")
Back to Journals » ImmunoTargets and Therapy » Volume 4
Enhancement of antibody-dependent cell mediated cytotoxicity: a new era in cancer treatment
Authors Rajasekaran N , Chester C, Yonezawa A, Zhao X, Kohrt H
Received 11 December 2014
Accepted for publication 11 February 2015
Published 15 May 2015 Volume 2015:4 Pages 91—100
DOI https://doi.org/10.2147/ITT.S61292
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Michael Shurin
Video abstract presented by Cariad Chester
Views: 1299
Narendiran Rajasekaran,1,* Cariad Chester,1,* Atsushi Yonezawa,1,2 Xing Zhao,1,3 Holbrook E Kohrt1
1Division of Oncology, Stanford School of Medicine, Stanford University, Stanford, CA, USA; 2Department of Clinical Pharmacology and Therapeutics, Kyoto University Hospital, Kyoto, Japan; 3Tissue Engineering and Stem Cells Research Center, Department of Immunology, Guiyang Medical University, Guiyang, Guizhou Province, People's Republic of China
*These authors contributed equally to this work
Abstract: The therapeutic efficacy of some anti-tumor monoclonal antibodies (mAbs) depends on the capacity of the mAb to recognize the tumor-associated antigen and induce cytotoxicity via a network of immune effector cells. This process of antibody-dependent cell-mediated cytotoxicity (ADCC) against tumor cells is triggered by the interaction of the fragment crystallizable (Fc) portion of the mAb with the Fc receptors on effector cells like natural killer cells, macrophages, γδ T cells, and dendritic cells. By augmenting ADCC, the antitumor activity of mAbs can be significantly increased. Currently, identifying and developing therapeutic agents that enhance ADCC is a growing area of research. Combining existing tumor-targeting mAbs and ADCC-promoting agents that stimulate effector cells will translate to greater clinical responses. In this review, we discuss strategies for enhancing ADCC and emphasize the potential of combination treatments that include US Food and Drug Administration-approved mAbs and immunostimulatory therapeutics.
Keywords: ADCC, NK cell, reovirus, TLR, CD137
Introduction
In the last decade, therapeutic antibodies have become a backbone of routine treatment strategies for a variety of different tumor types (Table 1). Currently, 18 different antibodies have obtained US Food and Drug Administration (FDA) approval for use in oncologic settings.1 In addition to triggering direct induction of cell death, tumor-targeting antibodies exert antitumor properties through three important innate effector mechanisms: complement-dependent cytotoxicity, antibody-dependent cellular phagocytosis (ADCP), and antibody-dependent cell-mediated cytotoxicity (ADCC). Complement-dependent cytotoxicity is the result of the fragment crystallizable (Fc) region of an antigen–immunoglobulin complex triggering a cascade of more than 30 proteins that culminates in the formation of the membrane-attack complex, an amalgam of subunits that functions to perforate the phospholipid bilayer of the target cell and induce lysis.2 ADCP describes target cell elimination by the innate network of phagocytic cells, primarily neutrophils, monocytes, and macrophages. ADCP and ADCC are understood as complementary processes that emphasize the importance of innate immune cells in the therapeutic efficacy of some monoclonal antibodies (mAbs).
