Back to Journals » International Journal of Nanomedicine » Volume 16
Enzyme-Functionalized Mesoporous Silica Nanoparticles to Target Staphylococcus aureus and Disperse Biofilms
Authors Devlin H ![]() , Fulaz S
, Fulaz S ![]() , Hiebner DW, O'Gara JP
, Hiebner DW, O'Gara JP ![]() , Casey E
, Casey E ![]()
Received 20 November 2020
Accepted for publication 9 February 2021
Published 8 March 2021 Volume 2021:16 Pages 1929—1942
DOI https://doi.org/10.2147/IJN.S293190
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Henry Devlin,1,* Stephanie Fulaz,1,* Dishon Wayne Hiebner,1 James P O’Gara,2 Eoin Casey1
1UCD School of Chemical and Bioprocess Engineering, University College Dublin, Dublin, Ireland; 2Department of Microbiology, School of Natural Sciences, National University of Ireland, Galway, Ireland
*These authors contributed equally to this work
Correspondence: Eoin Casey
UCD School of Chemical and Bioprocess Engineering, University College Dublin, Belfield, Dublin, Ireland
Email [email protected]
Background: Staphylococcus aureus biofilms pose a unique challenge in healthcare due to their tolerance to a wide range of antimicrobial agents. The high cost and lengthy timeline to develop novel therapeutic agents have pushed researchers to investigate the use of nanomaterials to deliver antibiofilm agents and target biofilm infections more efficiently. Previous studies have concentrated on improving the efficacy of antibiotics by deploying nanoparticles as nanocarriers. However, the dispersal of the extracellular polymeric substance (EPS) matrix in biofilm-associated infections is also critical to the development of novel nanoparticle-based therapies.
Methods: This study evaluated the efficacy of enzyme-functionalized mesoporous silica nanoparticles (MSNs) against methicillin-resistant S. aureus (MRSA) and methicillin-sensitive S. aureus (MSSA) biofilms. MSNs were functionalized with the enzyme lysostaphin, which causes cell lysis of S. aureus bacteria. This was combined with two other enzyme functionalized MSNs, serrapeptase and DNase I which will degrade protein and eDNA in the EPS matrix, to enhance eradication of the biofilm. Cell viability after treatment with enzyme-functionalized MSNs was assessed using a MTT assay and CLSM, while crystal violet staining was used to assess EPS removal.
Results: The efficacy of all three enzymes against S. aureus cells and biofilms was significantly improved when they were immobilized onto MSNs. Treatment efficacy was further enhanced when the three enzymes were used in combination against both MRSA and MSSA. Regardless of biofilm maturity (24 or 48 h), near-complete dispersal and killing of MRSA biofilms were observed after treatment with the enzyme-functionalized MSNs. Disruption of mature MSSA biofilms with a polysaccharide EPS was less efficient, but cell viability was significantly reduced.
Conclusion: The combination of these three enzymes and their functionalization onto nanoparticles might extend the therapeutic options for the treatment of S. aureus infections, particularly those with a biofilm component.
Keywords: MRSA, lysostaphin, antimicrobial, antibiofilm, EPS matrix
Introduction
Staphylococcus aureus is a Gram-positive opportunistic pathogen carried by about 20–50% of the human population.1–3 It is one of the leading causes of both hospital, and community-acquired infections, ranging from mild skin irritations to life-threatening diseases such as endocarditis, pneumonia and sepsis.4,5 Management of S. aureus infections often involves prolonged therapy with antibiotics such as beta-lactams, vancomycin, clindamycin or doxycycline. Treatment options are complicated by antibiotic resistance,6–9 as well as biofilms and subpopulations of persister cells,10–12 both of which exhibit high tolerance towards conventional antibiotics and are associated with antibiotic treatment failures and relapsing infections. In order to overcome this issue, novel anti-staphylococcal therapies are required.
The use of enzymes as a treatment for S. aureus biofilm eradication is an area of research which holds much potential. Previous studies have used a range of enzymes to treat biofilms formed by both methicillin-resistant S. aureus (MRSA) and methicillin-sensitive S. aureus (MSSA) biofilms;13,14 however, a major issue which has hindered the development of novel antibiofilm treatments is the complex composition of the self-produced, extracellular polymeric substance (EPS) matrix.15 The matrix formed by MRSA biofilms is primarily composed of proteins and eDNA, while MSSA biofilm matrices are mainly composed of polysaccharides.15–17 Therefore, identifying an enzyme treatment regime that would be effective against these different biofilm matrices could enable a more rapid response following the onset of an infection. The use of nanoparticles (NPs) as carriers of antimicrobial/antibiofilm agents is an area which has gained much interest over the last two decades.18–21 NPs can penetrate the deepest regions of the biofilm to specifically target cells, proteins or polysaccharides, thus increasing the local concentration and shielding the antimicrobial agent from deactivation within the matrix.21–23
One such agent with significant potential is the lytic enzyme, lysostaphin, which is derived from Staphylococcus simulans. This enzyme cleaves the pentaglycine interbridges of S. aureus peptidoglycan cell wall undermining cell integrity.24 Lysostaphin is active against both metabolically inactive and active cells, thus ensuring its effectiveness against persister cells and biofilm cells which are often unaffected by antibiotic treatment.25 In previous tests, lysostaphin was demonstrated to have higher antimicrobial activity than commonly used antibiotics such as vancomycin.26 Hogan et al 2017,13 compared the effectiveness of several enzymatic agents against MRSA and MSSA biofilms and reported that lysostaphin was the most effective antibacterial agent. Combining this enzyme with positively charged nanocarriers (such as amine-functionalized mesoporous silica nanoparticles (MSNs)), which have been reported to localize preferentially around the bacterial cell wall,21 will help to increase the local concentration of the enzyme and improve the treatment of S. aureus biofilm infections.27–29 MSNs have been extensively researched and are one of the most important porous materials, widely investigated as a drug-carrier, mainly due to their stable structure, easily functionalisable surface chemistry, including enzyme immobilization, as well as increased biocompatibility and safety.27,29–34 Furthermore, MSNs have also been shown to penetrate to the deepest regions of biofilm samples and so provide an attractive candidate for biofilm targeting.35 Lysostaphin has been shown to target S. aureus bacteria and biofilms effectively; however, their widespread use has been limited due to the high production cost.36 The biofilm can also inhibit enzyme treatments through reduced penetration or deactivation in the EPS matrix.23 It is hypothesized that loading the enzyme onto nanoparticles, which are known to localize and surround bacterial cells21 will increase the enzyme’s efficacy against biofilm samples. Through immobilization onto NPs, lysostaphin’s effectiveness may be enhanced further with targeted delivery to the bacterial cells within biofilms, thus expanding its therapeutic potential for S. aureus infections.
The removal of the biofilm EPS matrix is a crucial step when considering novel antibiofilm treatments. If the matrix is not removed, it provides an ideal environment for future bacterial growth and biofilm formation.23,37 The enzymes DNase I, α-amylase, proteinase K, serrapeptase, plasmin and dispersin B have all been shown to degrade the biofilm matrix in different bacteria.13,14,38,39 These enzymes can act by breaking up the EPS components such as proteins, polysaccharides, fibrin or eDNA. Targeting eDNA and protein adhesins present in MRSA biofilms, and to a lesser extent, MSSA biofilms could have broad activity in different types of S. aureus infections. eDNA is critical in the development of MRSA biofilms and is also known to interact with poly-N-acetylglucosamine in MSSA biofilms,40 thus providing a common target.
