Back to Journals » International Journal of Nanomedicine » Volume 19
Enhancing Dendritic Cell Activation Through Manganese-Coated Nanovaccine Targeting the cGAS-STING Pathway
Authors Wang Q ![]() , Gao Y, Li Q
, Gao Y, Li Q ![]() , He A, Xu Q, Mou Y
, He A, Xu Q, Mou Y
Received 28 September 2023
Accepted for publication 5 January 2024
Published 11 January 2024 Volume 2024:19 Pages 263—280
DOI https://doi.org/10.2147/IJN.S438359
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Jie Huang
Qiyu Wang,1,* Ying Gao,2,* Qiang Li,1,* Ao He,1 Qinglin Xu,1 Yongbin Mou1
1Department of Oral Implantology, Nanjing Stomatological Hospital, Affiliated Hospital of Medical School, Research Institute of Stomatology, Nanjing University, Nanjing, 210008, People’s Republic of China; 2Department of Stomatology, the 964 Hospital, Changchun, Jilin, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qiang Li; Yongbin Mou, Department of Oral Implantology, Nanjing Stomatological Hospital, Affiliated Hospital of Medical School, Research Institute of Stomatology, Nanjing University, #30 Zhongyang Road, Nanjing, 210008, People’s Republic of China, Tel +86 25 83620236, Fax +86 25 83620202, Email [email protected]; [email protected]
Background: Nanovaccines have emerged as a promising vaccination strategy, exhibiting their capacity to deliver antigens and adjuvants to elicit specific immune responses. Despite this potential, optimizing the design and delivery of nanovaccines remains a challenge.
Methods: In this study, we engineered a dendritic mesoporous silica-based nanocarrier enveloped in a metal-phenolic network (MPN) layer containing divalent manganese ions and tannic acid (MSN@MT). This nanocarrier was tailored for antigen loading to serve as a nanovaccine, aiming to activate the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway in dendritic cells (DCs). Our experimental approach encompassed both cellular assays and mouse immunizations, allowing a comprehensive evaluation of the nanovaccine’s impact on DC activation and its influence on the generation of antigen-specific T-cell responses.
Results: MSN@MT demonstrated a remarkable enhancement in humoral and cellular immune responses in mice compared to control groups. This highlights the potential of MSN@MT to effectively trigger the cGAS-STING pathway in DCs, resulting in robust immune responses.
Conclusion: Our study introduces MSN@MT, a unique nanocarrier incorporating divalent manganese ions and tannic acid, showcasing its exceptional ability to amplify immune responses by activating the cGAS-STING pathway in DCs. This innovation signifies a stride in refining nanovaccine design for potent immune activation.
Keywords: nanocarrier, metal-phenolic network, manganese ions, tannic acid
Introduction
Nanovaccines represent a promising advancement in the field of vaccination, which uses nanoparticles as carriers and adjuvants to enhance antigen delivery and presentation within the immune system. Among the key players in this process, DCs play a critical role as primary antigen-presenting cells (APCs), initiating and orchestrating adaptive immune responses through antigen capture, processing, and presentation to T cells. The effective activation of DCs by nanovaccines is essential for the induction of potent and protective immune responses against diverse pathogens and tumors.1,2 However, current nanovaccine strategies encounter challenges related to DC activation, including limited uptake efficiency, rapid clearance, and intracellular degradation of antigens, resulting in suboptimal antigen cross-presentation and attenuated immune signaling.
To address these challenges, novel strategies have been proposed in the design of nanovaccines to achieve more efficient antigen delivery. These approaches encompass encapsulating antigens to enhance the stability of the carriers, preventing antigen degradation before uptake by DCs, and modifying targeting molecules to augment DC uptake.3,4 Lysosomal escape is a critical mechanism that enables nanoparticles to circumvent degradation within the acidic lysosomal environment, thereby releasing antigens into the cytosol—an indispensable process for effective cross-presentation by DCs.5 Chemical compounds with lysosomal escape capabilities, such as polyethyleneimine, have been developed.6 However, the design of these compounds often presents challenges including biological toxicity, biodegradability, and non-specificity for DCs.7 Hence, current research is directed towards pH-responsive, photo-sensitive, or thermo-sensitive biomaterials for achieving lysosomal escape.8–12 Apart from achieving proficient antigen delivery, the immunogenicity and adjuvant potential of nanovaccine are of paramount importance.13 The cGAS-STING pathway emerges as a focal point in the design of nanovaccines.14–17 Serving as a cytoplasmic DNA sensing pathway, it triggers the production of type I interferons (IFN) and other pro-inflammatory cytokines, facilitating DC maturation and antigen presentation. Through both effective antigen delivery and inherent immunogenicity, nanovaccines can provoke robust enough immune responses, making them promising candidates to combat a variety of diseases.
In this study, we have engineered a novel nanocarrier utilizing dendritic mesoporous silica nanoparticles (MSN) as the core platform for antigen loading, coated by a metal-phenolic network. This coating is formed by the complexation of tannic acid (TA) and manganese ions (Mn2+) (Figure 1a).18,19 The surface of the MSN is amine-functionalized, enabling efficient adsorption of the amount of antigens.20 As a polydentate ligand substance, TA can undergo coordination reactions with manganese ions, forming MPN coatings which self-assemble under a neutral environment and undergo degradation within the acidic environment of cellular lysosomes.21 The degradation of this coating is accompanied by the influx of water molecules, causing lysosomal rupture and facilitating lysosomal escape.22 Moreover, a substantial amount of Mn2+ is released into the DC cytoplasm. These Mn2+ possess the unique ability to independently activate the cGAS-STING pathway, even in the absence of dsDNA antigens.16,23 The orchestrated combination of MSN as an efficient antigen adsorbent, the pH-responsive coating for lysosomal escape, and the involvement of released Mn2+ for cGAS-STING activation collectively contribute to the enhanced immunogenicity and antigen presentation capability of our nanocarrier design (Figure 1b).
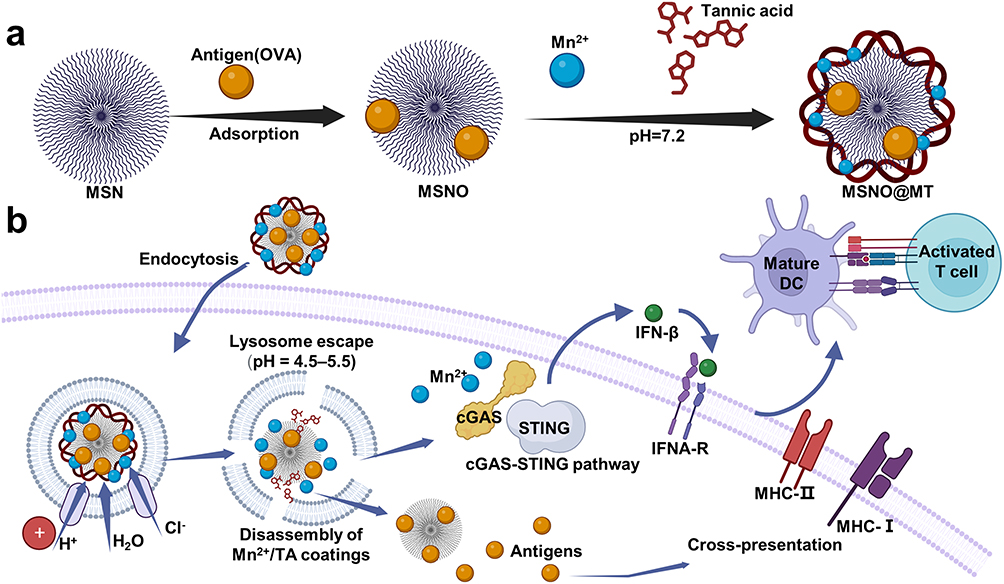 |
Figure 1 Enhancing immune response through pH-modulated self-assembly of nanocarriers targeting DCs. (a). The preparation process of antigen-loaded MSN@MT nanocarriers. coating of aminated dendritic mesoporous silica loaded with antigen via tannic acid and Mn2+ complexation under neutral pH conditions. Following model antigen OVA loading, the uncoated and coated MSNs are denoted as MSNO and MSNO@MT, respectively. (b). Schematic illustration of antigen delivery to DCs by MSN@MT. MSN@MT enhances DC uptake capacity, utilizing its lysosomal escape capability to heighten antigen delivery efficiency. Through Mn2+ activation, it triggers the cGAS-STING pathway, leading to the release of IFNβ, subsequently facilitating antigen cross-presentation and DC maturation. Consequently, this process elevates antigen-specific T-cell immune responses. |
Materials and Methods
Chemicals and Materials
Dendritic mesoporous silica nanoparticles were purchased from Zhengzhou Feynman Biotechnology Co., Ltd. (Zhengzhou, China). 3-Aminopropyltrimethoxysilane (APTES), ovalbumin (OVA), MnCl2, and TA were obtained from Sigma-Aldrich (MO, USA). DMEM culture medium, RPM 1640 culture medium, and phosphate-buffered saline (PBS) were purchased from HyClone (UT, USA). Fetal bovine serum (FBS) was obtained from Gibco (CA, USA). Granulocyte-macrophage colony-stimulating factor (GM-CSF) was purchased from PeproTech (NJ, USA). Fluorescein Isothiocyanate (FITC) -conjugated OVA was obtained from Solabo Biotechnology Co., Ltd. (Beijing, China). Flow cytometry (FCM) antibodies, including anti-CD80, anti-CD86, anti-CD40, anti-SIINFEKL-H2Kb, anti-CD11c, anti- major histocompatibility complex class II (MHC-II), anti-CD3, anti-CD4, anti-CD8, and anti-IFN-γ, were purchased from eBioscience (CA, USA). Western Blot (WB) antibodies, including β-actin, STING, Phospho-STING (p-STING), TANK-binding kinase 1 (TBK1), and Phospho-TBK1 (p-TBK1), Interferon regulatory Factor 3 (IRF3), Phospho-IRF-3 (p-IRF3) were purchased from Cell Signaling Technology (Denver, USA). PageRuler Prestained Protein Ladder (Thermo Scientific) was utilized as the molecular mass marker.
