Back to Journals » International Journal of Nanomedicine » Volume 16
Enhancing of Nanocatalyst-Driven Chemodynaminc Therapy for Endometrial Cancer Cells Through Inhibition of PINK1/Parkin-Mediated Mitophagy
Authors Gong X, Pu X ![]() , Wang J, Yang L, Cui Y
, Wang J, Yang L, Cui Y ![]() , Li L
, Li L ![]() , Sun X, Liu J, Bai J, Wang Y
, Sun X, Liu J, Bai J, Wang Y
Received 14 July 2021
Accepted for publication 13 September 2021
Published 29 September 2021 Volume 2021:16 Pages 6661—6679
DOI https://doi.org/10.2147/IJN.S329341
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Xiaodi Gong,1,* Xin Pu,2,* Jing Wang,1 Linlin Yang,1 Yunxia Cui,1 Lijuan Li,1 Xiao Sun,1 Jichang Liu,2 Jingfeng Bai,3 Yudong Wang1,4
1Department of Gynecologic Oncology, The International Peace Maternity and Child Health Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, People’s Republic of China; 2State Key Laboratory of Chemical Engineering, School of Chemical Engineering, East China University of Science and Technology, Shanghai, People’s Republic of China; 3Biomedical Instrument Institute, School of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai, People’s Republic of China; 4Shanghai Municipal Key Clinical Specialty, Female Tumor Reproductive Specialty, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yudong Wang
Department of Gynecologic Oncology, The International Peace Maternity and Child Health Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200030, People’s Republic of China
Tel +86-21-64070434-18602
Email [email protected]
Jingfeng Bai
Biomedical Instrument Institute, School of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai, 200030, People’s Republic of China
Tel +86-21-62933209-601
Email [email protected]
Purpose: Iron-based nanomaterials have recently been developed as excellent and potent Fenton reagents to reactive oxygen species (ROS) during chemodynamic therapy (CDT). The performance of the materials, however, can be impaired by the intrinsic antioxidant defense mechanism in organisms, such as autophagy.
Methods: The nanoscale metal-organic frameworks (nMOFs), nMIL-100 (Fe), were exploited and characterized. Also, the Fenton-like catalytic characteristics, anti-endometrial cancer (EC) effects and potential mechanisms of nMIL-100 (Fe) nanoparticles were investigated in vitro.
Results: The synthesized nMIL-100 (Fe) nanocatalyst catalyzed hydroxyl radicals (·OH) production in the presence of hydrogen peroxide (H2O2) and simultaneously depleted intracellular glutathione (GSH). Combining with H2O2, nMIL-100 (Fe) nanoparticles exhibited enhanced cytotoxicity for EC cells, especially for progesterone treatment-insensitive KLE cells, probably due to relatively lower expression of the catalase gene. The accumulated ·OH initiated PTEN induced putative kinase 1 (PINK1)/E3 ubiquitin-protein ligase Parkin-mediated cytoprotective mitophagy in turn to partially rescue ·OH-induced cell apoptosis. Furthermore, both pretreatments of EC cells with siRNA-mediated Parkin knockdown and Mdivi-1 (a mitophagy inhibitor) addition were sufficient to ensure nMIL-100 (Fe) synergizing with H2O2-induced oxidative damages.
Conclusion: These results suggest that the degree of mitophagy should be taken into consideration to optimize therapeutic efficiency when developing ROS based-CDT for EC cancer therapies. Therefore, a nMIL-100 (Fe)-guided, elevated ROS and overwhelmed mitophagy-mediated therapeutic strategy may have greater promise for EC therapy compared with current treatment modalities.
Keywords: mitophagy, chemodynamic therapy, metal-organic frameworks, nanocatalyst, endometrial cancer
Graphical Abstract:

Introduction
Endometrial cancer (EC) is the most common gynecologic malignancy of the female genital tract in developed countries. The incidence of EC increase by approximately 1% every year as individual ages potentially due to the declining fertility rate and increasing body weight.1 It was estimated that there will be 66, 570 new uterine cancer cases, with 12,940 deaths eventually succumb to this disease in the United States in 2021.2 However, since the mid-1970s, survival rates of most common cancer types except uterine cancers have been improved,3 in which EC-related mortality has increased from 0.3% (1997–2008) to 1.9% (2008–2018), being 2-fold higher of that in occurrence.4,5 Although hormonal therapy using progestational agents has demonstrated promising clinical outcomes, with ~68–85.3% of response rates, particularly in patients with estrogen receptor (ER)/progesterone receptor (PR)-positive disease.6 Unfortunately, 33.1–49.8% of patients who were initially sensitive to progesterone gradually become resistant to that, which lead to the disease recurrence and progression.7 Thus, many patients, especially those of childbearing age, eventually will have to undergo hysterectomies and permanently be deprived of reproductive function. Despite the increased number of non-endometrioid carcinoma subtype may contribute to the worse prognosis,8 an undeniable fact is that over the past decades little treatment advances have been substantially achieved.9 Therefore, novel therapeutic strategies are urgently needed to improve treatment outcomes.
Oxygen-containing chemical species with high reactivity are termed to ROS mainly including singlet oxygen (1O2), superoxide radicals (O2•−), ·OH and peroxides (O22−).10 The regulation of intracellular ROS levels is fundamentally important to maintain cellular homeostasis.11 Actually, ROS is a double-edged sword. At lower levels, ROS serves as second messengers in modulating various physiological processes and stimulating stress-response signaling pathways.12 However, excessive ROS activates the higher level of oxidative stress and further induces cytotoxicity through triggering peroxidation of protein, lipids and DNA,13 which further reinforces efficacy of chemotherapy and radiotherapy to achieve excellent synergistic antitumor outcome.14
Recently, multiple nanoplatforms incorporating endogenous and exogenous stimuli-responsive ROS production have been applied to cancer therapy, such as photodynamic therapy (PDT), sonodynamic therapy (SDT) and CDT, have been designed and developed. Among those, CDT has been a spotlight of investigations. Specifically, based on the intrinsic biochemical properties of special tumor microenvironment (TME) (e.g. weak acidity,15 high H2O2 level16 and elevated GSH concentration17), CDT could convert intra-tumoral H2O2 into higher oxidative ·OH against tumors by triggering and facilitating various catalytic reactions such as Fenton or Fenton-like chemistry in-situ TME. The targeted therapy can effectively avoid the tissue penetration of ROS generated by exogenous triggering mechanism (eg, light, ultrasound, and X-ray). Yet few reports in this attractive and feasible CDT strategy for EC have been performed.
Spontaneous Fenton reactions occur naturally in organisms and play a significant role in sustaining ROS steady-state without any therapeutic benefits. This is primarily due to ROS accumulation below the toxicity threshold. Thus, many pH-sensitive self-catalyzed peroxidase mimics (represented by iron-based nanomaterial) as exogenous Fenton reagents have been introduced into acidic-TEM to trigger excessive cytotoxic ·OH generation for cancer therapy.18 Fe3O4 is one of the most prominent nanocatalysts with inherent horseradish peroxidase (HRP)-like activity and can efficiently catalyze substrate oxidation to occur chromogenic reaction as that of natural enzymes in the presence of H2O2.19–22 Iron oxide nanoparticles (IONPs) have a pH-sensitive dual enzyme-like potential, which could exert peroxidase-like activity at the mildly acidic pH of TME, and also exhibit catalase-like activity at the neutral pH in normal tissues. This versatile nanozyme displayed highly selective antitumor implication.23 Moreover, a FeOx-engineered hollow mesoporous silica nanoparticles (HMSNs) showed both tumor-specific lysosomal ·OH generation and high biodegradability by an “iron-extraction” approach in a high-protein tumor environment.24,25
In fact, the endogenous redox-defense mechanisms dedicated to scavenging oxidative threat will be correspondingly activated to reach a new redox equilibrium state.12 It is commonly accepted that ROS stress can induce cellular autophagy (specifically refer to Macroautophagy) in a direct or indirect manner (eg, Adenosine Monophosphate Activated Protein Kinase: AMPK, transcription factors as nuclear factor kappa-B: NF-κB),26–28 which is an evolutionarily highly conserved catabolic process and has been used to describe to be intracellular self-digestion processes since 1960, in which self-impaired, denatured and aging macromolecules as well as organelles undergo a lysosomes-mediated degradation.29 As a result, aberrant autophagy will elicit the onset and progression of diabetes, dysfunctional immunity, cancer and neurodegenerative disorders.30–32 In established tumors, it exerts the dual role in redox regulation associated with not only the duration and dose of ROS exposure but also the cell types. Moderate ROS-induced autophagy allows cells to escape from oxidative damages, whereas excessive or persistent ROS will cause autophagic cell death.33,34 Notably, this phenomenon occurred only in cancer cells, not untransformed counterparts.35 However, the impact of Fenton nanocatalyst-induced ROS cytotoxicity on EC and the role of mitophagy in CDT have rarely been reported.
Herein, the nMOFs, nMIL-100 (Fe), with highly available Fe active sites was firstly selected as Fenton nanocatalyst to escalate intracellular high reactive ·OH stress in EC cells (Scheme 1). The inherent peroxidase-like activity of nMIL-100 (Fe) and the inductive effect on autophagy, especially mitophagy, were adequately evaluated and documented in the presence of H2O2. Moreover, further studies had demonstrated that nMIL-100 (Fe)-amplified highly toxic ·OH could initiate classical PINK1/Parkin pathway mediated protective mitophagy. In turn, mitophagy acted as an obstacle to the full effect of ·OH oxidative damage. Our results suggested that mitophagy was endogenous negative feedback for nMIL-100 (Fe) nanocatalyst-driven CDT strategy. It is thus necessary to timely counteract passive upregulation of mitophagy to ensure efficient antineoplastic treatments for EC cancer.