Similarly, production of fibrin in S. aureus biofilms via the activity of coagulase on the abundant host extracellular matrix glycoprotein fibrinogen represents an important target for antibiofilm enzymes.13,21,38,39 Serrapeptase is a proteolytic enzyme with the potential to degrade fibrin,41–43 and has been shown to increase the efficacy of antibiotic agents when used for the eradication of biofilm-associated infections.44 Artini, 2011,45 showed that serrapeptase did not affect the viability of planktonic S. aureus cells, despite being able to prevent biofilm formation.
In this study, three different enzymes, lysostaphin, serrapeptase and DNase I, were individually immobilized on the surface of MSNs, and enzyme-functionalized MSNs were combined to develop a novel treatment strategy for MSSA and MRSA biofilms. Amine-functionalized MSNs were used as enzyme-carriers, to improve enzyme stability and increase enzyme penetration in the biofilm and to investigate the antibacterial and EPS matrix dispersal efficacy of enzyme-loaded MSNs against MRSA and MSSA biofilms as shown in Figure 1. The activity of the enzyme-functionalized MSNs was compared to the free enzymes. The data presented provides new insights into the development of broad-spectrum antibiofilm treatments using enzyme functionalized MSNs.
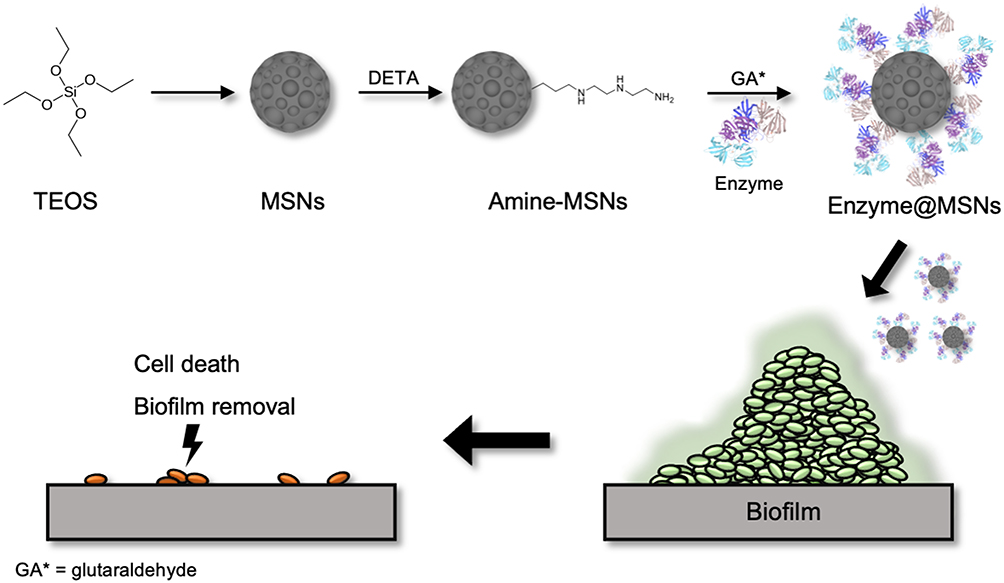 |
Figure 1 Schematic representing a simplified reaction of MSNs synthesis, surface functionalization with amino groups and enzyme immobilization. It also includes a representation of the proposed mechanism of action, indicating the biofilm treatment with enzyme-functionalized MSNs led to the cell death and biofilm dispersal. |
Materials and Methods
Materials
Hexadecyltrimethylammonium bromide (CTAB, 98%), tetraethyl orthosilicate (TEOS, 99%), triethanolamine (TEA, 99%), acetic acid,N1-(3-trimethoxysilylpropyl) diethylenetriamine (DETA), glycerol, tryptic soy agar and tryptic soy broth (TSB), cell proliferation kit 1 (MTT), NaCl, glucose, PBS and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich. Ethanol (99.5%) and hydrochloric acid (HCl, 37%) were obtained from Merck. Crystal Violet (CV) was purchased from Honeywell Riedel-de-Haën™. LIVE/DEAD® BacLight™ Bacterial Viability Kit was purchased from Life Technologies Carlsbad, California. All reagents were used as received without further purification. Clear 96-well plates were purchased from SARSTEDT, Germany. Costar® Black 96-well optically clear plates were purchased from Corning, UK.
Synthesis, Functionalization and Enzyme Immobilization on MSNs
The synthesis of MSNs was performed using an adapted Stöber method as previously described.35 The amine-functionalization of MSNs was carried out using DETA as the aminated silane precursor as previously described.21 For the enzyme immobilization, 150 mg of amine-functionalized MSNs was dispersed in 30 mL of water using an ultrasonic bath. To this suspension, 1 mL of glutaraldehyde 50% (v/v) was added, and the mixture was stirred at room temperature for 3 hours. The obtained product was washed three times with water and once with appropriate buffer (sodium acetate, pH 4.5 for lysostaphin and PBS for serrapeptase and DNase I) using centrifugation (9,000 rpm, 15 min). The nanoparticles were suspended in the appropriate buffer, and the enzyme was added; 5 mg of lysostaphin (from Staphylococcus simulans, Sigma Aldrich) for Lys@MSNs, 380 mg of serrapeptase (250 000 SPU per dose, Arthur Andrew Medical) for Ser@MSNs and 16 mg of DNase I (from bovine pancreas grade II, Roche) for DN@MSNs. The suspensions were stirred overnight at room temperature, and then, washed with corresponding buffers.
MSNs Characterization
Zeta potential of nanoparticles suspension in water (1 mg mL−1) was measured in a folded capillary zeta cell using a Zetasizer Nano-ZS apparatus (Malvern Instruments). All measurements were done in triplicates with 15 scans each. TEM analysis was performed using a FEI Tecnai G2 on samples dispersed in ethanol and deposited on carbon-coated copper grids. Average size and size distribution of nanoparticles were analyzed by Fiji software46 using TEM images, analyzing a minimum of 150 nanoparticles per sample. N2 adsorption-desorption isotherms were recorded in a BET Nova 2400e (Quantachrome, UK) apparatus at 77 K under continuous adsorption conditions. Samples were outgassed at 110 oC for 16 h prior to experiments. The surface area was obtained by the BET (Brunauer-Emmett-Teller) analysis. The pore volume and pore size were obtained using the BJH (Barrett-Joyner-Halenda) model on adsorption. FT-IR was performed in a Bruker Vertex 70 Spectrophotometer. Samples in ethanol dispersion were deposited onto NaCl FT-IR cards and dried at 70 oC. Spectrum was obtained using a resolution of 4 cm−1, 64 scans and a spectral window from 4000 to 400 cm−1. Dissolution 1H NMR was used to dissolve the MSNs and obtain a clear spectrum of the surface groups. Approximately 10–15 mg of MSNs were dissolved in 662 μL of deuterium oxide (D2O) and 38 μL sodium deuteroxide (NaOD). The mixture was incubated overnight at 37 oC under stirring. The spectrum was obtained in a Varian Inova 300 MHz Spectrometer using 128 scans. In order to evaluate enzyme immobilization on MSNs, the enzyme-MSNs suspensions, the supernatant and the washing solutions were collected and analyzed using the Lowry assay. To further estimate enzyme immobilization onto MSNs, thermogravimetric analysis (TGA) was performed on powder specimen using a Rheometric Scientific STA 1500 simultaneous thermal analyzer (Rheometric Scientific Ltd., Piscataway, New Jersey, USA). Samples were tested in matched platinum/rhodium crucibles in a flowing nitrogen atmosphere with a flow rate of 50 cm3 min−1. A sample mass of 15 mg ± 10 mg and a heating rate of 10 °C min−1 was used for all experiments.
