Back to Journals » International Journal of Nanomedicine » Volume 20
Exosome-Mediated Mitochondrial Regulation: A Promising Therapeutic Tool for Alzheimer’s Disease and Parkinson’s Disease
Authors Jung YH, Jo HY, Kim DH, Oh YJ, Kim M ![]() , Na S, Song HY, Lee HJ
, Na S, Song HY, Lee HJ
Received 5 January 2025
Accepted for publication 7 April 2025
Published 17 April 2025 Volume 2025:20 Pages 4903—4917
DOI https://doi.org/10.2147/IJN.S513816
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Young Hyun Jung,1,* Hyo Youn Jo,2,3,* Dae Hyun Kim,2,3 Yeon Ju Oh,2,3 Minsoo Kim,1 Seunghyun Na,1 Ho-Yeon Song,4 Hyun Jik Lee2,3
1Department of Physiology, College of Medicine, Soonchunhyang University, Cheonan, 31151, Republic of Korea; 2Laboratory of Veterinary Physiology, College of Veterinary Medicine and Veterinary Medicine Center, Chungbuk National University, Cheongju 28644, Republic of Korea; 3Institute for Stem Cell & Regenerative Medicine (ISCRM), Chungbuk National University, Cheongju, 28644, Republic of Korea; 4Department of Microbiology and Immunology, School of Medicine, Soonchunhyang University, Cheonan-si, 31151, Republic of Korea
*These authors contributed equally to this work
Correspondence: Hyun Jik Lee, College of Veterinary Medicine, Chungbuk National University, Cheongju, 28644, Republic of Korea, Tel +82-43-261-2597, Email [email protected]
Abstract: Alzheimer’s disease (AD) and Parkinson’s disease (PD) are representative neurodegenerative diseases with abnormal energy metabolism and altered distribution and deformation of mitochondria within neurons, particularly in brain regions such as the hippocampus and substantia nigra. Neurons have high energy demands; thus, maintaining a healthy mitochondrial population is important for their biological function. Recently, exosomes have been reported to have mitochondrial regulatory potential and antineurodegenerative properties. This review presents the mitochondrial abnormalities in brain cells associated with AD and PD and the potential of exosomes to treat these diseases. Specifically, it recapitulates research on the molecular mechanisms whereby exosomes regulate mitochondrial biogenesis, fusion/fission dynamics, mitochondrial transport, and mitophagy. Furthermore, this review discusses exosome-triggered signaling pathways that regulate nuclear factor (erythroid-derived 2)-like 2-dependent mitochondrial antioxidation and hypoxia inducible factor 1α-dependent metabolic reprogramming. In summary, this review aims to provide a profound understanding of the regulatory effect of exosomes on mitochondrial function in neurons and to propose exosome-mediated mitochondrial regulation as a promising strategy for AD and PD.
Keywords: neurodegenerative disease, Alzheimer’s disease, Parkinson’s disease, exosome, mitochondria
Graphical Abstract:

Introduction
Neurodegenerative diseases (NDs), including Alzheimer’s (AD) and Parkinson’s disease (PD), are characterized by progressive neuron degeneration, primarily within the central nervous system (CNS).1 These debilitating NDs are a major global health challenge because they are growing at epidemic proportions, and have only a few treatment options.2 AD and PD are associated with different pathological characteristics in their clinical manifestations. AD is the most common ND and is characterized by progressive cognitive impairment. Such impairment is associated with the accumulation of amyloid-beta peptide (Aβ) plaques and neurofibrillary tangles formed by hyperphosphorylated tau protein in the hippocampus and cerebral cortex, which results in neuronal death and brain atrophy.3 The amyloid cascade hypothesis suggests that Aβ accumulation causes tau pathology, neuroinflammation and oxidative stress.4 By contrast, PD is characterized by motor symptoms such as bradykinesia, rigidity, tremor, and postural instability, which occur when α-synuclein forms Lewy bodies in dopaminergic neurons in the substantia nigra.5 These distinctive pathologies are closely associated with the signs of mitochondrial dysfunction, such as increased oxidative stress and impaired energy metabolism, which are the main drivers of NDs and result in severe disability and reduced quality of life.6 The number of patients affected by dementia is estimated to increase to 130 million by 2050.7 Further, the global economic cost of AD-associated dementia and PD is estimated to reach US$2 trillion by 2030 and US$79 billion by 2037, respectively.8,9 However, despite extensive research and clinical efforts, developing effective therapeutic strategies to reverse the pathogenesis of AD and PD remains challenging.
Roles of Mitochondrial Dysfunction in AD and PD
Mitochondria are central organelles that play a key role in the cellular metabolism of all mammalian cells, including neurons.10 As neurons are particularly high energy demanding cells, which they need for synaptic formation and functional homeostasis, mitochondrial dysfunction is closely associated with energy depletion, oxidative stress and Ca2+ overload, leading to neurodegeneration.11,12 Further, imbalanced mitochondrial dynamics and dysfunction are significantly related to loss of neuron function in AD and PD.13 In AD, mitochondrial dysfunction and reactive oxygen species (ROS) overproduction are key in the processing of the amyloid precursor peptide (APP), Aβ accumulation, and formation of tau neurofibrillary tangles.14 A previous report in AD cybrids showed that mitochondrial ROS reduction reversed upregulated AD markers such as Aβ levels, apoptosis, and mitochondrial fission.15 In addition, inhibition of mitochondrial energetics results in neuron loss in the substantia nigra and striatum with tau hyperphosphorylation and aggregation.16,17 Moreover, age-related mitophagy dysregulation impairs lysosomal maturation and clearance of defective mitochondria.18 This failure in mitochondrial quality control (MQC) results in abnormal mitochondrial trafficking in neurons, leading to synaptic dysfunction.18 In PD, nigral neurons with high density of mitochondria are susceptible to mitochondrial dysfunction, eventually resulting in extensive neuronal degeneration.19 In addition, pesticides that stimulate mitochondrial fragmentation by inhibiting the ubiquitin protease system induced a PD-like phenotype in SH-SY5Y neuron cell models.20 Competitive inhibition of the interaction between nuclear factor (erythroid-derived 2)-like 2 (Nrf2) and Kelch-like ECH-associated protein 1 (Keap1) by 6-hydroxydopamine (6-OHDA), a structural analog of dopamine, is important for mitochondrial ROS-dependent neuronal apoptosis.21 Severe mitochondrial DNA damage has been observed in postmortem brain samples from PD patients.22 Further, previous studies demonstrated that sporadic PD is closely linked to a deficiency of mitochondrial complex I, with decreased ATP levels and excessive ROS generation.23–25 Mutations in mitophagy-regulating molecules such as Parkin and PTEN-induced kinase 1 (PINK1) are considered key players in familial PD pathogenesis.13 In fact, Parkin or PINK1 mutation in various in vivo models impairs mitochondrial complex I and III-mediated respiration, leading to significant loss of dopaminergic neurons.26–28
Exosomes
Given the crucial involvement of mitochondrial dysfunction in NDs, elucidating intercellular communication that can govern mitochondrial function is essential.13 Exosomes are a type of extracellular vesicle typically 40–100 nm in diameter.29 Exosomes are formed through inward budding of the plasma membrane within multivesicular bodies and subsequently released into the extracellular space by exocytosis.30,31 Exosomes are found in nearly all biological fluids, including blood, urine, saliva, cerebrospinal fluid, and breast milk,29,32 and play a crucial role in intercellular communication by delivering exogenous substances such as proteins, mRNAs, microRNAs, and lipids.29 Importantly, exosomes can transfer mitochondrial DNA, proteins, and metabolites which are able to alter mitochondrial function in the recipient cells and may rescue mitochondrial abnormalities found in NDs.33 In the CNS, the exosomes have attracted attention due to their therapeutic potential in NDs such as AD, PD, traumatic brain injury and brain stroke, as well as in intercellular communication.34 Brain-derived exosomes (BDEs) are a crucial mediator of intercellular communications and waste control between CNS cells, including neurons, glial cells, and connective tissues.34 Therefore, these findings suggest that BDE-regulated neurogenic niches including the hippocampus, are important for neurogenesis and cognitive improvement in NDs.35 Exosomes are able to regulate MQC pathways such as mitophagy and biogenesis, which could be exploited as a therapeutic strategy for the mitochondrial dysfunction seen in AD and PD pathology.36 In addition, exosomes can be used as non-invasive diagnostic biomarkers for AD and PD.37,38 Exosomes have potential as a drug delivery system because of their easily engineered membrane and high permeability to tissue and the blood-brain barrier (BBB).39,40 As natural nanoparticles with low immunogenicity, exosomes have a lower risk of causing an immune response when crossing the BBB. Further, they can be selectively targeted through surface modification, for instance, by attaching ligands that bind to BBB-associated receptors.30,41 This review will introduce the regulatory effects of exosomes on mitochondrial biogenesis, dynamics, mitophagy, antioxidant systems, and metabolic reprogramming, and highlight the potential of exosome-based therapeutics targeting mitochondria in AD and PD.