Animals and Cells
Female C57BL/6 mice aged 6–8 weeks were purchased from Jiangsu Jicuiyaokang Biotechnology Co., Ltd. (Jiangsu, China). The mice were fed and cared for according to the guidelines of the Institutional Animal Care and Use Committee of Nanjing University Medical School. DC2.4 cells were obtained from Wuhan Punoisi Life Technology Co., Ltd. (Wuhan, China) and were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% FBS. Bone marrow-derived dendritic cells (BMDCs) were extracted and induced using the Lutz method. Initially, 6–8-week-old C57BL/6 mice were selected, and long bones (tibia and femur) were used as the source of bone marrow. The bone marrow cells were flushed and filtered using RPMI 1640 medium, and the bone marrow cell suspension was seeded at a density of 1×106 cells in non-adherent culture dishes (RPMI 1640, 10% FBS, 20 ng/mL GM-CSF), 10 mL, designated as day 0. On day 3, an additional 10 mL of complete medium was added. On day 6 and day 8, half of the medium was replaced, and BMDCs were harvested on day 10. DC2.4 cells are cultured in RPMI1640 medium with 10% FBS and 1% Penicillin-Streptomycin solution, while B3Z cells are cultured in RPMI1640 medium with 8% FBS, 0.05 mM 2-mercaptoethanol, and 2 mM L-glutamine.
Preparation of MSN and MSN@MT
Ten mg of MS are taken and dispersed in 20 mL of anhydrous ethanol under sonication. Subsequently, the dispersed silica solution is added to a flask, and 10 μL of APTES is added dropwise while stirring at 60 °C for 24 hours. Following this, centrifugation is performed at 15,000 rpm, the supernatant is separated, and the precipitate is resuspended in anhydrous ethanol under sonication. This process is repeated three times, resulting in the animation modification of the MS surface, which is denoted as MSN afterward. One mg of antigen-loaded MSN is dissolved in 5 mL of deionized water (ddH2O) and subjected to sonication for 2 minutes. Then, 10 μL of TA (40 mg/mL) and 10 μL of MnCl2 (10 mg/mL) are added, followed by stirring for 5 minutes. Subsequently, 100 μL of PBS (500 mM, pH 7.0) is added, and stirring continues for 2 hours. Finally, the above liquid is collected and washed three times with ddH2O, and MSN@MT is obtained. For later antigen delivery experiments, the model antigen OVA was loaded onto MSN at various concentrations. The uncoated and coated MSNs are denoted as MSNO and MSNO@MT.
Characterization of Nanocarrier Size and Surface Charge
The hydration particle size was measured using the BT-90 Nanoparticle Size Analyzer (DanDong Bettersize Instruments Ltd., China), and transmission electron microscopy (TEM) and elemental mapping images were acquired using Talos F200X G2 (FEI, United States). Zeta potential was measured using the Malvern Zetasizer Nano ZS90 (Malvern Panalytical Ltd., United Kingdom).
Detection of the Loaded OVA Antigen of Nanocarrier
MSNs stored in anhydrous ethanol were centrifuged and then resuspended in ddH2O to form a 1 mg/mL nanoparticle suspension. OVA was added to the suspension, followed by sonication for 1 minute and stirring at room temperature for 2 hours. After stirring, the suspension was centrifuged at 14,800 rpm for 10 minutes. The remaining OVA content in the supernatant was determined using a bicinchoninic acid (BCA) Pierce assay kit (Thermo Fisher Scientific, USA). The amount of OVA loaded onto the MSN was calculated by subtracting the remaining OVA content in the supernatant from the initial OVA content in the system. The loading efficiency of MSN with OVA was then determined using the formula:
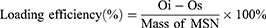
(Oi: initial OVA content in the reaction system; Os: OVA content in the supernatant after stirring)
Detection of the Release of OVA and Mn2+
To assess the release of OVA from MSNO@MT under neutral and acidic conditions, MSNO@MT was dispersed in PBS buffers with pH values of 5.5 and 7.4, followed by incubation in a 37°C water bath. Supernatants were centrifuged and collected at 2, 4, 12, 24, and 48 hours. After collection of the supernatant, PBS with the same pH is replenished. The protein concentration in the supernatant was measured using the BCA method, and the cumulative release of OVA was calculated. To assess the release of Mn2+ from MSNO@MT under neutral and acidic conditions, MSNO@MT was dispersed in PBS buffers with pH values of 5.5 and 7.4, followed by incubation in a 37°C water bath. Supernatants were collected at 2, 4, 8, 12, 24, 48, and 72 hours. After collection of the supernatant, PBS with the same pH is replenished. The manganese concentration in the supernatant was determined using Inductively Coupled Plasma Mass Spectrometry (ICP-MS) with the Agilent 7800 (United States), and the cumulative release of manganese ions was calculated.
Ultraviolet-Visible (UV-Vis) Absorption
The UV-Vis spectra of MSN and MSN@MT were measured in the range of 200~1000 nm using an M3 Multimode Microplate Reader (Molecular Devices; USA), with an equivalent MSN concentration of 1 mg/mL.
Cell Viability Assay
To investigate the biocompatibility of the nanovaccine, DC2.4 cells were seeded at a density of 1×105 cells per well in a 96-well plate and incubated overnight until they adhered to the plate. The samples, MSN and MSN@MT, were maintained at the same MSN concentration (10, 20, 50, and 100 μg/mL), along with PBS and OVA at equivalent concentrations, and co-incubated with the cells for 24 hours. Cell viability was evaluated using the cell counting kit-8 (CCK-8) assay following the experimental protocol provided by the manufacturer (Tokyo Chemical Industry Co., Ltd.; Japan). Specifically, the culture medium in all test wells was replaced with a complete medium containing 10% CCK-8 solution and incubated at 37°C for 1~4 hours. The supernatant was then used to measure the absorbance at 450 nm using an M3 Multimode Microplate Reader (Molecular Devices; USA). Cell viability was calculated using the following formula:
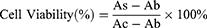
(As: Absorbance of the experimental well-containing medium, CCK-8, and the sample; Ac: Absorbance of the control well-containing medium and CCK-8 without the sample; Ab: Absorbance of the blank well-containing medium and CCK-8 without cells).
Hemolysis Assay
To assess the blood compatibility of the nanovaccine, we employed freshly collected mouse blood samples. Utilizing techniques including centrifugation, we isolated red blood cells from the whole blood. Diverse concentrations of nanovaccine samples were blended with the separated red blood cells. Notably, both positive and negative controls were introduced—a positive control recognized for inducing hemolysis (H2O) and a negative control that elicits no hemolytic effect (PBS). Subsequently, the samples underwent a 2-hour incubation under appropriate conditions to incite the hemolytic response. Following this, we gauged the absorbance of the samples at 570 nm with a microplate reader to gauge the extent of hemolysis based on the optical density (OD). Hemolysis is calculated with the following formula:

FCM Analysis of DC Uptake of Nanovaccine in vitro
To investigate the uptake of nanovaccine by DCs after in vitro stimulation, FITC-conjugated OVA protein was loaded onto the nanovaccine. DCs (1×106) were seeded in a 12-well plate and incubated overnight. Subsequently, they were stimulated with PBS, OVA-FITC, MSNO-FITC, and MSNO@MT-FITC with the equivalent OVA concentration at 20 μg/mL except for the PBS group. The cells were incubated at 37 °C, and at 1, 4, and 12 hours, DCs from each group were collected. After washing the cells three times with an FCM buffer, FCM analysis was performed to measure the mean fluorescence intensity (MFI) of FITC and the percentage of FITC-positive cells in the different treatment groups.