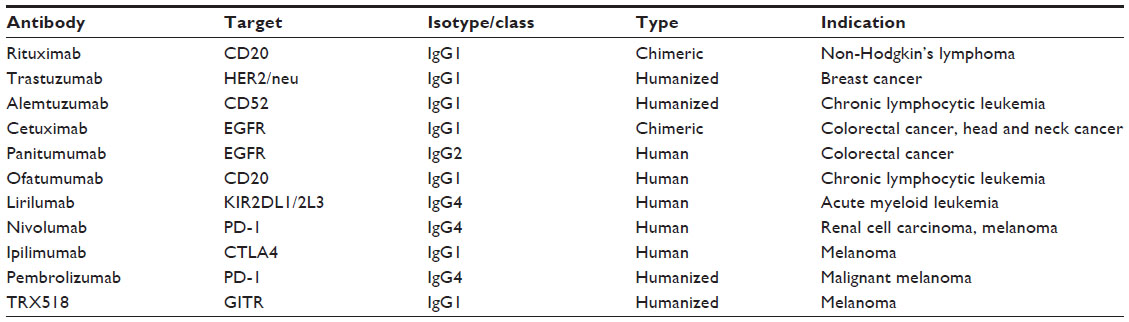 | Table 1 List of antibodies mentioned in the review |
Recent work has validated the importance of ADCC in tumor clearance and focused on enhancing ADCC in the cancer setting.3–6 Originally, ADCC was described as the mechanism by which effector immune cells lyse antibody-coated target cells through the release of cytotoxic molecules like perforin and granzyme.7 However, ADCC is now understood as a multitiered process that invokes a network of coordinated immune cells, including monocytes, macrophages, dendritic cells (DCs), and granulocytes. For example, Fc-gamma receptor (FcγR) ligation on natural killer (NK) cells can induce the secretion of proinflammatory cytokines like interferon gamma (IFN-γ), which can accelerate DC maturation.8,9 Mature DCs enhance antigen presentation and train tumor-specific lymphocytes, producing an immunological memory response.
The specificity of ADCC is conferred by the binding of the antibody through its fragment antigen-binding portion to the tumor-associated antigen on the target cell.10,11 Expression of FcγRs that recognize the Fc portion of the bound antibodies on the cytotoxic effector cells then initiate ADCC.3 The magnitude of the cytotoxic response is regulated by different classes of activating and inhibiting Fc receptors.6 Of the different FcRs: FcγR binds to immunoglobulin G (IgG), FcαR binds to immunoglobulin A, FcεR binds to immunoglobulin E, and Fcμ/αR binds both immunoglobulin M and immunoglobulin A. Of these, FcγRs are the predominant mediators of ADCC.11 Most hematopoietic cells, except T cells, can express FcγRs.12 There are three types of FcγRs, and each type of FcγR recognizes IgG subclasses with different affinities. FcγRI (cluster of differentiation [CD]64), expressed on macrophages and neutrophils, is an activating receptor that binds to human IgG1 and IgG3 with high affinity and mediates phagocytosis of target cells. Signaling from the activating receptors on the phagocytes is attenuated by FcγRIIa (CD32A), a low-affinity receptor, and FcγRIIb (CD32B), an inhibitory receptor.11,13 FcγRIIIb is a low-affinity activating receptor expressed in neutrophils. FcγRIIIa (CD16) is an activating low-affinity receptor expressed on NK cells and macrophages.13 However, unlike the other hematopoietic cells, NK cells do not express the inhibitory FcγRIIb receptors. Without the influence of Fc-mediated inhibitory signaling, NK cells are free to act as key mediators of ADCC in the presence of antibody-coated tumor targets.11
Human NK cells comprise around 5% of lymphocytes circulating in the blood and are defined by a CD14−CD19−CD3−CD56+ phenotype.14,15 They are further subdivided into two subsets defined by their expression of CD16: CD56lowCD16+ NK cells and CD56hiCD16− NK cells. The CD56lowCD16+ is the predominant subset in the peripheral blood and displays early cytolytic functions, while the CD56hiCD16− cells are distributed in the tissues and secondary lymphoid organs and display a late response, secreting primarily IFN-γ and tumor necrosis factor (TNF)-α. However, recent data also shows that the dichotomous subset functionality may not be completely polarized: a novel CD56lowCD16− subset exhibits cytokine secretory functions very early after activation and thereby aids in cytolytic activity.16,17
NK cell activation is tightly controlled by combinatorial signaling via a network of activating and inhibitory receptors. The NKp receptors and leukocyte immunoglobulin-like receptors are solely activating receptors, while the killer cell immunoglobulin-like receptors (KIRs) and CD94-NKG2A receptor family contain both inhibitory and activating receptors.18 The interplay of these activating and inhibiting receptors regulates the responses of NK cells when they encounter potential target cells. Efficient cytolytic activity of the NK cells depends on the high-avidity binding of the FcγRIIIa to antibodies that are bound to multimeric antigen targets. This binding results in a strong activation signal that overcomes the inhibitory signals, leading to a cytotoxic and cytokine response.