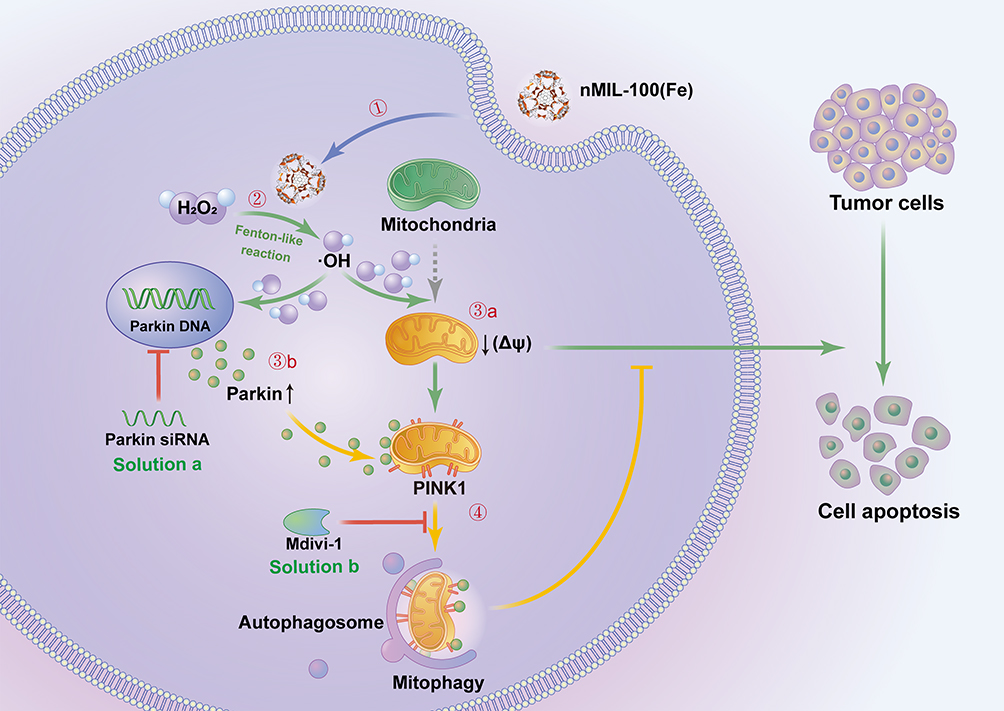 |
Scheme 1 Illustration of nMIL-100 (Fe) nanocatalyst for tumor chemodynamic therapy and the underlying mechanism of mitophagy. The nMIL-100 (Fe) NPs were internalized into the cytoplasm of EC cells (Step 1) and acted as Fenton reagents catalyzed H2O2 to generate highly oxidative ·OH in slightly acid TME (Step 2). Owing to the excessive toxic ·OH around normal mitochondria, the depolarization of mitochondrial membrane potential (Δψ) was subsequently initiated, which further induced mitochondrial oxidative damages and ultimate cell apoptosis (Step 3a). During this process the loss of Δψ forced PINK1 to be stagnated on the outer membrane of mitochondria (OMM). Step 3b) PINK1 further recruited the upregulated cytosolic Parkin in response to ·OH pressure and eventually activated mitophagy for cellular rejuvenation (Step 4). As a solution, suppressing mitophagy using both Parkin siRNA transfection and Mdivi-1 addition (a mitophagy inhibitor) observably amplified ·OH therapeutic effects and exacerbated cell damage. |
Materials and Methods
Chemicals
Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) was from GE Healthcare Life Sciences HyClone Laboratories (Beijing, China), Epithelial Cell Medium (ECM) from ScienCell (Carlsbad, CA, USA). Fetal bovine serum (FBS), penicillin–streptomycin and Lipofectamine™ 3000 Transfection Reagent were from Thermo Fisher Scientific (Beijing, China). H2O2 was obtained from Sinopharm Chemical Reagents Co., Ltd. Enhanced Cell Counting Kit-8 (C0042), Lyso-Tracker Red (C1046), Mito-Tracker Red CMXRos (C1035), Antifade Mounting Medium with DAPI (P0131), Calcein AM Cell Fluorescence Counting Kit (C2013FS), Reactive Oxygen Species Assay Kit (S0033S) and BeyoClick™ EdU-594 (C00788L) were from Beyotime Biotechnology (Jiangsu, China). Micro Reduced Glutathione (GSH) Assay Kit (BC1175) was from Solarbio Life Science (Beijing, China). Iron (III) chloride hexahydrate (FeCl3·6H2O, AR), 1,3,5-Benzenetricarboxylic acid (H3BTC, 99%), 2,2ʹ-Azino-bis (3-ethylbenzothiazoline −6-sulfonic Acid Ammonium Salt) (ABTS), 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) and 3-Methyladenine (3-MA) were purchased from Aladdin (Shanghai, China). Mitochondrial division inhibitor 1 (Mdivi-1) was from MedChemExpress. JC-1 MitoMP Detection Kit (MT09) was obtained from Dojindo Laboratories (Shanghai, China). 3,3ʹ, 5.5ʹ-Tetramethyl benzidine (TMB), Glutathione reduced (GSH), 1.10-Phenanthroline monohydrate, Fluorescein isothiocyanate, Isomer I (FITC) (A642107-0500), Cytoplasmic and Mitochondrial Protein Extraction Kit (C500051-0050) were from Sangon Biotech (Shanghai, China). Antibodies were purchased from Cell Signaling Technology as follows: anti-rabbit IgG, HRP-linked antibody (#7074S), anti-mouse IgG, HRP-linked antibody (#7076S), rabbit anti-Parkin (#4221S), rabbit anti-SQSTM1/P62 (#8025T), rabbit anti-Cleaved Caspase-3 (9662S), rabbit anti-Cleaved PARP (5625T), rabbit anti-Atg7 (#383498), rabbit anti-LC3B (#382687), rabbit anti-COX IV (250135), rabbit anti-Mitofusin1 (Mfn1, 509880), rabbit anti-Mitofusin2 (Mfn2, 340604) and rabbit anti-PINK1 (385376) were from Zen BioScience (Chengdu, China). Rabbit anti-Miro1 (A5838) was from ABclonal (shanghai, China). Hoechst 33342-PropidiμMIodide (PI), HRP-conjugated mouse anti-GAPDH (#30203ES10) were from Yeasen Biotech (Shanghai, China).
Preparations of nMIL-100 (Fe)
Synthesis of nMIL-100 (Fe) was adapted from a previously protocol described in the literature with minor modification. In a typical procedure, iron (III) nitrate nonahydrate (484 mg, 1.2 mmol) and 1,3,5-benzenetricarboxylic acid (210 mg, 1.0 mmol) were dissolved in 5 mL of deionized water (DI) under stirring at room temperature. Then, the as-prepared solution was transferred into a Teflon-lined stainless-steel autoclave, which was subsequently sealed and maintained at 180 °C for 12 h. The autoclave was then cooled down to room temperature naturally. The yellow precipitate was collected by filtration and washed with DI and menthol three times. Finally, the nMIL-100 (Fe) was recovered by further drying for 12 h in a vacuum oven at 60 °C.
Characterizations
SEM measurements and atomic EDS mapping were carried out on a Gemini SEM 500 working at 15 kV acceleration voltage.
X-ray diffraction (XRD) of the samples was performed on a Rigaku-3014 diffractometer with a monochromator using Cu Kα (λ=0.154 nm) radiation. The diffractograms were recorded in the 2θ range 3–50° in steps of 0.04° with a scan rate of 2° per min. The UV−vis diffuse reflectance spectrum (UV−vis DRS) was obtained with a UV−vis spectrophotometer (Varian Cary 500).
The surface element states were determined by X-ray photoelectron spectroscopy (XPS, Thermo Scientific EscaLab250Xi) with Al Kα radiation (1486.6 eV) and all binding energies were corrected by the C 1s line at 284.6 eV.
Catalytic Activity Measurement
In order to investigate the catalytic activity for TMB oxidation, the absorbance changes at 652 nm were measured under various experimental conditions including different concentration of H2O2 (25, 50, and 100 μM), nMIL-100 (Fe) (12.5, 25, 50 and 100 μg/mL), as well different pH values (PH=5.0, 6.5, 7.4). Among these, 100μg/mL nMIL-100 (Fe) and 50 μM H2O2 in deionized (DI) water (PH=6.0) followed by the addition of 1mM TMB was a major catalytic reaction system in follow-up experiments.
ESR Detection
A 20 mM DMPO, a spin trapping probe, was added into DI water (pH=6.0) containing 100 μg/mL nMIL-100 (Fe) and 50 μM H2O2 at ambient temperature for 1 min. The reaction solution was immediately transferred to a quartz capillary tube. The ESR spectra was recorded on a Bruker EMX-8/2.7 Electron Spin Resonance Spectrometer.
Reaction Kinetic Measurement
To assess the peroxidase-like activity, the kinetic analyses were performed at a fixed concentration of nMIL-100 (Fe) (100 μg/mL) using H2O2 (100 μM) or TMB (1 mM) as the substrate followed by adding varying concentration of TMB (0.1 to 1.6 mM) or H2O2 (0.05 to 0.8 mM), respectively. The absorbance changes of the reaction solution at 652 nm were read by a UV spectrophotometer in a kinetic mode. The measured absorbance values were then transformed into the corresponding concentration of oxidized TMB according to the Beer–Lambert Law:
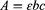
In which A is the absorption value at 652 nm, b stands for the optical length (1.0 cm), ε represents the molar absorption coefficient of oxidized TMB (39,000 M−1cm−1) and c indicates concentration for oxidized TMB.
The kinetic curve of nMIL-100 (Fe) was obtained based on the Michaelis−Menten equation:
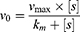
In which v0 is the initial velocity, Vmax is the maximal rate of the reaction, [S] represents the substrate concentration, and Km denotes the Michaelis−Menten constant. Vmax and Km were calculated via Lineweaver–Burk plots on the basis of the following equation:
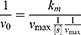
GSH-Depletion Ability
In order to address the consumption of GSH, the absorbance variations of the solution including 50 μM H2O2, 0.2 mM Phe and 100 μg/mL nMIL-100 (Fe) with/without GSH (10 mM) at 525 nm were detected by UV−vis spectrophotometer. Moreover, to investigate GSH-depletion ability of oxidized TMB, the UV absorbance changes at 625 nm of the reaction solution containing 50 μM H2O2, 1 mM TMB and 100 μg/mL nMIL-100 (Fe) in the presence/absence of GSH (10 mM) were immediately monitored. Subsequently, quantitative analysis of intracellular GSH level after nMIL-100 (Fe) treatment was tested by reduced GSH assay kit.
Cell Lines and Culture
Human endometrial carcinoma cancer cell lines KLE and Ishikawa were obtained from the American Type Culture Collection (ATCC) and incubated in DMEM-F12 medium added with 10% FBS, 1% penicillin and streptomycin at 37 °C in a 5% CO2 atmosphere. The cells were pretreated with 3-MA and Mdivi-1 for 2 h.
Human endometrial epithelial cells (hEEC; MZ-2408) were purchased from Mingzhou Biotechnology Co., Ltd. (Ningbo, China). The cells were maintained in ECM medium supplemented with 2% FBS, 1% epithelial cell growth supplement and 1% penicillin-streptomycin at 37 °C in an incubator with 5% CO2. The cell viability of hEEC treated with nMIL-100 (Fe) or H2O2 for 24 h was assessed, respectively.
Cell Uptake
To perform endocytosis analysis of nMIL-100 (Fe), KLE cells were cultured in 6-well plates at a density of 2×105 cells well−1. After 24 h, 100 μg/mL FITC-nMIL-100 (Fe) diluted with complete cell medium was introduced into every well for an additional 6 h. Thereafter the cells were stained with Lyso-Tracker Red dye, shielded from light for 30 min at 37 °C, and then washed with PBS three times. Finally, DAPI was added into each well for nuclear staining in the dark for 10 min. Subsequently, the cells were immediately monitored using fluorescence microscopy and red/green fluorescence co-localization was analyzed by the ImageJ 1.8.0 (USA) software.
Cell Viability Assays
To identify cell mortality induced by different treatment regimens, KLE and ISK cells were cultured in 96-well plate at a density of 5×103 cells well−1 supplemented 200 ul DMEM-F12 complete medium overnight. Cell culture medium was replaced with fresh DMEM-F12 medium containing various dose of nMIL-100 (Fe) or H2O2 alone or together for a further 24 h or 48 h in 5% CO2 at 37 °C. The same amount of PBS was used as negative control. Cell viability was determined by an enhanced CCK-8 assay. Briefly, a 100 μL medium with 10μL CCK-8 solution was introduced to each well for 1 h and then the absorbance values at 450 nm were detected with a microplate reader (EL800, Bio-Tek Instrument, USA). The detection steps of hEEC cell viability were the same as the above.