CLSM Observation on Lysosomal Escape Effect
To investigate the lysosomal escape effect of the nanovaccine, FITC-conjugated OVA was loaded onto the MSN. DC2.4 cells (1×105) were seeded in confocal dishes and incubated at 37°C overnight to allow the cells to adhere. The cells were then treated with OVA-FITC, MSNO-FITC, and MSNO@MT-FITC, all with the equivalent OVA-FITC concentration of 50 μg/mL. After 6 hours, Lysotracker Red (Beyotime) working solution was added to the cells and incubated for 0.5 hours at 37°C to stain the lysosomes. After rinsing with PBS, cells were fixed with cell fixation solution for 30 minutes. Subsequently, the dish was treated with a DAPI staining solution, followed by observation using confocal laser scanning microscopy (CLSM). Nine random fields were selected, and the average FITC fluorescence intensity and Pearson’s coefficient were calculated using ImageJ software to analyze the co-localization of FITC, and Lysotracker Red signals.22
FCM Analysis of DC Maturation and Antigen Presentation in vitro
To investigate the regulation of DC maturation and antigen presentation by the nanovaccine after in vitro stimulation, 1×106 BMDCs were cultured in a 12-well plate overnight. The cells were then stimulated with PBS, OVA, MSNO, and MSNO@MT. MSN and MSN@MT were loaded with the equivalent OVA concentration at 20 μg/mL except for the PBS group. After 24 hours of incubation at 37°C, the cells were collected and stained with FITC-conjugated anti-CD11c, PE-conjugated anti-CD80, and APC-conjugated anti-CD86, following the protocol provided by the manufacturer (eBioscience; USA). FCM was used to analyze the expression levels of CD80 and CD86 in CD11c-positive cells in the different treatment groups. Similar procedures were followed to detect the expression levels of CD40, SIINFEKL-H-2Kb, and MHC-II.
Antigen Presentation Assay
B3Z cells are a type of T cell hybridoma that express a specific T cell receptor for a peptide from OVA (SIINFEKL) presented by MHC class I molecules. They also have a reporter gene that produces β-galactosidase, an enzyme that can break down certain substrates, when they are activated by SIINFEKL. Chlorophenol red-β-d-galactopyranoside (CPRG) is one of the substrates that can be hydrolyzed by β-galactosidase to produce a red-colored product. In simple terms, BMDCs (1×106/mL) were co-cultured separately with OVA, MSNO, and MSNO@MT for 6 hours (with a consistent OVA concentration of 50 μg/mL). Following this, B3Z T cells (1×106/mL) were introduced for a 16-hour co-culture. PBS was employed as the negative control, and SIINFEKL (1 μg /mL) served as the positive control. After washed with PBS and resuspended in 100μL CPRG working solution (PBS containing 0.15 mM CPRG, 1 mM MgCl2, and 0.125% NP-40), cells incubate at 37 °C for 6~8 hours. Finally, the extent of antigen presentation was assessed by measuring absorbance at 595 nm using a microplate reader.
WB Analysis of DC Intracellular Pathway Activation
To investigate the regulation of gene expression in BMDCs after in vitro stimulation with the nanovaccine, 1×106 BMDCs were cultured in a 12-well plate overnight. The cells were then stimulated with PBS, OVA, MSNO, and MSNO@MT. MSNO and MSNO@MT were loaded with the same amount of OVA as in the OVA group, which was 20 μg/mL. After 24 hours of incubation, the cells were lysed using RIPA buffer, and the supernatant was collected to obtain total protein. The protein concentration was measured using a BCA protein assay kit and adjusted to the same protein concentration. Total protein from different treatment groups was separated using Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a Polyvinylidene fluoride (PVDF) membrane. The PVDF membrane was blocked with a blocking buffer (5% bovine serum albumin), followed by sequential incubation with primary antibodies and horseradish peroxidase (HRP)-conjugated secondary antibodies. Finally, the protein expression levels of β-actin, STING, p-STING, TBK1, p-TBK1, IRF3, and p-IRF3 were detected using a chemiluminescence imaging system (Tanon; China).
qPCR Analysis of DC Intracellular Pathway Activation
To investigate the regulation of gene expression in BMDCs after in vitro stimulation with the nanovaccine, 1×106 BMDCs were cultured in a 12-well plate overnight and then stimulated with PBS, OVA, MSNO, and MSNO@MT. The loading of OVA on MSN and MSN@MT was consistent with the OVA group, at 20 μg/mL. After 24 hours of incubation, total RNA was extracted from the cells using the Trizol method, and RNA content was assessed using a NanoDrop nucleic acid and protein detector. cDNA was synthesized using a reverse transcription reagent kit (Novogene; China). Quantitative real-time PCR (qPCR) was performed to analyze the gene expression levels of interferon regulatory factor 3 (IRF3), interleukin (IL)-6, IL-12b, tumor necrosis factor α (TNFα), IFNb1, C-X-C motif chemokine ligand 10 (CXCL10), and interferon-Induced protein with tetratricopeptide repeats 1 (IFIT1) in each group, with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the reference gene. The primer sequences used for qPCR amplification of the target gene were as follows: IRF3-F (GAGAGCCGAACGAGGTTCAG), IRF3-R (CTTCCAGGTTGACACGTCCG), IFNb1-F (TGGGTGGAATGAGACTATTGTTG), IFNb1-R (CTCCCACGTCAATCTTTCCTC), IL6-F (TAGTCCTTCCTACCCCAATTTCC), IL6-R (TTGGTCCTTAGCCACTCCTTC), TNFα-F (CCCTCACACTCAGATCATCTTCT), TNFα-R (GCTACGACGTGGGCTACAG), IL12b-F (GCTGCAGAAGACCTTCCAAGA), IL12b-R (TGGAGTTCCTGGTGTTGGTG), CXCL10-F (TGCTGCTGCTGCTGTCTTCT), CXCL10-R (TCCCTCAGCCAGATGCAATA), IFIT1-F (TGGCAAGAAGGAAGAAGAAGG), IFIT1-R (TGCTGCTGTTCTTCCTCTTC), GAPDH-F (AGGTCGGTGTGAACGGATTTG), GAPDH-R (TGTAGACCATGTAGTTGAGGTCA). The primers were designed using Primer Bank and synthesized by Genescript company.
ELISA of Cytokine Release
To investigate the production of cytokines by BMDCs after in vitro stimulation with the nanovaccine, 1×106 BMDCs were cultured in a 12-well plate overnight and then stimulated with PBS, OVA, MSNO, and MSNO@MT. After 24 hours of incubation, the cell culture supernatants were collected and centrifuged at 3,000 rpm for storage. The levels of cytokines, including IL-6, TNFα, and IFN-γ, in the samples were assessed using Enzyme-linked immunosorbent assay (ELISA) kits (Lianke; China). This analysis allowed the quantification of cytokine secretion from DCs in response to different nanovaccine treatments, providing valuable insights into the immune response elicited by the nanovaccine.
Construction of Animal Model and Evaluation of Immunological Effects
Female C57BL/6 mice aged 6–8 weeks were randomly divided into four groups (n=5). The animal experiments involved in this study were approved by the Animal Ethics and Welfare Committee of Nanjing University and all experimental procedures were performed in accordance with National Institutes of Health guidelines. On days 0, 7, and 14, each group received a subcutaneous injection of 25μL PBS, OVA, MSNO, or MSNO@MT into the left footpad. The OVA-loaded MSNO and MSNO@MT had the same OVA content as the OVA group, at 25 μg. After the final immunization on day 14, peripheral blood, inguinal lymph nodes (iLNs) on the treated side, and spleen were collected to assess the immune response. Additionally, major organs were sectioned and subjected to hematoxylin and eosin (H&E) staining to evaluate the in vivo toxicity of the nanovaccine.24
Peripheral blood was allowed to clot at 37 °C for 2 hours, followed by overnight refrigeration at 4°C. The blood samples were then centrifuged at 3000 rpm for 10 minutes at 4 °C to collect the serum. The serum OVA-specific Immunoglobulin G (IgG) levels were measured using ELISA, where the OD value at 450 nm was proportional to the antibody concentration.25 The relative titers of the experimental groups were calculated by dividing their OD values by the average OD value of the PBS group. To assess IFN-γ levels in mouse serum, a small mouse IFN-γ ELISA kit (MultiSciences) was employed following the manufacturer’s instructions.
To evaluate the maturity of inguinal lymph node DCs, lymph nodes adjacent to the treated side were ground into single-cell suspensions and subjected to FCM analysis using the same staining method as the in vitro DC maturation and antigen presentation level analysis. The spleens of immunized mice were ground and treated with red blood cell lysis buffer. The following approaches were used: (1) spleen single-cell suspensions were cultured in 96-well plates at a concentration of 1×106 cells per well in RPMI-1640 containing 10% FBS and 10μg/mL SIINFEKL peptide for 12 hours, followed by brefeldin A/ionomycin incubation for 6 hours.26,27 The stimulated spleen cells were then surface stained with anti-CD3, anti-CD4, and anti-CD8 antibodies, fixed and permeabilized using BD Cytofix/Cytoperm kit (BD biosciences, USA), and intracellularly stained with an anti-IFN-γ antibody using BD Perm/Wash buffer to detect the activation of OVA-specific cytotoxic T cells. (2) Spleen single-cell suspensions were seeded into pre-coated ELISPOT plates at a concentration of 2×105 cells per well. Using the mouse IFN-γ ELISPOT kit (Abcam, USA) and following the manufacturer’s instructions, the secretion of IFN-γ by immunized mice was assessed. Using the mouse IFN-γ ELISPOT kit (Abcam, USA) and following the manufacturer’s instructions, the secretion of IFN-γ by immunized mice was assessed with an ELISPOT reader. The current standards for spot detection are based on parameters optimized by the instrument following Mabtech guidelines.