Bacterial Culture
The bacterial strains which were selected for this study were Methicillin-resistant S. aureus BH1CC (MRSA) and Methicillin-sensitive S. aureus 8325–4 (MSSA). Bacterial stock cultures were stored in 25% (v/v) glycerol at -80 °C. These cultures were streaked on Tryptic soy agar plates and incubated at 37 °C for 24 h. A single bacterial colony was used to inoculate 50 mL of sterile Tryptic soy broth in a 250 mL. Erlenmeyer flask and incubated overnight at 37 °C with shaking at 200 rpm. For biofilm formation, the overnight culture was diluted to a final optical density (OD600) of 0.001.
MIC (Minimum Inhibitory Concentration) and MBIC (Minimum Biofilm Inhibitory Concentration) Determination
The antimicrobial and antibiofilm properties of the MSNs, the enzyme-functionalized MSNs and the free enzymes were tested against both MRSA and MSSA strains. Overnight cultures were prepared as described and incubated in a 96-well plate. The enzymes’ antimicrobial and antibiofilm activity were tested over various concentrations (0.05–500 µg mL−1). The unloaded MSNs were tested over a concentration range of 0–1000 µg mL−1. After incubation for 24 h at 37 °C under static conditions, the absorbance values (600 nm) of the suspensions were recorded to determine the MIC using a plate reader (SpectraMax iD3, Molecular devices). To determine the MBIC value, the spent culture media was removed, and the wells were washed three times with sterile water to remove planktonic cells. Crystal violet (0.1%) was used to determine biofilm formation. The MBIC was determined as the lowest concentration where no biofilm formation was observed.
Biofilm Formation
Biofilms were formed in clear 96-well plates (SARSTEDT). The MRSA strain BH1CC was cultured in TSB supplemented with 2% glucose to promote biofilm formation.17 For MSSA biofilm formation, the culture media was supplemented with 4% NaCl.16 Biofilms were cultured at 37 °C for 24 and 48 h under static conditions to assess the difference in activity against early-stage biofilms and more mature biofilm samples. For 48 h biofilm formation, the media was changed after 24 h to ensure optimal culture conditions were maintained.
Exposure of S. aureus Biofilms to Enzyme Loaded MSNs and Free Enzymes
To assess the antibiofilm effects of the MSNs, the enzyme-functionalized MSNs, and free enzymes, S. aureus biofilms were cultured for 24 h and 48 h under static conditions. Following biofilm formation, the spent media was removed, and the biofilms were washed three times with sterile water to remove planktonic cells before being treated with either the enzyme-loaded MSNs or the free enzymes for 24 h. Following exposure, the solution was removed, and the biofilms were washed three times with sterile water to remove residual nanoparticles or enzymes. To assess bacterial cell viability after exposure to either the MSNs, the enzyme-functionalized MSNs, or the free enzymes, a MTT assay was utilized. Briefly, following the removal of planktonic cells and residual MSNs, 150 µL of PBS and 50 µL of the MTT solution was added to each well, and the plate was incubated for 2 h at 37 °C under static conditions. Following this incubation, the MTT – PBS solution was removed, and 200 µL of dimethyl sulfoxide was added to solubilize the MTT solution, which was metabolized by the live bacterial cells into an insoluble purple compound (formazan). The plate was incubated for 15 min at 37 °C under static conditions before an absorbance reading was taken at 550 nm, using a plate reader (SpectraMax iD3, Molecular Devices) to determine cell viability within the biofilm samples.47,48 To assess the EPS matrix removal activity, biofilms were cultured and exposed as described above. Following treatment, the biofilms were washed three times with sterile water to remove planktonic cells and residual MSNs and enzymes. Following this, 200 µL of crystal violet (0.1%) was added to each well, and the plate was incubated at 37 °C for 25 min in the dark under static conditions. Following this, the crystal violet solution was removed, and the biofilms were washed at least five times to remove unbound dye. Acetic acid solution (200 µL at 33%, v/v) was added to each well, and the plate was once again incubated at 37 °C for 20 mins under static conditions. Finally, the solubilized dye was transferred to a new 96-well plate, and absorbance readings (600 nm) were recorded using a plate reader (SpectraMax iD3, Molecular Devices) to assess EPS matrix removal. All experiments were carried out in triplicate, with a minimum of 24 independent replicates per run.
Confocal Laser Scanning Microscopy
To further assess the antibacterial activity of the enzyme-loaded MSNs, biofilms of MRSA and MSSA were cultured in optically clear bottom 96-well plates (Costar®) for 24 or 48 h under static conditions at 37 °C. Following biofilm formation, the spent culture solution was removed, and the biofilms washed three times with sterile water to remove planktonic cells and spent media. The biofilms were then exposed to the enzyme-loaded MSNs (0.25, 0.33 and 0.5 mg mL−1) and incubated. After 24 h the MSN solution was removed, and the biofilms were washed three times with sterile water to remove unbound MSNs. The biofilms were then stained for 30 min in the dark by adding a mixture of SYTO 9 (6.0 μM) and propidium iodide (30 μM) (LIVE/DEAD® BacLightTM Bacterial Viability Kit, Life Technologies, Carlsbad, California, USA). Biofilm images were acquired with an Olympus FluoView FV1000 Confocal Laser Scanning Microscope. SYTO 9 signals were detected using a multi-wavelength argon laser (excitation wavelength 488 nm and an emission wavelength range of 500–550 nm). Propidium iodide signals were detected with a laser wavelength of 561 nm and an emission range of 675–750 nm.
Statistical Analysis
Statistical significance was determined using a Student’s t-test (two-tailed) for all experiments. A value of p < 0.05 was considered significant. All experiments were carried out in triplicate (n = 3). Error bars were identified using standard error of the mean. All statistical analyses were performed using Excel (Microsoft Office).
Results and Discussion
Synthesis and Characterization of Enzyme-Functionalized MSNs
MSNs were prepared using an adapted Stöber method.21 The characteristic porosity was evaluated using the Brunauer-Emmett-Teller (BET) method (Figure S1(a)) and a typical type IV adsorption-desorption isotherm was observed with a high surface area (1308 m2 g−1). The capillary condensation was observed at a relative pressure (P/P0) between 0.4 and 0.5, indicating the presence of mesoporous channels. The pore size was determined by the Barret-Joyner-Halenda (BJH) method (Figure S1(b)), with an average pore size of 2.5 nm, and a pore volume of 0.930 cc g−1. Following MSNs functionalization with amine groups, FT-IR (Figure S2) and 1H NMR (Figure S3) characterization was performed to confirm the surface functionalization. In the FT-IR spectrum is possible to observe the Si-O symmetrical (800 cm−1) and asymmetrical (1070 cm−1) stretching, characteristic from the silica core. The C-N stretching (1235 cm−1) and N-H bending (1417 cm−1) from primary amines and N-H stretching (3685 cm−1) from secondary amines as well as the alkane stretching bands (2910 and 2987 cm−1) indicate that surface functionalization was successful.49 Dissolution 1H NMR helped to confirm surface functionalization further, allowing the assignment of the peaks to the protons found in the aminated silane DETA, used for the amine-functionalization.