Moreover, cytotoxicity assays were also evaluated using Calcein AM Cell Viability Assay Kit combined with Propidium Iodide (PI) double staining, in which the numbers of live (green fluorescence) and dead cells (red fluorescence) were counted by Image J 1.8.0 (USA) software. In brief, the 100 μg/mL nMIL-100 (Fe) or 50 μM H2O2 or both treated cells were incubated with Calcein AM working buffer (1×) in the dark at room temperature for 30 min followed by PI staining for 10 min. Subsequently, cells were observed by a FV1000 confocal fluorescence microscope.
Besides, to assess the effect of Parkin expression on cell proliferation, EdU Cell Proliferation Kit assay was performed according to manufacturer’s protocols. Briefly, 10 µM EdU prewarmed at 37 °C was added into the treated cells and the incubation continued for another 2 h. After that, the medium was removed and cells were then fixed for 15 min (Immunol Staining Fix Solution, Beyotime P0098), washed 3 times for 5 min (3% BSA/PBS) and permeabilized for 10 min (Enhanced Immunostaining Permeabilization Buffer, Beyotime P0097). After washing again, cells were supplemented with a 0.5 mL click reaction buffer incubated at room temperature for 30 min, shielded from light. Afterwards, Hoechst 33342 was used with nuclear counterstain for 10 min. Cells immediately were detected with a FV1000 confocal fluorescence microscope and subsequently the red and green intensity ratio was analyzed by the ImageJ 1.8.0 (USA) software.
ROS Detection
The intracellular ROS was detected with a 2′, 7′-dichlorofluorescein diacetate (DCFH-DA) fluorescent probe according to the instructions. KLE and ISK cells were incubated in 6-well plates, respectively, with a density of 2×105 cells/well overnight. Afterward, cells were added with 100 μg/mL nMIL-100 (Fe) or 50 μM H2O2, or both all for 24 h. Meanwhile, cells cultured in complete medium as control. Then, the mediums were replaced by a mixture containing 10 μM DCFH-DA probe. After incubation at 37 °C for 30 min, the cells were rinsed with serum-free media three times and subsequently visualized via a FV1000 confocal fluorescence microscope. The green fluorescence intensity in each group was calculated by the ImageJ 1.8.0 (USA) software.
Autophagy Analysis
Immunofluorescence
A 2×105 cells/well KLE cells were seeded in 6-well plates and allowed to attach overnight. Cells were transfected with pEGFP-LC3B (OBiO Technology Shanghai Corp., Ltd.) by Lipofectamine™ 3000 transfection reagent. After 24 h of transfection, the medium was changed by fresh culture media containing 100 μg/mL nMIL-100 (Fe) or 50 μM H2O2, or combination for 4 h or 6 h. Afterward, the cells were washed with pre-warmed PBS three times and fresh media containing Lyso-Tracker Red or Mito-Tracker Red fluorescent dyes were further added. After incubation at 37 °C for 30 min, cells were rinsed again gently and then observed using a FV1000 confocal fluorescence microscope. Subsequently, the co-localization of green LC3 puncta with red lysosomes or mitochondria was, respectively, evaluated by the ImageJ 1.8.0 (USA) software.
Bio-TEM
Above 106 KLE cells were collected with a cell scraper and centrifuged at 3000 rpm for 10 min, then the supernatant was discarded. A 2.5% glutaraldehyde solution precooled at 4 °C was slowly added along the wall of the tube and then placed for at least 12 h at 4 °C (The fixed solution was required to be filled to the centrifuge tube, so that the samples were completely immersed in it), and placed it at 4°C overnight. The fixed solution was poured out and the samples were rinsed three times with phosphate buffer saline (PBS, 0.1 M, pH 7.0) for 15 min each time. Further, 1% osmium acid solution was used to fix the sample for another 2 h, then the waste fixed solution was carefully taken out, and the samples were washed again with PBS (0.1 M, pH 7.0) three times for 15 min each time. Next, the samples were dehydrated with ethanol solution of gradient concentration (including 30%, 50%, 70%, 80%, 90% and 95%) for 15 min at each concentration, and then treated with 100% ethanol for 20 min. Finally, the samples were transferred to pure acetone for 20 min. The sample was treated with a mixture of embedding agent (EPON 812 resin) and acetone (V/V=1/1) for 1 h and at another ratio (V/V=3/1) for an additional 3 h. After that, the samples were embedded in pure EPON 812 resin overnight. The embedded samples were obtained by embedding the permeated samples and heating them overnight at 70 °C. The samples were sliced with Leica EM UC7 ultrathin slicer, and slices at 70–90 nm were obtained. The slices were stained with lead citrate solution and 50% ethanol saturated solution of uranium dioxide acetate for 10 min, respectively, and then observed under transmission electron microscope (JEOL 1010).
Immunoblotting
KLE and ISK cells were, respectively, treated with 100 μg/mL nMIL-100 (Fe) or 50 μM H2O2, or both all in the presence or absence of 3-MA (2 mM, 2 h) pretreatment for 24 h or 48 h. Afterward, cytotoxicity was firstly determined by CCK-8 assay. Secondly, whole-cell extracts lysed with RIPA lysis buffer were prepared for Western blotting analysis as follows. Protein lysates were separated by 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and then transblotted onto nitrocellulose (NC) membranes. Subsequently, the NC membranes were blocked in 5% BSA at room temperature for 2 h and further incubated with anti-LC3B, anti-P62, anti-PINK1, anti-Parkin, anti-cCaspase-3, anti-cPARP and HRP-conjugated mouse anti-GAPDH (diluted to 1:1000) at 4 °C overnight. After washing in Tris-buffered saline with 0.1% Tween-20 (TBST) three times for 5 min each on the shaker, the membranes were incubated with corresponding horseradish peroxidase (HRP)-labeled secondary antibodies (diluted to 1:5000) at room temperature for 1 h. The protein signals were visualized using enhanced chemiluminescence (ECL) reagent on an Image Quant LAS4000 system (GE Healthcare), and were subjected to semi-quantitative assays by the ImageJ 1.8.0 (USA) software followed by normalized to background.
KLE and ISK cells were transiently transfected with negative control (NC) siRNA or ATG7, PRKN-targeting siRNAs as well pcDNA3.1-NeoGreen-P2A-PRKN-3xFLAG or empty vector plasmids using Lipofectamine Transfection Reagent according to the manufacturer’s instructions at 24 h before treated with 100 μg/mL nMIL-100 (Fe) and 50 μM H2O2. On one hand, the cell viability was detected by CCK-8 kit or EdU incorporation assay and other, the whole-cell lysates were collected for Western blot analysis. The primary antibodies used included anti-ATG 7, anti-Parkin, anti-cCaspase-3, anti-cPARP, anti-Mfn1, anti-Mfn2, anti-Miro1 and HRP-conjugated mouse anti-GAPDH. All primary antibodies were diluted to 1:1000.
Mitochondrial Translocation of Parkin
Immunoblotting
Total cell lysates of KLE and ISK cells were harvested at different time points after the combination treatment containing 100 μg/mL nMIL-100 (Fe) and 50 μM H2O2. Furthermore, to establish whether ·OH induced by nMIL-100 (Fe) and H2O2 involved Fenton-like reaction stimulated PINK1/Parkin pathway mediated-mitophagy, the associated protein expression levels were estimated with Western blotting analysis.
Detection of Mitochondrial Membrane Potential
In order to investigate the effects of ROS on mitochondrial damage, variations in mitochondrial membrane potential between different treatment groups were verified staining with JC-1 fluorescent probe according to manufacturer’s protocols, in which mitochondrial depolarization was assessed by measuring the red/green ratio of fluorescence intensity after 6 h of treatments using ImageJ 1.8.0 (USA) software.
The Enrichment of Mitochondrial Components and Immunoblotting
KLE and ISK cells were incubated in 10 cm dishes to a density of 1×107 cells/dish and then treated by 100 μg/mL nMIL-100 (Fe) combined with 50 μM H2O2 for 6 h. Cells were collected and rinsed twice in pre-chilled PBS. The mitochondrial and cytosol fractionation isolation from both cells were respectively carried out as follows: Cells were sequentially centrifuged at 3000 rpm for 5 min and then resuspended with 1 mL cytoplasmic fraction extracts buffer followed by being homogenized on ice 50 times. After that, the obtained homogenate was vortexed vigorously for 15 s, rested on ice for 15 min and centrifuged at 3000 rpm for 10 min at 4 °C. The supernatant was harvested directly and concentrated at 12,000 rpm for 30 min at 4 °C. As a result, the purified supernatant was the cytosolic proteins. Immediately, the precipitation was resuspended again with 0.1 mL cytoplasmic fraction extract buffer, vortexed for 30 s, and centrifuged at 13,000 rpm for 30 min at 4 °C. Finally, the obtained precipitation was further added with 0.1 mL mitochondria lysate buffer and the centrifugation procedure in previous step was repeated. The supernatants were considered as the denatured mitochondria proteins. The mitochondrial and cytosol fractionations were subjected to Western blot analysis.
Immunofluorescence
After the addition of 100 μg/mL nMIL-100 (Fe) and 50 μM H2O2 for 6 h, ISK cells fixed in prechilled methanol/acetone (7:3) mixture for 10 min, washed twice for 5 min in PBS, and blocked with 3% BSA for 30 min. Then, the cells were incubated with the Parkin primary antibody (1:200 dilution) at 4 °C overnight followed by rinsing in PBS twice for 5 min on a shaker, the Alexa Fluor 488 goat anti-mouse secondary antibody (1:1000 dilution, Jackson ImmunoResearch, 115-545-003) was added for 1 h at 37 °C. Afterwards, Mito-Tracker Red and Hoechst 33342 dyes were applied successively for mitochondria and nuclear labeling. Fluorescence images of different channels were recorded by fluorescence microscopy and the mean fluorescence intensity value of Parkin and its co-localization with red mitochondria were measured by ImageJ 1.8.0 (USA) software.
Statistical Analysis
All the experiments were performed independently at least in triplicate. All the data were expressed as mean ± standard deviation (SD). Statistical analyses were carried out with the GraphPad Prism software version 8.0. The unpaired two-tailed Student’s t-test was used for comparison between two groups. Multiple group comparisons were performed using one-way ANOVA or two-way ANOVA test. P < 0.05 was considered statistically significant.
