Back to Journals » OncoTargets and Therapy » Volume 14
Expression Profiles of tRNA-Derived Fragments and Their Potential Roles in Multiple Myeloma
Received 18 January 2021
Accepted for publication 16 March 2021
Published 22 April 2021 Volume 2021:14 Pages 2805—2814
DOI https://doi.org/10.2147/OTT.S302594
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Federico Perche
Cong Xu,1 Yunfeng Fu2
1Department of Hematology, The Third Xiangya Hospital of Central South University, Changsha, 410000, People’s Republic of China; 2Department of Blood Transfusion, The Third Xiangya Hospital of Central South University, Changsha, 410000, People’s Republic of China
Correspondence: Yunfeng Fu Tel/Fax + 86-73188618511
Email [email protected]
Background: Multiple myeloma is an incurable hematologic malignancy. The discovery of mechanisms may help to find new therapeutic targets and prolong survival. tRNA-related fragments (tRF) and tRNA halves (tiRNA) are small RNA derived from tRNAs and implicated in a wide variety of pathological processes, including cancer initiation and progression. However, there are almost no research reporting the role of these tRNA derived fragments in myeloma as far as we know.
Methods: In this study, we proposed the expression profiles of tRFs/tiRNAs in myeloma through RNA-sequencing in new diagnosed myeloma and healthy donors. The expression of selected tRFs/tiRNAs was further validated in clinical specimens by qPCR. Bioinformatic analysis was performed to predict their potential roles in multiple myeloma.
Results: A total of 33 upregulated tRFs/tiRNAs and 22 downregulated tRFs/tiRNAs were identified. GO enrichment and KEGG pathway analysis were performed to analyze the functions of one significantly up-regulated and one significantly down-regulated tRNA-derived fragments. tRFs/tiRNAs may be involved in MM initiation and progression.
Conclusion: We concluded that tRFs/tiRNAs were dysregulated and could be potential biomarkers for multiple myeloma.
Keywords: multiple myeloma, tRNA related fragments, tRNA halves, cancer initiation
Introduction
Multiple myeloma (MM) is the second most common hematological malignancy and is characterized by the accumulation of malignant plasma cells in the bone marrow.1 With the advent of new drugs, such as proteasome inhibitors and immunomodulatory agents, the outcomes of patients with MM have steadily improved. Despite improvements in response and survival rates, MM remains a costly and incurable disease.2,3 Elucidating the mechanisms governing MM development is pivotal to optimizing the clinical efficacy and prolonging survival. Myeloma cells are dependent on multiple factors at all stages of tumor development and progression. Among these factors, noncoding RNAs (ncRNAs) play critical roles.4,5
The largest portion of the mammalian genome is transcribed into RNA species with little or no coding potential, which are known as ncRNAs. An explosion of interest in RNA biology has identified a variety of regulatory functions for ncRNAs.6,7 With the rapid development of high-throughput sequencing in recent years, a new class of small ncRNAs derived from tRNAs has attracted increasing attention from researchers.
According to the cleavage site, these tRNA derived fragments can be broadly classified into two main groups: tRNA related fragments (tRFs) generated from mature or precursor tRNA and tRNA halves (tiRNAs) generated by specific cleavage in the anticodon loops of mature tRNA.8 On the basis of mapped positions on the precursor or mature tRNA transcript, tRFs/tiRNAs are subdivided into five types: tRF-5, tRF-3, tRF-1, tRF-2 and tiRNA. tRFs/tiRNAs are involved in various molecular processes through translation regulation, epigenetic regulation and RNA silencing.9,10
Accumulating evidence supports the significance of tRFs/tiRNAs in multiple human diseases, including cancer. For example, some sex hormone-dependent tRNA-derived RNAs were found to enhance cell proliferation in breast cancer cells and to act as oncogenes in breast cancer.11 tRF-1001, which is present in the cytoplasm and produced by the tRNA 3ʹ-endonuclease ELAC2, is essential for the proliferation of prostate cancer cells and colorectal cancer cells.12 Upregulated or downregulated tRFs/tiRNAs are both involved in the occurrence and development of solid tumors.13,14 In hematological malignancies, limited data have demonstrated the potential roles played by tRFs/tiRNAs in leukemia and lymphoma.15,16 To the best of our knowledge, few studies have described the roles played by tRFs/tiRNAs in the mechanism underlying MM. One study showed that smoldering multiple myeloma patients with lower expression levels of 3′-tRF-LeuAAG/TAG, i-tRF-GluCTC, and i-tRF-ProTGG were at great risk of suffering from MM.17 However, the expression profile of tRFs/tiRNAs in myeloma patients compared to healthy people has not been determined.