Statistical Analysis
All experiments were independently repeated three times or more unless otherwise stated. The sample size and statistical data are indicated in the respective figure legends. All results are presented as mean ± standard deviation (S.D.). Statistical analysis was performed using students’ t-test (Group = 2) or one-way analysis of variance (one-way ANOVA) (Group ≥ 3) with multiple comparisons adjusted using Tukey’s test unless otherwise stated in GraphPad Prism 9.0 (GraphPad Software, USA). Significance levels are classified as ns (no significance, P > 0.05), * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001.
Results
Construction and Characterization of Nanocarriers
The MS is firstly modified with APTES for animation, which enhances its pH stability and dispersibility in aqueous environments and facilitates the binding of antigens, denoted as MSN afterward.28,29 Following antigen loading, Mn2+ and TA self-assemble to form a coating on MSN under a neutral pH environment, denoted as MSN@MT. Energy dispersive X-ray (EDX) elemental mapping illustrates the uniform distribution of amino and manganese elements on the silica spheres. Scanning transmission electron microscopy-high-angle annular dark-field (STEM-HAADF) imaging reveals the coating structure of unloaded MSN@MT (Figure 2a), and transmission electron microscopy (TEM) observation confirms the presence of the coating structure on both MSN and MSN@MT (Figure 2b). The UV-Vis spectroscopic analysis further reveals an absorption peak at 300 nm for MSN@MT, as compared to MSN (Figure 2c). After multiple washes with ddH2O and centrifugation, the precipitate of MSN@MT exhibits a consistent brownish-yellow color, which is distinct from the colorless uncoated MSN (Figure S1a). These findings collectively demonstrate the successful modification of MSN with Mn2+/TA coating. Dynamic light scattering (DLS) was employed to measure the hydrodynamic diameters of MS, MSN, MSNO@MT, and MSN@MT, yielding diameters at 336 ± 67.09, 340 ± 47.64, 345 ± 48.61 and 347 ± 49.90 nm, respectively (Figure 2d). These diameters are higher than those obtained from TEM images (259.8 nm of MSN and 262.0 nm of MSN@MT), attributed to the presence of the hydration layer (Figure S1b). The zeta potentials of dispersed particles, including MS, MSN, MSNO, MSNO@MT, and MSN@MT, were measured as shown in Figure 2e. The shift in surface charge from negative to positive for MS to MSN indicates the successful modification with APTES. Conversely, the shift from positive to negative for MSNO to MSNO@MT confirms the successful formation of the Mn2+/TA coating. OVA was loaded onto MSN at various concentrations. The uncoated and coated MSNs are denoted as MSNO and MSNO@MT, respectively. By quantifying the remaining protein content in the supernatant after adsorption, we determined that MSN possesses excellent antigen-loading capacity to form MSNO (Figure 2f). In addition, the loading capacities of tannic acid and manganese ions on dendritic mesoporous silica were quantified by measuring the component concentrations in the supernatant before and after the synthesis using a Spectrophotometer and ICP-MS, respectively. The amounts of OVA, tannic acid, and manganese ions loaded per mg of MSN are illustrated in Table S1. In the following experiments, the amount of OVA is 500 μg per mg of MSN unless otherwise specified. To validate the pH responsiveness of MSNO@MT, the cumulative release of OVA and Mn2+ was measured at 37 °C in PBS at pH 7.4 and 5.5 (simulating the acidic environment within lysosomes). As shown in Figure 2g and h, MSNO@MT exhibited a faster release rate and a greater amount of OVA and manganese ions at pH 5.5 compared to pH 7.4. This confirms that the Mn2+/TA coating can dissociate within endosomes, leading to the release of antigen and Mn2+.
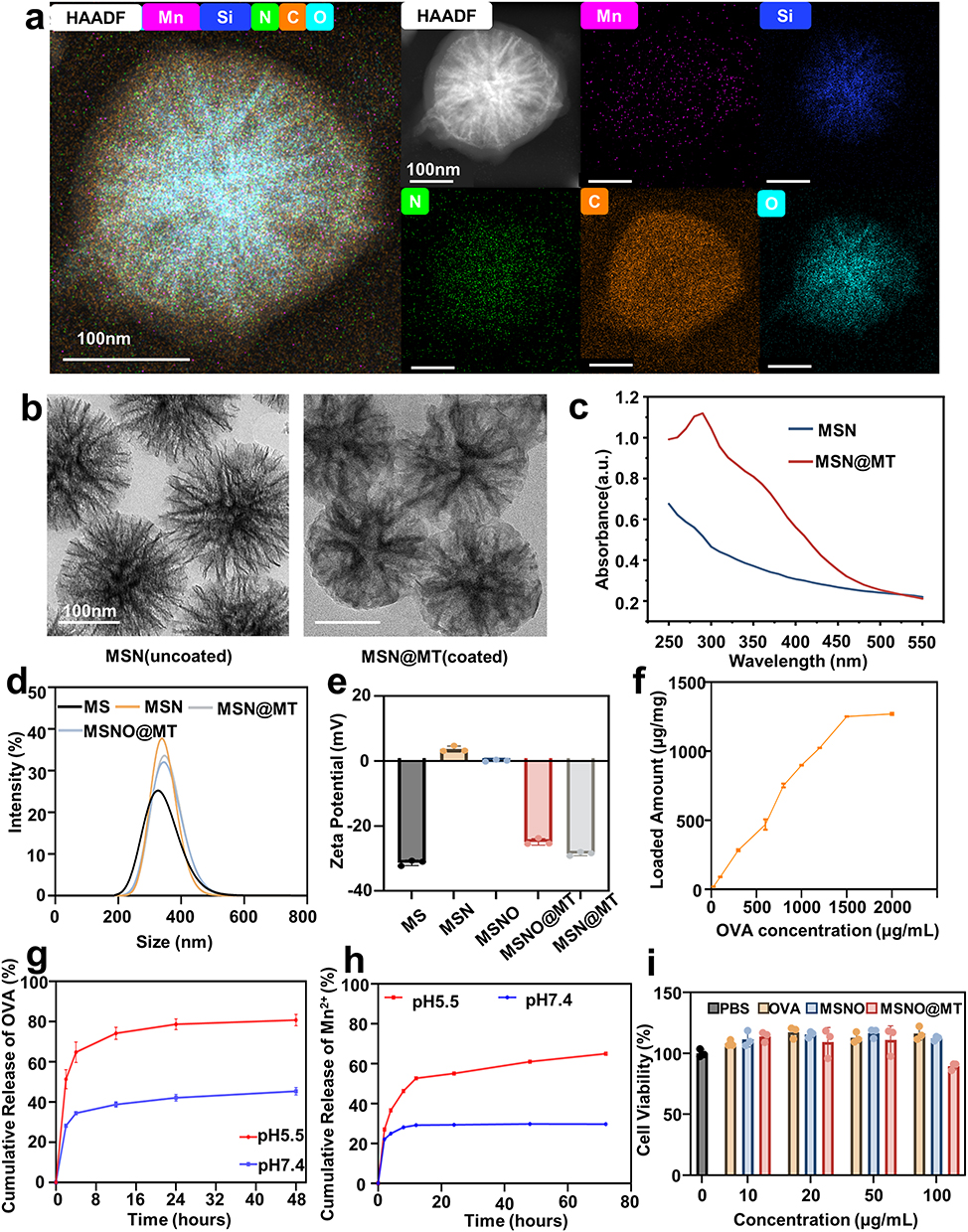 |
Figure 2 Characterization of the nanovaccine carrier MSN@MT. (a) STEM-HAADF images and EDX elemental mapping images of MSN@MT. Scale bars: 100 nm. Purple, blue, green, Orange, and cyan represent Mn, Si, N, C, and O elements, respectively. (b) TEM images of MSN and MSN@MT. (c) UV-Vis analysis comparison between MSN and MSN@MT. (d) Hydrodynamic diameters of MS, MSN, MSNO, MSNO@MT, and MSN@MT, as measured by dynamic light scattering (DLS). (e) zeta potential of MS, MSN, MSNO, MSNO@MT MSN@MT. (f) The ability of MSN to adsorb model antigen OVA at different initial concentrations (25~2000 μg/mL). (g) Cumulative release of OVA from MSNO@MT in pH 5.5 and 7.4. (h) Cumulative release of Mn2+ from MSNO@MT in pH 5.5 and 7.4. (i) Cell viability analysis of OVA, MSNO, and MSNO@MT co-cultured with DC2.4 cells at the illustrated concentrations (n = 3). |
Prior to conducting biological experiments, we further investigated the biocompatibility of the nanocarrier. Using the CCK-8 assay, we assessed the cell viability of DC2.4 cells after 24-hour co-culture with different concentrations of OVA, MSNO, and MSNO@MT, with PBS as the negative control. As shown in Figure 2i, even at concentrations as high as 100 μg/mL, MSNO@MT exhibited no toxicity (cell viability > 80%). Additionally, in the hemolysis assay, wherein fresh mouse red blood cells were co-cultured with the nanocarriers for 2 hours, both MSNO and MSNO@MT displayed negligible hemolysis even at a concentration of 200 μg/mL, underscoring their excellent blood compatibility (Figure S1c and d). The above results demonstrated our successful development of the designed nanocarrier, MSN@MT, which features an Mn2+/TA coating and excellent biocompatibility.
