")
Back to Journals » OncoTargets and Therapy » Volume 13
Estrogen-Related Receptor α (ERRα) and G Protein-Coupled Estrogen Receptor (GPER) Synergistically Indicate Poor Prognosis in Patients with Triple-Negative Breast Cancer
Authors Ye S, Xu Y, Wang L, Zhou K, He J, Lu J, Huang Q, Sun P, Wang T
Received 31 May 2020
Accepted for publication 24 July 2020
Published 7 September 2020 Volume 2020:13 Pages 8887—8899
DOI https://doi.org/10.2147/OTT.S265372
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Shuang Ye,1,* Yuanyuan Xu,1,* Ling Wang,1 Kewen Zhou,1 Jiehua He,2 Jiabin Lu,2 Qitao Huang,2 Peng Sun,2 Tinghuai Wang1
1Department of Physiology, Zhongshan School of Medicine, Sun Yat-Sen University, Guangzhou 510080, People’s Republic of China; 2State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Department of Pathology, Sun Yat-Sen University Cancer Center, Guangzhou 510060, People’s Republic of China
*These authors contributed equally to this work.
Correspondence: Peng Sun; Tinghuai Wang Email [email protected]; [email protected]
Purpose: The present study aims to demonstrate the correlation between estrogen-related receptor α (ERRα) and G protein-coupled estrogen receptor (GPER) expression and its predictive role in the prognosis of patients with triple-negative breast cancer (TNBC).
Methods: A retrospective review of 199 cases of TNBC was conducted to assess the GPER and ERRα expression, and its clinicopathologic and prognostic implications. Subsequently, the effects of ERRα and GPER on cell viability, migration, and invasion induced by estrogen were also investigated in vitro.
Results: Compared to TNBCs with ERRα low expression, ERRα-high patients exhibited higher nuclear grade, more frequent lymph nodal metastasis, a higher rate of local recurrence, and distant metastasis. Survival analyses revealed that ERRα-high patients had decreased overall survival (OS), local recurrence-free survival (LRFS), and distant disease-free survival (DDFS) than ERRα-low patients. The GPER expression level positively correlated with ERRα (R=0.167, P=0.18), and TNBCs with ERRα-low/GPER-low demonstrated the best survival outcomes among groups. In vitro, E2 significantly enhanced cell viability, migration, and invasion in BT-549 and MDA-MB-231 cell lines, which was associated with the increased expression of ERRα. Moreover, the overexpression of ERRα induced by estrogen and G1 (GPER agonist) was reversed by knocking down of GPER and blocking the MAPK signaling with PD98059 in both cell lines.
Conclusion: Our findings suggest that ERRα and GPER synergistically predict unfavorable prognosis in TNBCs. Mechanically, GPER mediates the upregulation expression of ERRα induced by estrogen and promotes cell viability, migration, and invasion.
Keywords: triple-negative breast cancer, GPER, ERRα, estrogen, prognosis
Introduction
Identification of molecular subtypes of breast cancer (BC) has led to a better understanding of its biologic behavior, which provides valuable information for the individualized treatment for BCs. Triple-negative breast cancer (TNBC) is an aggressive subtype of breast cancer (BC), which is negative for estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2). TNBC accounts for 15–20% of all BC, which is always poorly differentiated and predominantly occurs in young women.1–4 Despite the development of novel clinical therapies in TNBCs, such as Poly-ADP-ribose polymerase inhibitors and immunotherapy, however, TNBC still demonstrates worse prognosis than non-TNBC, with a median OS of 28 months, approximately.5,6 Recurrence and distant metastasis are the leading causes of mortality in patients with TNBC,4,7,8 and recent studies suggest that younger patients with TNBC may have a higher incidence of recurrence or metastasis when compared with their older counterparts. However, the underlying mechanism is still unclear.9–11 One of the hypotheses is that female patients with TNBC at a younger age exhibit higher estrogen levels, suggesting that estrogen may still affect the tumor biological behavior. Given the lack of activated ER signaling in TNBC, it is generally believed that estrogen may indirectly be involved in the regulation of malignant phenotype in TNBC by activating effectors related to ER signaling or binding other receptors.
Tumorous aggressive behaviors are closely related to high-energy metabolism.12–15 Estrogen-related receptor α (ERRα) is an orphan nuclear receptor, which is also highly homologous with classic estrogen receptor α (ERα). ERRα expression is found in tissues requiring high energy metabolism as well as various tumorous tissues,16,17 which has been reported to act as a metabolic regulator in vitro.18 Clinicopathological studies have revealed that high expression of ERRα is associated with increased TNM stage of breast cancer, more frequent recurrence, and related poor survival outcome.19,20 Growing evidences indicate that the ERR family promotes cell migration and invasion by regulating the Warburg effect and metabolism-related genes17,21–23 Although estrogen is not a natural ligand for the ERR family, ERRs do share target genes, coregulatory proteins, ligands, and sites of action with the ERs.24 Li et al25 have found that physiological levels of 17beta-estradiol (E2) regulate the expression of ERRα in SKBR3 breast cancer cells which suggest that ERRs can actively influence the estrogenic response, and the pharmacological modulation of ERR family activity may be clinically valuable in estrogen-related cancer, especially in breast cancer.
On the other hand, G protein-coupled estrogen receptor (GPER) has been identified as a novel estrogen membrane receptor.26 Estrogen binds to and activates GPER in the plasma membrane, and ligand-bound GPER induces activations of adenylyl cyclase, cAMP, calcium mobilization, and mitogen-activated protein kinase (MAPK).26,27 Previous studies by other investigators and us have revealed that GPER is commonly expressed in TNBC, and estrogen promotes cell migration and invasion in TNBC via GPER signaling.28–30 Clinically, TNBCs with high expression of GPER demonstrate worse OS, LRFS, and DDFS than in the GPER-low group, especially in the premenopausal setting.31,32 Interestingly, a study by Li et al25 also demonstrates that GPER agonists, including G1 and tamoxifen, as well as overexpression of GPER, could lead to an increase of ERRα mRNA level, suggesting that GPER/ERRα-mediated signaling may be relevant to breast cancer cells escaping inhibitory control by tamoxifen. A recent study by Kotula-Balak et al33 also implies the existence of an interaction between GPER and ERRα in tumorous Leydig cells.
Therefore, the current study aimed to demonstrate the correlation between ERRα and GPER expression and its clinicopathologic and prognostic implications in patients with TNBC. Moreover, we also determine the role of ERRα/GPER in breast cancer cell proliferation, migration, and invasion and explore the preliminary mechanism of the interactions between GPER and ERRα in vitro.
Materials and Methods
Clinicopathologic Features Analysis
Clinicopathologic data of patients diagnosed as invasive breast carcinoma at the Sun Yat-sen University Cancer Center (SYSUCC) during the year 2000–2010 were retrieved. All patients were operated at SYSUCC and did not receive neoadjuvant therapy. Formalin-fixed paraffin-embedded (FFPE) tissue specimens were stained routinely with hematoxylin and eosin (H&E). ER, PR, and HER2 status were determined on immunohistochemical (IHC) staining. ER, and PR status were classified as negative using a cut-off of 1%, according to the American Society of Clinical Oncology/College of American Pathologists (ASCO/CAP) guidelines.34 HER2 status was defined as negative with 0, 1+ as well as 2+ on IHC without HER2 gene amplification on fluorescence in situ hybridization (FISH).35 The H&E and IHC slides from a total of 199 cases were retrospectively reviewed by two pathologists (PS, JH) to reconfirm the diagnosis of TNBC. Clinicopathological features, including age at diagnosis, menopausal status, laterality of tumor, nuclear grade, tumor size, lymph node metastasis, TNM stage, were analyzed. The tumor staging was based on the TNM stage was assessed according to the criteria established by the 8th edition American Joint Committee on Cancer (AJCC 8th) staging manual for breast cancer.