In this study, we investigated the expression profiles of tRFs/tiRNAs in paired newly diagnosed MM and healthy donor samples through RNA-sequencing and quantitative real-time PCR (qPCR) verification. Next, using bioinformatics analysis, we characterized the functions of these RNAs in the initiation and progression of MM. These findings may provide potential biomarkers and therapeutic targets for MM.
Materials and Methods
Clinical Samples
This study was approved by ethics committee of the third Xiangya hospital. Twenty new diagnosed MM(NDMM) patients and 18 healthy donors were enrolled, respectively. The diagnosis of NDMM was based on the diagnostic criteria of symptomatic multiple myeloma defined by National Comprehensive Cancer Network (NCCN). Bone marrow was obtained from NDMM patients and healthy donors after informed consent.
RNA Extraction and Quantitative RT-PCR Validation
Firstly, plasma cells were enriched from bone marrow samples using the anti-human CD138 magnetic-activated cell sorting (MACS) beads (Miltenyi, Germany). The purity of separated plasma cells was above 90% (identified by flow cytometry). Total RNA was then extracted from enriched plasma cells using TRIzol (Invitrogen, USA) following the instruction manual. Prepared RNA was preserved at −80°C. RNA samples were qualified and quantified by agarose gel electrophoresis and NanoDrop ND-1000 (NanoDrop, USA). qRT-PCR was implemented using ViiA 7 Real-time PCR System (Applied Biosystems) and 2 × PCR Master Mix. U6 small nuclear RNA (snRNA) was used as internal reference. The fold changes were calculated and normalized by U6. The primers used are listed in Table 1. RNA quality is shown in Supplementary Table 1. RNA integrity number (RIN) was evaluated by Agilent BioAnalyzer 2100 and was all above 7.
 |
Table 1 Primers for qRT-PCR Validation of Candidate tRFs/tiRNAs |
Library Preparation and Sequencing
Firstly, total RNA samples were pretreated to remove RNA modifications that interfere with small RNA-seq library construction. The libraries then were denatured as single-stranded DNA molecules, captured on Illumina flow cells, amplified in situ as sequencing clusters and sequenced for 50 cycles on Illumina NextSeq 500 system following the manufacturer’s instructions. Image analysis and base calling were conducted by Solexa pipeline v1.8 (Off-Line Base Caller software, v1.8).
Sequencing quality was examined by FastQC. The abundance of tRFs/tiRNAs was evaluated by sequencing counts and normalized as counts per million of total aligned reads (CPM). R package edgeR was used to calculate the differentially expressed tRFs/tiRNAs. Hierarchical clustering, Volcano plots, Pie plots and Venn plots were constructed by R or Perl.
Bioinformatic Analysis of tRFs/tiRNAs
We used TargetScan algorithms to predict gene targets of tRFs/tiRNAs.18 Gene Ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) function enrichment analysis were conducted using DAVID Bioinformatics Resources 6.8.19 Graphing was performed with Cytoscape 3.7.2 and GraphPad Prism 8.0.1.
Statistical Analysis
Analysis of data was performed with two-tail unpaired Student t-test using SPSS 23.0 software. qPCR validation was presented as mean ± standard deviation. The statistical significance level was set at P value < 0.05.