Therapeutic Effect of Exosomes on AD and PD
The therapeutic effects of different kinds of exosomes from various cell origins, including mesenchymal stem cells (MSCs), neural cells, and immune cells, have been reported in AD and PD.34 In AD, MSC-derived exosomes are emerging as a promising therapeutic strategy for amyloidogenesis and neurodegeneration. MSC-derived exosomes increased α‐secretase expression but decreased beta-secretase 1 (BACE1) expression, suppressing Aβ accumulation and microglia-regulated neuroinflammation.42,43 Exosomes derived from hypoxia-preconditioned MSCs improved synaptic dysfunction and anti-inflammation through anti- and pro-inflammatory cytokine regulation in APP/presenilin 1 (PS1) AD mice.44 In addition, exosome injection into the lateral ventricle of AD mice increased BDNF expression, a key factor in synapse formation and neurogenesis.45 Previous studies using animal behavioral tests, including the Morris water maze, open field, and novel object recognition tests, supported the neuronal recovery and cognitive improvement produced by MSC-derived exosomes.43,45,46 In addition, exosomes from curcumin-pretreated MSCs could inactivate the protein kinase B (Akt)/GSK3β pathway and ameliorate cognitive dysfunction by inhibiting tau hyper-phosphorylation.47 Intravenous administration of MSC exosomes modified with CNS-specific rabies virus glycoprotein for brain targeting significantly reduced Aβ accumulation and inactivated astrocytes.48 In addition, those modified exosomes improved cognitive function in APP/PS1 mice better than control exosomes.48 Emerging data indicate that exosomes are involved in AD pathology through regulation of Aβ neurotoxicity and tau hyperphosphorylation. In 6-OHDA-induced PD rats, intranasal administration of exosomes derived from human dental stem cells normalized tyrosine hydroxylase levels in the substantia nigra and striatum, and it improved spatial recognition and memory.49 Microglial exosomes have been shown to play a critical role in α-synuclein transmission and its pathology in PD.50 In fact, exosomes from microglia containing α-synuclein preformed fibrils stimulated protein aggregation in recipient neurons, suppressing autophagic flux and resulting in lysosome-associated membrane protein 2 (LAMP2) degradation.50
Exosomes and Mitochondrial Regulation in AD and PD
Mitochondrial dysfunction results in lower energy production and increased ROS, which contributes to oxidative stress, leading to metabolic dysregulation. This is marked by a reduction in glucose uptake and the tricarboxylic acid cycle, impairments in oxidative phosphorylation, and a reduction in the energetic support provided to neurons by astrocytes and oligodendrocytes.51,52 Exosomes have emerged as critical regulators of mitochondrial function, offering new insights into the pathogenesis and potential treatment of NDs. These exosomes influence various aspects of mitochondrial function and dynamics.53,54 By delivering specific cargo molecules, exosomes modulate the expression of genes and proteins crucial for mitochondrial homeostasis and their proper function. This review describes the intricate relationship between exosomes and mitochondrial regulation in diverse pathologic aspects of AD and PD.
Regulatory Effect of Exosomes on Mitochondrial Biogenesis
Recent studies have revealed a close relationship between mitochondrial dysfunction and NDs such as AD and PD, leading to increased interest in therapeutic approaches targeting mitochondrial biogenesis,55 a process in which exosomes play a crucial role. Extensive research on mitochondrial biogenesis and the Peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α), nuclear respiratory factor 1 (NRF1), and mitochondrial transcription factor A (TFAM) pathways has shown that PGC1α activation in the cell nucleus which is triggered by stimuli such as AMP-activated protein kinase (AMPK), sirtuin (SIRT), P38 mitogen activated protein kinase (P38 MAPK), and Akt, initiates transcription of NRF1 and TFAM, driving mitochondrial biogenesis.56,57 In a PD mouse model, a significant loss of dopamine neurons was observed in mice harboring a conditional knockout of PGC1α in the ventral midbrain.58 Accordingly, researchers are currently investigating how exosomes from different cell types can influence mitochondrial biogenesis by transferring these regulatory factors. For example, Feng et al reported that STIM-activating enhancer (STIMATE)-positive type II alveolar epithelial cell-derived exosomes enhanced mitochondrial biogenesis by activating the PGC1α pathway in conditional knockout mice lacking STIMATE, which helped reduce lung damage.59 Further, exosomes derived from stem cells from human exfoliated deciduous teeth could deliver TFAM mRNA to dental pulp stem cells, boosting mitochondrial biogenesis and enhancing bone regeneration.60 Moreover, transfer of circRNA from oxaliplatin-resistant cells via exosomes could revert the miR-30e-5p-mediated inhibition of PGC1α activity, increasing mitochondrial biogenesis and reprogramming the cells to favor oxidative phosphorylation.61 Similarly, a rat model for investigating potential treatments for liver ischemia-reperfusion injury received exosomes from adipose tissue-derived MSCs, increasing expression levels of PGC1α, NRF1, and TFAM, all related to mitochondrial biogenesis, along with improved liver function.62 Considering the depletion of factors related to mitochondrial biogenesis in NDs, using exosomes to supplement them can have therapeutic benefits. In fact, in a neural system-specific Sirt1 conditional knockout APP/PS1 mouse model of AD, neural stem cell-derived exosomes significantly increased SIRT1-dependent PGC1α signaling and improved mitochondrial biogenesis by increasing NRF1 and cytochrome c oxidase subunit 4 (COXIV) synthesis.63 These findings suggest that certain exosomes can enhance mitochondrial biogenesis in neurons, potentially offering a treatment for AD (Figure 1).
 |
Figure 1 Illustration showing the effect of molecules delivered by exosomes on mitochondrial biosynthesis in neurons. STIMATE, TFAM mRNA, SIRT1, and various factors delivered via exosomes upregulate PGC1α/NRF1/TFAM signaling. Exosomes also transfer other factors that downregulate miR-30e-5p, which inhibits PGC1α signaling. |
Regulatory Effect of Exosomes on Mitochondrial Dynamics
Mitochondria are dynamic intracellular organelles that continuously change their number and morphology. Here, mitochondrial dynamics will be categorized into fusion, fission, and transport. Mitochondrial dynamics is critical for maintaining mitochondrial homeostasis and regulates several cellular processes such as cell cycle progression, apoptosis, cell migration, mitophagy, and mitochondrial ROS production.64,65 Closely related to mitochondrial biogenesis, mitochondrial dynamics plays a crucial role in the pathogenesis and progression of NDs. The imbalance in mitochondrial dynamics is evident in AD and PD.66,67 For example, in vulnerable neurons in AD, this imbalance causes excessive oxidative stress and creates an environment likely to lead to energy depletion.68
Mitochondrial Fusion
Mitochondrial fusion involves the connection of inner and outer membranes between two mitochondria, facilitating the exchange of genetic material and metabolites.69 This process increases the oxidative phosphorylation capacity and allows redistribution of mitochondrial DNA between damaged and healthy mitochondria.70 Mitochondrial fusion comprises two main steps: outer mitochondrial membrane (OMM) fusion and inner mitochondrial membrane (IMM) fusion. OMM fusion is primarily mediated by mitofusin 1 and 2 (Mfn1 and Mfn2), which are transmembrane GTPases, while IMM fusion is facilitated by optic atrophy-1 (OPA1), a dynamin-related protein in the IMM.71 In AD, Aβ accumulation impairs mitochondrial function, reducing membrane potential and ATP production in hippocampal neurons and astrocytes.72,73 In mouse hippocampal neurons, inhibition of miR-195, a negative regulator of mitochondrial dynamics which targets Mfn2 gene, enhanced memory by ameliorating mitochondrial structural damage and reducing synaptic degradation.74 Regarding PD, α-synuclein overexpression inhibits mitochondrial fusion by reducing the expression of Mfn1, Mfn2, and OPA1.75 A recent study demonstrated the potential of exosome-mediated mitochondrial fusion regulator delivery which stimulates mitochondrial fusion. For example, trophoblast stem cell-derived exosomes were found to ameliorate doxorubicin-induced cardiotoxicity by increasing Mfn2 expression in mouse cardiomyocytes, thereby enhancing mitochondrial fusion.76 These findings highlight the therapeutic potential of targeting mitochondrial fusion proteins in AD and PD and that exosomes could be effective delivery vehicles for such interventions (Figure 2). Despite the limited studies focusing on engineering exosomes to improve mitochondrial dynamics for treating AD and PD, the potential applications of exosomes in these NDs require further research.