Results and Discussion
Synthesis and Characterization of nMIL-100 (Fe) Nanoparticles
The structure of MIL-100 (Fe) is established from trimers inorganic structural units of FeO6 octahedra joined with a common vertex μ3-O, which are further linked by 1,3,5-benzenetricarboxylic acid (1,3,5-BTC) ligands to form the hybrid supertetrahedra.36 As shown in Figure 1A, the nanoscale MIL-100 (Fe) nanoparticles [nMIL-100 (Fe) NPs] were fabricated via a typical solvothermal reaction with Fe (NO3)3.9H2O and BTC as previously reported.37 Figure 1B was SEM images of the nMIL-100 (Fe) NPs, which showed highly homogeneous dispersity and exhibited an average diameter of approximately 100 nm. The morphologies were similar to those of previously published investigations.38,39 Moreover, the X-ray diffraction (XRD) pattern of as-prepared nMIL-100 (Fe) NPs matched well with the diffraction peaks of the simulated Fe (III)-based MIL-100 structure from the crystallographic information file (Figure 1C). The atomic EDS mapping showed the distribution of carbon, oxygen and iron corresponded to the inset images of the area enclosed by the white rectangles. The distribution of iron marked with blue color revealed a large amount of iron is uniformly distributed on the surface of nMIL-100 (Fe) (Figure 1D). The X-ray spectroscopy (XPS) spectra showed that nMIL-100 contained the Fe element. Further, the Fe 2p spectrums could be deconvoluted into two peaks centered at 724.6 and 711.5 eV, corresponding to the peaks of Fe 2p1/2 and Fe 2p3/2, respectively, which indicated that the iron atoms in the nMIL-100 (Fe) were presented as Fe3+ (Figure 1E). The above results illustrated the synthesized nanoparticles were in accord with the canonical MIL-100 (Fe) structures.
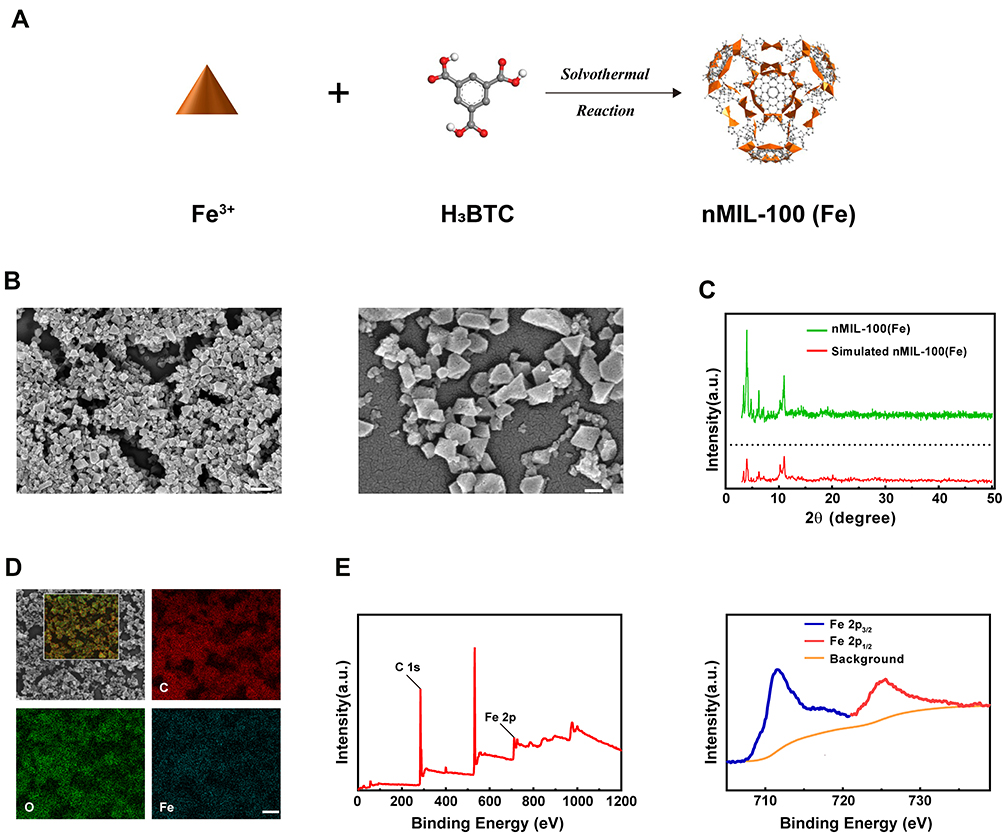 |
Figure 1 Preparations and characterizations of nMIL-100 (Fe) Nanoparticles. (A) Schematic representation of nMIL-100 (Fe) synthesis process. (B) SEM images of the nMIL-100 (Fe) NPs (scale bars are 200 nm and 50 nm, respectively). (C) Powder XRD pattern s of nMIL-100 (Fe) NPs and simulation. (D) SEM images of nMIL-100 (Fe) and atomic EDS mapping of the inset region: C: carbon, O: oxygen, Fe: iron (scale bar is 100 nm). (E) XPS spectra of nMIL-100 (Fe). |
Catalytic Activity and GSH-Depletion Capability of the nMIL-100 (Fe)
To investigate whether the as-synthesized nMIL-100 (Fe) NPs in our study exert a Fenton nanozyme effect, the catalytic property of nMIL-100 (Fe) NPs was assessed via oxidizing the substrate 3,3ʹ, 5,5ʹ-tetramethylbenzidine (TMB) into oxidized TMB (oxTMB) in the presence of H2O2. As is presented in Figure 2A, only in nMIL-100 (Fe) NPs combined with H2O2 (hereafter referred to as “M+H”) group the colorless TMB turned into blue oxTMB with the maximum absorption peak at 652 nm, which were precisely attributed to nMIL-100 (Fe) Fenton-reagents catalyzing H2O2 conversion into high reactive ·OH. Besides, we also evaluated the peroxidase-like activity of nMIL-100 (Fe) NPs with 2,2′-azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) as substrate (Figure S1).
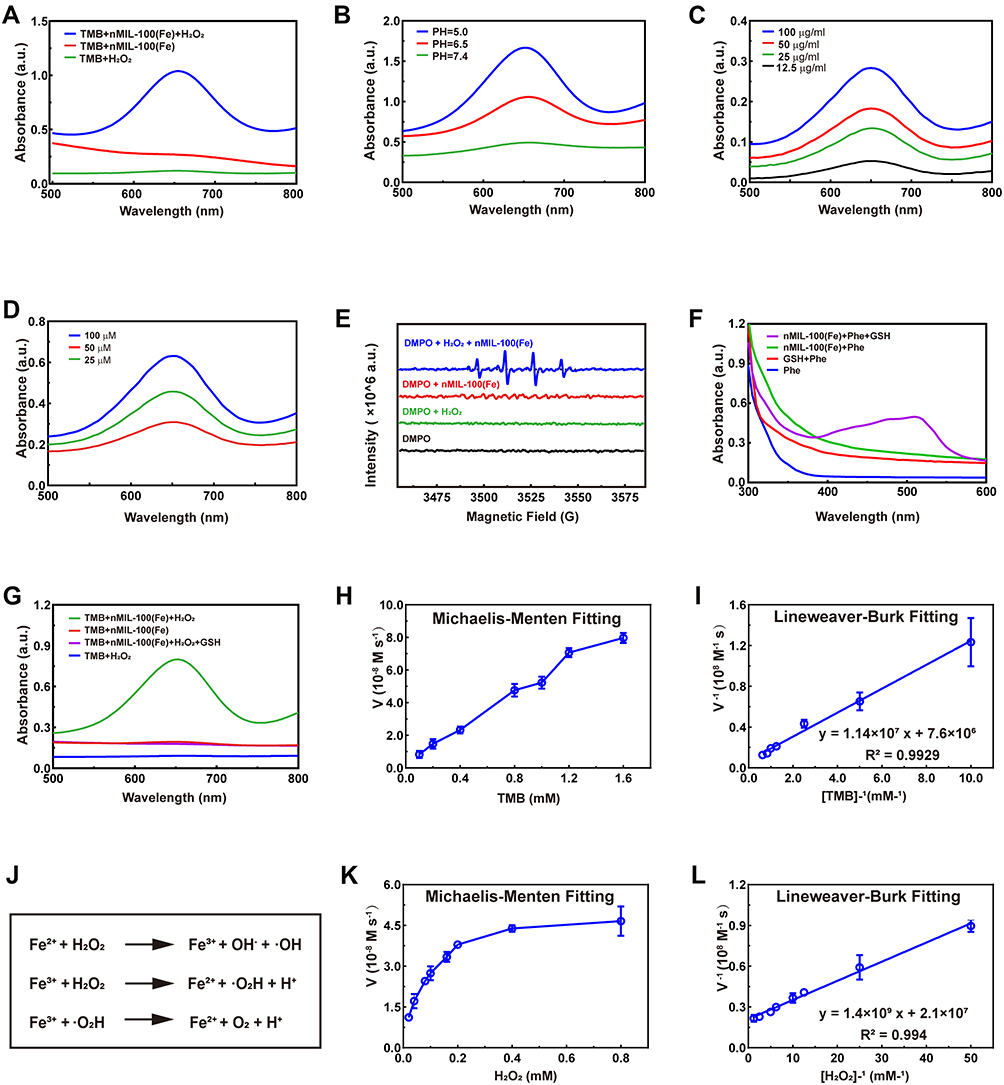 |
Figure 2 Catalytic activity and GSH-depletion Capability of the nMIL-100 (Fe) NPs. (A and B) UV-vis absorption of TMB at 652 nm of TMB/H2O2, TMB/nMIL-100 (Fe) and TMB/H2O2/nMIL-100 (Fe). (C and D) PH values, nMIL-100 (Fe) and H2O2 concentration-dependent catalytic activity of nMIL-100 (Fe) to TMB. (E) ESR spectra for DMPO/·OH adducts detection in the presence of DMPO, DMPO/H2O2, DMPO/nMIL-100 (Fe), DMPO/nMIL-100 (Fe)/H2O2. (F) UV-vis absorption spectra of GSH/Phe/nMIL-100 (Fe) at 525 nm. (G) UV-vis absorption spectra ox-TMB and ox-TMB/GSH. (H and K) Michaelis-Menten kinetic assays of nMIL-100 (Fe): (H) TMB as substrates; (K) H2O2 as substrates. (I and L) Lineweaver-Burk plotting of nMIL-100 (Fe) based on (H and K) respectively. (J) Fenton reaction mechanism. [T]=TMB 1×10−3 M, [H]= H2O2 50×10−6 M, [M]=nMIL-100 (Fe) ×100 μg mL−1, [DMPO]=50 ×10−3 M, [GSH]=10 ×10−6 M, [Phe]=0.2 ×10−6 M. Data were represented as mean ± SD (n=3). |
Additionally, the optimum pH value for effective Fenton action was pH=3–4 as previously reported,40 while the pH in TME varied from approximately 5.0 (inner lysosome) to 7.4. Therefore, it is necessary to further explore the effect of various pH (simulated mild acidic TME) on catalytic activity of nMIL-100 (Fe). Notably, the catalytic activity of nMIL-100 (Fe) NPs in pH 5.0 was superior to that in pH 7.4 in our reaction system according to the oxTMB absorbance maximum of 650 nm (Figures 2B and S2). Also, the UV–vis absorption spectra demonstrated that the peroxidase activity of nMIL-100 (Fe) NPs was concentration-dependent with nMIL-100 (Fe) NPs and H2O2 (Figure 2C and D).