Results
General Features of tRFs/tiRNAs Expression in MM
We performed tRFs/tiRNAs sequencing on bone marrow specimens from five MM patients and five normal donors. Clinical characteristics of MM patients and healthy donors are shown in Table 2. After Illumina quality control and 5ʹ, 3ʹ-adaptor trimmed, reads with length 14nt ~ 40nt were retained. tRNA isodecoders share the same anticodon but have differences in their body sequence. All the mature tRNAs and pre‐tRNAs were found to produce tDRs in our profiles. We counted the number of significantly differentially expressed subtype tRFs/tiRNAs against tRNA isodecoders which the CPM of the sample is not less than 20 (Figure 1A and B). tRFs/tiRNAs may be produced from different tRNAs by cleavage into the fragments with identical sequences. The number of tRF-3a and tRF-3b, which mainly occur in the cytoplasm, decreased in myeloma patients. The pie charts depict the distribution of the number for each subtype tRFs/tiRNAs (Figure 1C and D).
 |
Table 2 Baseline Characteristics of MM Patients and Healthy Donors |
 |
Figure 1 Categories of tRFs/tiRNAs in MM patients and healthy donors. (A) The number of up-regulated tRFs/tiRNAs against tRNA isodecoders. (B) The number of down-regulated tRFs/tiRNAs against tRNA isodecoders. (C) The distribution of up-regulated tRFs/tiRNAs subtypes. (D) The distribution of down-regulated tRFs/tiRNAs subtypes. |
Differentially Expressed tRFs/tiRNAs Between MM and Normal Groups
Venn diagram shows the commonly expressed and specifically expressed tRFs/tiRNAs (Figure 2A). The “commonly expression” means CPM values ≥ 20 in both two groups, and the “specifically expression” means CPM values ≥ 20 in one group and <20 in the other group. We identified 354 upregulated tRFs/tiRNAs and 515 downregulated tRFs/tiRNAs in this project in total. After limiting the condition with fold change ≥1.5 and P < 0.05, there remained 33 up-regulated and 22 down-regulated tRFs/tiRNAs. The heatmap shows hierarchical clustering of significantly differentially expressed tRFs/tiRNAs (Figure 2B). We present the overall tRFs/tiRNAs expression variation between the two groups in the volcano plots (Figure 2C).
 |
Figure 2 Expression profiles of tRFs/tiRNAs in paired healthy donors and MM patients. (A) Venn diagram based on number of commonly expressed and specifically expressed tRFs/tiRNAs between healthy donors and MM patients. (B) The hierarchical clustering heatmap for significantly differentially expressed tRFs/tiRNAs. (C) The volcano plot of tRFs/tiRNAs (MM vs healthy donors). |
tiRNA-1:34-Glu-TTC-2 Was Upregulated and tRF-60:76-Arg-ACG-1-M2 Was Downregulated in MM
We performed qPCR to detect the expression of three significantly upregulated or downregulated tRFs/tiRNAs in paired healthy donors and MM samples. The expression of all these six tRFs/tiRNAs was aligned to the sequencing data, while the most significantly upregulated and downregulated tRFs/tiRNAs were tiRNA-1:34-Glu-TTC-2 and tRF-60:76-Arg-ACG-1-M2, respectively (Figure 3A). Then, we expanded sample size for further verification of tiRNA-1:34-Glu-TTC-2 and tRF-60:76-Arg-ACG-1-M2 expression in 18 healthy donors and 20 MM patients. At last, we confirmed that tiRNA-1:34-Glu-TTC-2 was upregulated and tRF-60:76-Arg-ACG-1-M2 was downregulated in MM (Figure 3B).