Herein, four families of MSNs were investigated: amine-functionalized (amine-MSNs), lysostaphin-functionalized MSNs (Lys@MSN), serrapeptase-functionalized MSNs (Ser@MSN) and DNase I-functionalized MSNs (DN@MSN). Representative TEM images of all synthesized nanoparticles are represented in Figure 2, whereas the size and surface charge are summarized in Table 1. The synthesized nanoparticles are spherical with a diameter of approximately 36 nm and have a clear porous structure. The enzyme functionalization significantly altered the nanoparticles’ charge and had no significant impact on nanoparticles size and morphology as seen in TEM images. To further evaluate the enzyme-immobilization onto the MSNs, the protein quantification by Lowry assay and TGA analysis were both used, and the results are shown in Table 1. From the TGA data, it was possible to estimate the number of enzymes immobilized per MSN. Each enzyme-immobilized nanoparticle had a different number of enzyme chains immobilized on its surface, as it would be expected, considering the differences in the steric hindrance of the enzymes, the number of amine-groups available for the formation of the covalent bond with the MSNs surface, and also the initial concentration of enzymes in the reaction.
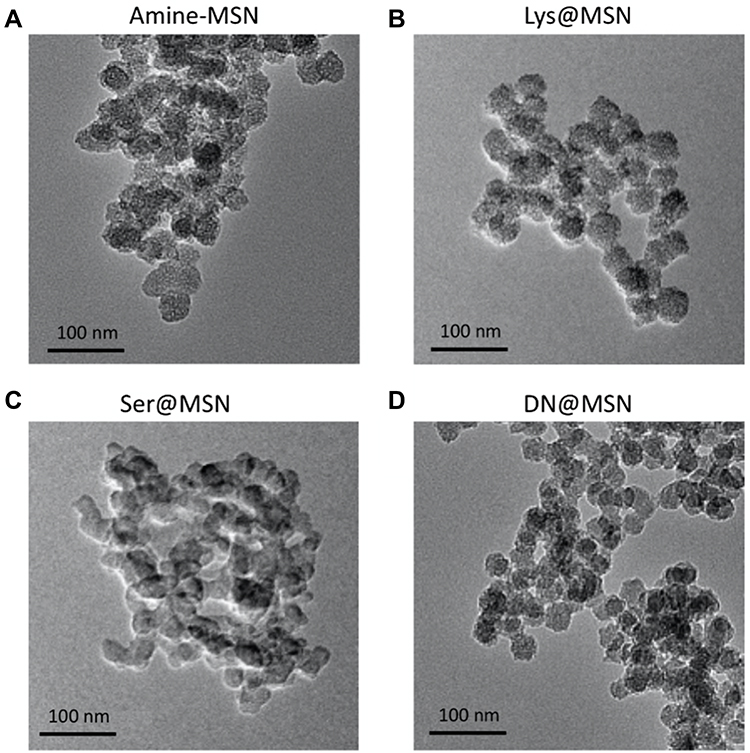 |
Figure 2 Representative TEM images of (A) amine-MSN, (B) Lys@MSN, (C) Ser@MSN and (D) DN@MSN. Scale bars represent 100 nm. |
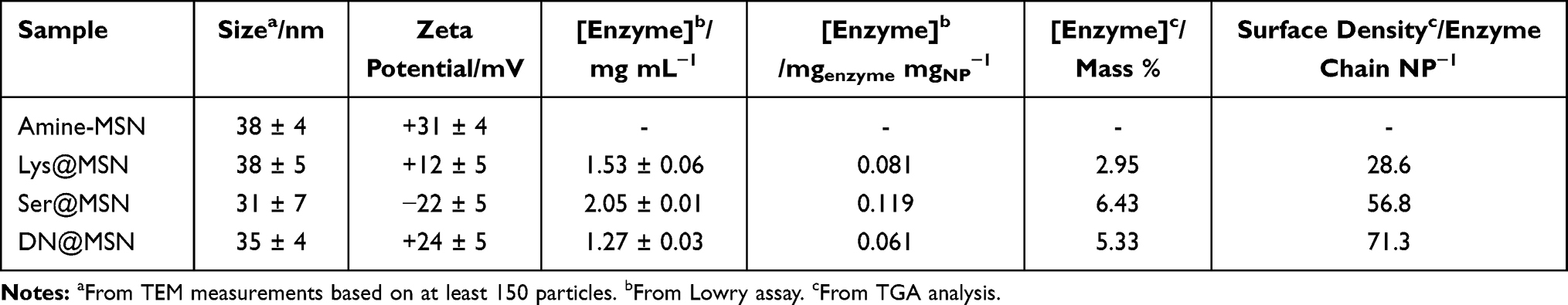 |
Table 1 Physicochemical Characterization of Functionalized MSNs (Amine-MSN, Lys@MSN, Ser@MSN and DN@MSN) and Protein Quantification |
Biofilm Dispersal and Inactivation
New approaches to the treatment of biofilm infections should aim to both kill cells and disperse the EPS. Here we investigated the enzyme lysostaphin (Lys), an excellent anti-staphylococcal agent50,51 that can rapidly lyse S. aureus52 by cleaving a region between the third and fourth glycine residues of the pentaglycine cross bridges of the peptidoglycan in the cell wall regardless of metabolic activity.25 To disperse the biofilm EPS, we investigated serrapeptase and DNase I to target protein and eDNA adhesins present in MRSA biofilms.17,21,53,54 MSSA biofilms, in which polysaccharide intercellular adhesin is the major EPS component, were also investigated for control purposes.
For antimicrobial and biofilm eradication experiments, MSNs were functionalized separately with lysostaphin, serrapeptase and DNase I. MSNs nanoparticles were used as carriers for the enzymes to improve enzyme stability and increase enzyme penetration into the biofilm. Amine-functionalized MSNs have been previously reported to localize around the S. aureus cells, enabling an increase in antimicrobial efficacy.21 When used in combination, the Lys@MSNs would target the bacterial cells causing cell lysis while the DN@MSNs and Ser@MSNs would disrupt the MRSA EPS matrix by targeting two main components, eDNA and proteins. The MICs and MBICs the MSNs, the enzyme-functionalized MSNs and free enzymes were measured against MRSA and MSSA and are shown in Table S1. It was found that the unloaded MSNs had no inherent antibacterial activity over the tested range. From the MIC results, it was evident that the Lys@MSNs showed improved efficacy against S. aureus bacterial cells when compared to the free enzyme. There was a 7.5-fold decrease in the MIC for MRSA bacteria treated with Lys-functionalized MSNs compared to free Lys, while a 5-fold decrease in MBIC was measured. Similar results were observed for MSSA bacteria with a 5-fold decrease in the MIC and a 3.75-fold decrease in the MBIC values with Lys-functionalized MSNs compared to free Lys. It is hypothesized that the overall positive charge of these functionalized MSNs leads to a preferential interaction with the negatively charged bacterial cell wall, thus reducing the overall concentration of enzyme required to inactivate the bacteria. Inhibition of biofilm formation was improved when serrapeptase and DNase I were functionalized to the surface of the MSNs. These results once again confirm that the use of nanoparticles can improve the efficacy of antimicrobial agents, consistent with previous reports.21,55,56 Recent studies have shown that through nanoparticle immobilization, enzymatic stability, specificity, pH and temperature responses can be improved compared to free enzymes.57–59
Biofilms previously grown for 24 h and 48 h were treated with MSNs, the enzyme-functionalized MSNs and the free enzymes for 24 h to observe the impact on biofilm structure and dispersal. Cell viability (Figure S4) and biomass removal (Figure S5) were evaluated. A slight reduction in cell viability and biomass of both S. aureus biofilms was observed when biofilms were treated with MSNs. Although statistical analysis indicates this difference is non-significant (p>0.05), this may result from changes in the metabolic activity (ie a shift of the bacteria to a dormant state) in response to the presence of the MSNs.60 When free Lys and Lys@MSNs are used to treat pre-formed biofilms, it is possible to note a significant reduction in cell viability (Figure S4) when the enzyme is immobilized on the MSNs. For the other two enzymes, the immobilization onto MSNs did not significantly affect their role in reducing bacterial cell viability. Additionally, when biomass reduction (Figure S5) is considered, the enzyme immobilization onto MSNs did not significantly impact their activity.