Considering the short half-life of ·OH (10−9 s),41 Electron spin resonance spectrum (ESR) was conducted to detect and validate the generation of ·OH, which was trapped by adding 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) to form relatively long-lived DMPO/·OH adducts. As performed in Figure 2E, the characteristic 1:2:2:1 spectrum of DMPO/·OH could be only observed in M+H groups, while no signal was detected in nMIL-100 (Fe) NPs, H2O2 and the control groups, which suggested the existence of H2O2 was necessary for nMIL-100 (Fe) NPs to exert its catalytic activity observed. The above ESR data was the direct evidence that nMIL-100 (Fe) catalyzed H2O2 to convert into highly oxidative ·OH in mildly acidic TME as shown in Figure 2J.
As known, in response to augmented oxidative stress, enormous antioxidant defense mechanisms will be resorted to offset the risk factor in vivo. Glutathione (GSH), one of the most important endogenous antioxidants, plays a well-established role in maintaining cellular redox homeostasis. However, it is reluctant to accept that the protective function is constantly amplified in cancer due to the elevated levels of GSH, which is inevitable to conferring therapeutic resistance to tumor cells.42,43 Recently, many experiments have confirmed that Fe-based nanoparticles can deplete GSH via transforming the reduced state into the oxidized glutathione (GSSG).44,45 In this study, we also found nMIL-100 (Fe) NPs could be reduced by GSH to form orange metal ion complexes with 1.10-Phenanthroline monohydrate (Phe), appearing a concomitant increase in absorbance at 525 nm (Figure 2F). As confirmed in Figure 1A, only in the presence of nMIL-100 (Fe) and H2O2, TMB can be oxidized to oxTMB, which has a characteristic absorption peak at 525 nm. However, when GSH was added to the system, the characteristic absorption peak completely disappeared, indicating that oxTMB was reduced into TMB by GSH, and the reduced GSH was depleted and converted to GSSG (Figure 2G). Altogether, our as-prepared nMIL-100 (Fe) NPs acted as a peroxidase-like nanocatalysts, which catalyzed the formation of ·OH, simultaneously consumed the radical scavenger-GSH via Fenton-like reaction, eventually exacerbated the accumulation of ROS.
Afterwards, the peroxidase-like catalytic activity of nMIL-100 (Fe) NPs were also evaluated by determining Michaelis–Menten steady-state kinetics. Using diverse concentrations of TMB and H2O2 as substrates, respectively, in pH =6 reaction system, the corresponding plots of initial reaction velocities for oxidized TMB product unfold typical Michaelis−Menten curves (Figure 2H and K). The maximum initial velocity (Vmax) and Michaelis–Menten constant (Km) were calculated using Lineweaver–Burk fitting by the reciprocal transition of the abscissa and ordinate of Michaelis−Menten curves (Figure 2I and L). Learning from enzymology research, the smaller the Km value, the higher the enzyme binding affinity to substrates was. The apparent Km value of nMIL-100 (Fe) NPs with either TMB or H2O2 as the substrate was lower than that of horseradish peroxidase (HRP),40 indicating that nMIL-100 (Fe) had relatively higher catalytic activity even though in pH =6 mild acidic buffer (Table 1). That may be attributed to the regular meso- and micro-porous hybrid nanostructure of nMIL-100 (Fe) NPs with higher porosity, leading to the Fe (III) in its active center contacting more guest molecules (eg TMB, H2O2) compared to natural HRP, and in turn, increasing the nanozyme–substrate interaction. Therefore, nMIL-100 (Fe) NPs will be a sharp object for tumor treatment by oxidative damage, especially in tumor tissue with high expression of H2O2.
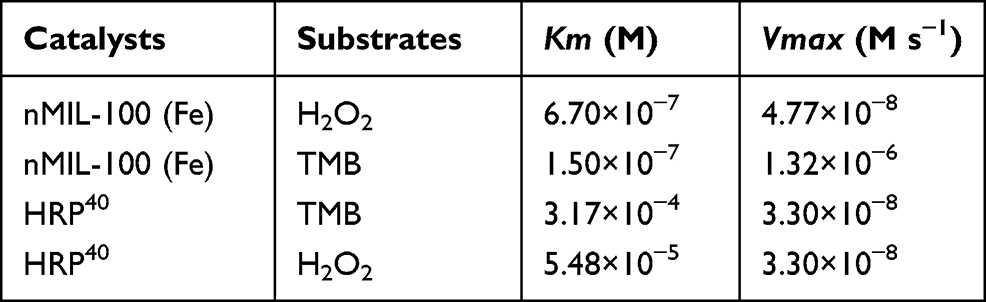 |
Table 1 Comparison of the Kinetic Parameters TMB Oxidation by nMIL-100 (Fe) |
Cytotoxicity of nMIL-100 (Fe) and H2O2 Nanosystem
The preceding experiments have established NPs mediated Fenton-like reaction to convert H2O2 into higher oxidation activities ·OH. In an attempt to confirm the tumoricidal potential of nMIL-100 (Fe) NPs - H2O2 system in vitro, we first assessed the cellular internalization efficiency of nanoparticles using progesterone-insensitive human endometrial adenocarcinoma cell line KLE as a model. Currently, progestin is used as the first-line therapy for inoperable EC patients. A high response rate is achieved in patients who are in initial use, however a higher recurrence rate (19.2–33.8%) was followed due to the occurrence of the acquired resistance to progestin.5,6 Therefore, a new replacement therapy need to be developed to overcome insensitivity and resistance to progestin treatment. In our study, after 2 hours of incubation, the nMIL-100 (Fe) NPs (brighter green spots) were successfully internalized into the KLE cells and co-localized within the lysosomes (red spots), forming yellow fluorescence in merge images compared with that of the control group (Figure 3A). In the merge channel, white arrows indicated the colocalization of nMIL-100 (Fe) and lysosomes. Furthermore, to better quantify the relative position relationship, Pearson’s correlation coefficient (PCC) and Manders’ Overlap coefficient (MOC) were calculated respectively by ImageJ software plug-in, Colocalization-Finder. The colocalization was confirmed as PCC rate ≥ 0.5 and meanwhile MOC rate ≥ 0.6. As displayed in Figure 3B, compared with nMIL-100 (Fe) or H2O2 treated alone groups, the combination group including nMIL-100 (Fe) and H2O2 exhibited brighter green H2DCFDA fluorescence in both KLE and Ishikawa cells (ISK, progesterone-sensitive human endometrial adenocarcinoma cell lines). The results suggested that the nMIL-100 (Fe) and H2O2 synergistic treatments mediated-Fenton chemistry evoked intracellular ROS production independently of cell type. Intriguingly, the intracellular GSH also could be depleted by nMIL-100 (Fe) NPs in a concentration dependent manner (Figure 3C). The “attack-defense” mechanisms imbalances inevitably led to excessive accumulation of ROS, which was a vital prerequisite for cellular constant oxidative damage. Accordingly, no significant cytotoxicity of nMIL-100 (Fe) NPs was observed in the absence of H2O2. Low cytotoxicity was induced by H2O2 at the concentration range within 200 μM (Figure S3). While in the nMIL-100 (Fe) and H2O2 combined treatment group, cell viability began to show signs of suppression when nMIL-100 (Fe) NPs concentration was just greater than 25 μg/mL (Figure 3D). That was probably owing to the ·OH accumulation accompanied with the GSH reduction with increasing incubation duration. Afterward, a combination of 100 μg/mL nMIL-100 (Fe) and 50 μM H2O2, which hereafter was abbreviated as “M+H” for simplicity, was selected for subsequent experiments unless otherwise special stated. One embarrassing problem must be acknowledged: The M+H system induced cytotoxicity was suboptimal. Specifically, the inhibition rate of ISK cell proliferation was only less than 50% for 24 h after treatment and the percent of apoptotic cells did not exceed 10% regardless of the duration of treatment (Figure S4). That may be attributed to intrinsic differential expression of estrogen receptor (ER) and progesterone receptor (PR) in EC cells. Specifically, ISK was double-positive cells (ER+/PR+) compared to the double-negative expression in KLE (ER-/PR-). Meanwhile, we learned from the TCGA database in EC patients there was a positive correlation between the expression of ER and catalase (CAT),46 a critical antioxidant enzyme (Figure S5A and B). Thus, it was reasonable to speculate that catalase was highly expressed in ISK cells as we had confirmed in Figure S5B. Precisely owing to the conversion of H2O2 into H2O and O2 by catalase, the amount of H2O2 involved in nMIL-100 (Fe) mediated Fenton-like reaction was significantly diminished. That made ·OH, generated through the above catalytic reaction and with inherent short half-life, fail to effectively accumulate in the local environment, which ultimately mitigated the toxic effects of M+H Fenton nanocatalyst. Besides, other radical scavenger mechanisms such as superoxide dismutase (SOD),9 nuclear factor erythroid 2-related factor 2 (NRF2)47 and NADPH48 except for GSH may counteract the disruption induced by ·OH. Additionally, the LIVE/DEAD kit was applied to repetitively visualize the KLE and ISK cells killing effect of different additions, in which co-staining with calcein-AM (green) for live cells and PI (red) for dead and apoptotic cells. In line with the CCK-8 results, the red fluorescence in cells was readily detected in both KLE and ISK cells after being treated with M+H for 24 h. Furthermore, cell shrinkage and cell volume reduction were remarkably observed in this group (Figure 3E). Taken together, after successful internalization, nMIL-100 (Fe) next acted as Fenton reagents to catalyze intracellular H2O2 into highly oxidative ·OH. However, the effects of its oxidative damage to EC cells were not satisfactory, which could be partially due to different cell types, even though it simultaneously consumed intracellular antioxidants-GSH. Thus, other resistance mechanisms irrespective of cell types were likely to exist, which contributed to counteract cytotoxic effect of ·OH.