 |
Figure 3 qPCR validation of tRFs/tiRNAs expression. (A) Relative expression of three significantly upregulated tRFs/tiRNAs and downregulated tRFs/tiRNAs in paired healthy donors and MM samples. (B) Relative expression of tiRNA-1:34-Glu-TTC-2 and tRF-60:76-Arg-ACG-1-M2 18 healthy donors and 20 MM patients. **P<0.01, ***P<0.001. |
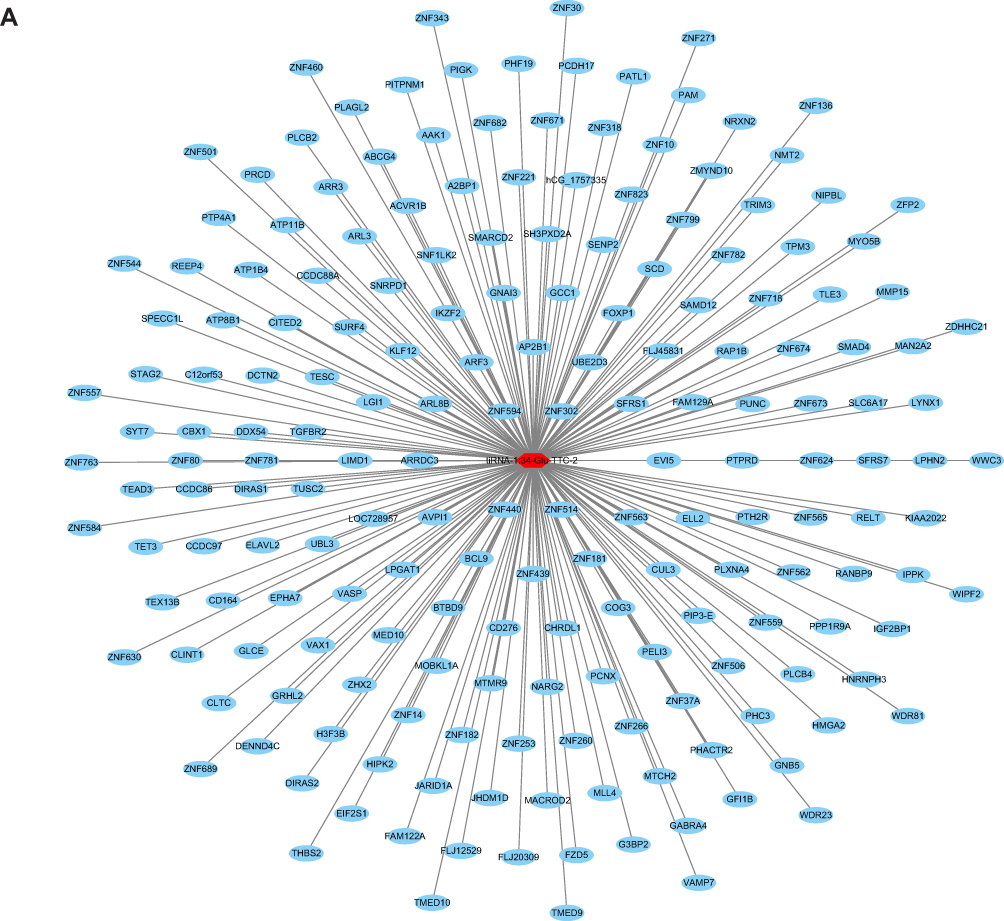 |
Figure 4 Continue. |
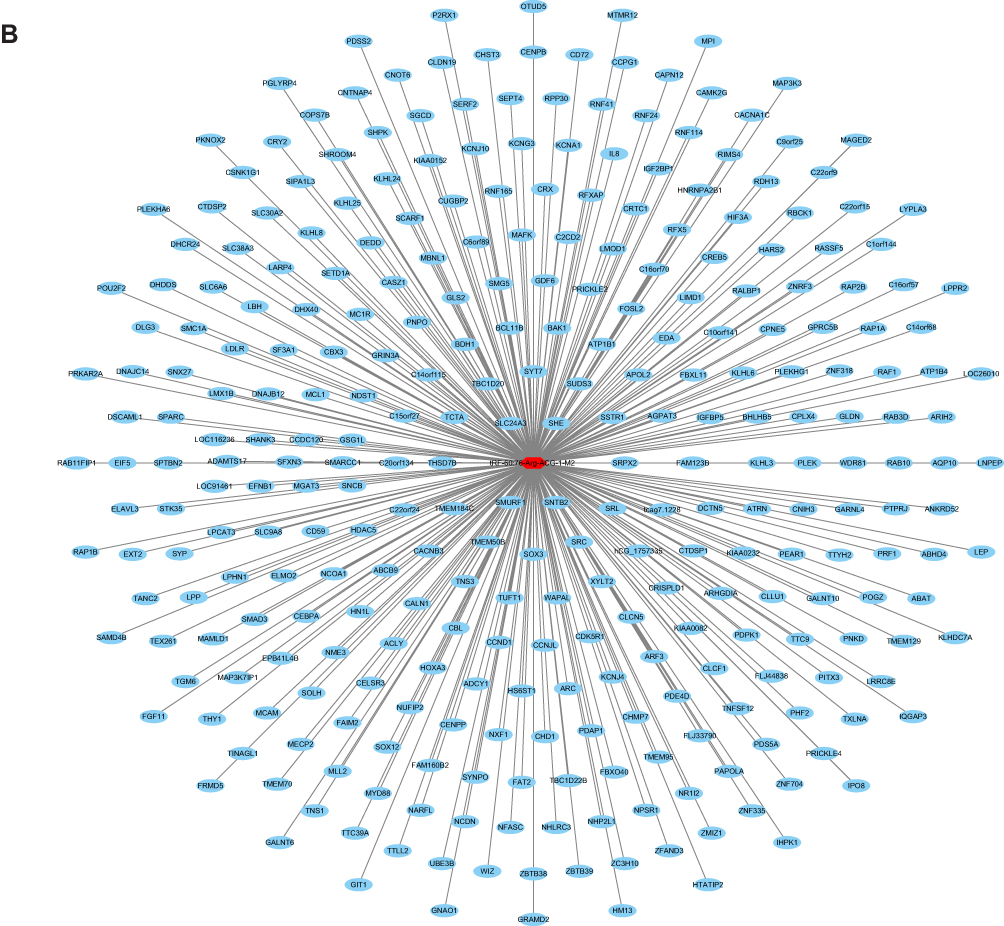 |
Figure 4 Target gene prediction of tRFs/tiRNAs. (A) Target genes of tiRNA-1:34-Glu-TTC-2. (B) Target genes of tRF-60:76-Arg-ACG-1-M2. |
Target Gene Prediction with Bioinformatics Tool
We used TargetScan algorithms to explore the putative roles of tiRNA-1:34-Glu-TTC-2 and tRF-60:76-Arg-ACG-1-M2 in MM. Through this strategy, we predicted 193 conserved targets and 310 conserved targets, respectively. The target genes were presented in the network diagrams constructed by Cytoscape (Figure 4A and B).
GO/KEGG Enrichment Analysis
We performed GO and KEGG enrichment analysis for target genes of tiRNA-1:34-Glu-TTC-2 and tRF-60:76-Arg-ACG-1-M2 using DAVID database. GO analysis showed that 37 and 58 GO terms were enriched (P < 0.05) for target genes of tiRNA-1:34-Glu-TTC-2 and tRF-60:76-Arg-ACG-1-M2, respectively. The most enriched terms of target genes of tiRNA-1:34-Glu-TTC-2 were “transcription, DNA-templated” and “regulation of transcription, DNA-templated” in biological process (BP) category, “nucleus” and “intracellular” in cellular component (CC) category, “protein binding” and “metal ion binding” in molecular function (MF) category (Figure 5A). Whereas, the most enriched terms of target