Rituximab was the first mAb approved in 1997 for the treatment of non-Hodgkin’s lymphoma. Following rituximab, several mAbs have become standard of care for the treatment of both solid tumors and hematological malignancies, including trastuzumab, alemtuzumab, cetuximab, panitumumab, and ofatumumab.6,19–23 Though clinical trials with mAbs have produced significant clinical benefits, there is a need to increase the potency of these therapies to obtain durable clinical remissions and an increase in survival. In this review, we discuss some of the promising novel strategies that could potentially enhance ADCC of tumor targets.
Modifying the antibody: Fc polymorphism and protein engineering
By modulating the strength of the synapse between the antibody Fc and the FcγR, the potency of ADCC can be enhanced. One variant of the FcγRIII, FcγRIIIa, is polymorphic for binding of human IgG. A valine at position 158 on the FcγRIIIa results in a greater affinity for IgG relative to receptors with a phenylalanine in the same position.24 Upon stimulus with IgG, NK cells with a valine-positive FcγRIIIa demonstrate a greater calcium influx and upregulation of surface interleukin (IL)-2 receptor (CD25), a canonical marker of NK cell activation.25 To investigate the effect of these findings clinically, multiple studies stratified patients by FcγRIIIa polymorphism and looked for differing outcomes. As suspected, patients with the high-affinity FcγRIII show increased clinical benefit from mAb therapy.26,27 These results suggest that NK cells with the valine-positive FcγRIIIa are advantageous for, and a predictive biomarker of, mAb-mediated cancer immunotherapy.
In contrast to the FcγRIIIa polymorphism, a relatively immutable mediator of ADCC, the glycosylation state of the antibody Fc fragment provides a parameter that can be synthetically optimized to enhance ADCC. Increasingly, posttranslational modifications of protein-based biopharmaceuticals are being utilized to enhance therapeutic activity.28 A common modification, glycosylation, is known to play a critical role in immune cell detection of antibodies. Specifically, glycosylation of the asparagine 297 residue in the CH2 domain of the antibody Fc region is accepted as integral to effective FcγRIIIa binding.29 Recent work has investigated the effects of Fc region fucosylation, sialylation, and galactosylation on FcR binding and ADCC within different immune effector subtypes. Generally, low levels of Fc fucosylation enhance mononuclear cell ADCC, while polymorphonuclear cells preferentially kill via highly fucosylated antibodies.30 These insights have led to the generation of glycoengineered antibodies that can elicit up to a tenfold increase in ADCC against cancer cell lines.31 Fc-optimization in antibodies against B-cell and ovarian cancer targets has been shown to improve ADCC in vitro and improve survival and tumor-cell depletion in murine xenograft tumor models.32 Recently, a glycoengineered antibody against CD37, a current antibody target in B cell malignancies, dramatically outperformed an earlier, nonengineered anti-CD37 antibody that is currently in Phase II clinical trials.33 Obinutuzumab (GA101), a glycoengineered, anti-CD20 mAb, recently outperformed rituximab in a head-to-head comparison in patients with chronic lymphocytic leukemia.34 Obinutuzumab, now approved by the FDA, was designed to have multiple oligosaccharides attached to asparagine 297 in the Fc region.35 The additional glycosylation leads to a greater binding affinity with FcγRIIIa and results in elevated potency and efficacy of ADCC and B-cell depletion.36
Protein engineering focused on optimizing the capacity of the Fc region of the mAb to bind FcγRs is another route to enhancing ADCC effector functions. By generating Fc variants with greater affinity to FcγRs, tumor-targeting antibodies can transmit a more-potent activating signal to NK and phagocytic cells. By utilizing computational designs algorithms and high-throughput screening assays, Fc region mutations have been identified that increase the binding of alemtuzumab, trastuzumab, rituximab, and cetuximab to human FcγRIIIa.37 The designed Fc variants provide substantial enhancement of ADCC relative to nonengineered antibodies and in macaques, a double-mutant S239D/I332E variant of an anti-CD20 mAb proved superior at depleting B cells relative to its nonengineered counterpart. Fc optimization has also been shown to improve survival and enhance tumor-cell depletion in mouse xenograft tumor models for ovarian cancer.32 The Fc-optimized anti-human epidermal growth factor receptor 2 antibody margetuximab has now entered clinical testing in patients with relapsed or refractory advanced breast cancer (NCT01828021).