Since novel studies for biofilm eradication need to focus on killing bacterial cells and dispersing the EPS matrix, to avoid future recolonization, the enzymes and enzyme-functionalized MSNs were, henceforward, used in combination. Lys@MSNs and Ser@MSNs were tested in combination (0.25 and 0.5 mg mL−1) along with Lys@MSNs and DN@MSNs (0.25 and 0.5 mg mL−1). Finally, Lys@MSNs, Ser@MSNs and DN@MSNs were assessed in combination (0.33 mg mL−1). Ser@MSNs and DN@MSNs were not used as a combination as neither showed antibacterial activity throughout the MIC identification assays, and this study aimed to target both the EPS matrix and the bacterial cells. Biofilm samples were also treated with the free enzymes at the concentration they are found in the nanoparticles to observe the role of the immobilization onto MSNs. These concentrations were chosen to ensure that the lysostaphin’s final concentration was above or equal to the MIC for all treatment combinations. Following the 24 h exposure, the antibacterial suspensions were removed from the wells, and the biofilms were washed three times with sterile water to remove unbound MSNs. Cell viability and biomass removal were assessed using a MTT assay (Figure 3) and CV staining (Figure 4). Biofilms were also grown, for 24 or 48 h in glass-bottom 96-well plates treated with the enzyme functionalized MSNs for 24 h and imaged using confocal microscopy (Figures 5, 6, S6 and S7) to confirm the antibacterial activity of the enzyme-functionalized MSNs.
 |
Figure 3 Cell viability of (A) 24 h MRSA, (B) 24 h MSSA, (C) 48 h MRSA and (D) 48 h MSSA biofilms after exposure to enzyme-loaded MSNs (all concentrations in mg mL−1), as determined by MTT assay. Error bars represent standard error of the mean (n = 3). Letters (a, b, c, d) represent a significant difference in antibacterial activity between test conditions, ns represents no significant difference between test conditions, p < 0.05. |
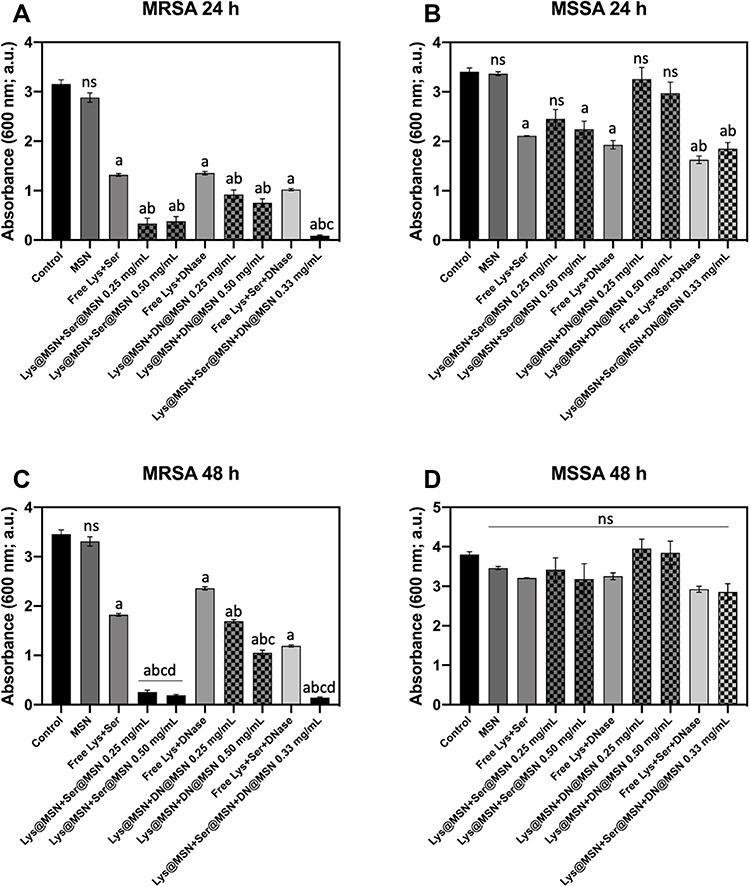 |
Figure 4 Biomass removal of (A) 24 h MRSA, (B) 24 h MSSA (C) 48 h MRSA and (D) 48 h MSSA biofilm cells after exposure enzyme-loaded MSNs (all concentrations in mg mL−1), as determined by CV staining. Error bars represent standard error of the mean (n = 3). Letters (a, b, c, d) represent a significant difference in antibacterial activity between test conditions, ns represents no significant difference between test conditions, p < 0.05. |
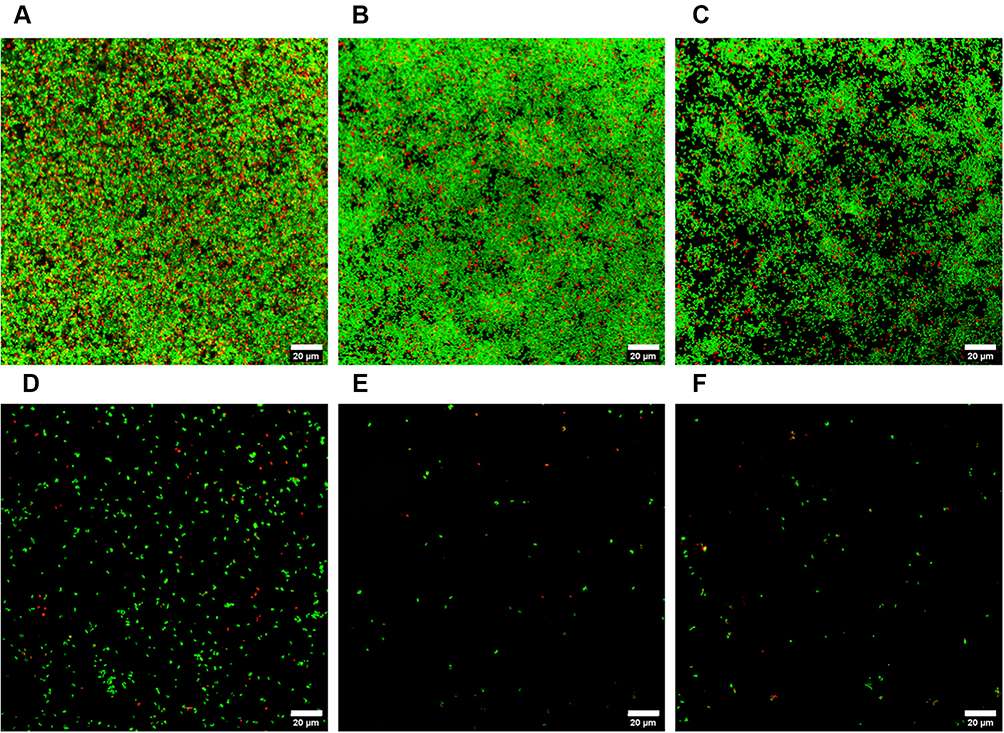 |
Figure 5 CLSM images of 24 h MRSA biofilms after exposure to enzyme-loaded MSNs (all concentrations in mg mL−1), for 24 h. (A) Control, (B) 0.25 (Lys + DNase I) MSNs, (C) 0.5 (Lys + DNase I) MSNs, (D) 0.25 (Lys + Ser) MSNs, (E) 0.5 (Lys + Ser) MSNs and (F) 0.33 (Lys + Ser + DNase I)) MSNs. Live bacterial cells (green) are stained using SYTO 9 while dead cells (red) are stained with Propidium iodide. Scale bars represent 20 μm. |
 |
Figure 6 CLSM images of 24 h MSSA biofilms after exposure to enzyme-loaded MSNs (all concentrations in mg mL−1), for 24 h. (A) Control, (B) 0.25 (Lys + DNase I) MSNs, (C) 0.5 (Lys + DNase I) MSNs, (D) 0.25 (Lys + Ser) MSNs, (E) 0.5 (Lys + Ser) MSNs and (F) 0.33 (Lys + Ser + DNase I) MSNs. Live bacterial cells (green) are stained using SYTO 9 while dead cells (red) are stained with Propidium iodide. Scale bars represent 20 μm.). |
The enzyme functionalized MSNs demonstrated a significant ability to reduce the viability of the bacterial cells within both the MRSA and MSSA biofilms (p < 0.05) (Figure 3). Bacterial cell viability decreased with increasing concentrations of Lys@MSNs. The most significant reduction in cell viability was measured when all three enzyme-functionalized MSNs were combined (Lys + Ser + DNase I at 0.33 mg mL−1) (p < 0.05), indicating that EPS matrix dispersal improved Lys-mediated killing of biofilm cells. Consistent with this, confocal imaging of biofilms after MSN treatment also revealed a visible decrease in viable cells as the concentration of Lys@MSNs increased, with the combination of all three enzymes showing the most potent activity (Figures 5 and 6). For MRSA, near-complete removal of the EPS matrix was observed in both 24 and 48 h biofilms, with few remaining viable cells (Figure 5 and S6). When compared to the activity of the free enzymes, a stark difference is observed. It is clear that when the S. aureus biofilms are treated with the free enzymes, either alone or in combination, the reduction in cellular viability is not as pronounced as when the enzyme-loaded MSNs are utilized (Figure 3 and S4). This may be a result of reduced penetration or inactivation of the enzymes within the EPS matrix.61 It has been previously reported that positively charged MSNs will preferentially localize around the bacterial cells,21 so by covalently binding the lysostaphin enzyme to the amine-MSNs a high local concentration around the bacterial cell was ensured, leading to a higher cell eradication rate.