Immunohistochemistry Staining
FFPE tumor tissues were deparaffinized by xylene and then rehydrated by a series of graded ethanol. Antigen retrieval was conducted using EDTA buffer (GPER, pH=9.0; ERRα, pH=8.0). Subsequently, tissues were incubated with anti-GPER primary antibody (1:100 dilution, Abcam, UK) and anti-ERRα antibody (1:100 dilution, Novus Biologicals, USA) for 50 min at 37°C. Secondary antibodies (Zhongshan Golden Bridge Biotechnology, China) were incubated for 30 min at 37°C, followed by the staining of DAB (Zhongshan Golden Bridge Biotechnology, China) and then hematoxylin. Endometrial carcinoma tissue was used as positive staining control, which has been demonstrated to express GPER and ERRα.36,37 Finally, the slides were mounted and semi-quantified by two experienced pathologists (PS, JH) blind to the outcome by consensus. The expression of GPER and ERRα was calculated as the product of dyeing intensity (grade as 0, negative; 1, weak; 2, moderate; and 3, strong staining) and positive-staining scope (0, <5%; 5%–24%; 25%–49%; 50%–74%; 75%–100%). For the division of high and low expression groups of GPER and ERRα, the receiver-operating characteristic (ROC) curves were plotted to generate the optimal cut-off values. The immunoreactive score of IRS=0.7 and 0.8 was used throughout the manuscript for GPER and ERRα, respectively.
Cell Culture
Human mammary epithelial cell MCF-10A and breast cancer cell lines BT-549, MDA-MB-231 were obtained from the Institute of Biochemistry and Cell Biology, Shanghai Institute of Biological Sciences (Shanghai, China). Cells were cultivated at 37°C in a humidified atmosphere with 5% CO2. MCF-10A and BT-549 were cultured in RPMI 1640 medium (Gibco, USA), and MDA-MB-231 was cultured in Leibovitz’s L-15 medium (Hyclone, USA). Both medium were supplemented with 10% FBS (Gibco, USA). Cells passaged more than five times were abandoned in our experiments. The information about associated drugs in this experiment were as follows: 17β-estradiol (E2, MilliporeSigma, Germany), G1 (MilliporeSigma, Germany), G15 (MilliporeSigma, Germany), the MEK inhibitor PD98059 (#167869-21-8, MedChemExpress, USA). The E2 concentration used in the present study was 100nM, which had been reported to show the most significant effects according to our previous time-concentration experiments.29
Protein Extraction and Western Blotting
Cells were lysed using Triton-X lysis buffer, and then the protein concentration was measured by the BCA kit (Thermo Fisher Scientific, USA). 30 μg of protein samples per lane was separated by polyacrylamide gel electrophoresis and then transferred onto PVDF membrane (Thermo Fisher Scientific, USA). 5% BSA (Thermo Fisher Scientific, USA) dissolved in TBST (Beijing Solarbio Science and Technology, China) was used to block the membrane. The band was incubated with the primary antibody overnight at 4°C. Primary antibodies were applied as follows: anti-GPER (1:5000 dilution, Abcam, UK), anti-ERRα (1:5000 dilution, Cell Signaling Technology, USA), anti-GAPDH (1:5000 dilution, Cell Signaling Technology, USA), and anti-β-actin (1:5000 dilution, Cell Signaling Technology). Secondary antibody (1:5000 dilution, Santa Cruz Biotechnology) incubation was carried out at room temperature for 2h. Protein bands were then detected using chemiluminescence reagent (Millipore, UK) on the ChemiDocTM imaging system (Biorad, USA) and analyzed semi-quantitatively using the Image lab software (Biorad, USA). β-actin and GAPDH were served as internal control.
RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
RNA was extracted using Trizol® (Thermo Fisher Scientific, USA) following the manufacturer’s construction and then quantified on Nanodrop™ spectrophotometer (Thermo Fisher Scientific, USA). For reverse transcription, 1 μg total RNA was added according to the PrimeScript RT reagent Kit (Takara Japan). Quantitative real-time PCR reaction was performed using the SYBR Premix ExTaqTM Kit (Takara, Japan) on the BIO-RAD CFX96 system. Cycling conditions were as follows: denaturation at 95°C for 5 min, followed by 40 cycles of 95°C for 15 s, 60°C for 30 s, 72°C for 30 s. Relative expression levels of the targeted gene were determined by the 2−ΔΔCt method and normalized to GAPDH or β-actin. All the primers involved in this study were designed and synthesized by Jierui (China), and sequence information is listed in Supplementary Table 1.
Wound-Healing Assay
To examine cell migration ability under different treatment conditions wound-healing assay was conducted. Briefly, 5 105 cells were seeded into a 6-well plate overnight to adhere. When cells reached 90–100% confluence, 10 µL sterile micropipette tip was used to scratch one gap vertically at the bottom of the culture plate. PBS was used to wash away cellular debris, and then serum-free and phenol red-free medium was added to eliminate the interference of E2. An inverted microscope captured the closure of the wound area after the scratch under 200
105 cells were seeded into a 6-well plate overnight to adhere. When cells reached 90–100% confluence, 10 µL sterile micropipette tip was used to scratch one gap vertically at the bottom of the culture plate. PBS was used to wash away cellular debris, and then serum-free and phenol red-free medium was added to eliminate the interference of E2. An inverted microscope captured the closure of the wound area after the scratch under 200 magnification at pointed time. Each experiment was triplicated. The wound-healing distance of cells was analyzed using ImageJ software.
magnification at pointed time. Each experiment was triplicated. The wound-healing distance of cells was analyzed using ImageJ software.
Cell Viability Assay
Cell Counting Kit-8 (CCK-8) was carried out to measure the cell viability. 1 103 cells were plated into a transparent 96-well plate and cultured at 37°C. After consequent drug treatment, 10 µL CCK-8 reagent (Dojindo, Japan) was added into each well and incubated for 1–4 h. The OD values at 450 nm were detected in a microplate spectrometer. Per group was set more than three replicate wells, and experiments were triplicated.
103 cells were plated into a transparent 96-well plate and cultured at 37°C. After consequent drug treatment, 10 µL CCK-8 reagent (Dojindo, Japan) was added into each well and incubated for 1–4 h. The OD values at 450 nm were detected in a microplate spectrometer. Per group was set more than three replicate wells, and experiments were triplicated.
Transwell Invasion Assay
1 104 cells suspended in 200 µL serum-free medium were seeded in 8 µm pore sized transwell upper chamber (Corning, USA) coated with 200 µg/mL Matrigel (BD Science, CA). The lower chamber was added 900 µL complete medium containing 10% FBS. After the incubation of 12 h, the inserts were washed with PBS and then fixed with 4% paraformaldehyde (Beijing Solarbio Science and Technology, China) for 10 min, stained by 0.1% of crystal violet solution (Beijing Solarbio Science and Technology, China) for 30 min. Cells in the upper surface were wiped out softly by cotton swabs. Random six fields per well were photographed under 200
104 cells suspended in 200 µL serum-free medium were seeded in 8 µm pore sized transwell upper chamber (Corning, USA) coated with 200 µg/mL Matrigel (BD Science, CA). The lower chamber was added 900 µL complete medium containing 10% FBS. After the incubation of 12 h, the inserts were washed with PBS and then fixed with 4% paraformaldehyde (Beijing Solarbio Science and Technology, China) for 10 min, stained by 0.1% of crystal violet solution (Beijing Solarbio Science and Technology, China) for 30 min. Cells in the upper surface were wiped out softly by cotton swabs. Random six fields per well were photographed under 200 magnification and counted by ImageJ software.
magnification and counted by ImageJ software.
Transfection of siRNA and Plasmid
Commercial siRNA of GPER and ERRα were purchased from RiboBio (Guangzhou, China), while GPER expression plasmid was synthesized by Sino Biological (Beijing, China). Transient transfection using Lipofectamine™ 3000 transfection reagent (Invitrogen, USA) was conducted under the manufacturer’s construction. For siRNA, a total of 125 µL Opti-MEM (Invitrogen, USA) was used to incubate 3.75 µL Lipofectamine 3000 reagent and 50 nM siRNA for 10 min and then added into FBS-free culture medium. 6–8 h later, the culture medium was replaced with complete medium containing 10% FBS. To overexpressing GPER, the P3000 reagent was added additionally into the transfection system according to the construction. Subsequent experiments were conducted after 48 h transfection.