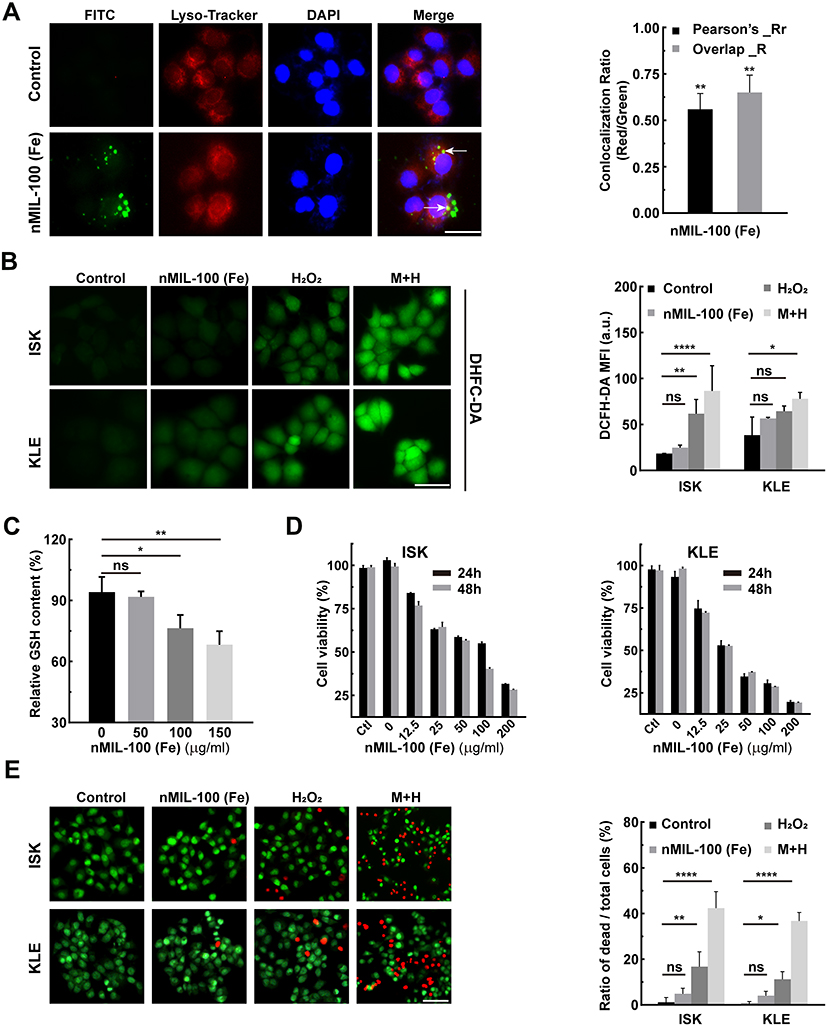 |
Figure 3 Cytotoxicity of nMIL-100 (Fe) and H2O2 nanosystem. (A) Confocal fluorescence images of nMIL-100 (Fe) NPs uptake by KLE cells treated with nMIL-100 (Fe) -FITC (green) for 2 h, and then incubated with Lyso-Tracker (red) and DAPI (blue) (scale bar is 20 µm). The colocalization (white arrows) between nMIL-100 (Fe)-FITC and Lyso-Tracker in (a) by Pearson’s coefficient and Manders overlap coefficients analysis. (B) ROS detection of ISK and KLE cells using fluorescent probe H2-DCFDA (scale bar is 20 µm), graph of mean fluorescence intensities (MFI) in different formulations (control, 100 µg mL−1 nMIL-100 (Fe) alone, 50×10−6 M H2O2 alone and or a combination of both) for 24 h. (C) Relative levels of GSH after treatment with (50,100,150 µg mL−1) nMIL-100 (Fe) for 24 h (scale bar is 50 µm). (D) Cell viability detection of ISK and KLE cells by CCK-8 assay after different administrations including Control, 50×10−6 M H2O2 alone and a combination of (0, 12.5, 25, 50, 100, 200 µg mL−1) nMIL-100 (Fe) and 50×10−6 M H2O2 for 24 h and 48 h. (E)The fluorescent images of ISK and KLE cells undergoing the same treatments as aforementioned (B). Both cells were stained with Live/Dead kit (scale bar is 100 µm). Living (green) and dead (red) cells were labeled and the relative ratio of dead to total cells was shown. [H2O2] = 50×10−6 M, [nMIL-100 (Fe)] = 100 µg mL−1, [M+H] = 100 µg mL−1 nMIL-100 (Fe) + 50×10−6 M H2O2, Data were represented as mean ± SD (n=3). *p < 0.05, **p < 0.01, ****P< 0.001, ns, not significant. |
Autophagy Induction by nMIL-100 (Fe) and H2O2 Nanosystem
Aberrant increased endogenous and/or exogenous ROS is a potent activator of autophagy primarily including macroautophagy and chaperone-mediated autophagy (CMA).49,50 Less is known about the influence of ROS on microautophagy. In light of this, KLE cells were first transient transfected with EGFP-LC3B plasmid. At 24 h after transfection, the combination of nMIL-100 (Fe) and H2O2 as well as nMIL-100 (Fe) or H2O2 alone were supplied for an additional 6 h. As expected, M+H treatment induced a significant aggregation of EGFP-LC3B puncta (green fluorescence) even being into plaque-like structure, indicating the increased autophagosome formation. Additionally, the colocalization of LC3B puncta with Lyso-Tracker (red fluorescence) was also observed, revealing the process of autophagosomes-lysosomes fusion into autolysosomes (autophagosome maturation) to facilitate further degradation of the internal contents (Figure 4A). In addition, bio-TEM was further used to observe the dynamic autophagic process (also known as autophagy flux) including initiation (isolation membrane generation and elongation indicated by white arrows), maturation (the formation of autophagosomes denoted by red arrows), fusion and cargo degradation (the formation of autolysosomes presented by blue arrows) (Figure 4B). The numbers of autophagosomes and autolysosomes were strongly increased after the co-treatment of nMIL-100 (Fe) and H2O2 than that of nMIL-100 (Fe), H2O2 treated alone and control group. Such TEM observations echoed the aforementioned immunofluorescence results, suggesting that nMIL-100 (Fe)-H2O2 catalytic nanosystems could induce autophagy in EC cells.
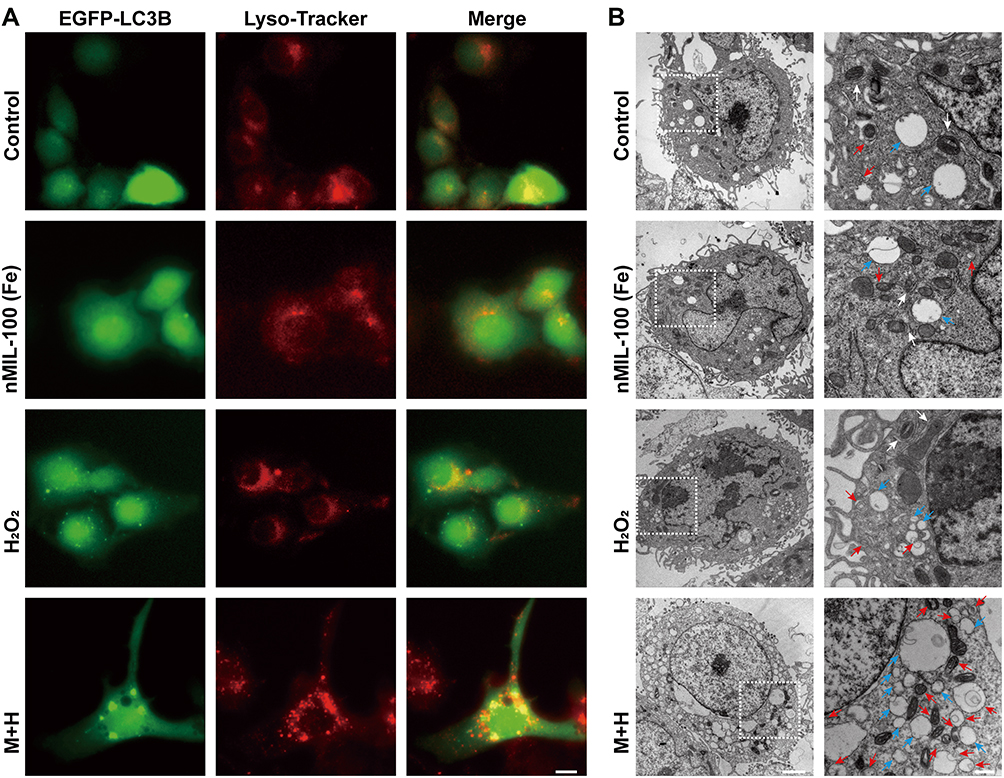 |
Figure 4 Autophagy induction by nMIL-100 (Fe) and H2O2 nanosystem. (A) Confocal fluorescence images of KLE cells transfected with EEGFP-LC3B plasmids followed by different treatments for 24 h. And the co-localization analysis of EEGFP-LC3B puncta (green) and lysosomes (red) after con-stained with Lyso-Tracker (red) (scale bar is 5 µm). (B) Bio-TEM images of KLE cells subjected to the same treatment as (A) for 24 h. White arrowheads indicate the formation of isolation membrane at early stage of autophagy; red arrows denote the mature autophagosome; blue arrowheads mark the autophagolysosome at the late stage of autophagy, respectively (left, scale bar, 2 µm; right, scale bar, 0.5 µm). |
The autophagic flux was regulated by the effect of ROS on the transcriptional activation of autophagy-related genes and degradation of proteins and organelles (eg mitochondria). Since 2% mitochondrial oxygen consumption was converted into superoxide, mitochondria were regarded as a major source of endogenous ROS.51,52 Excessive mitoROS can induce the opening of mitochondrial permeability transition pore (MPTP) and the inner membrane anion channel (IMAC), and further trigger the loss of mitochondrial membrane potential and sudden increase of ROS from mitochondrial electron transport chain (ETC), which is termed as ROS-induced ROS release (RIRR).53,54 As shown in Figure 5A, we visualized a remarkably increased number of autophagosomes and the overlap of EGFP-LC3B and Mito-Tracker red treated by M+H for 4 h or 6 h, indicating the existence of mitochondrial autophagy (mitophagy), which was consistent with the bio-TEM analysis that the damaged mitochondria were being wrapped by isolation membranes (white arrows indicated in Figure 4B). Moreover, mitochondrial membrane depolarization was evidenced by a decrease of the ratio of red to green JC-1 fluorescence after M+H addition, especially in the pretreatment with Mdivi-1 combination group (Figure 5B). The fluorescence images again highlighted cells with smaller volume and irregular morphology after M+H treatment (Figure S6). This may be owing to the aberrant accumulation of injured mitochondria, which failed to be removed by selective form of autophagy-mitophagy. The LC3BII is a well-established indicator for autophagosomes. To elucidate the phenomenon of autophagy in response to the ·OH production, KLE and ISK cells were, respectively, treated with M+H for different time intervals. The transformation of endogenous LC3BI to LC3BII was significantly increased in a time-dependent regardless of cell types, accompanied by up-regulation of critical nodal proteins in classic PINK1/Parkin mitophagy pathway (Figure 5C). In dysfunctional mitochondria, PINK1 was unable to be imported to the inner mitochondrial membrane (IMM) to undergo cleavage and degradation, and then was forced to stabilize on the damaged outer mitochondrial membrane (OMM).55,56 Otherwise, M+H treatment promoted the translocation of Parkin from the cytosol to the mitochondria compared with the control group (Figure 5D). Afterwards, the immunoblotting results were further verified by immunofluorescence staining in ISK cells expressing relatively higher Parkin. Despite the green fluorescence intensity of Parkin was apparently increased after M+H stimulation. A well-established colocalization of Parkin with Mito-Tracker was also validated; however, no significant differences of the PCC and MOC values were noted in both pre- and post-treatment group (white straight line in merge channel in Figures 5E and S7), which indicated a basal level of mitophagy occurred in ISK cells. It is now well appreciated that the translocation of Parkin activated by PINK1 kinase activity was a pivotal step for follow-up, ubiquitinating special substrates and autophagosome recruitment to instigate the autophagic clearance of damaged mitochondria.57,58 The above phenomenon reflected the fast self-renewal of EC cells via mitochondrial quality control. Taken together, nMIL-100 (Fe) and H2O2 mediated ·OH production induced a collapse of mitochondrial membrane potential via the RIRR process. The signals were further sensed and recognized by upregulated PINK1 kinase, resulting from retention on the dysfunctional OMM to escape from proteolysis. The latter recruited Parkin from cytoplasm to initiate the removal of dysfunctional and devastatingly damaged mitochondria.