Comparison of MRSA and MSSA biofilm biomass reduction, as measured by CV staining, revealed significant differences between the two strains (Figure 4). Regardless of biofilm maturity (24 vs 48 h), combining the three enzyme-functionalized MSNs led to an almost complete removal of the MRSA biofilm EPS matrix. In contrast, EPS dispersal in MSSA biofilms was significantly less effective. This was particularly evident when looking at more mature MSSA biofilms (48 h), where there was no significant reduction in EPS biomass compared to the control biofilm (Figure S7). These data are consistent with the different EPS components present in MRSA and MSSA biofilms. Because the MSSA EPS matrix is mainly composed of polysaccharides,21 targeting of eDNA and protein molecules should be less effective than in MRSA biofilms in which the matrix is composed of protein and eDNA adhesins.
Nevertheless, the combination of all three enzyme-functionalized MSNs led to the dispersal of approximately 50% of 24 h MSSA biofilms (p < 0.05) and a 25% reduction in 48 h MSSA biofilms (Figure 4B and D). Furthermore, Lys@MSNs maintained their effectiveness in reducing cellular viability as measured with both the MTT assay (Figure 3A and C) and particularly the CLSM microscopy (Figure 5 and S6). When observing the free enzymes’ results, it is evident that the activity is reduced compared to the loaded MSNs. This is particularly apparent when looking at the EPS removal of MRSA biofilms. In both 24 and 48 h samples a near-complete removal of the EPS matrix is achieved when treated with all three enzyme-loaded MSNs in combination; however, when the biofilms are treated with free enzymes in combination over 30% of the bacterial biomass remains compared to the untreated control (Figure 4). This indicated the vital role of nanoparticle functionalization in improving enzymes’ activity concerning bacterial cell eradication and EPS matrix removal.
S. aureus has been shown to readily enter into a nongrowing, dormant state to evade antibiotic treatment,12,62 during which biosynthetic processes will halt, and common antibiotic agents will not affect. Unlike many conventional antibiotic agents that target biosynthetic processes such as DNA, protein or cell wall synthesis,62,63 lysostaphin can target active cells and lyse dormant or persister cells,12,62 especially those within the biofilm. The removal of the biofilm EPS matrix may further help reduce the occurrence of reinfection or the formation of new infections by opportunistic pathogenic bacteria.64,65 A significant concern when utilizing enzyme-based biofilm treatments is the risk of dispersal of the pathogenic bacteria, spreading the pathogen and causing secondary infections.66 To counteract this issue, enzymatic treatments could be used in combination with systemic antibiotic agents to ensure secondary infections do not occur after biofilm dispersal.13
Conclusions
Infections caused by biofilm-forming S. aureus bacteria constitute a significant burden on health systems worldwide. As resistance to antimicrobial agents grows, there is an urgent requirement for developing novel therapeutic approaches. The use of enzymatic agents that can target both the bacterial cells, leading to cell death and causing the biofilm matrix’s dispersal is a promising solution. By functionalizing the enzymatic agents onto nanoparticles, their efficiency against both strains of S. aureus was significantly improved compared to the free enzymes, with a considerable reduction in MIC and MBIC values. This study showed that the combination of Lys@MSNs, Ser@MSNs and DN@MSNs led to near-complete eradication of the MRSA biofilm, EPS dispersal and a significant cell viability reduction. The enzyme-functionalized enzymes also exhibited significant activity against 24 h MSSA biofilms but were less effective in dispersing 48 h MSSA biofilms. Future studies will need to focus on improving the MSSA polysaccharide EPS matrix’s dispersal, which is not susceptible to serrapeptase or DNase I. This research has shown that the use of enzymatic agents functionalized on nanoparticles holds great potential to enhance current therapeutic approaches for the treatment of S. aureus biofilm-associated infections by increasing the efficacy of the enzymatic agent. Future work will need to examine the efficacy and safety in animal models.
Acknowledgments
We thank Science Foundation Ireland (SFI) for the support of this research under grant number 15/IA/3008. We also would like to thank Prof. Dr Kenneth Dawson from the Centre for BioNano Interactions (CBNI) UCD for the Zetasizer equipment.
Disclosure
The authors reports no conflicts of interest in this work.