Effector cell activation: oncolytic viruses
Oncolytic viruses (OVs) selectively kill and replicate within tumor cells but do not harm normal cells. They achieve this by direct oncolytic activity and by inducing an immune response against the infected tumor cells.38 OVs include a diversity of DNA and RNA viruses and fall into two types: 1) viruses that naturally replicate in cancer cells and are nonpathogenic to humans: reovirus, myxomavirus, Newcastle disease virus, and Seneca valley virus; and 2) viruses that are genetically modified to promote tumor selectivity: vesicular stomatitis virus, herpes simplex virus, and vaccine vectors like the vaccina virus, adenovirus, measles virus, and poliovirus. Because OV-based therapy targets multiple oncolytic pathways simultaneously, it has a lower probability of inducing resistance compared to therapeutics that individually target pathways (eg, small-molecule inhibitors). Preclinical studies in murine models have demonstrated that OVs act as powerful inducers of antitumor immunity by triggering strong inflammatory responses and a CD8+ T cell-mediated adaptive immune response.39–43 In clinical trials, OVs demonstrated limited toxicity and immune responses to the tumor, but overall antitumor efficacy has been limited.44,45 A major challenge in OV therapy is that the immune response elicited by OVs may also become detrimental to the therapeutic outcome. Viral neutralization by natural antibodies after systemic administration of the virus may lead to rapid clearance of the virus, thus impairing therapy.46 However, recent studies demonstrated that immune cells like NK cells can themselves be carriers of reovirus, thereby playing a vital role in enhancing tumor immunity. Here we discuss the possibility of combining the immunotherapy approaches with reovirus therapy to enhance the combined therapeutic potential of both the agents.
Reovirus
Reovirus is a naturally occurring double-stranded oncolytic virus. Reovirus commonly infects the respiratory and gastrointestinal tracts of humans but is not associated with any known human disease.47 Reovirus targets and kills tumor cells through selective replication in cells with an activated Ras pathway.48 Though Ras mutations occur in only 30% of human tumors, aberrant signaling in the Ras pathway is due to mutations occurring downstream of Ras, making non-Ras mutants suitable targets for reovirus therapy.49–53 The mechanism of antitumor activity of reovirus is due to its direct oncolytic activity on the tumor through induction of apoptotic pathways, as well as by activation of antitumor immunity.43,54–57 Recent studies have shown that reovirus administration causes secretion of proinflammatory cytokines and chemokines.57,58
Preclinical studies with reovirus as monotherapy have demonstrated the therapeutic benefits of reovirus in treating solid tumors like melanoma, glioma, and ovarian, breast, and colon cancers, as well as in a range of hematological malignancies like myelomas and lymphomas.48,58–61 Further, combination therapy of reovirus and vascular endothelial growth factor inhibitors in a mouse model of melanoma demonstrated that NK cells, CD4+ T cells, and CD8+ T cells were required for tumor regression. In another study, it was demonstrated that tumor infection by reovirus activates DCs that in turn induce NK cell recruitment, activation, and increased NK cell-mediated cytotoxicity.62,63 Recently, Hamano et al64 have demonstrated in a preclinical model that reovirus can augment trastuzumab-induced cytotoxicity in gastric cancer cells. Further, Adair et al65 interestingly show that reoviruses are protected from neutralizing mAbs by binding to NK cells, and these cells can also carry the reovirus to the targets facilitating direct cytotoxic killing of tumor cells. Further, NK cells, upon binding to reovirus, exhibit an upregulation of activation markers CD69 and CCR7, and killed tumor targets in a Type 1 interferon-dependent manner.66 Thus, systemic administration of reovirus by immune-cell carriage in combination with mAb therapies like rituximab or cetuximab can facilitate the delivery of reovirus directly to tumor target and result in increased NK-mediated ADCC.