 |
Figure 2 Illustration showing the way molecules delivered by exosomes regulate mitochondrial dynamics in neurons. In neurons, the balance between mitochondrial fusion and fission is important for MQC. Nrf2 delivered via exosomes inactivates Drp1, thereby reducing excessive mitochondrial fission. In addition, delivery of factors regulating the expression or transfer of Miro1 via exosomes are expected to transport healthy mitochondria to deteriorated neurons. |
Mitochondrial Fission
Mitochondrial fission occurs when mitochondria divide into two separate organelles. It happens during an increase in mitochondrial population or when damaged mitochondria are removed by mitophagy.77 Fission begins with ER tubules adhering to the site of impending mitochondrial division. Subsequently, dynamin-related protein 1 (Drp1) accumulates at this location, forming a ring-like structure. Proteins such as mitochondrial fission 1 (Fis1), Mff, MiD49, and MiD51, which are located in the OMM, interact with Drp1. Then, the ring structure formed by Drp1, and mediated by DNM2, cleaves mitochondria.78,79 Impaired mitochondrial fission has been reported in NDs, resulting in an increased number of elongated mitochondria due to relatively higher fusion rates.77,80 In AD, Aβ and tau interact with Drp1, causing excessive mitochondrial fragmentation, which leads to synaptic dysfunction, neuronal damage, and cognitive decline.81 Furthermore, Fis1 aggregation has been reported in AD and is associated with disease progression.82 In the striatum of PD brain, mitochondrial fragmentation and abnormal mitochondrial localization are observed as well.83,84 Several studies aimed at improving mitochondrial dynamics using exosomes. For example, injection of CAP-Nrf2-exoS, an exosome produced using an Nrf2 overexpression vector, into mice inhibited Drp1 phosphorylation in cells. This prevents excessive mitochondrial modification and migration, thereby inhibiting mitochondrial division and dysfunction.85 PD-related proteins such as α-synuclein, leucine-rich repeat kinase-2, PINK1, and Parkin are known to regulate Drp1 function. Further, some therapeutic approaches aim to partially inhibit Drp1 function in purpose of reducing neuron death in PD. These approaches include DNM1L (gene encoding Drp1) silencing using siRNA and Drp1 inhibition using compounds such as mdivi1, which have potential neuroprotective effects by reducing mitophagy impairment and protein aggregation caused by α-synuclein.86,87 These findings highlight the potential of targeting mitochondrial fission, particularly through exosome-mediated approaches, in the treatment of AD and PD (Figure 2).
Mitochondrial Transport
Mitochondrial transport is intracellularly and intercellularly crucial in neurons, where mitochondria migrate to specific areas such as the axon to meet localized energy demands.88 Moreover, mitochondria can traverse cell boundaries, facilitating the exchange of intracellular substances and contributing to recovery in response to CNS stimuli.89 A key protein involved in mitochondrial transport and anchoring is mitochondrial Rho GTPase 1 (Miro1). This OMM-bound protein typically functions in intracellular axonal transport of mitochondria. Miro1 binds to the OMM, interacts with TRAK1/2, and in turn binds to motor proteins such as dynein or kinesin, forming a motor complex. This complex adheres to microtubules, enabling intracellular movement.90 Impaired Miro1-dependent mitochondrial transport and associated astrocyte dysfunction play a role in the pathogenesis of both AD and PD.91–93 A previous study using iPSCs to model the characteristics of PD revealed that Miro1 defects are significantly more common in PD patients than in the healthy population.94 Further, Miro1 loss is associated with mitophagy dysfunction and mitochondrial hyperactivation in response to stress, potentially contributing to the development of PD in affected cells.95 In Miro1 knockout mouse models, there is significant mitochondrial absence, particularly in distal neuronal dendrites.96 Given the importance of proper mitochondrial distribution for meeting local energy demands and Ca2+ buffering, Miro1-mediated mitochondrial transport is a crucial mechanism in neurodegeneration.96 In addition, Miro1 overexpression could be useful for improving mitochondrial function. When Miro1-overexpressing MSCs were delivered to mice with ischemic damage, cell-to-cell communication was enhanced by transferring healthy mitochondria to damaged astrocytes, resulting in significantly reduced neurological deficits and bioenergetic restoration of astrocytes.97 Moreover, recent studies have demonstrated that exosomes can alter Miro1 levels in neurons.98,99 Mitochondria are transferred by exosomes to rescue neurons with mitochondrial dysfunction caused by NDs, but the role of exosome-mediated mitochondrial transport in ND pathogenesis is still understudied.98,99 Despite the limited studies on exosome-mediated regulation of Miro1 in NDs, the potential of exosomes to deliver or modulate proteins involved in mitochondrial transport offers an intriguing area for future research (Figure 2).
Mitophagy
Mitophagy is a subtype of autophagy that selectively degrades dysfunctional and impaired mitochondria.100 This process is crucial for maintaining mitochondrial quality and homeostasis.101 However, both excessive and insufficient mitophagy occur under abnormal conditions, potentially leading to cell death and neurodegeneration.102–104 Several mechanisms are responsible for mitophagy, each dependent on proteins such as PINK1, BCL2 interacting protein 3 (BNIP3), BNIP3-like (NIX) and FUNDC1.105 PINK1 is imported into the IMM and degraded by MG132-sensitive proteases. However, when mitochondria are damaged, proteasomal stress from MG132 causes proteasome dysfunction and PINK1 accumulation.106 Then, accumulated PINK1s are embedded into the OMM and recruit Parkin, which then activates the ubiquitin-proteasome system and starts the universal ubiquitylation and degradation of OMM proteins, eventually leading to mitophagy.107 Regulation of mitophagy by exosomal miRNAs and proteins is an effective strategy for MQC.108,109 Exosomes derived from astrocytes under oxygen-glucose deprivation/re-oxygenation conditions transfer miR-138-5p, which downregulates DNA methyltransferase 3A (DNMT3A), leading to decreased methylation of the promoter of Ras homolog enriched in brain like 1 (Rhebl1), ultimately increasing mitophagy in neurons.108 Other than regulating the DNMT3A/Rhebl1 axis, miR-138-5p also affect PINK1/Parkin signaling further enhancing mitophagy and reducing reperfusion injuries in middle cerebral artery occlusion.108 In addition, insulin-like growth factor 1 regulates mitophagy in astrocytes through exosomal miR-let-7e enhancing mitophagy with increased PINK1 and NIX expression in traumatic brain injury.110 In PD, dysregulated PINK1/parkin-mediated mitophagy reportedly contributes to the accumulation of damaged mitochondria and neuron dysfunction.111 In dopaminergic neurons of patients with PD, exosomes mediate the disposal of damaged mitochondria and mitochondrial-derived vesicles through the PINK1/Parkin pathway.112 In PD rodent models, the condition significantly improved after treatment with MSC-derived exosomes, which increased Parkin and DJ-1 levels in the brain compared to rats treated with rotenone.113 Similarly, M2 microglia-derived exosomes can regulate mitophagy in neurons via the PINK1/Parkin pathway, providing neuroprotection by restoring mitochondrial function and reducing ROS in AD.114 In most ND cases, exosomes are utilized to increase mitophagy and remove defective mitochondria. However, exosomes can also be used to reduce excessive mitophagy in NDs. Rotenone, a mitochondrial complex I inhibitor, induces excessive mitophagy, leading to neuron apoptosis in PD, but epicatechin gallate-loaded exosomes reverse this effect by suppressing PINK1 and Parkin expression.115 Taken together, exosome-mediated mitophagy facilitates the clearance of damaged mitochondria thus improving MQC in AD and PD (Figure 3). While enhanced mitophagy can be beneficial in clearing damaged mitochondria, excessive mitophagy can be detrimental. Thus, future research should focus on developing exosome-based therapies that fine-tune mitophagy to optimal levels in damaged neurons.
 |
Figure 3 Illustration showing that molecules delivered by exosomes regulate mitophagy in neurons. Numerous activators delivered by exosomes, such as BNIP3, miR-138-5p, and miR-let-7e, each send signals that ultimately increase mitophagy. However, epicatechin gallate delivered via exosomes alleviated the excessive apoptosis observed in neurodegenerative diseases. |
Exosome-Mediated Modulation of Nrf2 and Hypoxia Inducible Factor 1 (HIF1) Pathways
Exosomes are critical mediators in the therapeutic landscape of AD and PD due to their ability to modulate essential cellular pathways. Specifically, Nrf2 and HIF1 pathways are pivotal in modulating mitochondrial antioxidative defenses and metabolic reprogramming, respectively. By influencing these pathways, exosomes protect neurons from mitochondrial oxidative stress, promoting metabolic adaptations. This section discusses how exosome-mediated modulation of these pathways is a promising strategy for the treatment of AD and PD (Table 1).