Statistical Analyses
Data in this study were analyzed using the SPSS version 26.0 (IBM Inc., USA) and GraphPad Prism 7.0 (GraphPad Software Inc., USA). Eligible patients were considered as the unmatched cohort. To reduce bias, we also developed a 1:1 (ERRα-low: ERRα-high) matched cohort using propensity score matching (PSM) for age, nuclear grade, tumor size, LNM, and TNM stage with a caliper of 0.03. The clinicopathological variables were compared between groups using the Chi-square test. The Pearson correlation coefficient was used to analyze the correlation between the expression of ERRα and GPER. OS, LRFS, and DDFS curves were drawn using the Kaplan–Meier methods and were compared using Log rank tests. Univariate and multivariate analyses of the patients’ survival were performed using the Cox proportional hazards regression model and the logistic regression model. For experiments in vitro, the difference between groups was compared using the Student’s t-test or one-way ANOVA. Two-tailed p-value <0.05 was considered statistically significant.
Results
Association Between ERRα Expression and Clinicopathologic Features in TNBCs
The baseline clinicopathological features of 199 TNBCs are summarized in Table 1, of which 54.8% (109/199) was classified into ERRα-low group, while 45.2% (90/199) was classified into ERRα-high group. Examples of IHC staining for ERRα with different intensities have been shown in Figure 1. Compared to ERRα-low group, ERRα-high TNBCs exhibited higher nuclear grade (grade II, 12.2% vs 23.9%; grade III, 87.8% vs 76.1%; p=0.036), more frequent LNM (52.2% vs 40.4%; p=0.014), and higher TNM stage (p=0.007). No significant difference was observed in age, menopausal status, tumor size, and laterality between groups. Besides, 37.7% (75/199) of TNBCs had a high expression of GPER, and these cases were predominantly in the ERRα-high group than in the ERRα-low group (50.0% vs 27.5%; p=0.001). The expression of GPER was positively correlated to that of ERRα with Pearson correlation coefficient of 0.167 (p=0.018; Supplementary Figure 1).
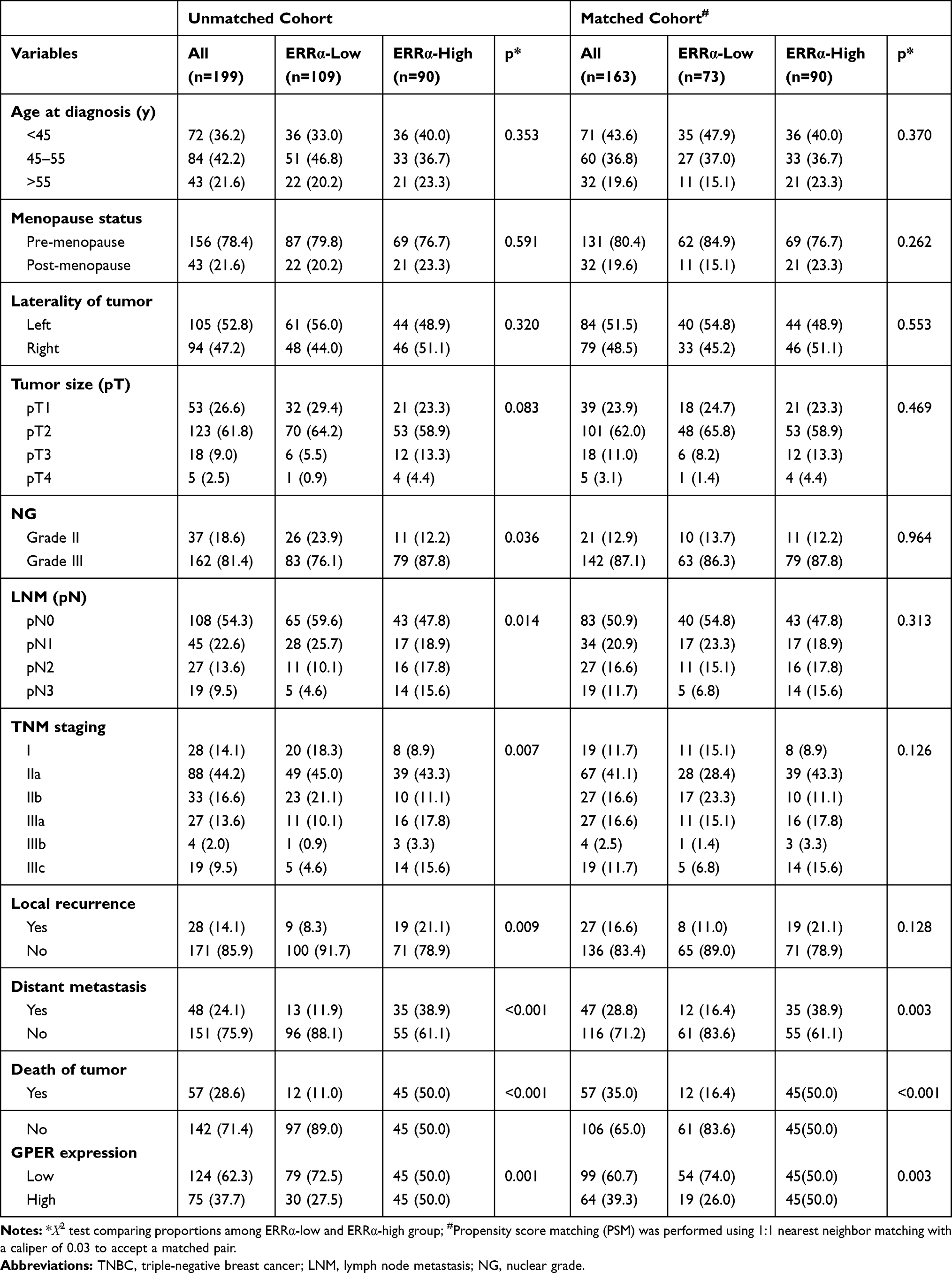 |
Table 1 Association Between ERRα Expression and Clinicopathologic Features in TNBCs |
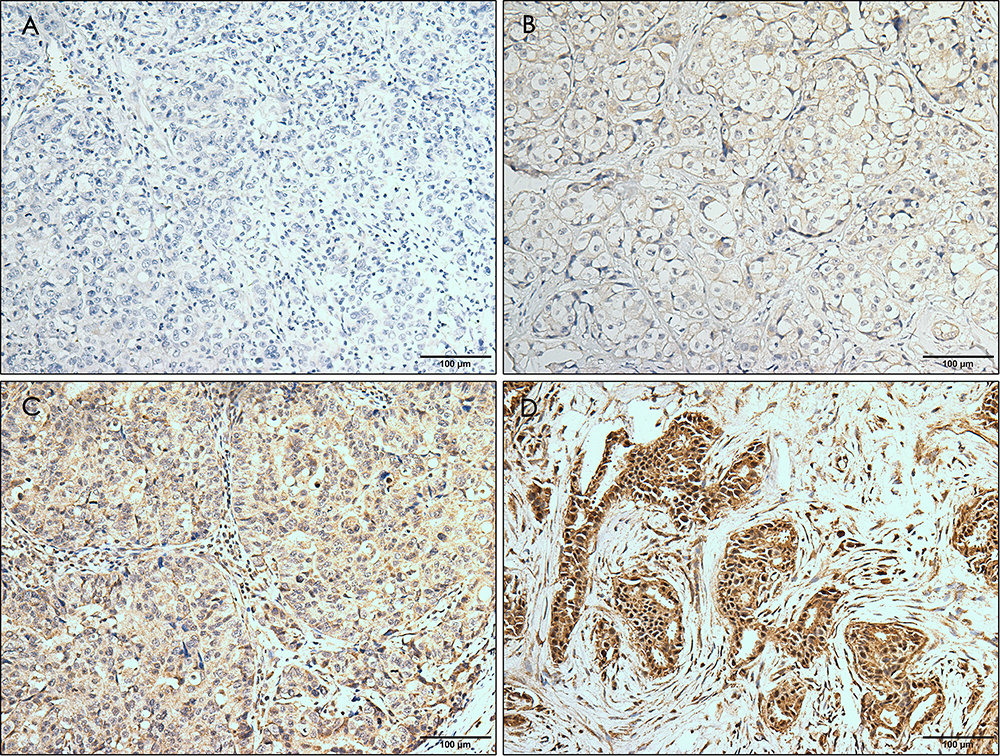 |
Figure 1 Immunohistochemical staining for ERRα with different intensity. The staining is graded as negative (A), weak (B), moderate (C), and strong (D). Magnification, ×10. |
Prognostic Implications of ERRα in TNBCs
The patients were followed-up for 2–205 months, with a median of 113 months. Compared to ERRα-low group, ERRα-high TNBCs demonstrated increased incidences of local recurrence (21.1% vs 8.3%; p=0.009), distant metastasis (38.9% vs 11.9%; p<0.001), and more patients with high ERRα died of tumor (50.0% vs 11.0%; p<0.001) in the unmatched cohort. Similarly, in the matched cohort, more ERRα-high TNBCs died of tumor (50.0% vs 16.4%, p<0.001) and exhibited distant metastasis (38.9% vs 16.4%, p=0.003) than TNBCs with low ERRα expression (Table 1). The survival curves are shown in Figure 2, ERRα-high TNBCs were associated with a decrease in OS, LRFS, and DDFS when comparing to ERRα-low group in both cohorts. Similar survival outcomes were observed between GPER-high and GPER-low groups (Supplementary Figure 2). Further survival analysis in the subgroups according to the co-expression status of ERRα and GPER revealed that in the unmatched cohort, ERRα-high/GPER-high subgroup was significantly associated with the worst survival outcomes of both OS, LRFS, and DDFS, while patients in ERRα-low/GPER-low exhibited a relative excellent survival (p<0.001). A decreased DDFS was observed in ERRα-low/GPER-high subgroup when comparing to ERRα-high/GPER-low subgroup, while the survival curve of OS or LRFS in these two groups was overlapping. In the matched cohort, ERRα-low/GPER-low TNBCs had the best OS, LRFS, and DDFS, followed by ERRα-high/GPER-low subgroup. ERRα-low/GPER-high subgroup displayed an increased OS than patients with ERRα-low/GPER-low. However, no significant difference was found between these two subgroups in LRFS and DDFS (Figure 3). A Cox proportional hazards regression model was then used to identify the biomarkers and clinicopathological factors affecting the prognosis of patients with TNBC. The results are summarized in Table 2. In the univariate model, LNM, as well as the expression of ERRα and GPER, were identified as prognostic indicators for OS, LRFS, and DDFS, while nuclear grade and tumor size were also associated with OS and DDFS, but not LRFS. Multivariate analysis proved that LNM and the expression of ERRα and GPER were independent factors for OS, LRFS, and DDFS. Besides, tumor size was also suggested as an independent factor OS.
