH2 in hepatocellular carcinoma: progression, immunity, and potential targeting therapies. Exp Hematol Oncol. 2023;12(1):52. doi:10.1186/s40164-023-00405-2
21. Zhang Q, Cheng S, Cao L, et al. LINC00978 promotes hepatocellular carcinoma carcinogenesis partly via activating the MAPK/ERK pathway. Biosci Rep. 2020;40(3):BSR20192790. doi:10.1042/BSR20192790
22. Xu X, Gu J, Ding X, et al. LINC00978 promotes the progression of hepatocellular carcinoma by regulating EZH2-mediated silencing of p21 and E-cadherin expression. Cell Death Dis. 2019;10(10):752. doi:10.1038/s41419-019-1990-6
23. Zhou X, Man M, Cui M, et al. Relationship between EZH2 expression and prognosis of patients with hepatocellular carcinoma using a pathomics predictive model. Heliyon. 2024;10(20):e38562. doi:10.1016/j.heliyon.2024.e38562
24. Zhang JJ, Chen JT, Hua L, et al. miR-98 inhibits hepatocellular carcinoma cell proliferation via targeting EZH2 and suppressing Wnt/β-catenin signaling pathway. Biomed Pharmacother. 2017;85:472–478. doi:10.1016/j.biopha.2016.11.053
25. Kong J, Qiu Y, Li Y, et al. TGF-β1 elevates P-gp and BCRP in hepatocellular carcinoma through HOTAIR/miR-145 axis. Biopharm Drug Dispos. 2019;40(2):70–80. doi:10.1002/bdd.2172
26. Zhou J, Che J, Xu L, et al. Enhancer of zeste homolog 2 promotes hepatocellular cancer progression and chemoresistance by enhancing protein kinase B activation through microRNA-381-mediated SET domain bifurcated 1. Bioengineered. 2022;13(3):5737–5755. doi:10.1080/21655979.2021.2023792
27. Han Y, Chen M, Wang A, et al. STAT3-induced upregulation of lncRNA CASC11 promotes the cell migration, invasion and epithelial-mesenchymal transition in hepatocellular carcinoma by epigenetically silencing PTEN and activating PI3K/AKT signaling pathway. Biochem Biophys Res Commun. 2019;508(2):472–479. doi:10.1016/j.bbrc.2018.11.092
28. Yang S, Ruan X, Hu B, et al. lncRNA SNHG9 enhances liver cancer stem cell self-renewal and tumorigenicity by negatively regulating PTEN expression via recruiting EZH2. Cell Tissue Res. 2023;394(3):441–453. doi:10.1007/s00441-023-03834-x
29. Varambally S, Cao Q, Mani RS, et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322(5908):1695–1699. doi:10.1126/science.1165395
30. Wang L, Zhang X, Jia LT, et al. c-Myc-mediated epigenetic silencing of MicroRNA-101 contributes to dysregulation of multiple pathways in hepatocellular carcinoma. Hepatology. 2014;59(5):1850–1863. doi:10.1002/hep.26720
31. Xu L, Beckebaum S, Iacob S, et al. MicroRNA-101 inhibits human hepatocellular carcinoma progression through EZH2 downregulation and increased cytostatic drug sensitivity. J Hepatol. 2014;60(3):590–598. doi:10.1016/j.jhep.2013.10.028
32. Chen C, Wang Z, Ding Y, et al. Tumor microenvironment-mediated immune evasion in hepatocellular carcinoma. Front Immunol. 2023;14:1133308. doi:10.3389/fimmu.2023.1133308
33. Cai L, Michelakos T, Yamada T, et al. Defective HLA class I antigen processing machinery in cancer. Cancer Immunol Immunother. 2018;67(6):999–1009. doi:10.1007/s00262-018-2131-2
34. Mu X, Wei Y, Fan X, et al. Aberrant activation of a miR-101-UBE2D1 axis contributes to the advanced progression and chemotherapy sensitivity in human hepatocellular carcinoma. Cell Death Discov. 2024;10(1):422. doi:10.1038/s41420-024-02193-y
35. Takami T, Terai S, Yokoyama Y, et al. Human homologue of maid is a useful marker protein in hepatocarcinogenesis. Gastroenterology. 2005;128(5):1369–1380. doi:10.1053/j.gastro.2005.03.014
36. Wang L, Zhu L, Liang C, et al. Targeting N6-methyladenosine reader YTHDF1 with siRNA boosts antitumor immunity in NASH-HCC by inhibiting EZH2-IL-6 axis. J Hepatol. 2023;79(5):1185–1200. doi:10.1016/j.jhep.2023.06.021