Effective DC Uptake and Lysosomal Escape of Nanovaccine in vitro
The efficiency of antigen uptake by DCs serves as a crucial criterion for assessing its antigen delivery efficacy. Therefore, we performed FCM cell-uptake analysis to evaluate the nanovaccine’s delivery efficacy into DCs. DC2.4 cells were cocultured with free OVA-FITC, MSN, and MSN@MT loaded with FITC-labeled OVA (referred to as MSNO-FITC and MSNO@MT-FITC respectively) and FCM analysis was conducted at 1, 4, and 12 hours post co-culturing. The results revealed that the MSNO@MT-FITC group exhibited a significantly enhanced average fluorescence intensity and a higher proportion of FITC-positive cells compared to the MSNO-FITC group (Figures 3a, b and S2a). Interestingly, in the MSNO@MT-FITC group, the average fluorescence intensity at 12 hours was slightly lower than that at 4 hours, possibly due to the metabolic and degradation processes of FITC-labeled OVA inside DCs. Moreover, CLSM showed a significant increase in FITC fluorescence intensity in the MSNO@MT group compared to the control group, aligning with the conclusions drawn from FCM analysis (Figure S2b and c). The results above proved that the Mn2+/TA coating significantly enhances the cellular uptake of the nanovaccines.
 |
Figure 3 MSN@MT facilitates DC cellular uptake and lysosomal escape. (a) Representative histograms and (b). mean fluorescence intensity of flow cytometric analysis of DC2.4 cells co-cultured with OVA-FITC, MSNO-FITC, and MSNO@MT-FITC for 1, 4, and 12 hours (n = 3). (c). Representative confocal microscopy images of DC2.4 cells co-cultured with OVA-FITC, MSNO-FITC, and MSNO@MT-FITC for 6 hours. The colors represent OVA-FITC (green), nucleus (blue), lysosomes (red), and co-localization of OVA-FITC and lysosomes (yellow). (d) The relative fluorescence intensity of red and green fluorescence in the MSNO-FITC and (e) MSNO@MT-FITC groups, represented by the white dashed line in the CLSM image in (a), measured in ImageJ software. (f) Pearson’s correlation coefficient values of MSNO and MSNO@MT, measured in ImageJ (n = 9). The indicated groups were analyzed using one-way ANOVA. ****P < 0.0001. |
To assess the nanovaccine’s ability to escape lysosomal degradation within DCs, we stained lysosomes in red and employed CLSM to observe the co-localization of antigens and lysosomes. In our observations, MSNO@MT-FITC (green) displayed a notable separation from lysosomes (red), while MSNO-FITC displayed a co-localization with lysosomes (yellow) (Figure 3c). Relative fluorescence intensity of antigens and lysosomes at the white dashed lines was measured and the MSNO@MT-FITC group exhibited fluorescence intensity separation compared to that of the MSNO-FITC group (Figure 3d and e). The Pearson correlation coefficient, which quantifies the degree of co-localization, was also calculated, and the result showed that MSNO@MT-FITC had a lower value compared to MSNO-FITC (Figure 3f). These results suggest that the metal-phenolic coating, formed through the intricate interaction between Mn2+ and TA, facilitates the successful escape of the nanovaccine from lysosomal confinement.
Induction of DC Maturation and Enhancement of Antigen Presentation by Nanovaccines
Mature dendritic cells (DCs) exhibit an upregulation of co-stimulatory molecules and release pro-inflammatory cytokines.30 This interplay between DCs and T cells is of paramount importance, as it shapes the interaction and communication between these two cell types.31 FCM analysis of BMDCs treated under various conditions revealed a significant upregulation of co-stimulatory molecules (CD80, CD86, and CD40) when co-cultured with MSNO@MT (Figure 4a–c). The proportion of CD80+ CD86+ DCs was 33%, surpassing the 16%, 18.5%, and 14.5% observed in the PBS, OVA, and MSNO treatment groups, respectively. CD80, CD86, and CD40 are essential co-stimulatory molecules for the interaction between BMDCs and T cells.32 Furthermore, the FCM analysis demonstrated that MSNO@MT treatment led to higher levels of MHC-II expression on DCs, consistent with the enhanced OVA uptake observed in this group (Figure 4d). The upregulation of MHC-II on MSNO@MT-treated DCs indicated the robust adjuvant effect of MSN@MT and the increased intracellular OVA level.
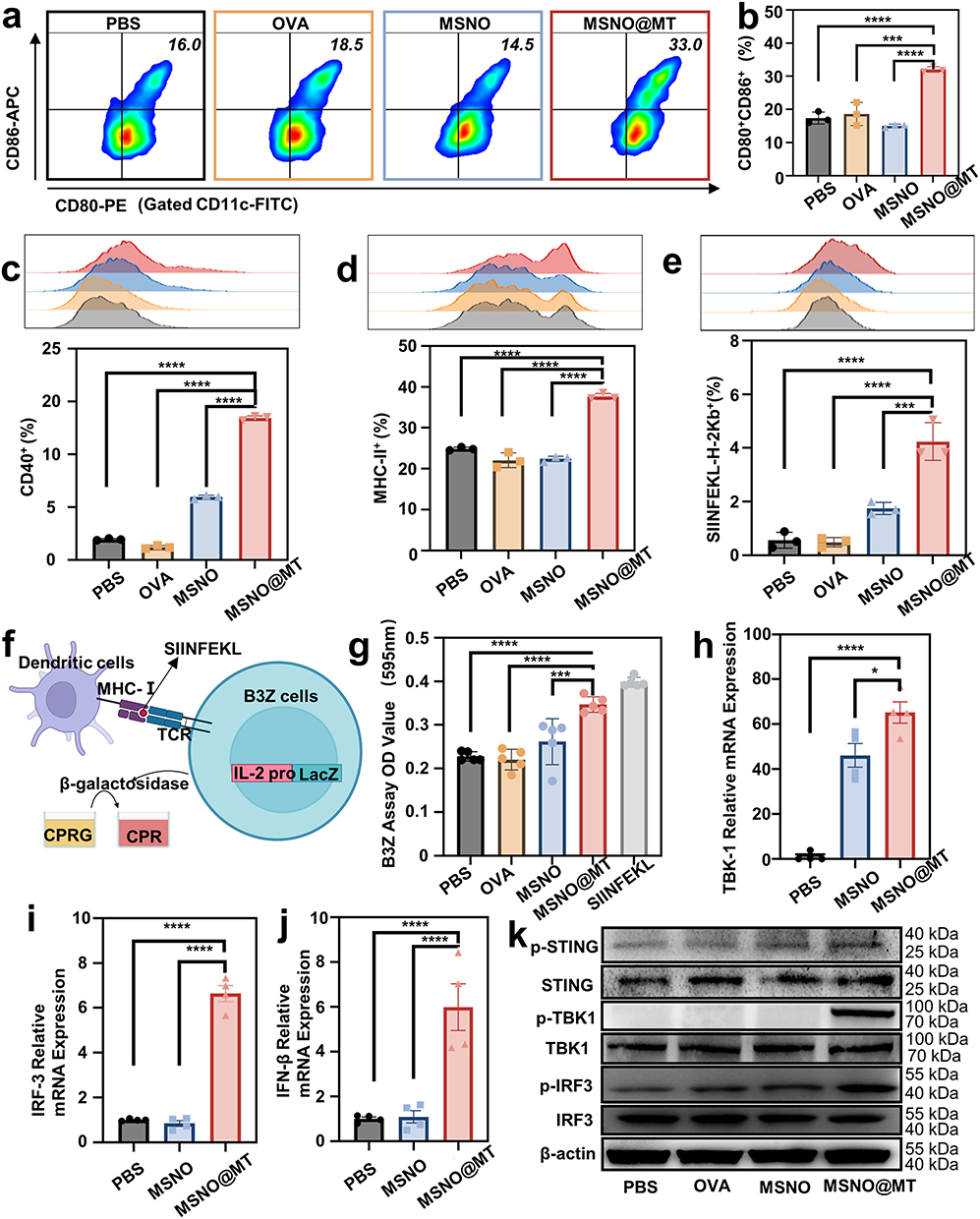 |
Figure 4 MSN@MT facilitates DC maturation and antigen cross-presentation while activating the cGAS-STING pathway. Representative (a) flow cytometry scatter plots and (b) proportions of cells positive for CD80+CD86+. Representative histograms and proportions of cells positive for (c) CD40+, and (d) MHC-II+ among BMDCs after 24 hours of co-culture with PBS, OVA, MSN, and MSN@MT, respectively (n = 3). (e). Histograms from flow cytometry illustrating the percentage of SIINFEKL+ cells among BMDCs after 24 hours of co-culture with PBS, OVA, MSNO, and MSNO@MT, respectively (n = 3). (f). Diagram illustrating the CPRG assay. (g). Absorbance at 595nm after 6 hours of co-culture of different treatment groups of B3Z cells with CPRG substrate (n=5). Relative mRNA expression levels of (h) TBK1, (i) IRF3, and (j) IFN-β in BMDCs after 24 hours of co-culture with PBS, OVA, MSNO, and MSNO@MT, respectively, analyzed by qPCR (n=4). (k). Protein expression levels of p-STING, p-TBK1, and p-IRF3 in BMDCs after 24 hours of co-culture with PBS, OVA, MSNO, and MSNO@MT, respectively, as analyzed through Western blot. The indicated groups were analyzed using one-way ANOVA. Significance levels are classified as *P < 0.05, ***P < 0.001, and ****P < 0.0001. |
The nanovaccines load OVA as a pathogen antigen, which DCs capture and process into small fragments, named SIINFEKL. These antigen fragments then bind to major histocompatibility complex class I (MHC-I) molecules on the surface of DCs, presenting MHC-I molecules (H-2Kb) bonded SIINFEKL.33 The interaction of CD8+ T cells with the antigen-MHC-I complex triggers their activation, proliferation, and differentiation into effector cells, such as cytotoxic T lymphocytes (CTLs).34 The FCM analysis revealed that, compared to OVA and MSNO-treated BMDCs, MSNO@MT-treated BMDCs showed approximately 8.72-fold and 2.43-fold upregulation of SIINFEKL-H-2Kb-positive cells (Figure 4e). Moreover, we utilized the CPRG assay to evaluate the in vitro cross-presentation of DC cells and the activation of T cells. B3Z T cells recognize the SIINFEKL-H-2Kb complex on DCs through their T-cell receptors (TCRs). Upon TCR activation, B3Z T cells produce β-galactosidase, which can be detected using CPRG (Figure 4f).35 Quantitative analysis revealed that in comparison to PBS, OVA, and MSNO, MSNO@MT induces a higher degree of cross-presentation by DCs, leading to enhanced T-cell activation (Figure 4g).