 |
Figure 5 PINK1/Parkin mediated-mitophagy activation by nMIL-100 (Fe) and H2O2 nanosystem. (A) The fluorescence images of GFP-LC3B-transfected ISK cells after treatment with 100 μg/mL nMIL-100 (Fe) and 50 μM H2O2 for 4 h or 6 h followed by Mito-Tracker (red) incubation (scale bar is 20 µm). The colocalization between green spots and red mitochondria tracker was elevated by Plot Profile tool. (B) Quantifications of the mitochondrial membrane potential (MMP) ΔΨm (JC-1 red/green fluorescence intensity ratio) after different treatment including control, 100 μg mL−1 nMIL-100 (Fe) alone, 50×10−6 M H2O2 alone, a combination of both or a pretreatment of Mdivi-1 (5×10−6 M, 1 h) before the both additions for 24 h. (C) Western blot analysis of PINK1, Parkin, LC3-I and LC3-II expression in ISK and KLE cells treated with 100 μg mL−1 nMIL-100 (Fe) and 50×10−6 M H2O2 for different time intervals, using GAPDH as loading control. (D) Parkin expression in mitochondrial and cytosolic enriched fractions of ISK and KLE cells after100 μg mL−1 nMIL-100 (Fe) and 50×10−6 M H2O2 treatment for 6 h was determined by Western blot, using GAPDH and COX IV as cytosolic and mitochondrial loading control respectively. (E) The images of immunofluorescence in ISK cells undergoing the same treatments as the aforementioned in (D) followed by co-staining with Mito-Tracker (red) and Hoechst 3342 (blue) (scale bar is 20 µm). The MFI of Parkin and the colocalization (white straight lines) between green Parkin and red mitochondria tracker was analyzed by Plot Profile tool. Data were represented as mean ± SD (n=3). *p < 0.05, **p < 0.01, ns, not significant. |
Effects of Mitophagy on nMIL-100 (Fe) and H2O2 Nanosystem Mediated Oxidative Damage
Substantial evidence has confirmed that autophagy is a protective mechanism against various stress-induced cell apoptosis. In our previous results, autophagy levels were remarkably upregulated after M+H addition, primarily manifested by an increase of conversion of LC3-I to LC3-II and also PINK1/Parkin activation. Autophagy inhibitor 3-Methyladenine (3-MA) pretreatment followed by M+H treatment partially suppressed autophagy. At the same time, the caspase 3 was activated and the cleaved PARP (c-PARP) was increased. It suggested the initiation of apoptotic processes (Figure 6A). Besides, the above combined treatment dramatically inhibited cell proliferation irrespective of cell types (Figure 6B). To further verify the role of autophagy in the M+H-induced cytotoxicity, key autophagy genes were knocked down by Atg7 Small interfering RNA (siRNA). As displayed in Figure 6C, the Atg7-siRNA3 sequence had a higher knockdown efficiency accompanied by significant down-regulation of Parkin expression, which was chosen for subsequent experiments. In line with the previous 3-MA pretreatment effects, Atg7 RNA silencing in KLE cells before the M+H addition remarkably elevated the expression of cPARP and cCaspase-3 and reduced cell viability compared with other groups in a time-dependent manner in both EC cells (Figure 6D and E). Interestingly, the proliferation ability was unaffected in cells transfected using ATG 7 siRNA alone. Also, the inhibitory efficiency of cell proliferation was not ideal only by M+H treatment for 24 h (Figure 3D). While the synergistic treatment with autophagy blockage and M+H addition significantly suppressed cell viability, indicating adequately that the combination of both nMIL-100 (Fe) and H2O2 was necessary to ensure the function of the nMIL-100 (Fe) nanocatalyst. Taken together, autophagy inhibition by pretreatment with 3-MA or Atg7 RNA silencing hampered repair of oxidative DNA damage mediated by M+H stress and then exacerbated cell apoptosis. That is in turn potential chemodynamic antitumor effects mediated by ·OH deriving from M+H involved-Fenton-like reaction, which further might be aggravated by depletion of intracellular GSH.
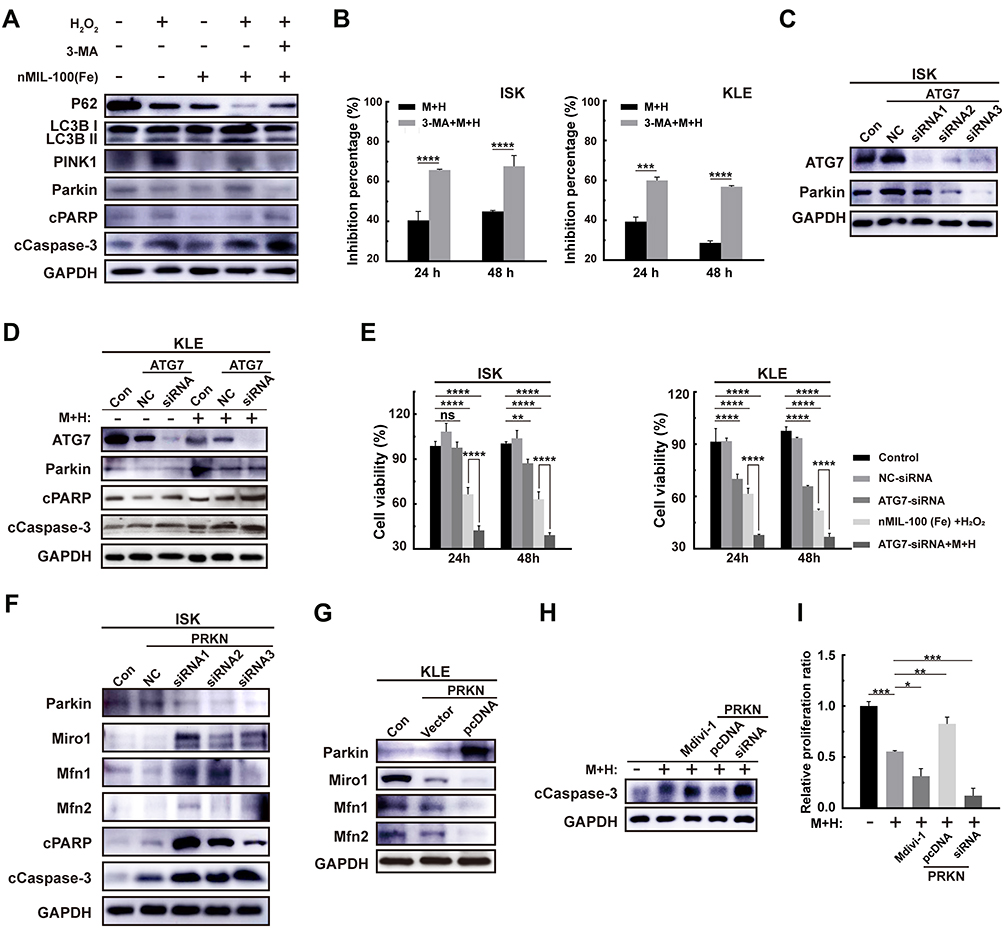 |
Figure 6 Effects of mitophagy on nMIL-100 (Fe) and H2O2 nanosystem mediated oxidative damage. (A) Western blot analysis of PINK1, Parkin, P62, cPARP, cCaspase-3, LC3-I and LC3-II expression in ISK and KLE cells treated with control, 100 μg mL−1 nMIL-100 (Fe) alone, 50×10−6 M H2O2 alone, a combination of both or a pretreatment of 3-MA (2×10−6 M, 1 h) accompanied by the both additions for 24 h, GAPDH protein were served as an internal control. (B) Relative cell growth inhibition percentage of indicated ISK and KLE cells on the 24 h and 48 h after the aforementioned combination or a 3-MA (2×10−6 M, 1 h) addition before the combination treatments. (C) Western blot for ATG 7-siRNA 1–3 knockdown efficiencies and the Parkin protein expression of ISK cells at 48 h after transfection. (D) The corresponding expression of apoptosis-related molecules in KLE cells transfected with or without control siRNA or ATG 7-siRNA followed by the aforementioned combination. (E) The relative cell viability of ISK and KLE cells after the same treatments as in (D) for 24 h or 48 h. (F) The protein levels of Parkin and its downstream molecules including Miro1, Mfn1 and Mfn2 after transient knockdown or overexpression its PRKN gene in ISK and KLE cells respectively. (G–I) Detection of active caspase 3 and relative cell growth inhibition ratio of ISK cells by the aforementioned combination additions for 24 h after different types pretreatments with Mdivi-1 (5×10−6 M, 1 h), Parkin-siRNA or Parkin-pcDNA transfection. Data were represented as mean ± SD (n=3). *p < 0.05, **p < 0.01, ***P< 0.005, ****P< 0.001, ns, not significant. |
Mitochondria are the important members in redox regulation of autophagy. A tight regulation of mitochondria quantity and quality is therefore essential for cellular homeostasis. As confirmed in Figure 5, M+H treatment initiated canonical PINK1/Parkin mitophagy pathway, in which increasing PINK1 triggered Parkin mitochondria translocation from cytoplasm and promoted the Parkin’s E3 ubiquitin ligase activity by phospho-ubiquitin chains. Then Parkin ubiquitinated more downstream mitochondrial substrates (such as Mfn1, Mfn2 or Miro1) in order to be recognized by autophagy receptors (e g p62 and OPTN). The interaction between mitochondria and autophagosomes launched selective autophagic clearance of damaged mitochondria.59 Therefore, to further assess the role of mitophagy in M+H-induced cell death, transient overexpression or knockdown of Parkin was conducted in ISK cells and KLE cells, respectively. Due to altered E3 ubiquitin ligase activity of Parkin, the corresponding change of substrate molecules including Mfn1, Mfn2 and Miro1 was observed (Figure 6F and G). Moreover, Parkin siRNA transfection in accordance with Mdivi-1 pretreatment in ISK cells markedly enhanced M+H-induced cytotoxicity by augmenting the expression of cCaspase-3 as well increased the inhibition of cell proliferation. However, Parkin gene overexpression apparently attenuated M+H-mediated oxidative stress cytotoxicity (Figure 6H and I). Overall, mitophagy, induced by nMIL-100 (Fe) and H2O2 nanosystem-elicited ·OH production, was proven to be a cytoprotective mechanism that allowed cell to survive under lethal oxidative stress via selective scavenging of damaged mitochondria to remodel cellular homeostasis. Therefore, it is necessary to identify whether therapeutic approaches (eg, chemotherapy, radiotherapy and nanotherapy)-induced autophagy exert a cytoprotective or cytotoxic role, whether autophagy enhances or weakens the therapeutic effect during treatments. As other studies confirmed, the inhibition of PINK1/Parkin- or Rab9a-involved mitophagy process acquired a radiosensitive effect on cancer cells.60,61 Also, downregulation expression of key genes including PINK1, FUNDC1 and AMBRA1 was considered as a profound chemosensitization approach for cancer therapy.62,63 However, ULK1-mediated mitophagy demonstrated an anti-cell proliferation property in colon cancer cell lines.64 Therefore, the results above reveal that the combination of Fenton reagents mediated-CDT and a mitophagy inhibitor like Mdivi-1 will exert synergistic antitumor effects, which is favorable to acquire high-efficacy cancer therapy. Next, our research efforts will focus on exploiting a versatile nanoplatform to implement the cooperation between CDT and chemotherapy in EC.