genes of tRF-60:76-Arg-ACG-1-M2 were “apoptotic process” and “regulation of transcription from RNA polymerase II promoter” in BP category, “cytoplasm” and “plasma membrane” in CC category, “protein binding” and “zinc ion binding” in MF category (Figure 5B).
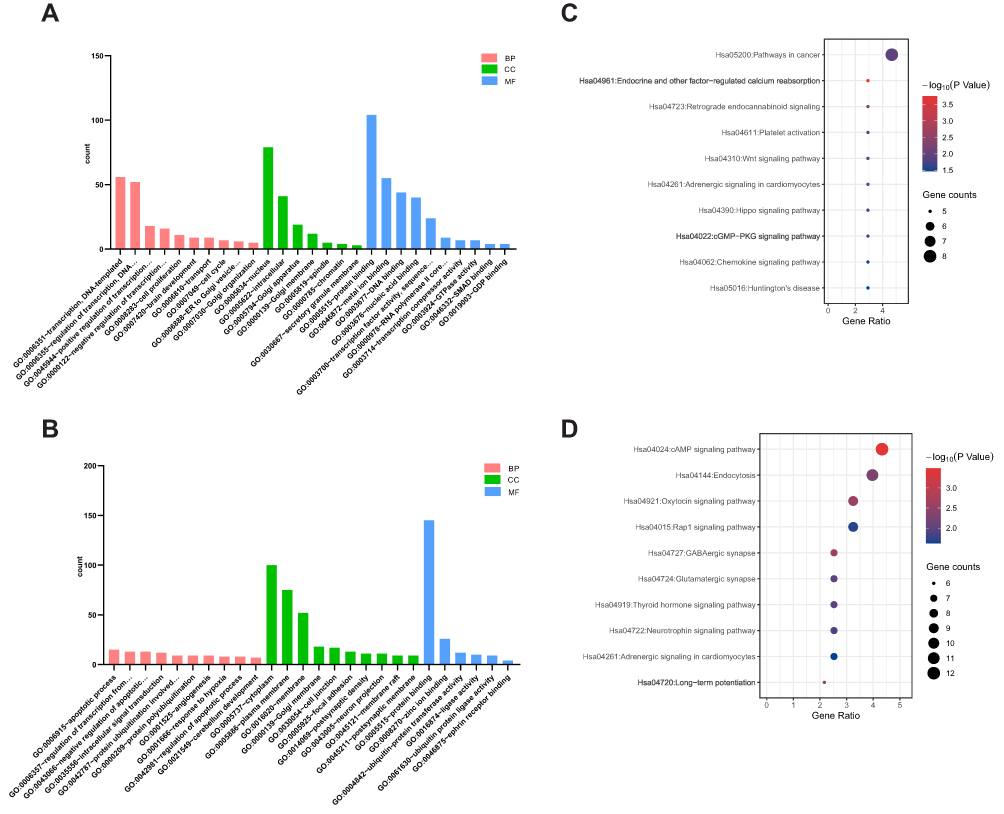 |
Figure 5 Functional analysis of tRFs/tiRNAs. (A) GO enrichment analysis of tiRNA-1:34-Glu-TTC-2. (B) GO enrichment analysis of tRF-60:76-Arg-ACG-1-M2. (C) KEGG pathway analysis of tiRNA-1:34-Glu-TTC-2. (D) KEGG pathway analysis of tRF-60:76-Arg-ACG-1-M2. |
KEGG pathway analysis indicated that tiRNA-1:34-Glu-TTC-2 might participate in cancer-related pathways such as Pathways in cancer, Wnt signaling pathway and Hippo signaling pathway (P < 0.05) (Figure 5C). While the most enriched pathways of tRF-60:76-Arg-ACG-1-M2 were cAMP signaling pathway, Endocytosis, Oxytocin signaling pathway and Rap1 signaling pathway (Figure 5D).
Discussion
Although the clinical outcome of MM has been considerably improved over the last several decades, MM is still an incurable type of hematological malignancy. The pathogenesis of MM is complex and multifactorial. Many genes or ncRNAs have been determined to be involved in the pathological process. However, whether tRFs/tiRNAs participate in MM initiation has not been elucidated. Therefore, we performed a preliminary evaluation of the role of tRFs/tiRNAs in MM.