References
1. Wertheim HFL, Melles DC, Vos MC, et al. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. 2005;5(12):751–762. doi:10.1016/s1473-3099(05)70295-4
2. Gorwitz RJ, Kruszon-Moran D, McAllister SK, et al. Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States, 2001-2004. J Infect Dis. 2008;197(9):1226–1234. doi:10.1086/533494
3. Kim W, Hendricks GL, Tori K, Fuchs BB, Mylonakis E. strategies against methicillin-resistant Staphylococcus aureus persisters. Future Med Chem. 2018;10(7):779–794. doi:10.4155/fmc-2017-0199
4. Hall-Stoodley L, Stoodley P, Kathju S, et al. Towards diagnostic guidelines for biofilm-associated infections. FEMS Immunol Med Microbiol. 2012;65(2):127–145. doi:10.1111/j.1574-695X.2012.00968.x
5. Tong SYC, Davis JS, Eichenberger E, Holland TL, Fowler VG. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. 2015;28(3):603–661. doi:10.1128/cmr.00134-14
6. Chambers HF, Deleo FR. Waves of resistance: staphylococcus aureus in the antibiotic era. Nat Rev Microbiol. 2009;7(9):629–641. doi:10.1038/nrmicro2200
7. Ambrosch A, Haefner S, Jude E, Lobmann R. Diabetic foot infections: microbiological aspects, current and future antibiotic therapy focusing on methicillin-resistant Staphylococcus aureus. Int Wound J. 2011;8(6):567–577. doi:10.1111/j.1742-481X.2011.00849.x
8. Morgan M. Treatment of MRSA soft tissue infections: an overview. Injury Int J Care Injured. 2011;42:S11–S17. doi:10.1016/s0020-1383(11)70127-9
9. Tuchscherr L, Kreis CA, Hoerr V, et al. Staphylococcus aureus develops increased resistance to antibiotics by forming dynamic small colony variants during chronic osteomyelitis. J Antimicrob Chemother. 2016;71(2):438–448. doi:10.1093/jac/dkv371
10. Allison KR, Brynildsen MP, Collins JJ. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature. 2011;473(7346):
11. Conlon BP, Nakayasu ES, Fleck LE, et al. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature. 2013;503(7476):
12. Helaine S, Kugelberg E. Bacterial persisters: formation, eradication, and experimental systems. Trends Microbiol. 2014;22(7):417–424. doi:10.1016/j.tim.2014.03.008
13. Hogan S, Zapotoczna M, Stevens NT, Humphreys H, O’Gara JP, O’Neill E. Potential use of targeted enzymatic agents in the treatment of Staphylococcus aureus biofilm-related infections. J Hosp Infect. 2017;96(2):177–182. doi:10.1016/j.jhin.2017.02.008
14. Watters CM, Burton T, Kirui DK, Millenbaugh NJ. Enzymatic degradation of in vitro Staphylococcus aureus biofilms supplemented with human plasma. Infect Drug Resist. 2016;9:71–78. doi:10.2147/idr.s103101
15. McCarthy H, Rudkin JK, Black NS, Gallagher L, O’Neill E, O’Gara JP. Methicillin resistance and the biofilm phenotype in Staphylococcus aureus. Front Cell Infect Microbiol. 2015;5:
16. O’Neill E, Pozzi C, Houston P, et al. Association between methicillin susceptibility and biofilm regulation in Staphylococcus aureus isolates from device-related infections. J Clin Microbiol. 2007;45(5):1379–1388. doi:10.1128/jcm.02280-06
17. O’Neill E, Pozzi C, Houston P, et al. A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB. J Bacteriol. 2008;190(11):3835–3850. doi:10.1128/jb.00167-08
18. Mishra B, Patel BB, Tiwari S. Colloidal nanocarriers: a review on formulation technology, types and applications toward targeted drug delivery. Nanomed Nanotechnol Biol Med. 2010;6(1):9–24. doi:10.1016/j.nano.2009.04.008
19. Wu SH, Hung Y, Mou CY. Mesoporous silica nanoparticles as nanocarriers. Chem Commun. 2011;47(36):9972–9985. doi:10.1039/c1cc11760b
20. Majumder J, Taratula O, Minko T. Nanocarrier-based systems for targeted and site specific therapeutic delivery. Adv Drug Deliv Rev. 2019;144:57–77. doi:10.1016/j.addr.2019.07.010
21. Fulaz S, Devlin H, Vitale S, Quinn L, O’Gara JP, Casey E. Tailoring nanoparticle-biofilm interactions to increase the efficacy of antimicrobial agents against Staphylococcus aureus. Int J Nanomedicine. 2020;15:4779–4791. doi:10.2147/ijn.s256227
22. Qayyum S, Khan AU. Nanoparticles vs. biofilms: a battle against another paradigm of antibiotic resistance. Medchemcomm. 2016;7(8):1479–1498. doi:10.1039/c6md00124f
23. Fulaz S, Vitale S, Quinn L, Casey E. Nanoparticle-biofilm interactions: the role of the EPS Matrix. Trends Microbiol. 2019;27(11):915–926. doi:10.1016/j.tim.2019.07.004
24. Raulinaitis V, Tossavainen H, Aitio O, et al. Identification and structural characterization of LytU, a unique peptidoglycan endopeptidase from the lysostaphin family. Sci Rep. 2017;7:6020. doi:10.1038/s41598-017-06135-w
25. Nahar S, Mizan MFR, Ha AJW, Ha SD. Advances and future prospects of enzyme-based biofilm prevention approaches in the food industry. Comprehensive Rev Food Sci Food Saf. 2018;17(6):1484–1502. doi:10.1111/1541-4337.12382
26. Yang XY, Li CR, Lou RH, et al. In vitro activity of recombinant lysostaphin against Staphylococcus aureus isolates from hospitals in Beijing, China. J Med Microbiol. 2007;56(1):71–76. doi:10.1099/jmm.0.46788-0
27. Kankala RK, Wang SB, Chen AZ. Nanoarchitecting Hierarchical Mesoporous Siliceous Frameworks: a New Way Forward. Iscience. 2020;23:
28. Kankala RK, Zhang HB, Liu CG, et al. Metal species-encapsulated mesoporous silica nanoparticles: current advancements and latest breakthroughs. Adv Funct Mater. 2019;29:
29. Kankala RK, Han YH, Na J, et al. Nanoarchitectured structure and surface biofunctionality of mesoporous silica nanoparticles. Adv Mater. 2020;32:
30. Kuthati Y, Kankala RK, Lin SX, Weng CF, Lee CH. pH-triggered controllable release of Silver-Indole-3 Acetic Acid Complexes from Mesoporous Silica Nanoparticles (IBN-4) for effectively killing malignant bacteria. Mol Pharm. 2015;12(7):2289–2304. doi:10.1021/mp500836w
31. Bhaysar D, Patel V, Sawant K. Systematic investigation of in vitro and in vivo safety, toxicity and degradation of mesoporous silica nanoparticles synthesized using commercial sodium silicate. Microporous Mesoporous Mater. 2019;284:343–352. doi:10.1016/j.micromeso.2019.04.050
32. Xu C, Yu MH, Noonan O, et al. Core-cone structured monodispersed mesoporous silica nanoparticles with ultra-large cavity for protein delivery. Small. 2015;11(44):5949–5955. doi:10.1002/smll.201501449
33. Xu C, Niu YT, Popat A, Jambhrunkar S, Karmakar S, Yu CZ. Rod-like mesoporous silica nanoparticles with rough surfaces for enhanced cellular delivery. J Mater Chem B. 2014;2(3):253–256. doi:10.1039/c3tb21431a
34. Chen Y, Chen HR, Shi JL. In vivo bio-safety evaluations and diagnostic/therapeutic applications of chemically designed mesoporous silica nanoparticles. Adv Mater. 2013;25(23):3144–3176. doi:10.1002/adma.201205292