Currently, reovirus is being used in clinical trials as monotherapy or in combination with other anticancer agents. Phase I trials performed using intratumoral administrations have proved that reovirus is well tolerated as monotherapy.67,68 Other Phase I/II trials tested the systemic administration of reovirus as monotherapy in patients with advanced malignancies and found that intravenous injections were safe and well tolerated and encouragingly showed tumor reduction.69–72 The majority of ongoing clinical trials with reovirus involve combinatorial treatment strategies. Reovirus is currently being tested in combination with radiotherapy or multiple-chemotherapy agents like docetaxel, apaclitaxel, and carboplatin.73–75 The latter studies demonstrated that reovirus administration does not increase toxicity associated with radiation and chemotherapies. These recent clinical data support the prospect of using reovirus in combination with mAbs for antitumor therapies. A summary of active clinical trials testing reovirus is presented in Table 2.
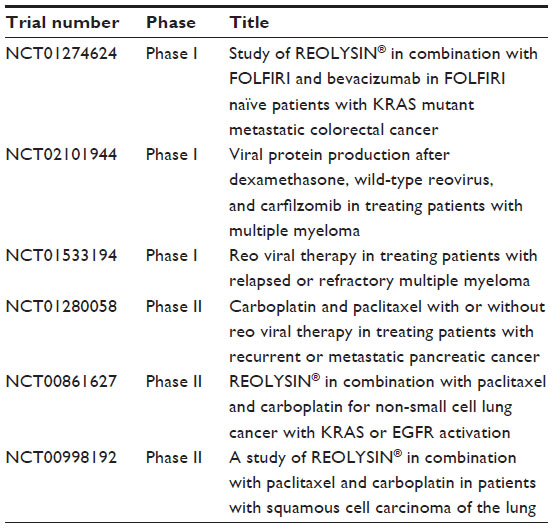 | Table 2 Ongoing active clinical trials with reovirus |
Effector cell activation: toll-like receptor agonists
Toll-like receptors (TLRs) are expressed by both effector cells of the immune system and cancer cells.76 TLR family of receptors (TLR1–TLR10) recognize highly conserved structural motifs known as pathogen-associated molecular patterns, which are exclusively expressed by microbial pathogens, or damage-associated molecular patterns, which are endogenous molecules released from necrotic or dying cells. On binding, they initiate signaling cascades that result in a variety of cellular responses, including proinflammatory cytokine expression.77 TLR agonists have immune stimulatory effects through the induction of costimulatory molecules like CD80 and CD86 on DCs and the production of inflammatory cytokines like TNF-α and IL-2. But they may also induce several immune suppressive factors like IL-10 and programmed cell-death protein-1 ligand (PD-L1) that makes them poor candidates as standalone tumor therapeutics.78,79
Only a few TLR agonists are currently licensed by the FDA for use in cancer patients: Bacillus Calmette–Guérin (TLR2/TLR4), monophosphoryl lipid A (TLR4), and imiquimod (TLR7).80,81 However, recent in vitro studies have demonstrated that TLR8 stimulation through its agonist VTX-2337 enhanced the activation and function of NK cells in the presence of cetuximab-coated head and neck cancer cells.82 Similarly the TLR3 ligand polyinosinic:polycytidylic acid (polyI:C) increased the cetuximab-dependent ADCC by NK cells against head and neck cancer cell lines. During cetuximab-induced ADCC, the percentage of activated NK cells (CD107a+granzyme B+) increased significantly in presence of both the agonist and cetuximab, compared to either of them alone.83 Thus these TLR agonists in combination with cetuximab can enhance cetuximab induced ADCC against head and neck cancer.