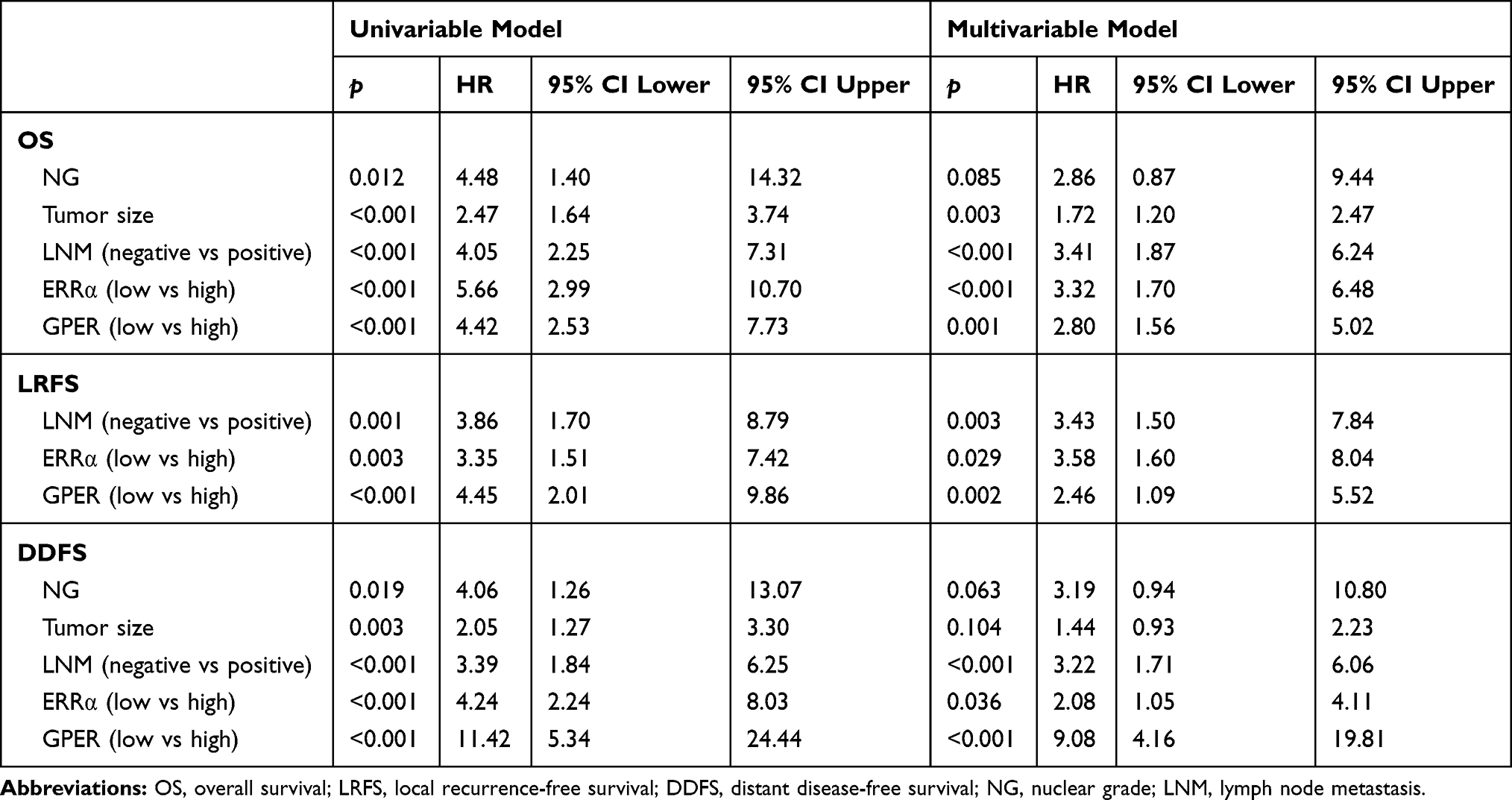 |
Table 2 Univariate and Multivariate Survival Analyses for OS, LRFS and DDFS |
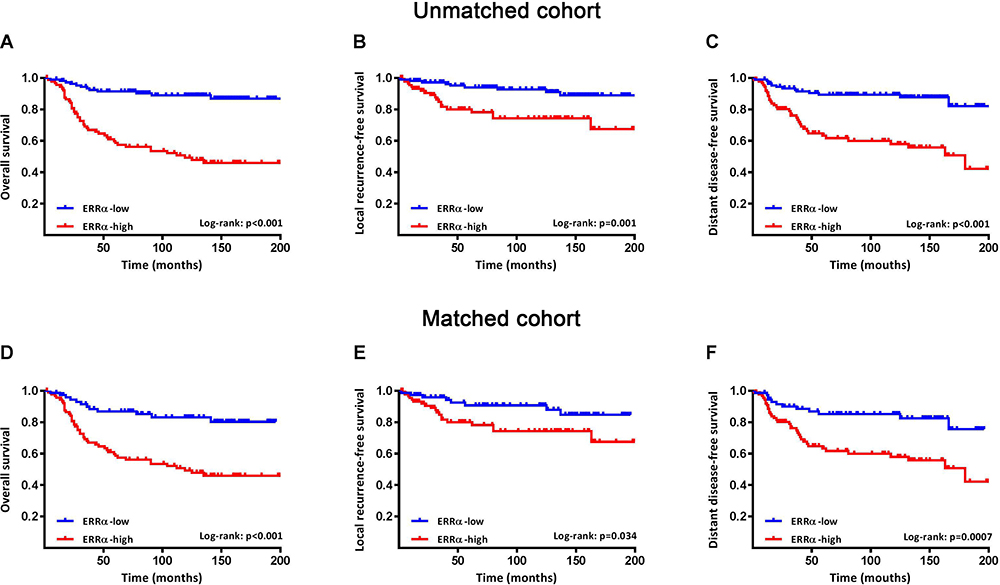 |
Figure 2 Survival curves of OS, LRFS, and DDFS in ERRα-low and ERRα-high groups. Kaplan-Meier curves of OS (A), LRFS (B), and DDFS (C) between groups in patients with TNBC in the unmatched cohort. Kaplan-Meier curves of OS (D), LRFS (E), and DDFS (F) between groups in patients with TNBC in a matched cohort using propensity score matching for age, nuclear grade, tumor size, LNM and TNM stage with a caliper of 0.03. |
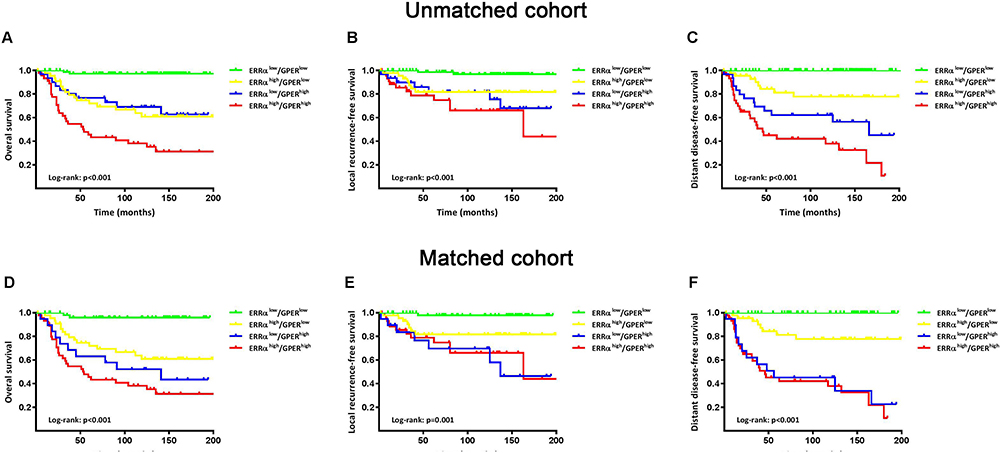 |
Figure 3 Survival curves of OS, LRFS, and DDFS based on the co-expression status of ERRα and GPER. Kaplan-Meier curves of OS (A), LRFS (B), and DDFS (C) among groups in patients with TNBC in the unmatched cohort. Kaplan-Meier curves of OS (D), LRFS (E), and DDFS (F) among groups in patients with TNBC in a matched cohort using propensity score matching for age, nuclear grade, tumor size, LNM and TNM stage with a caliper of 0.03. |
ERRα Mediates Malignant Phenotype of TNBC Induced by Estrogen
Our previous study suggests the overexpression of GPER is associated with poor prognosis, especially in pre-menopausal patients with TNBC.31 Thus, a Χ2 statistic of the Log rank test was used in the present study to calculate the survival discrimination based on the expression of ERRα and GPER in pre- and post-menopause patients (Table 3). Compared to post-menopausal patients, Χ2 statistic of pre-menopausal patients showed a larger and statistically significant difference between ERRα-high and ERRα-low group on OS (24.28, p<0.001 vs 10.54, p=0.012), LRFS (6.38, p=0.012 vs.3.88 p=0.049), and DDFS (18.07, p<0.001 vs 4.43, p=0.035). A larger Χ2 statistic between GPER-high and GPER-low group was also found on OS (27.72, p<0.001 vs 4.47, p=0.035), LRFS (19.17, p<0.001 vs 0.16 p=0.685), and DDFS (53.35, p<0.001 vs 10.15, p=0.001) in pre-menopausal TNBCs. These may imply the role of estrogen in the tumor progression of TNBC, affecting ERRα/GPER signaling. In vitro, treatment with 100nM E2 significantly enhanced the cell viability, migration, and invasion in both BT-549 and MDA-MB-231 cell lines as compared to the control (p<0.05; Figure 4). Compared to normal breast epithelial cell lines MCF-10A, the expression of ERRα was relatively higher in BT-549 and MDA-MB-231 cells (Figure 5A). We further verify the function of ERRα in TNBC by knocking down ERRα in BT-549 and MDA-MB-231 using RNA interference. Inhibition of