Furthermore, we observed that BMDCs treated with MSNO@MT secreted higher levels of IL-6 and TNF-α cytokines (Figure S3a and b). This finding was corroborated by qPCR results, which showed an upregulation of IL-6 and IL-12b mRNA expression in the MSNO@MT-treated group (Figure S3c and d). The secretion of IL-6 and TNF-α stimulates the activation and maturation of DCs, prompting their migration to lymph nodes, where they engage in interactions with T cells.36 The increased expression of IL-12b mRNA suggests that DCs treated with MSNO@MT are more capable of promoting the differentiation of naïve T cells into T helper type 1 cells (Th1 cells), thus enhancing cellular immune responses.37 These findings highlight the role of MSN@MT as a nanocarrier that promotes the maturation and antigen presentation of DCs.
Activation of the cGAS-STING Pathway and Upregulation of Downstream Immune Regulatory Signals by Nanovaccines
The cGAS-STING pathway is a pivotal signaling pathway that detects exogenous dsDNA and triggers the production of type I interferons.38 To validate our hypothesis that MSNO@MT, modified with Mn2+/TA, can activate the cGAS-STING pathway and trigger a potent immune response, we conducted qPCR analysis to examine mRNA expression levels in different treatment groups. Remarkably, the MSNO@MT-treated group exhibited notable upregulation of TBK1, IRF3 and IFNb1 levels compared to the PBS and MSNO groups (Figure 4h–j). TBK1 and IRF3, as integral components of the STING pathway, undergo reciprocal phosphorylation and activation, culminating in the transcriptional activation of interferons (IFNs) and IFN-stimulated genes (ISGs).39 Protein expression levels of p-STING, p-TBK1 and p-IRF3 were markedly elevated in the MSNO@MT-treated group, while the total STING, TBK1 and IRF3 protein levels remained unchanged (Figure 4k). IFNβ, a type I IFN, showcases antiviral, antiproliferative, and immunomodulatory properties.40 Binding to interferon-α/β receptor (IFNAR) and subsequent activation of the JAK-STAT pathway by IFNβ lead to the expression of Interferon-stimulated genes (ISGs), thus augmenting antigen presentation and T cell activation.31 Accordingly, we observed an upregulation in the mRNA expression of CXCL10 and IFIT1 in BMDCs treated with MSNO@MT (Figure S3e and f). These genes represent examples of ISGs with antiviral and immunomodulatory functions.38 Specifically, CXCL10 acts as a chemotactic factor, attracting T cells and natural killer cells (NK cells) to sites of infection, while IFIT1 is a protein that inhibits viral translation and replication.39
Nanovaccine-Mediated Immune Stimulation and Specific Immune Response Induction in vivo and ex vivo
To investigate the capacity of the nanocarrier MSN@MT to induce antigen-specific humoral and cellular immune responses in vivo, mice were grouped (n = 5 per group) and subcutaneously injected with PBS, OVA, MSNO, or MSNO@MT in the left footpad on days 0, 7, and 14. On day 21, serum, splenocytes, and iLNs cells were collected to evaluate the immunological effects of the nanovaccine (Figure 5a). We compared the body weight of mice during the immunization period (days 0, 7, 14, 21) and observed no differences among the treatment groups (Figure S4a). Histological examination of the major organs (heart, liver, spleen, lung, and kidney) from mice in each treatment group also revealed no apparent signs of inflammatory infiltration or toxic effects (Figure S4b). In the collected DCs from the iLNs, the proportion of co-stimulatory molecules CD80+CD86+ and CD40+ in the MSNO@MT-treated group was significantly higher than that in the control group (Figure 5b–d). Additionally, the MSNO@MT group exhibited a significantly higher population of antigen cross-presenting DCs (CD11c+SIINFEKL+ DCs) compared to that of the control group (Figure 5e). These findings are consistent with the in vitro experiments and indicate that MSN@MT-loaded antigens have a greater capacity to activate DCs, inducing their maturation and antigen presentation.
 |
Figure 5 MSN@MT enhances DC-mediated immune responses in mice. (a). Illustration of the mouse immunization model and experimental procedure. Nanovaccine is injected into the footpad of mice on days 0, 7, and 14. On day 21, serum, inguinal lymph nodes, and spleens are collected to assess humoral immunity, dendritic cell maturation, and T-cell activation within the mice. (b) Scatter plots and (c) the proportion of CD80+CD86+ DCs extracted from inguinal lymph nodes of mice in different treatment groups. (d). Proportion of CD40+ DCs extracted from inguinal lymph nodes of mice in different treatment groups, along with representative histograms. (e) Proportion of SIINFEKL+ DCs extracted from inguinal lymph nodes of mice in different treatment groups, along with representative histograms. (f) TNF-α and (g) IFN-γ levels in the serum of mice from different treatment groups measured by ELISA. (h). Relative titers of OVA-specific IgG and (i) the ratio of OVA-specific IgG1 to IgG2a in the serum of mice from different treatment groups (n = 5/group). The indicated groups were analyzed using one-way ANOVA. Significance levels are classified as ns (no significance, P>0.05), **P < 0.01, ***P < 0.001, and ****P < 0.0001. |
We further observed heightened levels of TNF-α and IFN-γ in MSNO@MT-treated mice serum, indicative of a cell-mediated immune response against intracellular pathogens (Figure 5f and g). Additionally, within the MSNO@MT-treated group, we observed an increased level of OVA-specific IgG antibodies in the serum (Figure 5h), suggesting that MSNO@MT effectively stimulates a heightened antigen-specific humoral response. Moreover, IgG subclasses, particularly the ratio of OVA-specific IgG1 to IgG2a, which reflects the type of responsive helper T cell, were measured. The ratio in MSNO@MT group was higher than 1, which indicates that MSNO@MT predominantly induced a T helper type 2 (Th2) response (Figure 5i).
To evaluate antigen-specific T-cell immunity, extracted splenocytes were restimulated with SIINFEKLE ex vivo. Utilizing the ELISPOT assay, we quantified the number of cells secreting IFN-γ, a signature of Th1 responses and CTL activity.41 Splenocytes from mice injected with MSN@MT-loaded OVA demonstrated increased IFN-γ spot forming units (SFU) upon secondary antigen exposure (Figure 6a and b). To delve into the specific T cell subsets responsible for IFN-γ secretion, we applied intracellular cytokine staining (ICS) in conjunction with FCM, revealing a notable increase in the proportion of IFN-γ-positive cells within the CD3+CD8+ and CD3+CD4+ T cell subsets of the MSNO@MT-treated group compared to that of the control group (Figure 6c–f). This underscores the notion that the MSNO@MT enhances the activation and amplification of CD8+ and CD4+ T cells producing IFN-γ. Collectively, these outcomes suggest that the nanovaccine elicits a robust Th1 response and cytotoxic T-cell response, which holds critical significance for the clearance of intracellular pathogens.
 |
Figure 6 MSN@MT induces specific T-cell immunity in mice. (a) Representative images and (b) data analysis of spleen cells measured using ELISPOT to detect cells producing IFN-γ spots upon restimulation. (c) Scatter plots and (d) data analysis showing the proportion of CD3+CD8+ cells secreting IFN-γ among spleen cells from immunized mice after in vitro restimulation. (e) Scatter plots and (f) data analysis depicting the proportion of CD3+CD4+ cells secreting IFN-γ among spleen cells from immunized mice after in vitro restimulation. The indicated groups were analyzed using one-way ANOVA. Significance levels are classified as **P < 0.01, ***P < 0.001, and ****P < 0.0001. |
Discussion
In addressing the current challenges in nanovaccine design, our study aimed to enhance antigen delivery efficiency and improve adjuvant performance. The development of the MSN@MT nanocarrier involved the complexation of manganese ions and tannic acid, resulting in a MPN coating on dendritic mesoporous silica. This novel nanocarrier was designed to overcome limitations in existing vaccine technologies.