37. Zeng T, Luo L, Huang Y, et al. Upregulation of miR-138 increases sensitivity to cisplatin in hepatocellular carcinoma by regulating EZH2. Biomed Res Int. 2021;2021(1):6665918. doi:10.1155/2021/6665918
38. Yang Y, Zhang Y, Cao J, et al. FGFR4 and EZH2 inhibitors synergistically induce hepatocellular carcinoma apoptosis via repressing YAP signaling. J Exp Clin Cancer Res. 2023;42(1):96. doi:10.1186/s13046-023-02659-4
39. Mansilla SF, de la Vega MB, Calzetta NL, et al. CDK-independent and PCNA-dependent functions of p21 in DNA replication. Genes. 2020;11(6):593. doi:10.3390/genes11060593
40. Zhou M, Zhang XY, Yu X. Overexpression of the long non-coding RNA SPRY4-IT1 promotes tumor cell proliferation and invasion by activating EZH2 in hepatocellular carcinoma. Biomed Pharmacother. 2017;85:348–354. doi:10.1016/j.biopha.2016.11.035
41. Su SG, Li QL, Zhang MF, et al. An E2F1/DDX11/EZH2 positive feedback loop promotes cell proliferation in hepatocellular carcinoma. Front Oncol. 2021;10:593293. doi:10.3389/fonc.2020.593293
42. Cheng D, Deng J, Zhang B, et al. LncRNA HOTAIR epigenetically suppresses miR-122 expression in hepatocellular carcinoma via DNA methylation. EBioMedicine. 2018;36:159–170. doi:10.1016/j.ebiom.2018.08.055
43. Liu Y, Cao Y, Cai W, et al. Aberrant expression of two miRNAs promotes proliferation, hepatitis B virus amplification, migration and invasion of hepatocellular carcinoma cells: evidence from bioinformatic analysis and experimental validation. PeerJ. 2020;8:e9100. doi:10.7717/peerj.9100
44. Mens MMJ, Ghanbari M. Cell cycle regulation of stem cells by MicroRNAs. Stem Cell Rev Rep. 2018;14(3):309–322. doi:10.1007/s12015-018-9808-y
45. Shen Q, Jiang S, Wu M, et al. LncRNA HEIH confers cell sorafenib resistance in hepatocellular carcinoma by regulating miR-98-5p/PI3K/AKT pathway. Cancer Manag Res. 2020;12:6585–6595. doi:10.2147/CMAR.S241383
46. Liu Y, Liang H, Jiang X. MiR-1297 promotes apoptosis and inhibits the proliferation and invasion of hepatocellular carcinoma cells by targeting HMGA2. Int J Mol Med. 2015;36(5):1345–1352. doi:10.3892/ijmm.2015.2341
47. Liu F, He Y, Shu R, et al. MicroRNA-1297 regulates hepatocellular carcinoma cell proliferation and apoptosis by targeting EZH2. Int J Clin Exp Pathol. 2015;8(5):4972–4980.
48. Mei J, Liu G, Li R, et al. LncRNA SNHG6 knockdown inhibits cisplatin resistance and progression of gastric cancer through miR-1297/BCL-2 axis. Biosci Rep. 2021;41(12):BSR20211885. doi:10.1042/BSR20211885
49. Wang C, Ke S, Li M, et al. Downregulation of LncRNA GAS5 promotes liver cancer proliferation and drug resistance by decreasing PTEN expression. Mol Genet Genomics. 2020;295(1):251–260. doi:10.1007/s00438-019-01620-5
50. Zeng Y, Jiang H, Chen Z, et al. Histone lactylation promotes multidrug resistance in hepatocellular carcinoma by forming a positive feedback loop with PTEN. Cell Death Dis. 2025;16(1):59. doi:10.1038/s41419-025-07359-9
51. Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58(5):235–263. doi:10.1002/em.22087
52. Yang SF, Chang CW, Wei RJ, et al. Involvement of DNA damage response pathways in hepatocellular carcinoma. Biomed Res Int. 2014;2014:153867. doi:10.1155/2014/153867
53. Niwa Y, Kamimura K, Ogawa K, et al. Cyclin D1 binding protein 1 responds to DNA damage through the ATM-CHK2 pathway. J Clin Med. 2022;11(3):851. doi:10.3390/jcm11030851
54. Tang T, Huang X, Zhang G, et al. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct Target Ther. 2021;6(1):72. doi:10.1038/s41392-020-00449-4
55. Calderaro J, Rousseau B, Amaddeo G, et al. Programmed death ligand 1 expression in hepatocellular carcinoma: relationship With clinical and pathological features. Hepatology. 2016;64(6):2038–2046. doi:10.1002/hep.28710
56. Zeng T, Zhao Q, Liu J, et al. Expression pattern of PD-1/PD-L1 in primary liver cancer with clinical correlation. Liver Int. 2023;43(9):1995–2001. doi:10.1111/liv.15666