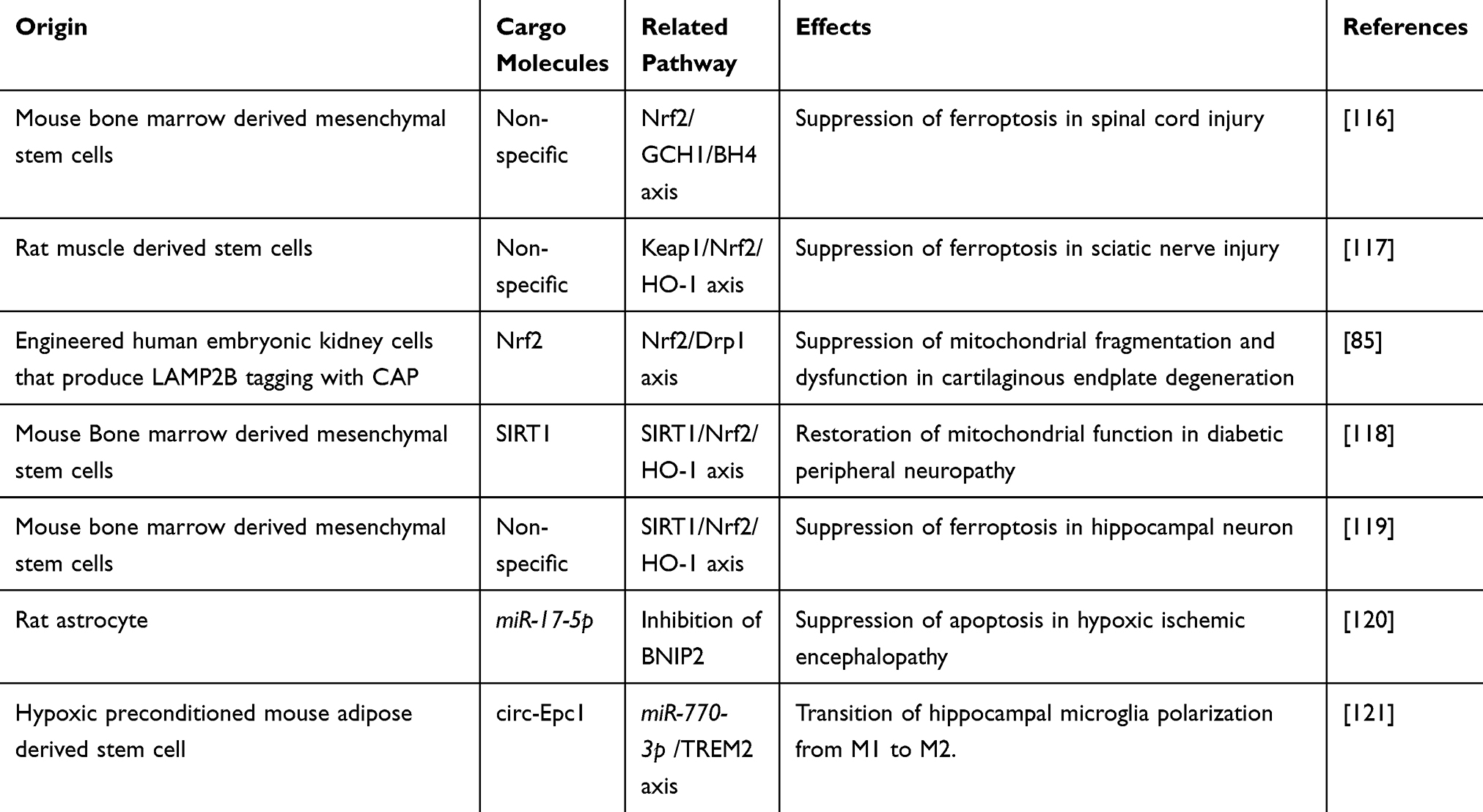 |
Table 1 Modulation of Nrf2 and HIF1 Signaling in Neurodegenerative Disease Using Exosomes Derived From Different Cells |
Exosome-Regulated Nrf2-Dependent Mitochondrial Antioxidant System
The transcriptional factor Nrf2 is involved in the control of neuronal antioxidation and protection against cellular damage in NDs, including AD and PD.122,123 Nrf2 regulates the expression of cytoprotective genes related to cellular antioxidant enzymes, glutathione synthesis, recycling enzymes, and iron sequestration proteins including superoxide dismutase 2, heme oxygenase 1 (HO-1), NAD(P)H quinone oxidoreductase 1, glutathione S-transferases, glutamate-cysteine ligase and glutathione reductase.124 The interactions between exosomes and Nrf2 not only facilitate the production of protective enzymes but also modulate mitochondrial dynamics. Recent research highlights the potential of exosomes as delivery vehicles for Nrf2 activators, which enhance the therapeutic efficacy of natural compounds and RNA-based treatments for AD.125 In recipient neurons, exosomes with Nrf2 activators alter Nrf2 signaling through direct transfer of Nrf2 protein or mRNA and transport of Keap1 inhibitors.125 The delivery of complementary sequences that inhibit NFE2L2 (gene encoding Nrf2) targeting-miRNAs increases Nrf2 expression.125 Accordingly, the therapeutic potential of exosome-regulated Nrf2 pathway for NDs has been investigated.126 MSC-derived exosomes have shown promise in mitigating ferroptosis during acute spinal cord injury, by activating the Nrf2/GTP cyclohydrolase I (GCH1)/5,6,7,8-tetrahydrobiopterin (BH4) axis.116 In addition, muscle-derived stem cell exosomes offer a potential therapeutic strategy for peripheral nerve injuries by suppressing ferroptosis in the sciatic nerve and dorsal root ganglion through activation of the Nrf2/HO-1 pathway, leading to improved post-injury recovery with enhanced nerve function and reduced muscle atrophy.117 Exosomes engineered to express chondrocyte affinity peptide carrying miR-140 and Nrf2 have demonstrated efficacy in the treatment of intervertebral disc degeneration.85,127 In addition, SIRT1-overexpressing exosomes derived from bone marrow MSCs improved diabetic peripheral neuropathy through Nrf2/HO-1-dependent antioxidation.118 Further, MSC-derived exosomes ameliorated cognitive impairment after exploratory laparotomy by inhibiting hippocampus ferroptosis in old mice with delayed neurocognitive recovery.119 Despite challenges in ensuring specific delivery into damaged neurons in NDs, it is possible to slow down ND progression through Nrf2-dependent mitochondrial antioxidation. Continued exploration of exosome-Nrf2 interactions holds great promise for advancing our understanding of disease pathogenesis and developing novel therapeutic strategies for AD and PD.
Exosome-Regulated HIF1-Dependent Metabolic Reprogramming
Like the Nrf2 pathway, HIF1-dependent metabolic reprogramming plays a crucial role in cellular stress adaptation responses in NDs such as AD and PD.128 HIF1 is a heterodimeric transcription factor composed of α (HIF1α) and β subunits (HIF1β). Under normoxic conditions, HIF1α is degraded through the ubiquitin-proteasome pathway.129 Prolyl hydroxylases hydroxylate proline residues in HIF1α, being recognized by the von Hippel-Lindau E3 ubiquitin ligase complex.130 Under hypoxic conditions, HIF-prolyl hydroxylases are inhibited leading to HIF1α stabilization. The stabilized HIF1α then translocates to the nucleus, where it dimerizes with HIF1β and binds to hypoxia-response elements.129 HIF1-dependent metabolic reprogramming under hypoxia affects glucose metabolism, mitochondrial function, antioxidant capacity, and neuron survival.131 HIF1 plays a critical role in facilitating cellular adaptation to stress, particularly in NDs. In addition to glucose metabolism and mitochondrial function, lipid and amino acid metabolism are modulated in a HIF1-dependent manner.132–134 HIF1α has neuroprotective properties by counteracting the detrimental effects of Aβ and preventing excessive tau phosphorylation, which highlights the potential of HIF1α as a therapeutic target for AD.135 The complex interplay between HIF1 signaling and exosome-mediated communication is identified as a pivotal factor in ND progression. Delivering HIF1 and other molecules specifically to the brain, exosomes can be used as a new therapeutic strategy for AD and PD.136 A previous study showed that astrocyte-derived exosomes protect neonatal rats from hypoxic-ischemic brain damage by inhibiting BNIP2 expression.120 This finding indicated that exosomes control HIF1-dependent metabolic pathways by delivering regulatory components such as mRNAs, proteins, and miRNAs. These components exert either a direct or indirect influence on HIF1 stability and functionality. A previous study showed that exosomes from adipose tissue-derived MSCs that underwent hypoxic preconditioning improved cognitive function in AD by delivering circ-Epc1.121 Then, circ-Epc1 regulated microglial polarization through miR-770-3p/TREM2 signaling, associated with shifting microglia from M1 to M2.121 Taken together, these findings suggest that exosome-regulated HIF1 signaling can be a promising therapeutic strategy for the treatment of AD and PD by controlling metabolic reprogramming and MQC.
Conclusion
In conclusion, this review emphasizes the role of exosomes in regulating mitochondrial function and their therapeutic potential for AD and PD. Exosomes modulate key aspects of mitochondrial biogenesis, fusion/fission balance, mitochondrial transport, and mitophagy, all crucial for neuron survival and function. Moreover, they play a key role in the regulation of Nrf2-dependent mitochondrial antioxidant responses and HIF1-dependent metabolic reprogramming, essential for MQC in neurons. Importantly, in the context of AD, exosomes have been investigated as a therapeutic tool for alleviating Aβ-induced mitochondrial toxicity and tau phosphorylation. Through exosome-mediated transfer of functional mitochondria or regulation of mitochondrial function in recipient neurons, exosomes hold significant potential in maintaining mitochondrial homeostasis. Consistently, in PD, exosomes prevent α-synuclein aggregation and mitochondrial dysfunction, further underscoring their role in disease progression. Despite these findings, detailed mechanisms underlying the interaction between exosomes and mitochondrial regulatory pathways, as well as their impact on AD and PD progression, remain unclear. To advance exosome-based therapeutic strategies for AD and PD, further research is needed to fully elucidate these mechanisms and enhance the engineering of exosome which targets mitochondrial dysfunction-induced NDs. Future studies should focus on optimizing exosome design to enhance mitochondrial function and associated regulatory pathways, ensuring precise delivery to affected brain regions and minimizing potential side effects.