In another study involving TLR9, it has been demonstrated that CpG-containing oligodeoxynucleotides (CpG ODN), the TLR9 agonist, can directly promote the secretion of cytokines by NK cells exposed to antibody-coated tumor cells by activating TLR9.84 Further, Sommariva et al85 have demonstrated in an in vivo advanced ovarian xenograft model that mice treated with a combination of CpG ODN and cetuximab had a significantly increased median survival rate relative to monotherapy with either agent. CpG ODNs can also activate NK cells through indirect activation of plasmacytoid DCs that stimulate IFN-γ production by T cells.86 CpG ODNs can also induce CD20 expression on malignant B cells.87 Thus the activating effect of CpG ODN on the effector cells as well as on the tumor cells can have a synergistic effect when used in combination with mAbs. It has been shown in preclinical studies that CpG ODNs enhance antitumor activity of rituximab in treating lymphomas88,89 and trastuzumab in treating breast cancer.87,90
Effector cell activation: agonistic and antagonistic mAbs
The importance of utilizing mAb therapy to elicit ADCC-mediated tumor clearance was initially established by studies exploring the mechanism of action of rituximab. One of the primary mechanisms by which rituximab exerts its antitumor effects is by making the CD20-expressing tumor a more attractive target for NK cell lysis. In the decades following the introduction of rituximab, subsequent mAbs have been developed that augment ADCC. A particularly promising strategy for enhancing ADCC via mAb therapy is targeting the costimulatory pathways that activate NK cell cytotoxicity. One molecule that has demonstrated strong preclinical success in this approach is CD137.
CD137
CD137 is a member of TNF receptor superfamily and is upregulated on NK cells after FcγRIIIa (CD16) ligation.91 Administration of agonistic anti-CD137 mAbs has been shown to amplify antitumor immune responses in a variety of different murine cancer models.92 On NK cells, activation of CD137 increases proliferation, degranulation, and IFN-γ secretion, leading to enhanced ADCC.93 The ability of anti-CD137 mAbs to enhance ADCC makes them ideal candidates for combination therapeutic strategies. We have previously demonstrated that targeting CD137 concomitantly with rituximab or trastuzumab administration accelerates tumor clearance in murine xenograft models of lymphoma and breast cancer.94,95 Recently, we combined cetuximab and anti-CD137 antibody therapy to obtain complete tumor resolution and prolonged survival in xenograft models of epidermal growth factor receptor-expressing cancer cells, head and neck cancer cells, and wild-type Kirsten rat sarcoma 2 viral oncogene homolog (KRAS-WT) and KRAS-mutant colorectal cancer.96 An anti-CD137 antibody, urelumab, is currently in clinical trials with rituximab for patients with non-Hodgkin’s lymphoma (NCT01775631) and with cetuximab in patients with colorectal cancer or head and neck cancer (NCT02110082).