Our rationale for creating MSN@MT centered on its unique design features. The MPN coating, responsive to pH changes, was intended to facilitate lysosomal escape, thereby enhancing the delivery of antigens to the cytoplasm of dendritic cells. Simultaneously, the inclusion of manganese ions aimed to activate the cGAS-STING pathway, further augmenting the adjuvant performance of the nanocarrier.
In our in vitro experiments, we successfully observed the desired lysosomal escape effect of MSN@MT. The activation of the cGAS-STING pathway was also validated. Furthermore, both in vitro and in vivo experiments substantiated the efficacy of MSN@MT loaded with the model antigen OVA. The nanocarrier promoted dendritic cell maturation and antigen presentation, leading to robust T-cell immune responses and humoral immune responses.
In the field of nanovaccine development, similar strategies of modifying dendritic mesoporous silica have been employed by other researchers. For instance, Chen et al used Fe(III) and tannic acid complexation to modify mesoporous silica nanoparticles, demonstrating the feasibility of the MPN coating strategy for lysosomal escape.22 Zhou et al utilized polyethyleneimine (PEI) and Fe (III)/TA MPN to enhance lysosomal escape in dendritic mesoporous silica, achieving effective tumor immunotherapy.42 Notably, our design, MSN@MT, avoided the use of toxic PEI and demonstrated good lysosomal escape effects without its presence.
Our results demonstrate the activation of the cGAS-STING pathway by MSNO@MT, leading to type I IFN secretion in DCs both in vitro (Figure 4h–k). Moreover, splenocytes from MSNO@MT-treated mice exhibited a substantial increase in IFN-γ secretion, including from CD4+ and CD8+ T cells, reinforcing the coherence of our findings (Figure 6a–f). Under physiological conditions, the increased levels of IFN-γ may exert an influence on the differentiation of both CD4+ and CD8+ T cells, favoring the Th1 cell phenotype. This Th1 bias could potentially contribute to heightened IgG2a production, thereby resulting in a relative increase in its proportion. The total OVA-specific IgG content in the serum of mice treated with MSNO@MT increased, indicating a stronger humoral immune response. However, the IgG1/IgG2a ratio greater than 1 often suggests a Th2-biased immune response (Figure 5i). Yet, IgG1 and IgG2a are generally associated with different immune responses and may be governed by different regulatory mechanisms. Nanovaccines may modulate the Th1/Th2 balance through pathways other than the activation of the cGAS-STING pathway. For example, studies on aluminum-based vaccines by Marichal et al revealed an IRF-independent mechanism for upregulating IgG1 and IRF3-dependent mechanisms for activating Th2 responses.43 In summary, mice immunized with MSNO@MT showed a significant increase in IFN-γ production, indicating the activation of cellular immunity and accompanied by increased antibody production to clear intracellular antigens. In conclusion, the substantial increase in interferon-gamma production in mice immunized with MSNO@MT signifies the activation of cellular immunity. This is accompanied by an augmented antibody response, indicating the potential efficacy of MSNO@MT as a nanocarrier and adjuvant in promoting both cellular and humoral immune responses.
The MSN@MT nanocarrier exhibits broad applicability in nanovaccine design. Its high antigen adsorption capacity, demonstrated with OVA, suggests versatility for various antigens. Moreover, the activation of the cGAS-STING pathway implies potential for nucleic acid-type antigens, particularly in the context of infectious diseases. However, the current design, relying on pH responsiveness alone, limits external control over vaccine behavior. Future research should explore the incorporation of new components to enable more precise modulation, considering factors such as light or sound stimuli.
In conclusion, our study introduces the MSN@MT nanocarrier as a promising platform for nanovaccine development. Its successful demonstration of efficient lysosomal escape, robust immune responses, and avoidance of toxic components positions it favorably for further exploration and potential clinical applications.
Conclusion
In summary, MSN@MT facilitated efficient uptake by DCs and enabled lysosomal escape upon cellular entry, resulting in enhanced antigen delivery. Subsequently, improved antigen cross-presentation was triggered, leading to activation of the cGAS-STING pathway and subsequent production of type I interferon, thereby promoting DC maturation. In addition to the protein-based vaccine demonstrated in this work, the MSN@MT platform can also be utilized for developing vaccines loaded with new antigens in the future, such as personalized nanovaccines carrying peptides and nucleic acids. These advancements hold promise for the continued development and optimization of the nanocarrier for diverse therapeutic applications.
Abbreviations
MSN, dendritic mesoporous silica nanoparticles; MSNO, dendritic mesoporous silicon adsorbed with ovalbumin; MSN@MT, dendritic mesoporous silica nanoparticles coated with the network of Mn2+ and tannic acid; MSNO@MT, dendritic mesoporous silica nanoparticles adsorbed with ovalbumin, coated with the network of Mn2+ and tannic acid; MSNO-FITC, dendritic mesoporous silicon adsorbed with fluorescein isothiocyanate-conjugated ovalbumin; MSNO@MT-FITC, dendritic mesoporous silicon adsorbed with fluorescein isothiocyanate-conjugated ovalbumin, coated with the network of Mn2+ and tannic acid; cGAS, cyclic GMP-AMP synthase; STING, stimulator of interferon genes; DC, dendritic cell; APCs, antigen-presenting cells; TA, tannic acid; IFN, interferons; APTES, 3-aminopropyltrimethoxysilane; PBS, phosphate-buffered saline; FBS, Fetal bovine serum; GMCSF, granulocyte-macrophage colony-stimulating factor; FITC, fluorescein isothiocyanate; OVA, ovalbumin; OVA-FITC, fluorescein isothiocyanate-conjugated ovalbumin; FCM, flow cytometry; MHCII, major histocompatibility complex class II; MHC-I, major histocompatibility complex class I; WB, Western blot; p-STING, phospho-stimulator of interferon genes; TBK1, TANK-binding kinase 1; p-TBK1, phospho-TANK-binding kinase 1; RPMI, Roswell Park Memorial Institute; BMDC, bone marrow-derived dendritic cell; ddH2O, deionized water; BCA, bicinchoninic acid; UV-Vis, ultraviolet-visible; CCK-8, cell counting kit-8; OD, optical density; MFI, mean fluorescence intensity; CLSM, confocal laser scanning microscopy; CPRG, chlorophenol red-β-d-galactopyranoside; PVDF, polyvinylidene fluoride; HRP, horseradish peroxidase; IL, Interleukin; TNFα, Tumor necrosis factor α; CXCL10, C-X-C motif chemokine ligand 10; IFIT1, interferon-induced protein with tetratricopeptide repeats 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ELISA, enzyme-linked immunosorbent assay; iLN, inguinal lymph node; H&E, hematoxylin and eosin; one way-ANOVA, one way-analysis of variance; EDX, energy dispersive X-ray; STEM-HAADF, scanning transmission electron microscopy-high-angle annular dark-field; TEM, transmission electron microscopy; DLS, dynamic light scattering; TCR, T-cell receptors; IFNAR, interferon-α/β receptor; ISGs, interferon-stimulated genes; NK cells, natural killer cells; IgG, Immunoglobulin G; CTLs, cytotoxic T lymphocytes; Th1 cells, T helper type 1 cells; Th2 cells, T helper type 2 cells.