Abbreviations
6-OHDA, 6-hydroxydopamine; AD, Alzheimer’s disease; Akt, Protein kinase B; Aβ, Amyloid beta; AMPK, AMP-activated protein kinase; APP, Amyloid precursor peptide; BACE1, beta-secretase 1; BBB, Blood-brain barrier; BDEs, Brain-derived exosomes; BDNF, Brain-derived neurotrophic factor; BH4, 5,6,7,8-tetrahydrobiopterin; BNIP3, Bcl2 interacting protein 3; CAP, Chondrocyte-affinity peptide; CNS, Central nervous system; COXIV, Cytochrome c oxidase subunit 4; DNMT3A, DNA methyltransferase 3A; Drp1, Dynamin-related protein 1; Fis1, Mitochondrial fission 1; FUNDC1, Fun14 domain containing 1; GCH1, GTP cyclohydrolase I; GSK3β, Glycogen synthase kinase 3β; HIF1, Hypoxia inducible factor 1; HIF1α, Hypoxia inducible factor 1 subunit α; HIF1β, Hypoxia inducible factor 1 subunit β; HO-1, Heme oxygenase 1; IMM, Inner mitochondrial membrane; iPSCs, Induced pluripotent stem cells; Keap1, Kelch-like ECH-associated protein 1; LAMP2, Lysosome-associated membrane protein 2; M1, Classical phenotype microglia; M2, Alternative phenotype microglia; Mff, Mitochondrial fission factor; Mfn1, Mitofusin 1; Mfn2, Mitofusin 2; MG-132, Carbobenzoxy-l-leucyl-l-leucyl-l-leucinal; MiD49, Mitochondrial dynamics proteins of 49kda; MiD51, Mitochondrial dynamics proteins of 51kda; Miro1, Mitochondrial rho GTPase 1; MQC, Mitochondrial quality control; MSCs, Mesenchymal stem cells; NAD(P)H, Nicotinamide adenine dinucleotide (phosphate) hydrogen; NDs, Neurodegenerative diseases; NIX, Bcl2 interacting protein 3-like; NRF1, Nuclear respiratory factor 1; Nrf2, Nuclear factor (erythroid-derived 2)-like 2; OMM, Outer mitochondrial membrane; OPA1, Optic atrophy 1; P38 MAPK, P38 mitogen activated protein kinase; PD, Parkinson’s disease; PGC1α, Proliferator-activated receptor gamma coactivator 1α; PINK1, PS1, presenilin 1; PTEN-induced kinase 1; Rhebl1, Ras homolog enriched in brain like 1; ROS, Reactive oxygen species; SIRT, Sirtuin; STIM, Stromal interaction molecule; STIMATE, Stim-activating enhancer; TFAM, Mitochondrial transcription factor A; TRAK1, Trafficking kinesin protein 1; TRAK2, Trafficking kinesin protein 2; TREM2, Triggering receptor expressed on myeloid cells 2.
Acknowledgments
The authors would like to thank Enago (www.enago.com) for the English language review. This research was supported by National R&D Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2021R1C1C1009595); Korea government (MSIT, No. RS-2023-00219563); Regional Innovation Strategy (RIS) through the NRF funded by the Ministry of Education (MOE, 2021RIS-001). This work was supported by the Soonchunhyang University Research Fund.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mukherjee A, Becerra Calixto AD, Chavez M, Delgado JP, Soto C. Mitochondrial transplant to replenish damaged mitochondria: a novel therapeutic strategy for neurodegenerative diseases? Prog mol Biol Transl Sci. 2021;177:49–63. doi:10.1016/bs.pmbts.2020.10.001
2. Wang S, Jiang Y, Yang A, Meng F, Zhang J. The expanding burden of neurodegenerative diseases: an unmet medical and social need. Aging Dis. 2024;2024:1. doi:10.14336/AD.2024.1071
3. Liu E, Zhang Y, Wang JZ. Updates in Alzheimer’s disease: from basic research to diagnosis and therapies. Transl Neurodegener. 2024;13(1):45. doi:10.1186/s40035-024-00432-x
4. Nasb M, Tao W, Chen N. Alzheimer’s disease puzzle: delving into pathogenesis hypotheses. Aging Dis. 2024;15(1):43–73. doi:10.14336/AD.2023.0608
5. Koeglsperger T, Rumpf SL, Schließer P, et al. Neuropathology of incidental Lewy body & prodromal Parkinson’s disease. mol Neurodegener. 2023;18(1):32. doi:10.1186/s13024-023-00622-7
6. Zhang X, Wang L, Li B, Shi J, Xu J, Yuan M. Targeting mitochondrial dysfunction in neurodegenerative diseases: expanding the therapeutic approaches by plant-derived natural products. Pharmaceuticals. 2023;16(2):277. doi:10.3390/ph16020277
7. Hansson O. Biomarkers for neurodegenerative diseases. Nat Med. 2021;27(6):954–963. doi:10.1038/s41591-021-01382-x
8. Tay LX, Ong SC, Tay LJ, Ng T, Parumasivam T. Economic burden of Alzheimer’s disease: a systematic review. Value Health Reg Issues. 2024;40:1–12. doi:10.1016/j.vhri.2023.09.008
9. Yang W, Hamilton JL, Kopil C, et al. Current and projected future economic burden of Parkinson’s disease in the U.S. NPJ Parkinsons Dis. 2020;6:15. doi:10.1038/s41531-020-0117-1
10. Xu J, Du W, Zhao Y, et al. Mitochondria targeting drugs for neurodegenerative diseases-design, mechanism and application. Acta Pharm Sin B. 2022;12(6):2778–2789. doi:10.1016/j.apsb.2022.03.001
11. Goyal S, Chaturvedi RK. Mitochondrial protein import dysfunction in pathogenesis of neurodegenerative diseases. mol Neurobiol. 2021;58(4):1418–1437. doi:10.1007/s12035-020-02200-0
12. Wang XL, Feng ST, Wang ZZ, Chen NH, Zhang Y. Role of mitophagy in mitochondrial quality control: mechanisms and potential implications for neurodegenerative diseases. Pharmacol Res. 2021;165:105433. doi:10.1016/j.phrs.2021.105433
13. Bustamante-Barrientos FA, Luque-Campos N, Araya MJ, et al. Mitochondrial dysfunction in neurodegenerative disorders: potential therapeutic application of mitochondrial transfer to central nervous system-residing cells. J Transl Med. 2023;21(1):613. doi:10.1186/s12967-023-04493-w
14. Swerdlow RH. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis. 2018;62(3):1403–1416. doi:10.3233/JAD-170585
15. Khan SM, Cassarino DS, Abramova NN, et al. Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann Neurol. 2000;48(2):148–155. doi:10.1002/1531-8249(200008)48:2<148::AID-ANA3>3.0.CO;2-7
16. Escobar-Khondiker M, Höllerhage M, Muriel MP, et al. Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J Neurosci. 2007;27(29):7827–7837. doi:10.1523/JNEUROSCI.1644-07.2007
17. Höglinger GU, Lannuzel A, Khondiker ME, et al. The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J Neurochem. 2005;95(4):930–939. doi:10.1111/j.1471-4159.2005.03493.x
18. Reddy PH, Oliver DM. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells. 2019;8(5):488. doi:10.3390/cells8050488
19. Macdonald R, Barnes K, Hastings C, Mortiboys H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: can mitochondria be targeted therapeutically? Biochem Soc Trans. 2018;46(4):891–909. doi:10.1042/BST20170501
20. Chen T, Tan J, Wan Z, et al. Effects of commonly used pesticides in China on the mitochondria and ubiquitin-proteasome system in Parkinson’s disease. Int J mol Sci. 2017;18(12):2507. doi:10.3390/ijms18122507
21. Ren J, Yuan L, Wang W, et al. Tricetin protects against 6-OHDA-induced neurotoxicity in Parkinson’s disease model by activating Nrf2/HO-1 signaling pathway and preventing mitochondria-dependent apoptosis pathway. Toxicol Appl Pharmacol. 2019;378:114617. doi:10.1016/j.taap.2019.114617
22. Sanders LH, McCoy J, Hu X, et al. Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson’s disease. Neurobiol Dis. 2014;70:214–223. doi:10.1016/j.nbd.2014.06.014
23. Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem. 1990;54(3):823–827. doi:10.1111/j.1471-4159.1990.tb02325.x
24. Parker WD Jr, Parks JK, Swerdlow RH. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008;1189:215–218. doi:10.1016/j.brainres.2007.10.061
25. Greenamyre JT, Sherer TB, Betarbet R, Panov AV. Complex I and Parkinson’s disease. IUBMB Life. 2001;52(3–5):135–141. doi:10.1080/15216540152845939
26. Flinn L, Mortiboys H, Volkmann K, Köster RW, Ingham PW, Bandmann O. Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain. 2009;132(Pt 6):1613–1623. doi:10.1093/brain/awp108
27. Flinn LJ, Keatinge M, Bretaud S, et al. TigarB causes mitochondrial dysfunction and neuronal loss in PINK1 deficiency. Ann Neurol. 2013;74(6):837–847. doi:10.1002/ana.23999
28. Stauch KL, Villeneuve LM, Purnell PR, Ottemann BM, Emanuel K, Fox HS. Loss of Pink1 modulates synaptic mitochondrial bioenergetics in the rat striatum prior to motor symptoms: concomitant complex I respiratory defects and increased complex II-mediated respiration. Proteomics Clin Appl. 2016;10(12):1205–1217. doi:10.1002/prca.201600005