ERRα significantly reduced cell viability, and suppressed the wound-healing and invasion in both BT-549 and MDA-MB-231 cells (Figure 5B–D). Moreover, despite the fact that ERRα is not a natural receptor for estrogen, Western blot and qRT-PCR exhibited that treatment with 100nM E2 upregulated ERRα at mRNA and protein level in both cell lines (Figure 5E). We also observed that enhanced cell viability, migration, and invasion induced by E2 were inhibited after transfecting with siEERα (Figure 5B–D and F). These results indicated that ERRα’s effects on trigger the malignant phenotype of TNBC might be regulated by E2.
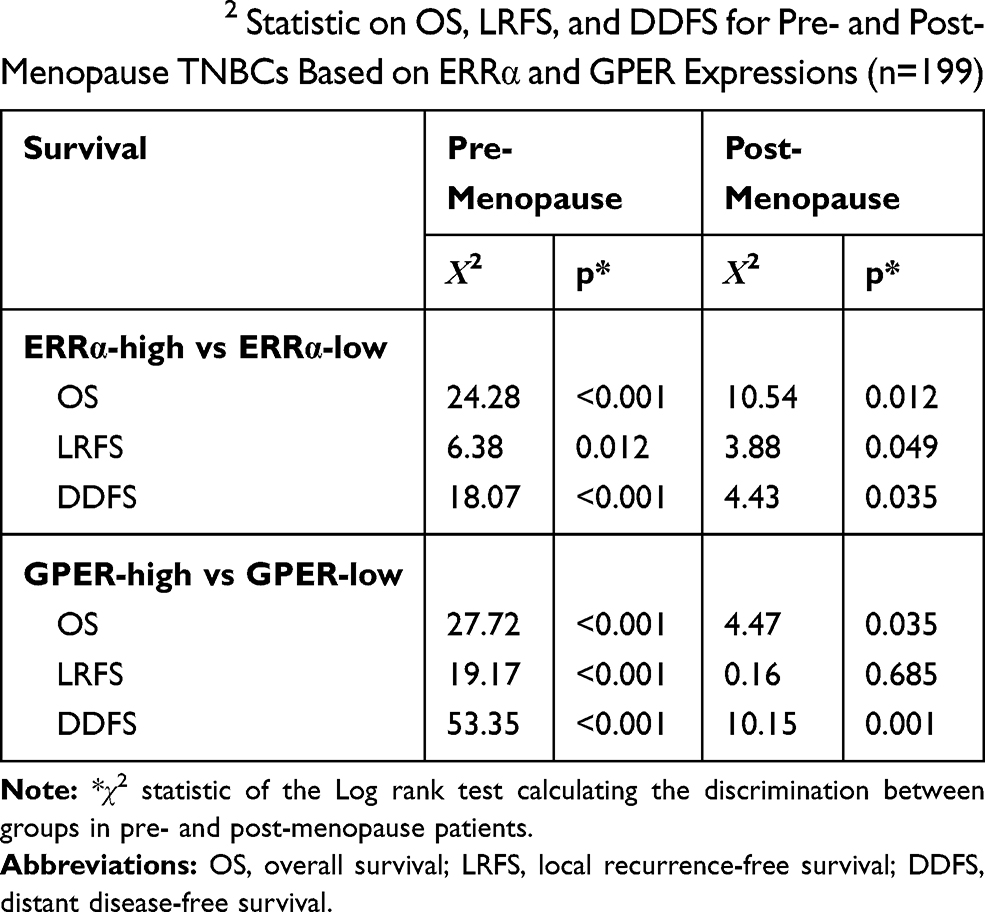 |
Table 3 The Χ2 Statistic on OS, LRFS, and DDFS for Pre- and Post-Menopause TNBCs Based on ERRα and GPER Expressions (n=199) |
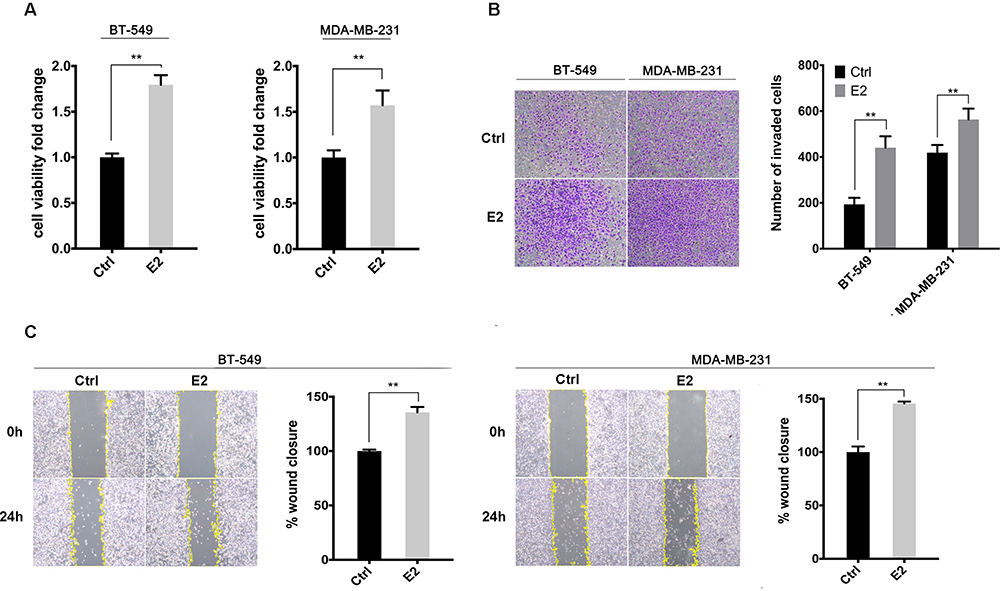 |
Figure 4 E2 enhanced cell viability, migration, and invasion in BT-549 and MDA-MB-231 cells. (A) Cell viability was detected using CCK-8 assay with the treatment of 100nM E2 or control. (B) Cell invasion was evaluated by the Transwell invasion assay with the treatment of 100nM E2 or control. The invasive cells were measured after 12h. (C) Cell migration was evaluated by a wound-healing assay with the treatment of 100nM E2 or control. The healing percentage was measured after 24h. **p < 0.01. |
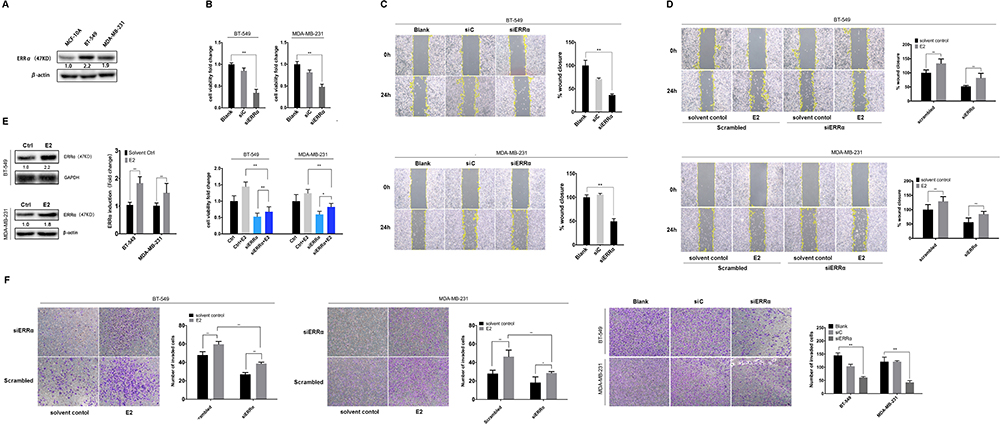 |
Figure 5 ERRα mediates the malignant phenotype of TNBC induced by estrogen. (A) Relative ERRα expression in normal mammary epithelial cells (MCF-10A) and TNBC cells (BT-549 and MDA-MB-231). (B) Cell viability was detected using CCK-8 assay with the siERRα and/or treatment of 100nM E2 in BT-549 and MDA-MB-231 cells. (C and D) Cell migration was evaluated by a wound-healing assay with the siERRα and/or treatment of 100nM E2 in BT-549 and MDA-MB-231 cells. The healing percentage was measured after 24h. (E) Relative ERRα expression in BT-549 and MDA-MB-231 cells incubated with 100nM E2 or control. (F) Cell invasion was evaluated by the Transwell invasion assay with the siERRα and/or treatment of 100nM E2 in BT-549 and MDA-MB-231 cells. The invasive cells were measured after 12h. *p < 0.05, **p < 0.01. |
GPER Intermediates the Regulation of ERRα by Estrogen