Conclusion
In conclusion, this study provided a potential therapeutic option based on special TME for EC patients. First, we successfully synthesized a nanoscale MIL-100 (Fe) and demonstrated its intrinsic peroxidase-like activity. Furthermore, significant cytotoxicity was elicited by nMIL-100 (Fe) involved CDT due to excellent catalysis and the property of intercellular GSH depletion. The abundant accumulated ROS during nMIL-100 (Fe)-CDT process induced cytoprotective mitophagy mediated by canonical PINK1/Parkin pathway, which in turn attenuated itself oxidative damages of EC cells. Overall, the current data highlights the importance of synergistic therapy of the inhibition of mitophagy and ROS-based treatment. Notwithstanding the present results was limited to in vitro levels, this work certainly offered valuable insights into how to establish more effective integrated therapeutic regimens. Therefore, extensive efforts will be devoted to developing such a nanocatalysis system that can accommodate both elevated oxidative attack and declined antioxidant defense, which is a feasible and potential strategy with tremendous clinical translation value for cancer therapy.
Acknowledgments
This study is supported by the Shanghai Municipal Key Clinical Specialty (No. shslczdzk06302), the Emerging frontier project of Municipal Hospital in Shanghai (SHDC12020130), National Natural Science Foundation of China (No. 81172477, 81402135), the Project of the Science and Technology Commission of Shanghai Municipality (No. 17441907400), and the Shanghai Jiao Tong University Medicine-Engineering Fund (No. YG2017MS41).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kasivisvanathan V, Rannikko AS, Borghi M, et al. MRI-targeted or standard biopsy for prostate-cancer diagnosis. N Engl J Med. 2018;378(19):1767–1777. doi:10.1056/NEJMoa1801993
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33.
3. Jemal A, Ward EM, Johnson CJ, et al. Annual report to the nation on the status of cancer, 1975–2014, featuring survival. J Natl Cancer Inst. 2017;109(9). doi:10.1093/jnci/djx030.
4. Henley SJ, Ward EM, Scott S, et al. Annual report to the nation on the status of cancer, part I: national cancer statistics. Cancer. 2020;126(10):2225–2249. doi:10.1002/cncr.32802
5. Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975–2017; 2020. National Cancer Institute. Available from: https://seer.cancer.gov/archive/csr/1975_2017/.
6. Park JY, Nam JH. Progestins in the fertility-sparing treatment and retreatment of patients with primary and recurrent endometrial cancer. Oncologist. 2015;20(3):270–278. doi:10.1634/theoncologist.2013-0445
7. Ciociola AA, Cohen LB, Kulkarni P. How drugs are developed and approved by the FDA: current process and future directions. Am J Gastroenterol. 2014;109(5):620–623. doi:10.1038/ajg.2013.407
8. Clarke MA, Devesa SS, Harvey SV, Wentzensen N. Hysterectomy-corrected uterine corpus cancer incidence trends and differences in relative survival reveal racial disparities and rising rates of nonendometrioid cancers. J Clin Oncol. 2019;37(22):1895–1908. doi:10.1200/JCO.19.00151
9. McAlpine JN, Temkin SM, Mackay HJ. Endometrial cancer: not your grandmother’s cancer. Cancer. 2016;122(18):2787–2798. doi:10.1002/cncr.30094
10. Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4(5):278–286. doi:10.1038/nchembio.85
11. Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol. 2013;13(5):349–361. doi:10.1038/nri3423
12. Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10(3):175–176. doi:10.1016/j.ccr.2006.08.015
13. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591. doi:10.1038/nrd2803
14. Fan W, Yung B, Huang P, Chen X. Nanotechnology for multimodal synergistic cancer therapy. Chem Rev. 2017;117(22):13566–13638. doi:10.1021/acs.chemrev.7b00258
15. Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11(9):671–677. doi:10.1038/nrc3110
16. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95. doi:10.1038/nrc2981
17. Lin LS, Song J, Song L, et al. Simultaneous Fenton-like ion delivery and glutathione depletion by MnO(2) -based nanoagent to enhance chemodynamic therapy. Angew Chem Int Ed. 2018;57(18):4902–4906. doi:10.1002/anie.201712027
18. Lin Y, Ren J, Qu X. Catalytically active nanomaterials: a promising candidate for artificial enzymes. Acc Chem Res. 2014;47(4):1097–1105. doi:10.1021/ar400250z
19. Yang BW, Chen Y, Shi JL. Nanozymes in catalytic cancer theranostics. Prog Biochem Biophys. 2018;45(2):237–255.
20. Gao L, Zhuang J, Nie L, et al. Intrinsic peroxidase-like activity of ferromagnetic nanoparticles. Nat Nanotechnol. 2007;2(9):577–583. doi:10.1038/nnano.2007.260
21. Huang DM, Hsiao JK, Chen YC, et al. The promotion of human mesenchymal stem cell proliferation by superparamagnetic iron oxide nanoparticles. Biomaterials. 2009;30(22):3645–3651. doi:10.1016/j.biomaterials.2009.03.032
22. Chang Q, Deng KJ, Zhu LH, Jiang GD, Yu C, Tang HQ. Determination of hydrogen peroxide with the aid of peroxidase-like Fe3O4 magnetic nanoparticles as the catalyst. Microchim Acta. 2009;165(3–4):299–305. doi:10.1007/s00604-008-0133-z
23. Chen Z, Yin JJ, Zhou YT, et al. Dual enzyme-like activities of iron oxide nanoparticles and their implication for diminishing cytotoxicity. ACS Nano. 2012;6(5):4001–4012. doi:10.1021/nn300291r
24. Fu J, Shao Y, Wang L, Zhu Y. Lysosome-controlled efficient ROS overproduction against cancer cells with a high pH-responsive catalytic nanosystem. Nanoscale. 2015;7(16):7275–7283. doi:10.1039/C5NR00706B
25. Wang L, Huo M, Chen Y, Shi J. Iron-engineered mesoporous silica nanocatalyst with biodegradable and catalytic framework for tumor-specific therapy. Biomaterials. 2018;163:1–13. doi:10.1016/j.biomaterials.2018.02.018
26. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26(7):1749–1760. doi:10.1038/sj.emboj.7601623
27. Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461. doi:10.1126/science.1196371
28. Djavaheri-Mergny M, Amelotti M, Mathieu J, et al. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem. 2006;281(41):30373–30382. doi:10.1074/jbc.M602097200
29. Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873. doi:10.1101/gad.1599207
30. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16(8):461–472. doi:10.1038/nrm4024
31. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi:10.1016/j.cell.2007.12.018
32. Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016;85(1):685–713. doi:10.1146/annurev-biochem-060815-014556
33. White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15(17):5308–5316. doi:10.1158/1078-0432.CCR-07-5023
34. Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9(12):1004–1010. doi:10.1038/nrm2529
35. Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008;15(1):171–182. doi:10.1038/sj.cdd.4402233
36. Horcajada P, Surblé S, Serre C, et al.Synthesis and catalytic properties of MIL-100(Fe), an iron (III) carboxylate with large pores. Chem Commun. 2007;(27):2820–2822. doi:10.1039/B704325B
37. Wang DK, Wang MT, Li ZH. Fe-based metal-organic frameworks for highly selective photocatalytic benzene hydroxylation to phenol. ACS Catal. 2015;5(11):6852–6857. doi:10.1021/acscatal.5b01949
38. Zhang Y, Lin L, Liu L, et al. Positive feedback nanoamplifier responded to tumor microenvironments for self-enhanced tumor imaging and therapy. Biomaterials. 2019;216:119255. doi:10.1016/j.biomaterials.2019.119255
39. Cai W, Gao H, Chu C, et al. Engineering phototheranostic nanoscale metal-organic frameworks for multimodal imaging-guided cancer therapy. ACS Appl Mater Interfaces. 2017;9(3):2040–2051. doi:10.1021/acsami.6b11579
40. Liu YL, Zhao XJ, Yang XX, Li YF. A nanosized metal-organic framework of Fe-MIL-88NH2 as a novel peroxidase mimic used for colorimetric detection of glucose. Analyst. 2013;138(16):4526–4531. doi:10.1039/c3an00560g
41. D’Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8(10):813–824. doi:10.1038/nrm2256
42. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30(1–2):42–59. doi:10.1016/j.mam.2008.05.005
43. Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134(3):489–492. doi:10.1093/jn/134.3.489
44. Liu F, Lin L, Zhang Y, et al. A tumor-microenvironment-activated nanozyme-mediated theranostic nanoreactor for imaging-guided combined tumor therapy. Adv Mater. 2019;31(40):e1902885. doi:10.1002/adma.201902885
45. Fu JK, Li T, Zhu YC, Hao YQ. Ultrasound-activated oxygen and ros generation nanosystem systematically modulates tumor microenvironment and sensitizes sonodynamic therapy for hypoxic solid tumors. Adv Funct Mater. 2019;29(51):16. doi:10.1002/adfm.201906195
46. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–247. doi:10.1038/35041687
47. Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009;48(2):91–104. doi:10.1002/mc.20465
48. Murphy MP. Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid Redox Signal. 2012;16(6):476–495. doi:10.1089/ars.2011.4289
49. Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36(1):30–38. doi:10.1016/j.tibs.2010.07.007
50. Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15(11):4829–4840. doi:10.1091/mbc.e04-06-0477
51. Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287(7):4434–4440. doi:10.1074/jbc.R111.271999
52. Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal. 2012;16(11):1323–1367. doi:10.1089/ars.2011.4123
53. Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192(7):1001–1014. doi:10.1084/jem.192.7.1001
54. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–950. doi:10.1152/physrev.00026.2013
55. Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191(5):933–942. doi:10.1083/jcb.201008084
56. Greene AW, Grenier K, Aguileta MA, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13(4):378–385. doi:10.1038/embor.2012.14
57. Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107(1):378–383. doi:10.1073/pnas.0911187107
58. Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–131.
59. Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111(42):E4439–4448. doi:10.1073/pnas.1405752111
60. Zheng R, Yao Q, Xie G, et al. TAT-ODD-p53 enhances the radiosensitivity of hypoxic breast cancer cells by inhibiting Parkin-mediated mitophagy. Oncotarget. 2015;6(19):17417–17429. doi:10.18632/oncotarget.4002
61. Wang J, Fang Y, Yan L, et al. Erythroleukemia cells acquire an alternative mitophagy capability. Sci Rep. 2016;6(1):24641. doi:10.1038/srep24641
62. MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol. 2005;7(6):591–600. doi:10.1038/ncb1258
63. Liu J, Chen Z, Guo J, Wang L, Liu X. Ambra1 induces autophagy and desensitizes human prostate cancer cells to cisplatin. Biosci Rep. 2019;39:8.
64. Boyle KA, Van Wickle J, Hill RB, Marchese A, Kalyanaraman B, Dwinell MB. Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation. J Biol Chem. 2018;293(38):14891–14904. doi:10.1074/jbc.RA117.001469
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.