29. Liang Y, Duan L, Lu J, Xia J. Engineering exosomes for targeted drug delivery. Theranostics. 2021;11(7):3183–3195. doi:10.7150/thno.52570
30. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478):eaau6977. doi:10.1126/science.aau6977
31. Jeppesen DK, Fenix AM, Franklin JL, et al. Reassessment of exosome composition. Cell. 2019;177(2):428–445.e18. doi:10.1016/j.cell.2019.02.029
32. Welton JL, Loveless S, Stone T, von Ruhland C, Robertson NP, Clayton A. Cerebrospinal fluid extracellular vesicle enrichment for protein biomarker discovery in neurological disease; multiple sclerosis. J Extracell Vesicles. 2017;6(1):1369805. doi:10.1080/20013078.2017.1369805
33. Zhang Y, Tan J, Miao Y, Zhang Q. The effect of extracellular vesicles on the regulation of mitochondria under hypoxia. Cell Death Dis. 2021;12(4):358. doi:10.1038/s41419-021-03640-9
34. Fallahi S, Zangbar HS, Farajdokht F, Rahbarghazi R, Mohaddes G, Ghiasi F. Exosomes as a therapeutic tool to promote neurorestoration and cognitive function in neurological conditions: achieve two ends with a single effort. CNS Neurosci Ther. 2024;30(5):e14752. doi:10.1111/cns.14752
35. Jovanovic VM, Salti A, Tilleman H, et al. BMP/SMAD pathway promotes neurogenesis of midbrain dopaminergic neurons in vivo and in human induced pluripotent and neural stem cells. J Neurosci. 2018;38(7):1662–1676. doi:10.1523/JNEUROSCI.1540-17.2018
36. Zong Y, Li H, Liao P, et al. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther. 2024;9(1):124. doi:10.1038/s41392-024-01839-8
37. Bir A, Ghosh A, Chauhan A, et al. Exosomal dynamics and brain redox imbalance: implications in Alzheimer’s disease pathology and diagnosis. Antioxidants. 2024;13(3):316. doi:10.3390/antiox13030316
38. Kim KY, Shin KY, Chang KA. Potential exosome biomarkers for Parkinson’s disease diagnosis: a systematic review and meta-analysis. Int J mol Sci. 2024;25(10):5307. doi:10.3390/ijms25105307
39. Salunkhe S, Chitkara D, Mittal A, Chitkara D, Mittal A. Surface functionalization of exosomes for target-specific delivery and in vivo imaging & tracking: strategies and significance. J Control Release. 2020;326:599–614. doi:10.1016/j.jconrel.2020.07.042
40. Barile L, Vassalli G. Exosomes: therapy delivery tools and biomarkers of diseases. Pharmacol Ther. 2017;174:63–78. doi:10.1016/j.pharmthera.2017.02.020
41. Choi H, Choi K, Kim DH, et al. Strategies for targeted delivery of exosomes to the brain: advantages and challenges. Pharmaceutics. 2022;14(3):672. doi:10.3390/pharmaceutics14030672
42. Yang L, Zhai Y, Hao Y, Zhu Z, Cheng G. The regulatory functionality of exosomes derived from hUMSCs in 3D culture for Alzheimer’s disease therapy. Small. 2020;16(3):e1906273. doi:10.1002/smll.201906273
43. Ding M, Shen Y, Wang P, et al. Exosomes isolated from human umbilical cord mesenchymal stem cells alleviate neuroinflammation and reduce amyloid-beta deposition by modulating microglial activation in Alzheimer’s disease. Neurochem Res. 2018;43(11):2165–2177. doi:10.1007/s11064-018-2641-5
44. Cui GH, Wu J, Mou FF, et al. Exosomes derived from hypoxia-preconditioned mesenchymal stromal cells ameliorate cognitive decline by rescuing synaptic dysfunction and regulating inflammatory responses in APP/PS1 mice. FASEB J. 2018;32(2):654–668. doi:10.1096/fj.201700600R
45. Liu S, Fan M, Xu JX, et al. Exosomes derived from bone-marrow mesenchymal stem cells alleviate cognitive decline in AD-like mice by improving BDNF-related neuropathology. J Neuroinflammation. 2022;19(1):35. doi:10.1186/s12974-022-02393-2
46. Reza-Zaldivar EE, Hernández-Sapiéns MA, Gutiérrez-Mercado YK, et al. Mesenchymal stem cell-derived exosomes promote neurogenesis and cognitive function recovery in a mouse model of Alzheimer’s disease. Neural Regen Res. 2019;14(9):1626–1634. doi:10.4103/1673-5374.255978
47. Wang H, Sui H, Zheng Y, et al. Curcumin-primed exosomes potently ameliorate cognitive function in AD mice by inhibiting hyperphosphorylation of the Tau protein through the AKT/GSK-3β pathway. Nanoscale. 2019;11(15):7481–7496. doi:10.1039/C9NR01255A
48. Cui GH, Guo HD, Li H, et al. RVG-modified exosomes derived from mesenchymal stem cells rescue memory deficits by regulating inflammatory responses in a mouse model of Alzheimer’s disease. Immun Ageing. 2019;16:10. doi:10.1186/s12979-019-0150-2
49. Narbute K, Pilipenko V, Pupure J, et al. Time-dependent memory and gait improvement by intranasally-administered extracellular vesicles in Parkinson’s disease model rats. Cell mol Neurobiol. 2021;41(3):605–613. doi:10.1007/s10571-020-00865-8
50. Guo M, Wang J, Zhao Y, et al. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain. 2020;143(5):1476–1497. doi:10.1093/brain/awaa090
51. Park MW, Cha HW, Kim J, et al. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. 2021;41:101947. doi:10.1016/j.redox.2021.101947
52. Rose J, Brian C, Woods J, et al. Mitochondrial dysfunction in glial cells: implications for neuronal homeostasis and survival. Toxicology. 2017;391:109–115. doi:10.1016/j.tox.2017.06.011
53. Yan X, Chen X, Shan Z, Bi L. Engineering exosomes to specifically target the mitochondria of brain cells. ACS Omega. 2023;8(51):48984–48993. doi:10.1021/acsomega.3c06617
54. Shukla S, Currim F, Singh J, et al. hsa-miR-320a mediated exosome release under PD stress conditions rescue mitochondrial ROS and cell death in the recipient neuronal and glial cells. Int J Biochem Cell Biol. 2023;162:106439. doi:10.1016/j.biocel.2023.106439
55. Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, Ahmadiani A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: pathogenesis and treatment. CNS Neurosci Ther. 2017;23(1):5–22. doi:10.1111/cns.12655
56. Chen L, Qin Y, Liu B, et al. PGC-1α-mediated mitochondrial quality control: molecular mechanisms and implications for heart failure. Front Cell Dev Biol. 2022;10:871357. doi:10.3389/fcell.2022.871357
57. Quan Y, Xin Y, Tian G, Zhou J, Liu X. Mitochondrial ROS-modulated mtDNA: a potential target for cardiac aging. Oxid Med Cell Longev. 2020;2020:9423593. doi:10.1155/2020/9423593
58. Jiang H, Kang SU, Zhang S, et al. Adult conditional knockout of PGC-1α leads to loss of dopamine neurons. eNeuro. 2016;3(4):ENEURO.0183–16. doi:10.1523/ENEURO.0183-16.2016
59. Feng Z, Jing Z, Li Q, et al. Exosomal STIMATE derived from type II alveolar epithelial cells controls metabolic reprogramming of tissue-resident alveolar macrophages. Theranostics. 2023;13(3):991–1009. doi:10.7150/thno.82552
60. Guo J, Zhou F, Liu Z, et al. Exosome-shuttled mitochondrial transcription factor A mRNA promotes the osteogenesis of dental pulp stem cells through mitochondrial oxidative phosphorylation activation. Cell Prolif. 2022;55(12):e13324. doi:10.1111/cpr.13324
61. Deng J, Pan T, Lv C, et al. Exosomal transfer leads to chemoresistance through oxidative phosphorylation-mediated stemness phenotype in colorectal cancer. Theranostics. 2023;13(14):5057–5074. doi:10.7150/thno.84937
62. Zhang Q, Piao C, Ma H, et al. Exosomes from adipose-derived mesenchymal stem cells alleviate liver ischaemia reperfusion injury subsequent to hepatectomy in rats by regulating mitochondrial dynamics and biogenesis. J Cell mol Med. 2021;25(21):10152–10163. doi:10.1111/jcmm.16952
63. Li B, Chen Y, Zhou Y, et al. Neural stem cell-derived exosomes promote mitochondrial biogenesis and restore abnormal protein distribution in a mouse model of Alzheimer’s disease. Neural Regen Res. 2024;19(7):1593–1601. doi:10.4103/1673-5374.385839
64. Liu AR, Lv Z, Yan ZW, et al. Association of mitochondrial homeostasis and dynamic balance with malignant biological behaviors of gastrointestinal cancer. J Transl Med. 2023;21(1):27. doi:10.1186/s12967-023-03878-1