First, we screened the expression profile of tRFs/tiRNAs in MM by RNA-sequencing. All tRFs/tiRNAs were named using the standardized tDR (tRNA-derived RNA) naming system (tDRnamer) from the Lowe Lab at the University of California Santa Cruz. The four parts in the name sequentially represent prefix, position, source tRNA and matching tRNA transcripts.20 tDRnamer provides a consistent, stable name and gives additional annotation for each submitted tDR sequence compared to tRFdb ID and MINTbase ID. The biogenesis of most of tRF-3s is unknown. The decrease in the number of tRF-3 may affect DNA replication, repair and damage response.10 Next, the two most differentially expressed tRFs/tiRNAs were verified by qPCR. To explore the possible biological functions of these RNAs, we predicted their target genes and performed GO/KEGG analysis. For tiRNA-1:34-Glu-TTC-2, its predicted target genes were primarily localized in the nucleus and implicated in transcription regulation and protein binding. However, the target genes of tRF-60:76-Arg-ACG-1-M2 were primarily located in the cytoplasm and participated in apoptotic processes and protein binding. This result is consistent with the common molecular functions of tRFs/tiRNAs in a previous report.12
tRFs/tiRNAs may regulate transcription and apoptotic processes through multiple molecular mechanisms. For example, Ivanov et al found that 5′-tiRNAAla and 5′-tiRNACys could repress global translation by displacing eIF4G and eIF4A (translation eukaryotic initiation factors) from mRNAs and displacing eIF4F from isolated m7G caps.21 Gkatza et al22 showed that the loss of NSUN2, a cytosine-5 RNA methyltransferase, could lead to impaired regulation of protein synthesis by altering the biogenesis of tiRNAs in response to stress.23 However, the mechanisms underlying how tRFs/tiRNAs regulate carcinogenesis have not been elucidated.