35. Fulaz S, Hiebner D, Barros CHN, et al. Ratiometric Imaging of the in Situ pH distribution of biofilms by use of fluorescent mesoporous silica nanosensors. ACS Appl Mater Interfaces. 2019;11(36):32679–32688. doi:10.1021/acsami.9b09978
36. Nour El-Din HT, Elhosseiny NM, El-Gendy MA, Mahmoud AA, Hussein MMM, Attia AS. A rapid lysostaphin production approach and a convenient novel lysostaphin loaded nano-emulgel; as a sustainable low-cost methicillin-resistant staphylococcus aureus combating platform. Biomolecules. 2020;10(3):
37. Koo H, Allan RN, Howlin RP, Stoodley P, Hall-Stoodley L. Targeting microbial biofilms: current and prospective therapeutic strategies. Nat Rev Microbiol. 2017;15(12):740–755. doi:10.1038/nrmicro.2017.99
38. Waryah CB, Wells K, Ulluwishewa D, et al. In vitro antimicrobial efficacy of tobramycin against staphylococcus aureus biofilms in combination with or without DNase I and/or Dispersin B: a Preliminary Investigation. Microbial Drug Resist. 2017;23(3):384–390. doi:10.1089/mdr.2016.0100
39. Zapotoczna M, McCarthy H, Rudkin JK, O’Gara JP, O’Neill E. An essential role for coagulase in staphylococcus aureus biofilm development reveals new therapeutic possibilities for device-related infections. J Infect Dis. 2015;212(12):1883–1893. doi:10.1093/infdis/jiv319
40. Mlynek KD, Bulock LL, Stone CJ, et al. Genetic and biochemical analysis of CodY-mediated cell aggregation in staphylococcus aureus reveals an interaction between Extracellular DNA and polysaccharide in the extracellular matrix. J Bacteriol. 2020;202(8):
41. Metkar SK, Girigoswami A, Murugesan R, Girigoswami K. In vitro and in vivo insulin amyloid degradation mediated by Serratiopeptidase. Mater Sci Eng C Mater Biol Appl. 2017;70:728–735. doi:10.1016/j.msec.2016.09.049
42. Tiwari M. The role of serratiopeptidase in the resolution of inflammation. Asian J Pharm Sci. 2017;12(3):209–215. doi:10.1016/j.ajps.2017.01.003
43. Srivastava V, Mishra S, Chaudhuri TK. Enhanced production of recombinant serratiopeptidase in Escherichia coli and its characterization as a potential biosimilar to native biotherapeutic counterpart. Microb Cell Fact. 2019;18:
44. Mecikoglu M, Saygi B, Yildirim Y, Karadag-Saygi E, Ramadan SS, Esemenli T. The effect of proteolytic enzyme serratiopeptidase in the treatment of experimental implant-related infection. J Bone Joint Surg Am. 2006;88A(6):1208–1214. doi:10.2106/jbjs.e.00007
45. Artini M, Scoarughi GL, Papa R, et al. A new anti-infective strategy to reduce adhesion-mediated virulence in staphylococcus aureus affecting surface proteins. Int J Immunopathol Pharmacol. 2011;24(3):661–672. doi:10.1177/039463201102400312
46. Schmid B, Schindelin J, Cardona A, Longair M, Heisenberg M. A high-level 3D visualization API for Java and ImageJ. BMC Bioinform. 2010;11:
47. Walencka E, Rozalska S, Sadowska B, Rozalska B. The influence of Lactobacillus acidophilus-derived surfactants on staphylococcal adhesion and biofilm formation. Folia Microbiol (Praha). 2008;53(1):61–66. doi:10.1007/s12223-008-0009-y
48. Wu YT, Zhu H, Willcox M, Stapleton F. Removal of biofilm from contact lens storage cases. Invest Ophthalmol Vis Sci. 2010;51(12):6329–6333. doi:10.1167/iovs.10-5796
49. Foschiera JL, Pizzolato TM, Benvenutti EV. FTIR thermal analysis on organofunctionalized silica gel. J Braz Chem Soc. 2001;12(2):159–164. doi:10.1590/s0103-50532001000200006
50. Szweda P, Schielmann M, Kotlowski R, Gorczyca G, Zalewska M, Milewski S. Peptidoglycan hydrolases-potential weapons against Staphylococcus aureus. Appl Microbiol Biotechnol. 2012;96(5):1157–1174. doi:10.1007/s00253-012-4484-3
51. Nithya S, Nimal TR, Baranwal G, et al. Preparation, characterization and efficacy of lysostaphin-chitosan gel against Staphylococcus aureus. Int J Biol Macromol. 2018;110:157–166. doi:10.1016/j.ijbiomac.2018.01.083
52. Lu JZQ, Fujiwara T, Komatsuzawa H, Sugai M, Sakon J. Cell wall-targeting domain of glycylglycine endopeptidase distinguishes among peptidoglycan cross-bridges. J Biol Chem. 2006;281(1):549–558. doi:10.1074/jbc.M509691200
53. O’Gara JP. ica and beyond: biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol Lett. 2007;270(2):179–188. doi:10.1111/j.1574-6968.2007.00688.x
54. Zapotoczna M, O’Neill E, O’Gara JP. Untangling the Diverse and redundant mechanisms of staphylococcus aureus biofilm formation. PLoS Pathog. 2016;12(7):
55. Lister JL, Horswill AR. Staphylococcus aureus biofilms: recent developments in biofilm dispersal. Front Cell Infect Microbiol. 2014;4:
56. Weldrick PJ, Hardman MJ, Paunov VN. Enhanced clearing of wound-related pathogenic bacterial biofilms using protease-functionalized antibiotic nanocarriers. ACS Appl Mater Interfaces. 2019;11(47):43902–43919. doi:10.1021/acsami.9b16119
57. Gunavadhi M. Nano-biocatalyst: bi-functionalization of protease and amylase on copper oxide nanoparticles. 2021;197.
58. Mateo C, Palomo JM, Fernandez-Lorente G, Guisan JM, Fernandez-Lafuente R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzyme Microb Technol. 2007;40(6):1451–1463. doi:10.1016/j.enzmictec.2007.01.018
59. Wu JC, Lee SS, Mahmood MMB, Chow Y, Talukder MMR, Choi WJ. Enhanced activity and stability of immobilized lipases by treatment with polar solvents prior to lyophilization. J Mol Catalysis B Enzymatic. 2007;45(3–4):108–112. doi:10.1016/j.molcatb.2007.01.003
60. Le TTA, Thuptimdang P, McEvoy J, Khan E. Phage shock protein and gene responses of Escherichia coli exposed to carbon nanotubes. Chemosphere. 2019;224:461–469. doi:10.1016/j.chemosphere.2019.02.159
61. Singh S, Singh SK, Chowdhury I, Singh R. Understanding the mechanism of bacterial biofilms resistance to antimicrobial agents. Open Microbiol J. 2017;11:53–62.
62. Lewis K. Persister Cells. In: Gottesman S, Harwood CS, editors. Annual Review of Microbiology, Vol 64, 2010. Annual Reviews. 2010:357–372.
63. Lewis K. Platforms for antibiotic discovery. Nat Rev Drug Discov. 2013;12(5):371–387. doi:10.1038/nrd3975
64. Flemming HC, Wingender J, Szewzyk U, Steinberg P, Rice SA, Kjelleberg S. Biofilms: an emergent form of bacterial life. Nat Rev Microbiol. 2016;14(9):563–575. doi:10.1038/nrmicro.2016.94
65. Pinto RM, Soares FA, Reis S, Nunes C, Van Dijck P. Innovative strategies toward the disassembly of the eps matrix in bacterial biofilms. Front Microbiol. 2020;11:
66. Fleming D, Rumbaugh K. The consequences of biofilm dispersal on the Host. Sci Rep. 2018;
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.