65. Ma Y, Wang L, Jia R. The role of mitochondrial dynamics in human cancers. Am J Cancer Res. 2020;10(5):1278–1293.
66. Wang X, Su B, Lee HG, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29(28):9090–9103. doi:10.1523/JNEUROSCI.1357-09.2009
67. Santos D, Esteves AR, Silva DF, Januário C, Cardoso SM. The impact of mitochondrial fusion and fission modulation in sporadic Parkinson’s disease. Mol Neurobiol. 2015;52(1):573–586. doi:10.1007/s12035-014-8893-4
68. Tarawneh R. Biomarkers: our path towards a cure for Alzheimer disease. Biomark Insights. 2020;15:1177271920976367. doi:10.1177/1177271920976367
69. Chen W, Zhao H, Li Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Signal Transduct Target Ther. 2023;8(1):333. doi:10.1038/s41392-023-01547-9
70. Labbé K, Murley A, Nunnari J. Determinants and functions of mitochondrial behavior. Annu Rev Cell Dev Biol. 2014;30:357–391. doi:10.1146/annurev-cellbio-101011-155756
71. Chiong M, Cartes-Saavedra B, Norambuena-Soto I, et al. Mitochondrial metabolism and the control of vascular smooth muscle cell proliferation. Front Cell Dev Biol. 2014;2:72. doi:10.3389/fcell.2014.00072
72. Cha MY, Han SH, Son SM, et al. Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS One. 2012;7(4):e34929. doi:10.1371/journal.pone.0034929
73. Sarkar P, Zaja I, Bienengraeber M, et al. Epoxyeicosatrienoic acids pretreatment improves amyloid β-induced mitochondrial dysfunction in cultured rat hippocampal astrocytes. Am J Physiol Heart Circ Physiol. 2014;306(4):H475–H484. doi:10.1152/ajpheart.00001.2013
74. Gao Z, Zhang R, Jiang L, et al. Administration of miR-195 inhibitor enhances memory function through improving synaptic degradation and mitochondrial dysfunction of the hippocampal neurons in SAMP8 mice. J Alzheimers Dis. 2022;85(4):1495–1509. doi:10.3233/JAD-215301
75. Kamp F, Exner N, Lutz AK, et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO j. 2010;29(20):3571–3589. doi:10.1038/emboj.2010.223
76. Duan J, Liu X, Shen S, et al. Trophoblast stem-cell-derived exosomes alleviate cardiotoxicity of doxorubicin via improving Mfn2-mediated mitochondrial fusion. Cardiovasc Toxicol. 2023;23(1):23–31. doi:10.1007/s12012-022-09774-2
77. Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev mol Cell Biol. 2010;11(12):872–884. doi:10.1038/nrm3013
78. Adebayo M, Singh S, Singh AP, Dasgupta S. Mitochondrial fusion and fission: the fine-tune balance for cellular homeostasis. FASEB J. 2021;35(6):e21620. doi:10.1096/fj.202100067R
79. Forte M, Schirone L, Ameri P, et al. The role of mitochondrial dynamics in cardiovascular diseases. Br J Pharmacol. 2021;178(10):2060–2076. doi:10.1111/bph.15068
80. Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi:10.1146/annurev.cellbio.22.010305.104638
81. Bera A, Lavanya G, Reshmi R, Dev K, Kumar R. Mechanistic and therapeutic role of Drp1 in the pathogenesis of Alzheimer’s disease. Eur J Neurosci. 2022;56(9):5516–5531. doi:10.1111/ejn.15611
82. Ihenacho UK, Meacham KA, Harwig MC, Widlansky ME, Hill RB. Mitochondrial fission protein 1: emerging roles in organellar form and function in health and disease. Front Endocrinol. 2021;12:660095. doi:10.3389/fendo.2021.660095
83. Arduíno DM, Esteves AR, Cortes L, et al. Mitochondrial metabolism in Parkinson’s disease impairs quality control autophagy by hampering microtubule-dependent traffic. Hum mol Genet. 2012;21(21):4680–4702. doi:10.1093/hmg/dds309
84. Henrich MT, Oertel WH, Surmeier DJ, Geibl FF. Mitochondrial dysfunction in Parkinson’s disease - a key disease hallmark with therapeutic potential. mol Neurodegener. 2023;18(1):83. doi:10.1186/s13024-023-00676-7
85. Lin Z, Xu G, Lu X, et al. Chondrocyte-targeted exosome-mediated delivery of Nrf2 alleviates cartilaginous endplate degeneration by modulating mitochondrial fission. J Nanobiotechnology. 2024;22(1):281. doi:10.1186/s12951-024-02517-1
86. Feng ST, Wang ZZ, Yuan YH, et al. Dynamin-related protein 1: a protein critical for mitochondrial fission, mitophagy, and neuronal death in Parkinson’s disease. Pharmacol Res. 2020;151:104553. doi:10.1016/j.phrs.2019.104553
87. Fan RZ, Guo M, Luo S, Cui M, Tieu K. Exosome release and neuropathology induced by α-synuclein: new insights into protective mechanisms of Drp1 inhibition. Acta Neuropathol Commun. 2019;7(1):184. doi:10.1186/s40478-019-0821-4
88. Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6–13. doi:10.1016/j.redox.2014.11.006
89. Shanmughapriya S, Langford D, Natarajaseenivasan K. Inter and intracellular mitochondrial trafficking in health and disease. Ageing Res Rev. 2020;62:101128. doi:10.1016/j.arr.2020.101128
90. Covill-Cooke C, Toncheva VS, Kittler JT. Regulation of peroxisomal trafficking and distribution. Cell mol Life Sci. 2021;78(5):1929–1941. doi:10.1007/s00018-020-03687-5
91. Flannery PJ, Trushina E. Mitochondrial dynamics and transport in Alzheimer’s disease. mol Cell Neurosci. 2019;98:109–120. doi:10.1016/j.mcn.2019.06.009
92. Phatnani H, Maniatis T. Astrocytes in neurodegenerative disease. Cold Spring Harb Perspect Biol. 2015;7(6):a020628. doi:10.1101/cshperspect.a020628
93. Grossmann D, Berenguer-Escuder C, Chemla A, Arena G, Krüger R. The emerging role of RHOT1/Miro1 in the pathogenesis of Parkinson’s disease. Front Neurol. 2020;11:587. doi:10.3389/fneur.2020.00587
94. Nguyen D, Bharat V, Conradson DM, Nandakishore P, Wang X. Miro1 impairment in a Parkinson’s at-risk cohort. Front mol Neurosci. 2021;14:734273. doi:10.3389/fnmol.2021.734273
95. López-Doménech G, Howden JH, Covill-Cooke C, et al. Loss of neuronal Miro1 disrupts mitophagy and induces hyperactivation of the integrated stress response. EMBO J. 2021;40(14):e100715. doi:10.15252/embj.2018100715
96. López-Doménech G, Higgs NF, Vaccaro V, et al. Loss of dendritic complexity precedes neurodegeneration in a mouse model with disrupted mitochondrial distribution in mature dendrites. Cell Rep. 2016;17(2):317–327. doi:10.1016/j.celrep.2016.09.004
97. Babenko VA, Silachev DN, Popkov VA, et al. Miro1 enhances mitochondria transfer from Multipotent Mesenchymal Stem Cells (MMSC) to neural cells and improves the efficacy of cell recovery. Molecules. 2018;23(3):687. doi:10.3390/molecules23030687
98. Peruzzotti-Jametti L, Bernstock JD, Willis CM, et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. PLoS Biol. 2021;19(4):e3001166. doi:10.1371/journal.pbio.3001166
99. Sharma A. Mitochondrial cargo export in exosomes: possible pathways and implication in disease biology. J Cell Physiol. 2023;238(4):687–697. doi:10.1002/jcp.30967
100. Li A, Gao M, Liu B, et al. Mitochondrial autophagy: molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022;13(5):444. doi:10.1038/s41419-022-04906-6
101. Um JH, Yun J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 2017;50(6):299–307. doi:10.5483/BMBRep.2017.50.6.056
102. Wang Y, Liu N, Lu B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci Ther. 2019;25(7):859–875. doi:10.1111/cns.13140
103. Huang CY, Kuo WW, Ho TJ, et al. Rab9-dependent autophagy is required for the IGF-IIR triggering mitophagy to eliminate damaged mitochondria. J Cell Physiol. 2018;233(9):7080–7091. doi:10.1002/jcp.26346
104. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333(6046):1109–1112. doi:10.1126/science.1201940
105. Belousov DM, Mikhaylenko EV, Somasundaram SG, Kirkland CE, Aliev G. The dawn of mitophagy: what do we know by now? Curr Neuropharmacol. 2021;19(2):170–192. doi:10.2174/1570159X18666200522202319
106. Um JW, Park HJ, Song J, et al. Formation of parkin aggregates and enhanced PINK1 accumulation during the pathogenesis of Parkinson’s disease. Biochem Biophys Res Commun. 2010;393(4):824–828. doi:10.1016/j.bbrc.2010.02.090