To explore whether tiRNA-1:34-Glu-TTC-2 and tRF-60:76-Arg-ACG-1-M2 are involved in the initiation and development of MM, we performed KEGG pathway analysis. Among the top ten enriched pathways, pathways in cancer, the Wnt signaling pathway, the Hippo signaling pathway and the Rap1 signaling pathway are widely known to be related to carcinogenesis in previous reports.23–25 These pathways are also associated with MM initiation and development. Harmen et al reported that aberrant Wnt signaling mediated the proliferation, migration, and drug resistance of MM cells and MM cells secreted Wnt antagonists that promoted the development of osteolytic lesions by impairing osteoblast differentiation.26 The Hippo pathway has also emerged as an important mediator of MM oncogenesis and is implicated in the pathogenesis of MM-induced bone disease.27,28 There are relatively few studies investigating the role played by the Rap1 pathway in MM. Taken together, tiRNA-1:34-Glu-TTC-2 and tRF-60:76-Arg-ACG-1-M2 may be involved in the pathological process of MM but these hypotheses required further validation.
Several study limitations warrant consideration. First, due to the short follow-up time, we have not obtained the efficacy and prognosis data of myeloma patients to date. Second, in this preliminary experiment, we did not collect specimens from cases of different stages of plasma cell disease (monoclonal gammopathy of undetermined significance, smoldering myeloma, multiple myeloma and plasma cell leukemia). The evaluation of the expression of tRFs/tiRNAs in these four stages requires further study. Third, although tRFs/tiRNAs are also involved in the silencing of microRNAs, there are few related data reports. The current bioinformatics databases do not provide information on the network of tRFs/tiRNAs-microRNA-mRNA. We look forward to realize this analysis basing on the in-depth research on tRFs/tiRNAs in the future.
In conclusion, we assessed the potential function of tRFs/tiRNAs in MM and the results establish a foundation for continued research. tRFs/tiRNAs may play important roles in the pathogenesis of myeloma and serve as clinical biomarkers and therapeutic targets in the future. Further experiments are warranted to explore how tRFs/tiRNAs regulate target genes and miRNAs in MM. We look forward to further research on tRFs/tiRNAs in MM.
Ethics Approval and Informed Consent
The studies involving human participants were reviewed and approved by ethics committee of the third Xiangya hospital. The approval ID was 2016121. The patients/participants provided their written informed consent to participate in this study. This study was conducted in accordance with the Declaration of Helsinki.
Acknowledgment
Authors would like to thank Ms. Ting Liang for her help in collecting some of the samples for this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by National Natural Science Foundation of China (Grant No. 82002452).
Disclosure
The authors declare no conflicts of interest.
References
1. Boise LH, Kaufman JL, Bahlis NJ, Lonial S, Lee KP. The Tao of myeloma. Blood. 2014;124(12):1873–1879. doi:10.1182/blood-2014-05-578732
2. Terpos E, Kleber M, Engelhardt M, et al. European myeloma network guidelines for the management of multiple myeloma-related complications. Haematologica. 2015;100(10):1254–1266. doi:10.3324/haematol.2014.117176
3. Gooding S, Olechnowicz SWZ, Morris EV, et al. Transcriptomic profiling of the myeloma bone-lining niche reveals BMP signalling inhibition to improve bone disease. Nat Commun. 2019;10(1):4533. doi:10.1038/s41467-019-12296-1
4. Gao M, Li C, Xiao H, et al. hsa_circ_0007841: a novel potential biomarker and drug resistance for multiple myeloma. Front Oncol. 2019;9:1261. doi:10.3389/fonc.2019.01261
5. Yuan X, Ma R, Yang S, et al. miR-520g and miR-520h overcome bortezomib resistance in multiple myeloma via suppressing APE1. Cell Cycle. 2019;18(14):1660–1669. doi:10.1080/15384101.2019.1632138
6. Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154(1):26–46. doi:10.1016/j.cell.2013.06.020
7. Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157(1):77–94. doi:10.1016/j.cell.2014.03.008
8. Anderson P, Ivanov P. tRNA fragments in human health and disease. FEBS Lett. 2014;588(23):4297–4304. doi:10.1016/j.febslet.2014.09.001
9. Guzzi N, Bellodi C. Novel insights into the emerging roles of tRNA-derived fragments in mammalian development. RNA Biol. 2020;17(8):1214–1222. doi:10.1080/15476286.2020.1732694
10. Kumar P, Kuscu C, Dutta A. Biogenesis and function of transfer RNA-related fragments (tRFs). Trends Biochem Sci. 2016;41(8):679–689. doi:10.1016/j.tibs.2016.05.004
11. Honda S, Loher P, Shigematsu M, et al. Sex hormone-dependent tRNA halves enhance cell proliferation in breast and prostate cancers. Proc Natl Acad Sci U S A. 2015;112(29):E3816–3825. doi:10.1073/pnas.1510077112
12. Yu M, Lu B, Zhang J, Ding J, Liu P, Lu Y. tRNA-derived RNA fragments in cancer: current status and future perspectives. J Hematol Oncol. 2020;13(1):121. doi:10.1186/s13045-020-00955-6
13. Huang B, Yang H, Cheng X, et al. tRF/miR-1280 suppresses stem cell-like cells and metastasis in colorectal cancer. Cancer Res. 2017;77(12):3194–3206. doi:10.1158/0008-5472.CAN-16-3146
14. Cui Y, Huang Y, Wu X, et al. Hypoxia-induced tRNA-derived fragments, novel regulatory factor for doxorubicin resistance in triple-negative breast cancer. J Cell Physiol. 2019;234(6):8740–8751. doi:10.1002/jcp.27533
15. Veneziano D, Tomasello L, Balatti V, et al. Dysregulation of different classes of tRNA fragments in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2019;116(48):24252–24258. doi:10.1073/pnas.1913695116
16. Maute RL, Schneider C, Sumazin P, et al. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc Natl Acad Sci U S A. 2013;110(4):1404–1409. doi:10.1073/pnas.1206761110
17. Karousi P, Papanota A-M, Artemaki PI, et al. A molecular signature of three tRNA-derived RNA fragments may discriminate smoldering from symptomatic multiple myeloma patients. Blood. 2019;134(Supplement_1):5528. doi:10.1182/blood-2019-129082
18. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4. doi:10.7554/eLife.05005
19. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi:10.1038/nprot.2008.211
20. DRnamer standardized naming for tRNA-derived RNAs[homepage on the Internet]. The Lowe Lab at University of California Santa Cruz; 2019.Available from: http://trna.ucsc.edu/tDRnamer/docs/naming/.
21. Ivanov P, Emara MM, Villen J, Gygi SP, Anderson P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell. 2011;43(4):613–623. doi:10.1016/j.molcel.2011.06.022
22. Gkatza NA, Castro C, Harvey RF, et al. Cytosine-5 RNA methylation links protein synthesis to cell metabolism. PLoS Biol. 2019;17(6):e3000297
23. Chng WJ, Gonzalez-Paz N, Price-Troska T, et al. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia. 2008;22(12):2280–2284. doi:10.1038/leu.2008.142
24. Han Y. Analysis of the role of the Hippo pathway in cancer. J Transl Med. 2019;17(1):116. doi:10.1186/s12967-019-1869-4
25. Zhang L, Cui M, Song L, Zhang M, Zhang J. Function, significance, and regulation of Rap1b in malignancy. Crit Rev Eukaryot Gene Expr. 2019;29(2):151–160. doi:10.1615/CritRevEukaryotGeneExpr.2019025997
26. Harmen VA, Kocemba KA, Spaargaren M, Pals ST. Aberrant Wnt signaling in multiple myeloma: molecular mechanisms and targeting options. Leukemia. 2019;33(5):1063–1075. doi:10.1038/s41375-019-0404-1
27. Fan S, Price T, Huang W, et al. PINK1-dependent mitophagy regulates the migration and homing of multiple myeloma cells via the MOB1B-mediated Hippo-YAP/TAZ pathway. Adv Sci (Weinh). 2020;7(5):1900860. doi:10.1002/advs.201900860
28. Kyriazoglou A, Ntanasis-Stathopoulos I, Terpos E, et al. Emerging insights into the role of the Hippo pathway in multiple myeloma and associated bone disease. Clin Lymphoma Myeloma Leuk. 2020;20(2):57–62. doi:10.1016/j.clml.2019.09.620
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.