57. Huang J, Tsang WY, Fang XN, et al. FASN inhibition decreases MHC-I degradation and synergizes with PD-L1 checkpoint blockade in hepatocellular carcinoma. Cancer Res. 2024;84(6):855–871. doi:10.1158/0008-5472.CAN-23-0966
58. Chen C, Zhao F, Peng J, et al. Soluble Tim-3 serves as a tumor prognostic marker and therapeutic target for CD8+ T cell exhaustion and anti-PD-1 resistance. Cell Rep Med. 2024;5(8):101686. doi:10.1016/j.xcrm.2024.101686
59. Soler MF, Abaurrea A, Azcoaga P, et al. New perspectives in cancer immunotherapy: targeting IL-6 cytokine family. J Immunother Cancer. 2023;11(11):e007530. doi:10.1136/jitc-2023-007530
60. Dai Z, Wang X, Peng R, et al. Induction of IL-6Rα by ATF3 enhances IL-6 mediated sorafenib and regorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2022;524:161–171. doi:10.1016/j.canlet.2021.10.024
61. Chen J, Sun HW, Wang RZ, et al. Glutamate promotes CCL2 expression to recruit tumor-associated macrophages by restraining EZH2-mediated histone methylation in hepatocellular carcinoma. Oncoimmunology. 2025;14(1):2497172. doi:10.1080/2162402X.2025.2497172
62. Liu H, Shen J, Lu K. IL-6 and PD-L1 blockade combination inhibits hepatocellular carcinoma cancer development in mouse model. Biochem Biophys Res Commun. 2017;486(2):239–244. doi:10.1016/j.bbrc.2017.02.128
63. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–629. doi:10.1038/nrclinonc.2017.44
64. Xu R, Lin L, Zhang B, et al. Identification of prognostic markers for hepatocellular carcinoma based on the epithelial-mesenchymal transition-related gene BIRC5. BMC Cancer. 2021;21(1):687. doi:10.1186/s12885-021-08390-7
65. Wang B, Li X, Zhao G, et al. miR-203 inhibits ovarian tumor metastasis by targeting BIRC5 and attenuating the TGFβ pathway. J Exp Clin Cancer Res. 2018;37(1):235. doi:10.1186/s13046-018-0906-0
66. Zhong T, Zhang W, Guo H, et al. The regulatory and modulatory roles of TRP family channels in malignant tumors and relevant therapeutic strategies. Acta Pharm Sin B. 2022;12(4):1761–1780. doi:10.1016/j.apsb.2021.11.001
67. Li S, Gao M, Li Z, et al. p53 and P-glycoprotein influence chemoresistance in hepatocellular carcinoma. Front Biosci. 2018;10(3):461–468. doi:10.2741/e833
68. Tang M, Huang Y, Liang X, et al. Sorafenib-loaded PLGA-TPGS Nanosystems enhance hepatocellular carcinoma therapy through reversing P-glycoprotein-mediated multidrug resistance. AAPS Pharm Sci Tech. 2022;23(5):130. doi:10.1208/s12249-022-02214-y
69. Carabias P, Espelt MV, Bacigalupo ML, et al. Galectin-1 confers resistance to doxorubicin in hepatocellular carcinoma cells through modulation of P-glycoprotein expression. Cell Death Dis. 2022;13(1):79. doi:10.1038/s41419-022-04520-6
70. Sun D, Liu J, Wang Y, et al. Co-administration of MDR1 and BCRP or EGFR/PI3K inhibitors overcomes lenvatinib resistance in hepatocellular carcinoma. Front Oncol. 2022;12:944537. doi:10.3389/fonc.2022.944537
71. Hoy SM. Tazemetostat: first Approval. Drugs. 2020;80(5):513–521. doi:10.1007/s40265-020-01288-x
72. Porazzi P, Nason S, Yang Z, et al. EZH1/EZH2 inhibition enhances adoptive T cell immunotherapy against multiple cancer models. Cancer Cell. 2025;43(3):537–551.e7. doi:10.1016/j.ccell.2025.01.013
73. Budagaga Y, Sabet Z, Zhang Y, et al. Tazemetostat synergistically combats multidrug resistance by the unique triple inhibition of ABCB1, ABCC1, and ABCG2 efflux transporters in vitro and ex vivo. Biochem Pharmacol. 2023;216:115769. doi:10.1016/j.bcp.2023.115769
74. Bao Q, Kumar A, Wu D, et al. Targeting EED as a key PRC2 complex mediator toward novel epigenetic therapeutics. Drug Discov Today. 2024;29(6):103986. doi:10.1016/j.drudis.2024.103986