GPER is reported to commonly expressed in ER-negative breast cancer.32,38 Western blotting showed that the expression of GPER was low in MDA-MB-231 cells and relatively high in BT-549 cells (Figure 6A). Thus, we knocked down GPER in BT-549 cells using siGPER, and overexpressed GPER in MDA-MB-231 cells by transfecting plasmid, respectively, to investigate whether GPER intermediate the interaction between E2 and ERRα. Our result revealed that both E2 and G1 (GPER agonist) significantly increased ERRα mRNA level as compared to control in both cell lines, especially in BT-549. This effect was suppressed in BT-549 cells by siGPER (Figure 6B). Consistently, overexpression of GPER notably increased ERRα mRNA level in MDA-MB-231 cells, while no change was observed with pretreatment of G15, which is regarded as a GPER antagonist (Figure 6B). Activation of the ERK1/2 MAPK signaling has been reported to be required for the activation of GPER ligands, such as E2 and G1.27,39 We also found that increased ERRα mRNA levels induced by E2 and G1 were markedly suppressed after the administration of ERK1/2 MAPK inhibitor PD98059 in both cell lines (Figure 6C).
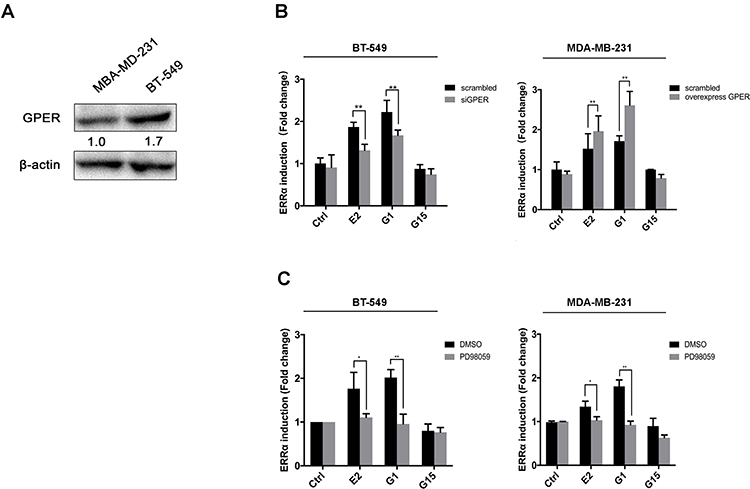 |
Figure 6 GPER intermediates the regulation of ERRα by estrogen. (A) Relative GPER expression in BT-549 and MDA-MB-231 cells. (B) Relative ERRα expression in BT-549 and MDA-MB-231 cells with treatment of 100nM E2, G1, or G15. (B) Relative ERRα expression in BT-549 and MDA-MB-231 cells overexpressing or knocking down of GPER with treatment of 100nM E2, G1, or G15. (C) Effects of blocking of ERK1/2 MAPKs with PD98059 on ERRα expression with treatment of 100nM E2, G1, or G15. *p < 0.05, **p < 0.01. |
Discussion
Estrogen receptor signaling is essential for the prognosis and treatment of ER-positive breast cancer. Application of endocrinotherapy, such as tamoxifen or toremifene, significantly improves the clinical outcome of patients with breast cancer. TNBC, which demonstrates a higher degree of malignancy and rapid progression,40,41 is proved to be lack of ER signaling activation. However, the clinical relevance of estrogen and TNBC remains controversial. A study by Contreras-Zárate et al found that estradiol induces BDNF/TrkB signaling in TNBC to promote brain metastases.42 Girgert et al suggested that 17β-estradiol significantly increased proliferation in TNBC in vitro, along with the activation of Src, EGFR, Cyclin D1, and CREB signaling.43 However, McNamara et al demonstrated that enzymes known to modulate levels of estrogens were detected in TNBC and were associated with longer disease-free survival, suggesting the presence of, and a potential protective effect of estrogens in TNBC.44 Current findings speculate the effect of estrogen in TNBC may be mediated by other activating effectors related to ER signaling or other receptors.