Acknowledgments
The authors acknowledge funding from the Natural Science Foundation of Jiangsu Province (BK20200710), and the Jiangsu Provincial Key Research and Development Program (BE2020629). The authors also want to thank BioRender (biorender.com) for providing drawing elements.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zhou J, Kroll A, Holay M, Fang R, Zhang L. Biomimetic nanotechnology toward personalized vaccines. Adv Mater. 2020;32(13):e1901255. doi:10.1002/adma.201901255
2. Dong H, Li Q, Zhang Y, Ding M, Teng Z, Mou Y. Biomaterials facilitating dendritic cell-mediated cancer immunotherapy. Adv Sci. 2023;10(18):e2301339. doi:10.1002/advs.202301339
3. Wang C, Ye Y, Hochu G, Sadeghifar H, Gu Z. Enhanced cancer immunotherapy by microneedle patch-assisted delivery of anti-PD1 antibody. Nano Lett. 2016;16(4):2334–2340. doi:10.1021/acs.nanolett.5b05030
4. Chattopadhyay S, Chen J, Chen H, Hu C. Nanoparticle vaccines adopting virus-like features for enhanced immune potentiation. Nanotheranostics. 2017;1(3):244–260. doi:10.7150/ntno.19796
5. Voltà-Durán E, Parladé E, Serna N, Villaverde A, Vazquez E, Unzueta U. Endosomal escape for cell-targeted proteins. Going out after going in. Biotechnol Adv. 2023;63:108103. doi:10.1016/j.biotechadv.2023.108103
6. Li N, Zhang T, Wang R, et al. Homotypic targeted nanoplatform enable efficient chemoimmunotherapy and reduced DOX cardiotoxicity in chemoresistant cancer via TGF-β1 blockade. J Control Release. 2023;361:147–160. doi:10.1016/j.jconrel.2023.07.063
7. Yang X, Wei Y, Zheng L, et al. Polyethyleneimine-based immunoadjuvants for designing cancer vaccines. J Mat Chem B. 2022;10(40):8166–8180. doi:10.1039/d2tb01358
8. Shen X, Dirisala A, Toyoda M, et al. pH-responsive polyzwitterion covered nanocarriers for DNA delivery. J Control Release. 2023;360:928–939. doi:10.1016/j.jconrel.2023.07.038
9. Pang X, Liang S, Wang T, et al. Engineering thermo-pH dual responsive hydrogel for enhanced tumor accumulation, penetration, and chemo-protein combination therapy. Int j Nanomed. 2020;15:4739–4752. doi:10.2147/ijn.S253990
10. Alkhazaleh A, Elfagih S, Chakka L, et al. Development of proanthocyanidin-loaded mesoporous silica nanoparticles for improving dental adhesion. Mol Pharmaceut. 2022;19(12):4675–4684. doi:10.1021/acs.molpharmaceut.2c00728
11. Yu H, Liu Y, Zheng F, Chen W, Wei K. Erianin-loaded photo-responsive dendrimer mesoporous silica nanoparticles: exploration of a psoriasis treatment method. Molecules. 2022;27(19). doi:10.3390/molecules27196328
12. Figari G, Gonçalves J, Diogo H, Dionísio M, Farinha J, Viciosa M. Understanding fenofibrate release from bare and modified mesoporous silica nanoparticles. Pharmaceutics. 2023;15(6):1624. doi:10.3390/pharmaceutics15061624
13. He A, Li X, Dai Z, et al. Nanovaccine-based strategies for lymph node targeted delivery and imaging in tumor immunotherapy. J Nanobiotechnol. 2023;21(1):236. doi:10.1186/s12951-023-01989-x
14. Vanpouille-Box C, Hoffmann J, Galluzzi L. Pharmacological modulation of nucleic acid sensors - therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2019;18(11):845–867. doi:10.1038/s41573-019-0043-2
15. Qiao N, Wang H, Xu Y, et al. A MnAl double adjuvant nanovaccine to induce strong humoral and cellular immune responses. J Control Release. 2023;358:190–203. doi:10.1016/j.jconrel.2023.04.036
16. Gu Y, Lin S, Wu Y, et al. Targeting STING activation by antigen-inspired MnO nanovaccines optimizes tumor radiotherapy. Adv Healthcare Mater. 2023;12(12):e2300028. doi:10.1002/adhm.202300028
17. Li J, Ren H, Qiu Q, et al. Manganese coordination micelles that activate stimulator of interferon genes and capture in situ tumor antigens for cancer metalloimmunotherapy. ACS Nano. 2022;16(10):16909–16923. doi:10.1021/acsnano.2c06926
18. Li Q, Teng Z, Tao J, et al. Elastic nanovaccine enhances dendritic cell-mediated tumor immunotherapy. Small. 2022;18(32):e2201108. doi:10.1002/smll.202201108
19. Xu C, Dobson H, Yu M, et al. STING agonist-loaded mesoporous manganese-silica nanoparticles for vaccine applications. J Control Release. 2023;357:84–93. doi:10.1016/j.jconrel.2023.03.036
20. Nguyen T, Choi Y, Kim J. Mesoporous silica as a versatile platform for cancer immunotherapy. Adv Mater. 2019;31(34):e1803953. doi:10.1002/adma.201803953
21. Ejima H, Richardson J, Liang K, et al. One-step assembly of coordination complexes for versatile film and particle engineering. Science. 2013;341(6142):154–157. doi:10.1126/science.1237265
22. Chen J, Li J, Zhou J, et al. Metal-phenolic coatings as a platform to trigger endosomal escape of nanoparticles. ACS nano. 2019;13(10):11653–11664. doi:10.1021/acsnano.9b05521
23. Zhao Z, Ma Z, Wang B, Guan Y, Su X, Jiang Z. Mn directly activates cGAS and structural analysis suggests Mn induces a noncanonical catalytic synthesis of 2ʹ3’-cGAMP. Cell Rep. 2020;32(7):108053. doi:10.1016/j.celrep.2020.108053
24. Zhang J, Fan B, Cao G, et al. Direct presentation of tumor-associated antigens to induce adaptive immunity by personalized dendritic cell-mimicking nanovaccines. Adv Mater. 2022;34(47):e2205950. doi:10.1002/adma.202205950
25. Tian D, Yang L, Wang S, et al. Double negative T cells mediate Lag3-dependent antigen-specific protection in allergic asthma. Nat Commun. 2019;10(1):4246. doi:10.1038/s41467-019-12243-0
26. Picker L, Singh M, Zdraveski Z, et al. Direct demonstration of cytokine synthesis heterogeneity among human memory/effector T cells by flow cytometry. Blood. 1995;86(4):1408–1419. doi:10.1182/blood.V86.4.1408.bloodjournal8641408
27. Zhang Y, Li Q, Ding M, et al. Endogenous/exogenous nanovaccines synergistically enhance dendritic cell-mediated tumor immunotherapy. Adv Healthcare Mater. 2023;12(17):e2203028. doi:10.1002/adhm.202203028
28. Zhang C, Dong Y, Gao J, Wang X, Jiang Y. Radial porous SiO2 nanoflowers potentiate the effect of antigen/adjuvant in antitumor immunotherapy. Front Chem Sci Eng. 2021;15(5):1296–1311. doi:10.1007/s11705-020-2034-6
29. Hao P, Peng B, Shan B-Q, Yang T-Q, Zhang K. Comprehensive understanding of the synthesis and formation mechanism of dendritic mesoporous silica nanospheres. Nanoscale Adv. 2020;2(5):1792–1810. doi:10.1039/D0NA00219D
30. Kim M, Kim J. Properties of immature and mature dendritic cells: phenotype, morphology, phagocytosis, and migration. RSC Adv. 2019;9(20):11230–11238. doi:10.1039/c9ra00818g
31. Luo M, Wang H, Wang Z, et al. A STING-activating nanovaccine for cancer immunotherapy. Nature Nanotechnol. 2017;12(7):648–654. doi:10.1038/nnano.2017.52
32. Edner NM, Carlesso G, Rush JS, Walker LSK. Targeting co-stimulatory molecules in autoimmune disease. Nat Rev Drug Discov. 2020;19(12):860–883. doi:10.1038/s41573-020-0081-9
33. Bonifaz L, Bonnyay D, Charalambous A, et al. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med. 2004;199(6):815–824. doi:10.1084/jem.20032220
34. Barry M, Bleackley R. Cytotoxic T lymphocytes: all roads lead to death. Nat Rev Immunol. 2002;2(6):401–409. doi:10.1038/nri819
35. Kanaseki T, Shastri N. Biochemical analysis of naturally processed antigenic peptides presented by MHC class I molecules. In: van Endert P, editor. Antigen Processing: Methods and Protocols. Springer New York; 2019:101–108.
36. Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9(4):271–285. doi:10.1038/nri2526
37. Schülke S. Induction of Interleukin-10 producing dendritic cells as a tool to suppress allergen-specific T helper 2 responses. Front Immunol. 2018;9:455. doi:10.3389/fimmu.2018.00455
38. Zhang J, Wu X, Hu Y, Chang M. A novel transcript isoform of TBK1 negatively regulates type I IFN production by promoting proteasomal degradation of TBK1 and lysosomal degradation of IRF3. Front Immunol. 2020;11:580864. doi:10.3389/fimmu.2020.580864
39. Yamashiro L, Wilson S, Morrison H, et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat Commun. 2020;11(1):3382. doi:10.1038/s41467-020-17156-x
40. Mazewski C, Perez R, Fish E, Platanias L. Type I Interferon (IFN)-regulated activation of canonical and non-canonical signaling pathways. Front Immunol. 2020;11:606456. doi:10.3389/fimmu.2020.606456
41. Ding H, Wang G, Yu Z, Sun H, Wang L. Role of interferon-gamma (IFN-γ) and IFN-γ receptor 1/2 (IFNγR1/2) in regulation of immunity, infection, and cancer development: IFN-γ-dependent or independent pathway. Biomed Pharmacother. 2022;155:113683. doi:10.1016/j.biopha.2022.113683
42. Zhou X, Su Q, Zhao H, Cao X, Yang Y, Xue W. Metal-phenolic network-encapsulated nanovaccine with pH and reduction dual responsiveness for enhanced cancer immunotherapy. Mol Pharmaceut. 2020;17(12):4603–4615. doi:10.1021/acs.molpharmaceut.0c00802
43. Marichal T, Ohata K, Bedoret D, et al. DNA released from dying host cells mediates aluminum adjuvant activity. Nature Med. 2011;17(8):996–1002. doi:10.1038/nm.2403
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.