75. Li X, Gera L, Zhang S, et al. Pharmacological inhibition of noncanonical EED-EZH2 signaling overcomes chemoresistance in prostate cancer. Theranostics. 2021;11(14):6873–6890. doi:10.7150/thno.49235
76. Qi W, Zhao K, Gu J, et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat Chem Biol. 2017;13(4):381–388. doi:10.1038/nchembio.2304
77. Straining R, Eighmy W. Tazemetostat: EZH2 Inhibitor. J Adv Pract Oncol. 2022;13(2):158–163. doi:10.6004/jadpro.2022.13.2.7
78. Wang Y, Liu D, Zhang T, et al. FGF/FGFR signaling in hepatocellular carcinoma: from carcinogenesis to recent therapeutic intervention. Cancers. 2021;13(6):1360. doi:10.3390/cancers13061360
79. Shi QS, Zhang YH, Long J, et al. SSH3 promotes malignant progression of HCC by activating FGF1-mediated FGF/FGFR pathway. Eur Rev Med Pharmacol Sci. 2020;24(22):11561–11568. doi:10.26355/eurrev_202011_23797
80. Hu J, Zhang J, Sun F, et al. Enhancer of zeste 2 polycomb repressive complex 2 subunit promotes sorafenib resistance of hepatocellular carcinoma though insulin-like growth factor 1 receptor. Anticancer Drugs. 2019;30(7):e0746. doi:10.1097/CAD.0000000000000746
81. Zhang H, Yu J, Zhang F, et al. BRD4 interacting genes as prognostic biomarkers in hepatocellular carcinoma for optimized treatment strategies. Sci Rep. 2025;15(1):5617. doi:10.1038/s41598-025-89614-9
82. Huang J, Yin Q, Wang Y, et al. EZH2 inhibition enhances PD-L1 protein stability through USP22-mediated deubiquitination in colorectal cancer. Adv Sci. 2024;11(23):e2308045. doi:10.1002/advs.202308045
83. Qin Y, Vasilatos SN, Chen L, et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene. 2019;38(3):390–405. doi:10.1038/s41388-018-0451-5
84. Cao R, Ni J, Zhang X, et al. Recent advances in enhancer of zeste homolog 2 inhibitors: structural insights and therapeutic applications. Bioorg Chem. 2025;154:108070. doi:10.1016/j.bioorg.2024.108070
85. Vogelmann A, Robaa D, Sippl W, et al. Proteolysis targeting chimeras (PROTACs) for epigenetics research. Curr Opin Chem Biol. 2020;57:8–16. doi:10.1016/j.cbpa.2020.01.010
86. Fu M, Wang Y, Ge M, et al. Chemically induced degradation of PRC2 complex by EZH2-targeted PROTACs via a ubiquitin-proteasome pathway. Bioorg Med Chem Lett. 2024;113:129968. doi:10.1016/j.bmcl.2024.129968
87. Velez J, Dale B, Park KS, et al. Discovery of a novel, highly potent EZH2 PROTAC degrader for targeting non-canonical oncogenic functions of EZH2. Eur J Med Chem. 2024;267:116154. doi:10.1016/j.ejmech.2024.116154
88. Mei H, Wu H, Yang J, et al. Discovery of IHMT-337 as a potent irreversible EZH2 inhibitor targeting CDK4 transcription for malignancies. Signal Transduct Target Ther. 2023;8(1):18. doi:10.1038/s41392-022-01240-3
89. Qi SM, Dong J, Xu ZY, et al. PROTAC: an effective targeted protein degradation strategy for cancer therapy. Front Pharmacol. 2021;12:692574. doi:10.3389/fphar.2021.692574
90. Huang X, Yan J, Zhang M, et al. Targeting epigenetic crosstalk as a therapeutic strategy for EZH2-aberrant solid tumors. Cell. 2018;175(1):186–199.e19. doi:10.1016/j.cell.2018.08.058
91. Yang J, Weisberg EL, Qi S, et al. Inhibition of the deubiquitinating enzyme USP47 as a novel targeted therapy for hematologic malignancies expressing mutant EZH2. Leukemia. 2022;36(4):1048–1057. doi:10.1038/s41375-021-01494-w
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
A Promising Future for Precision Epigenetic Therapy for Follicular and Diffuse Large B-Cell Lymphoma?
Chung C
Blood and Lymphatic Cancer: Targets and Therapy 2022, 12:99-106
Published Date: 4 August 2022
The Synergistic Mechanisms and Prospects of Transarterial Chemoembolization Combined with Immunotherapy for Hepatocellular Carcinoma
Chen QF, Chen S, Zhao M
Journal of Hepatocellular Carcinoma 2025, 12:841-854
Published Date: 30 April 2025