Consistent with previous studies,19,20 we observed that high expression of ERRα was associated with aggressive clinicopathologic features in TNBC, including high NG, frequent LNM, and advanced TNM stage, as well as poor survival outcomes. Age and menopausal status were neither associated with ERRα expression nor affecting the survival of TNBCs in this cohort. However, larger discrimination in survival curves between ERRα-high (or GPER-high) and ERRα-low (or GPER-low) group was observed in premenopausal patients than postmenopausal patients, which consistent with our previous study.31 These findings implied that ERRα and GPER might mediate the tumor-promoting effect of estrogen in TNBC. Notably, we also reveal a positive correlation between ERRα and GPER expressions, and the co-expression status of ERRα/GPER could further discriminate the survival tendency, which may provide a novel estrogen-related model for predicting the prognosis of TNBCs.
ERRα has been reported as tumorigenic by reprogramming the energy metabolism of tumor cells, leading to the production of various biosynthetic precursors, and creating a micromovement for tumor proliferation.12,13 Kallen et al45 have studied the structure of the ligand-binding domain (LBD) of ERRα by X-ray diffraction and found that the LBD of ERRα is almost entirely occupied by amino acid side chain, which is only possible to bind a ligand with less than 4 carbon atoms. Thus, estrogen is clearly not a natural ligand for ERRα. However, our data showed that the malignant phenotypes induced by E2 in TNBC cells were significantly suppressed by ERRα. Moreover, E2 treatment increased the protein and mRNA level of ERRα in vitro, suggesting the indirect interactions between E2 and ERRα. Interestingly, studies by Li et al25 and Kotula-Balak et al33 showed that the activation of GPER lead to.an increased ERRα level in HER2-positive breast cancer cells and tumorous Leydig cells, respectively. Similarly, we demonstrated that GPER agonists and overexpression of GPER also resulted in an increased ERRα level in TNBC, while E2 induced increased expression of ERRα was inhibited by knocking down of GPER as well as ERK1/2 MAPK signaling inhibitor. These results indicate that GPER may intermediate the regulation of ERRα by E2, and resulted in promoting cell proliferation, migration, and invasion in TNBC, which is consistent with the findings in clinicopathological and prognostic analyses.
The effect of estrogen in breast cancer is no longer rely on the expression of ER (predominantly ERα). The role of other subtypes of estrogen receptors, such as ERβ29,46,47 and ERα36,48,49 as well as other receptors or effectors related to ER signaling, such as GPER and ERR family, are raising concern in TNBC. However, the cross-talk among different ER subtypes or effectors related to ER signaling could be extremely complicated. The present study suggests the prognostic role of ERRα in TNBCs and provides a new idea for the understanding of estrogenic effects on the malignant phenotype of TNBC by ERRα-GPER interactions. The underlying mechanism interpreting the interaction between GPER and ERRα should be involved in further investigations. Moreover, given the lack of effective conventional therapies for the treatment of TNBC, Wisinski’s team has attempted to conduct a Phase 2 study evaluating single-agent high-dose estradiol targeting ERβ in patients with advanced TNBC,50 while limited efficacy was found. Promising therapeutic options targeting estrogen-related agents, such as GPER, ERR family, ERβ, and ERα36, may be warranted in further clinical trials.
Abbreviations
ERRα, estrogen-related receptor α; GPER, G protein-coupled estrogen receptor; TNBC, triple-negative breast cancer; E2, 17β-estradiol; OS, overall survival; LNM, lymph nodal metastasis; NG, nuclear grade; ER, estrogen receptor; PR, progesterone receptor; HER2, human epidermal growth factor receptor 2; LRFS, local recurrence-free survival; DDFS, distant disease-free survival; FFPE, formalin fixed paraffin-embedded; H&E, haematoxylin and eosin; FISH, fluorescence in situ hybridization; IHC, immunohistochemical staining; qRT-PCR, quantitative real-time PCR; PSM, propensity score matching; ROC, receiver operating characteristic curve; MAPK, mitogen-activated protein kinase; ERK1/2, extracellular regulated kinase 1/2.
Data Sharing Statement
The datasets generated and analyzed during the current study are also available from the corresponding author (Tinghuai Wang) on reasonable request.
Ethics Approval and Consent to Participate
All procedures performed in studies involving human participants were approved by the Ethics Committee and Research Committee of the Sun Yat-sen University Cancer Center (SYSUCC) and followed the ethical standards of the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The formal written informed consent was obtained from all individual participants included in the study.
Disclosure
The authors declare that they have no competing interests for this work.
References
1. Garcia Fernandez A, Gimenez N, Fraile M, et al. Survival and clinicopathological characteristics of breast cancer patient according to different tumour subtypes as determined by hormone receptor and Her2 immunohistochemistry. A single institution survey spanning 1998 to 2010. Breast. 2012;21:366–373. doi:10.1016/j.breast.2012.03.004
2. Guney Eskiler G, Cecener G, Egeli U, Tunca B. Triple negative breast cancer: new therapeutic approaches and BRCA status. APMIS. 2018;126:371–379. doi:10.1111/apm.12836
3. Rastelli F, Biancanelli S, Falzetta A, et al. Triple-negative breast cancer: current state of the art. Tumori. 2010;96:875–888. doi:10.1177/548.6505
4. Sharma P. Update on the treatment of early-stage triple-negative breast cancer. Curr Treat Options Oncol. 2018;19:22. doi:10.1007/s11864-018-0539-8
5. He J, Tsang JY, Xu X, et al. AJCC 8th edition prognostic staging provides no better discriminatory ability in prognosis than anatomical staging in triple negative breast cancer. BMC Cancer. 2020;20:18. doi:10.1186/s12885-019-6494-3
6. Li X, Yang J, Peng L, et al. Triple-negative breast cancer has worse overall survival and cause-specific survival than non-triple-negative breast cancer. Breast Cancer Res Treat. 2017;161:279–287. doi:10.1007/s10549-016-4059-6
7. Sihto H, Lundin J, Lundin M, et al. Breast cancer biological subtypes and protein expression predict for the preferential distant metastasis sites: a nationwide cohort study. Breast Cancer Res. 2011;13:R87. doi:10.1186/bcr2944
8. Lin NU, Claus E, Sohl J, Razzak AR, Arnaout A, Winer EP. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer. 2008;113:2638–2645. doi:10.1002/cncr.23930
9. Aapro M, Wildiers H. Triple-negative breast cancer in the older population. Ann Oncol. 2012;23(Suppl 6):52–55. doi:10.1093/annonc/mds189
10. Anderson WF, Jatoi I, Devesa SS. Distinct breast cancer incidence and prognostic patterns in the NCI’s SEER program: suggesting a possible link between etiology and outcome. Breast Cancer Res Treat. 2005;90(2):127–137. doi:10.1007/s10549-004-3777-3
11. Millikan RC, Newman B, Tse CK, et al. Epidemiology of basal-like breast cancer. Breast Cancer Res Treat. 2008;109:123–139. doi:10.1007/s10549-007-9632-6
12. De Santis MC, Porporato PE, Martini M, Morandi A. Signaling pathways regulating redox balance in cancer metabolism. Front Oncol. 2018;8:126. doi:10.3389/fonc.2018.00126
13. Heiden MGV, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi:10.1126/science.1160809
14. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi:10.1016/j.ccr.2012.02.014