107. Chan NC, Salazar AM, Pham AH, et al. Broad activation of the ubiquitin-proteasome system by parkin is critical for mitophagy. Hum mol Genet. 2011;20(9):1726–1737. doi:10.1093/hmg/ddr048
108. Zhu X, Liu Q, Zhu F, et al. An engineered cellular carrier delivers miR-138-5p to enhance mitophagy and protect hypoxic-injured neurons via the DNMT3A/Rhebl1 axis. Acta Biomater. 2024;186:424–438. doi:10.1016/j.actbio.2024.07.059
109. Jin Y, Wu O, Chen Q, et al. Hypoxia-preconditioned BMSC-derived exosomes induce mitophagy via the BNIP3-ANAX2 axis to alleviate intervertebral disc degeneration. Adv Sci. 2024;11(34):e2404275. doi:10.1002/advs.202404275
110. Dabin R, Wei C, Liang S, Ke C, Zhihan W, Ping Z. Astrocytic IGF-1 and IGF-1R orchestrate mitophagy in traumatic brain injury via exosomal miR-let-7e. Oxid Med Cell Longev. 2022;2022:3504279. doi:10.1155/2022/3504279
111. Jin SM, Youle RJ. PINK1- and parkin-mediated mitophagy at a glance. J Cell Sci. 2012;125(4):795–799. doi:10.1242/jcs.093849
112. Picca A, Guerra F, Calvani R, et al. mitochondrial-derived vesicles as candidate biomarkers in Parkinson’s disease: rationale, design and methods of the EXosomes in PArkiNson Disease (EXPAND) study. Int J mol Sci. 2019;20(10):2373. doi:10.3390/ijms20102373
113. Mohamed AS, Abdel-Fattah DS, Abdel-Aleem GA, El-Sheikh TF, Elbatch MM. Biochemical study of the effect of mesenchymal stem cells-derived exosome versus L-dopa in experimentally induced Parkinson’s disease in rats. mol Cell Biochem. 2023;478(12):2795–2811. doi:10.1007/s11010-023-04700-8
114. Li N, Shu J, Yang X, Wei W, Yan A. Exosomes derived from M2 microglia cells attenuates neuronal impairment and mitochondrial dysfunction in Alzheimer’s disease through the PINK1/parkin pathway. Front Cell Neurosci. 2022;16:874102. doi:10.3389/fncel.2022.874102
115. Luo S, Sun X, Huang M, Ma Q, Du L, Cui Y. Enhanced neuroprotective effects of epicatechin gallate encapsulated by bovine milk-derived exosomes against Parkinson’s disease through antiapoptosis and antimitophagy. J Agric Food Chem. 2021;69(17):5134–5143. doi:10.1021/acs.jafc.0c07658
116. Chen Y, Li B, Quan J, Li Z, Li Y, Tang Y. Inhibition of ferroptosis by mesenchymal stem cell-derived exosomes in acute spinal cord injury: role of Nrf2/GCH1/BH4 axis. Neurospine. 2024;21(2):642–655. doi:10.14245/ns.2448038.019
117. Liu Z, Zeng X, Bian W, et al. Exosomes from muscle-derived stem cells repair peripheral nerve injury by inhibiting ferroptosis via the keap1-Nrf2-Ho-1 axis. J Cell Biochem. 2024;125(8):e30614. doi:10.1002/jcb.30614
118. Shan L, Zhan F, Lin D, et al. SIRT1-enriched exosomes derived from bone marrow mesenchymal stromal cells alleviate peripheral neuropathy via conserving mitochondrial function. J mol Neurosci. 2022;72(12):2507–2516. doi:10.1007/s12031-022-02091-x
119. Liu J, Huang J, Zhang Z, et al. Mesenchymal stem cell-derived exosomes ameliorate delayed neurocognitive recovery in aged mice by inhibiting hippocampus ferroptosis via activating SIRT1/Nrf2/HO-1 signaling pathway. Oxid Med Cell Longev. 2022;2022:3593294. doi:10.1155/2022/3593294
120. Du L, Jiang Y, Sun Y. Astrocyte-derived exosomes carry microRNA-17-5p to protect neonatal rats from hypoxic-ischemic brain damage via inhibiting BNIP-2 expression. Neurotoxicology. 2021;83:28–39. doi:10.1016/j.neuro.2020.12.006
121. Liu H, Jin M, Ji M, Zhang W, Liu A, Wang T. Hypoxic pretreatment of adipose-derived stem cell exosomes improved cognition by delivery of circ-Epc1 and shifting microglial M1/M2 polarization in an Alzheimer’s disease mice model. Aging. 2022;14(7):3070–3083. doi:10.18632/aging.203989
122. Calkins MJ, Johnson DA, Townsend JA, et al. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid Redox Signal. 2009;11(3):497–508. doi:10.1089/ars.2008.2242
123. Villavicencio Tejo F, Quintanilla RA. Contribution of the Nrf2 pathway on oxidative damage and mitochondrial failure in Parkinson and Alzheimer’s disease. Antioxidants. 2021;10(7):1069. doi:10.3390/antiox10071069
124. Butterfield DA, Boyd-Kimball D, Reed TT. Cellular stress response (hormesis) in response to bioactive nutraceuticals with relevance to Alzheimer disease. Antioxid Redox Signal. 2023;38(7–9):643–669. doi:10.1089/ars.2022.0214
125. De Plano LM, Calabrese G, Rizzo MG, Oddo S, Caccamo A. The role of the transcription factor Nrf2 in Alzheimer’s disease: therapeutic opportunities. Biomolecules. 2023;13(3):549. doi:10.3390/biom13030549
126. Nouri Z, Barfar A, Perseh S, et al. Exosomes as therapeutic and drug delivery vehicle for neurodegenerative diseases. J Nanobiotechnology. 2024;22(1):463. doi:10.1186/s12951-024-02681-4
127. Liang Y, Xu X, Li X, et al. Chondrocyte-targeted MicroRNA delivery by engineered exosomes toward a cell-free osteoarthritis therapy. ACS Appl Mater Interfaces. 2020;12(33):36938–36947. doi:10.1021/acsami.0c10458
128. Chand Dakal T, Choudhary K, Tiwari I, Yadav V, Kumar Maurya P, Kumar Sharma N. Unraveling the triad: hypoxia, oxidative stress and inflammation in neurodegenerative disorders. Neuroscience. 2024;552:126–141. doi:10.1016/j.neuroscience.2024.06.021
129. Koh MY, Darnay BG, Powis G. Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1alpha, leading to its oxygen-independent degradation. Mol Cell Biol. 2008;28(23):7081–7095. doi:10.1128/MCB.00773-08
130. Marxsen JH, Stengel P, Doege K, et al. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J. 2004;381(Pt 3):761–767. doi:10.1042/BJ20040620
131. Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol. 2021;599(1):23–37. doi:10.1113/JP280572
132. Hamilton LK, Dufresne M, Joppé SE, et al. Aberrant lipid metabolism in the forebrain niche suppresses adult neural stem cell proliferation in an animal model of Alzheimer’s disease. Cell Stem Cell. 2015;17(4):397–411. doi:10.1016/j.stem.2015.08.001
133. Li K, Zheng Y, Wang X. The potential relationship between HIF-1α and amino acid metabolism after hypoxic ischemia and dual effects on neurons. Front Neurosci. 2021;15:676553. doi:10.3389/fnins.2021.676553
134. Andersen JV, Schousboe A. Milestone review: metabolic dynamics of glutamate and gaba mediated neurotransmission - the essential roles of astrocytes. J Neurochem. 2023;166(2):109–137. doi:10.1111/jnc.15811
135. Wang YY, Huang ZT, Yuan MH, et al. Role of hypoxia inducible factor-1α in Alzheimer’s disease. J Alzheimers Dis. 2021;80(3):949–961. doi:10.3233/JAD-201448
136. Tian Y, Fu C, Wu Y, Lu Y, Liu X, Zhang Y. Central nervous system cell-derived exosomes in neurodegenerative diseases. Oxid Med Cell Longev. 2021;2021:9965564. doi:10.1155/2021/9965564
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Serum Exosomal miRNA-125b and miRNA-451a are Potential Diagnostic Biomarker for Alzheimer’s Diseases
Duan X, Zheng Q, Liang L, Zhou L
Degenerative Neurological and Neuromuscular Disease 2024, 14:21-31
Published Date: 8 April 2024
Extracellular Vesicles Derived from FGF2-Primed Astrocytes Against Mitochondrial and Synaptic Toxicities in Parkinson’s Disease

Wen X, Cao W, Ding H, Chen A, Sun Z, Wang Y, Xi Y, Wu S
International Journal of Nanomedicine 2025, 20:4627-4644
Published Date: 13 April 2025
Exosomes Regulate the NLRP3/Caspase-1/IL-1β Signaling Pathway in Parkinson’s Disease: Mechanisms of Neuroinflammation Modulation and α-Synuclein Propagation
Zhang T, Du H, Wang S
Neuropsychiatric Disease and Treatment 2026, 22:587802
Published Date: 28 February 2026
Insulin Resistance Across Cerebrovascular and Related Disorders: Mechanisms, Measurement, Genetics, and Clinical Implications
Chen K, Nong Y, Liu Y, Ye Z
Neuropsychiatric Disease and Treatment 2026, 22:575306
Published Date: 30 April 2026
Evaluating the Role of AI Assistants in Accelerating Neurodegenerative Disease Research: Opportunities and Translational Limitations
Yu X, Yao Q
Neuropsychiatric Disease and Treatment 2026, 22:614952
Published Date: 30 May 2026