15. Lane AN, Tan J, Wang Y, Yan J, Higashi RM, Fan TW. Probing the metabolic phenotype of breast cancer cells by multiple tracer stable isotope resolved metabolomics. Metab Eng. 2017;43:125–136. doi:10.1016/j.ymben.2017.01.010
16. Giguere V, Yang N, Segui P, Evans RM. Identification of a new class of steroid hormone receptors. Nature. 1988;331:91–94. doi:10.1038/331091a0
17. Xu Z, Liu J, Gu L, Ma X, Huang B, Pan X. Research progress on the reproductive and non-reproductive endocrine tumors by estrogen-related receptors. J Steroid Biochem Mol Biol. 2016;158:22–30. doi:10.1016/j.jsbmb.2016.01.008
18. Deblois G, Giguere V. Oestrogen-related receptors in breast cancer: control of cellular metabolism and beyond. Nat Rev Cancer. 2013;13:27–36. doi:10.1038/nrc3396
19. Chang CY, Kazmin D, Jasper JS, Kunder R, Zuercher WJ, McDonnell DP. The metabolic regulator ERRα, a downstream target of HER2/IGF-1R, as a therapeutic target in breast cancer. Cancer Cell. 2011;20:500–510. doi:10.1016/j.ccr.2011.08.023
20. Cavallini A, Notarnicola M, Giannini R, et al. Oestrogen receptor-related receptor alpha (ERRα) and oestrogen receptors (ERα and ERβ) exhibit different gene expression in human colorectal tumour progression. Eur J Cancer. 2005;41:1487–1494. doi:10.1016/j.ejca.2005.04.008
21. Tam IS, Giguere V. There and back again: the journey of the estrogen-related receptors in the cancer realm. J Steroid Biochem Mol Biol. 2016;157:13–19. doi:10.1016/j.jsbmb.2015.06.009
22. Zhang S, Liu X, Liu J, Guo H, Xu H, Zhang G. PGC-1 alpha interacts with microRNA-217 to functionally regulate breast cancer cell proliferation. Biomed Pharmacother. 2017;85:541–548. doi:10.1016/j.biopha.2016.11.062
23. Audet-Walsh E, Papadopoli DJ, Gravel SP, et al. The PGC-1α/ERRα axis represses one-carbon metabolism and promotes sensitivity to anti-folate therapy in breast cancer. Cell Rep. 2016;14:920–931. doi:10.1016/j.celrep.2015.12.086
24. Giguère V. To ERR in the estrogen pathway. Trends Endocrinol Metab. 2002;13:220–225. doi:10.1016/S1043-2760(02)00592-1
25. Li Y, Birnbaumer L, Teng CT. Regulation of ERRα gene expression by estrogen receptor agonists and antagonists in SKBR3 breast cancer cells: differential molecular mechanisms mediated by G protein-coupled receptor GPR30/GPER-1. Mol Endocrinol. 2010;24:969–980. doi:10.1210/me.2009-0148
26. Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632. doi:10.1210/en.2004-1064
27. Filardo EJ, Quinn JA, Frackelton AR, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16:70–84. doi:10.1210/mend.16.1.0758
28. Zhou X, Wang S, Wang Z, et al. Estrogen regulates Hippo signaling via GPER in breast cancer. J Clin Invest. 2015;125:2123–2135. doi:10.1172/JCI79573
29. Zhou K, Sun P, Zhang Y, You X, Li P, Wang T. Estrogen stimulated migration and invasion of estrogen receptor-negative breast cancer cells involves an ezrin-dependent crosstalk between G protein-coupled receptor 30 and estrogen receptor beta signaling. Steroids. 2016;111:113–120. doi:10.1016/j.steroids.2016.01.021
30. Yu T, Liu M, Luo H, et al. GPER mediates enhanced cell viability and motility via non-genomic signaling induced by 17β-estradiol in triple-negative breast cancer cells. J Steroid Biochem Mol Biol. 2014;143:392–403. doi:10.1016/j.jsbmb.2014.05.003
31. Ye S, Xu Y, Li J, Zheng S, Sun P, Wang T. Prognostic role of GPER/Ezrin in triple-negative breast cancer is associated with menopausal status. Endocr Connect. 2019;8:661–671. doi:10.1530/EC-19-0164
32. Steiman J, Peralta EA, Louis S, Kamel O. Biology of the estrogen receptor, GPR30, in triple negative breast cancer. Am J Surg. 2013;206:698–703. doi:10.1016/j.amjsurg.2013.07.014
33. Kotula-Balak M, Milon A, Pawlicki P, et al. Insights into the role of estrogen-related receptors α, β and γ in tumor Leydig cells. Tissue Cell. 2018;52:78–91. doi:10.1016/j.tice.2018.04.003
34. Hammond MEH, Hayes DF, Dowsett M, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28:2784–2795. doi:10.1200/JCO.2009.25.6529
35. Wolff AC, Hammond MEH, Allison KH, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline focused update. J Clin Oncol. 2018;36:2105–2122. doi:10.1200/JCO.2018.77.8738
36. Skrzypczak M, Schuler S, Lattrich C, Ignatov A, Ortmann O, Treeck O. G protein-coupled estrogen receptor (GPER) expression in endometrial adenocarcinoma and effect of agonist G-1 on growth of endometrial adenocarcinoma cell lines. Steroids. 2013;78:1087–1091. doi:10.1016/j.steroids.2013.07.007
37. Watanabe A, Kinoshita Y, Hosokawa K, Mori T, Yamaguchi T, Honjo H. Function of estrogen-related receptor alpha in human endometrial cancer. J Clin Endocrinol Metab. 2006;91:1573–1577. doi:10.1210/jc.2005-1990
38. Luo HJ, Luo P, Yang GL, Peng QL, Liu MR, Tu G. G-protein coupled estrogen receptor 1 expression in primary breast cancers and its correlation with clinicopathological variables. J Breast Cancer. 2011;14:185–190. doi:10.4048/jbc.2011.14.3.185
39. Gaudet HM, Cheng SB, Christensen EM, Filardo EJ. The G-protein coupled estrogen receptor, GPER: the inside and inside-out story. Mol Cell Endocrinol. 2015;418(3):207–219. doi:10.1016/j.mce.2015.07.016
40. Dent R, Trudeau M, Pritchard KI, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–4434. doi:10.1158/1078-0432.CCR-06-3045
41. Gluz O, Liedtke C, Gottschalk N, Pusztai L, Nitz U, Harbeck N. Triple-negative breast cancer–current status and future directions. Ann Oncol. 2009;20:1913–1927. doi:10.1093/annonc/mdp492
42. Contreras-Zárate MJ, Day NL, Ormond DR, et al. Estradiol induces BDNF/TrkB signaling in triple-negative breast cancer to promote brain metastases. Oncogene. 2019;38:4685–4699. doi:10.1038/s41388-019-0756-z
43. Girgert R, Emons G, Gründker C. 17β-estradiol-induced growth of triple-negative breast cancer cells is prevented by the reduction of GPER expression after treatment with gefitinib. Oncol Rep. 2017;37:1212–1218. doi:10.3892/or.2016.5306
44. McNamara KM, Oguro S, Omata F, et al. The presence and impact of estrogen metabolism on the biology of triple-negative breast cancer. Breast Cancer Res Treat. 2017;161:213–227. doi:10.1007/s10549-016-4050-2
45. Kallen J, Lattmann R, Beerli R, et al. Crystal structure of human estrogen-related receptor alpha in complex with a synthetic inverse agonist reveals its novel molecular mechanism. J Biol Chem. 2007;282:23231–23239. doi:10.1074/jbc.M703337200
46. Reese JM, Bruinsma ES, Nelson AW, et al. ERβ-mediated induction of cystatins results in suppression of TGFβ signaling and inhibition of triple-negative breast cancer metastasis. Proc Natl Acad Sci U S A. 2018;115:E9580–E9589. doi:10.1073/pnas.1807751115
47. Zhao L, Huang S, Mei S, et al. Pharmacological activation of estrogen receptor beta augments innate immunity to suppress cancer metastasis. Proc Natl Acad Sci U S A. 2018;115:E3673–E3681. doi:10.1073/pnas.1803291115
48. Wang X, Zheng N, Dong J, Wang X, Liu L, Huang J. Estrogen receptor-α36 is involved in icaritin induced growth inhibition of triple-negative breast cancer cells. J Steroid Biochem Mol Biol. 2017;171:318–327. doi:10.1016/j.jsbmb.2017.05.009
49. Hu B, Yan W, Wang M, et al. Huaier polysaccharide inhibits the stem-like characteristics of ERα-36(high) triple negative breast cancer cells via inactivation of the ERα-36 signaling pathway. Int J Biol Sci. 2019;15:1358–1367. doi:10.7150/ijbs.27360
50. Wisinski KB, Xu W, Tevaarwerk AJ, et al. Targeting estrogen receptor beta in a phase 2 study of high-dose estradiol in metastatic triple-negative breast cancer: a wisconsin oncology network study. Clin Breast Cancer. 2016;16:256–261. doi:10.1016/j.clbc.2016.03.005
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.