KIR signaling
The killer cell immunoglobulin-like receptor (KIR) family constitutes one of the key mediators of NK cell activation. Inhibitory KIR molecules bind to the self-major histocompatibility complex class I ligands (HLA-A, HLA-B, HLA-C) and upon binding transduce inhibitory signals that abrogate the effects of activating receptors.97 Because major histocompatibility complex class I is expressed on virtually all healthy cells, KIR molecules are considered to be one of the primary mechanisms responsible for NK cell tolerance to self. Reducing KIR-mediated inhibitory signaling in NK cells via antibody blockade has been shown to increase NK cell cytotoxicity and survival of leukemia-bearing mice.98 A fully human mAb that binds KIR2DL1, KIR2DL2, and KIR2DL3 receptors enhanced NK cell-mediated lysis of tumor cells, including autologous acute myeloid leukemia (AML) blasts, but did not induce killing of normal peripheral blood mononuclear cells.99
Based on these results, a KIR-blocking mAb, lirilumab (IPH2102/BMS-986015), was developed and is currently being tested in clinical trials. Early-phase clinical trials of lirilumab in patients with multiple myeloma demonstrated increased patient-derived NK cell cytotoxicity ex vivo but failed to produce any objective responses.100 A trial of lirilumab in patients with AML in first complete remission further validated anti-KIR therapy as safe and tolerable, but only produced transient NK activation.101
There is also interest in using anti-KIR antibodies in combinatorial checkpoint blockade strategies. Immune checkpoints are inhibitory pathways that downregulate activated immune cells. During tumor genesis, cancer cells express proteins that activate immune checkpoint pathways and induce immune suppression, thereby evading targeting and removal by the immune system. Checkpoint blockade is a therapeutic strategy by which these inhibitory signals are blocked by antibodies. With the immune suppression in the tumor microenvironment removed, the tumor is targeted for clearance. A trial testing lirilumab and nivolumab, a fully human IgG4 antibody that inhibits the checkpoint marker programmed cell-death protein 1 (PD-1), is currently recruiting patients with advanced solid tumors (NCT01714739). Lirilumab is also being tested in combination with the anti-CTLA4 antibody, ipilimumab in patients with advanced melanoma, non-small cell lung cancer, and castrate-resistant prostate cancer (NCT01750580).
PD-1/PD-L1 axis
PD-1 and its ligands (PD-L1 and PD-L2) constitute another important signaling pathway in regulating NK cell activation and downstream ADCC. PD-1 and PD-L1 blockade is currently thought to primarily influence T-cell subsets, but following activation, PD-1 is expressed on NK cells and ligation of PD-1 transmits a negative regulatory signal, limiting NK cell cytotoxicity.102 In histologically diverse tumors, PD-L1 can be upregulated, providing a potent mechanism of immunosuppression in the tumor microenvironment.103 Tumors can also induce PD-L1 expression on immune subsets. In murine cancer models, tumor-derived interleukin IL-18 was shown to induce PD-L1 expression on a subset of immature NK cells, which then trafficked to lymph nodes and suppressed the DC/NK cell crosstalk necessary for the development of mature, cytotoxic NK cells.104 Preclinically, antibodies that block PD-1 signaling have been shown to enhance NK cell cytotoxicity against autologous, primary multiple myeloma tumors.105 In late 2014, the FDA approved the first anti-PD-1 mAb (pembrolizumab) based on a study that reported an overall response rate of 26% in ipilimumab-refractory advanced melanoma patients.106 Currently, clinical trials are being considered that combine PD-1 blockade and therapeutic, tumor-targeting antibodies.104
GITR/GITRL
Glucocorticoid-induced TNF receptor (GITR) ligand (GITRL) is frequently expressed on leukemia cells in AML and chronic lymphocytic leukemia, and impairs the reactivity of NK cells that express GITR and upregulate its expression following activation.107,108 GITRL also inhibits the rituximab-induced ADCC of NK cells.109 The anti-GITR mAb TRX518 blocks the interaction of GITR, expressed on NK cells, and its ligand GITRL, thereby increasing the cytotoxicity of NK cells. Thus, TRX518 is a promising candidate for combination with other mAbs where it can augment NK cell-mediated ADCC. A Phase I study with TRX518 (NCT01239134) is being conducted in patients with stage 2 or stage 4 melanoma.
Conclusion
The combination of tumor-targeting mAbs and ADCC-enhancing immunomodulators is a promising treatment strategy for oncology patients. As more tumor-associated antigens are identified and immune effectors’ activating pathways are better understood, the applicability of this approach will only increase. Though preliminary data from work with oncolytic viruses, TLR agonists, engineered mAbs, and immunostimulatory mAbs is encouraging, substantial clinical evidence is needed to validate these therapies. The results of ongoing clinical trials are eagerly awaited (Table 3). Hopefully, clinical studies validate this novel therapeutic strategy and lead to increased patient survival.
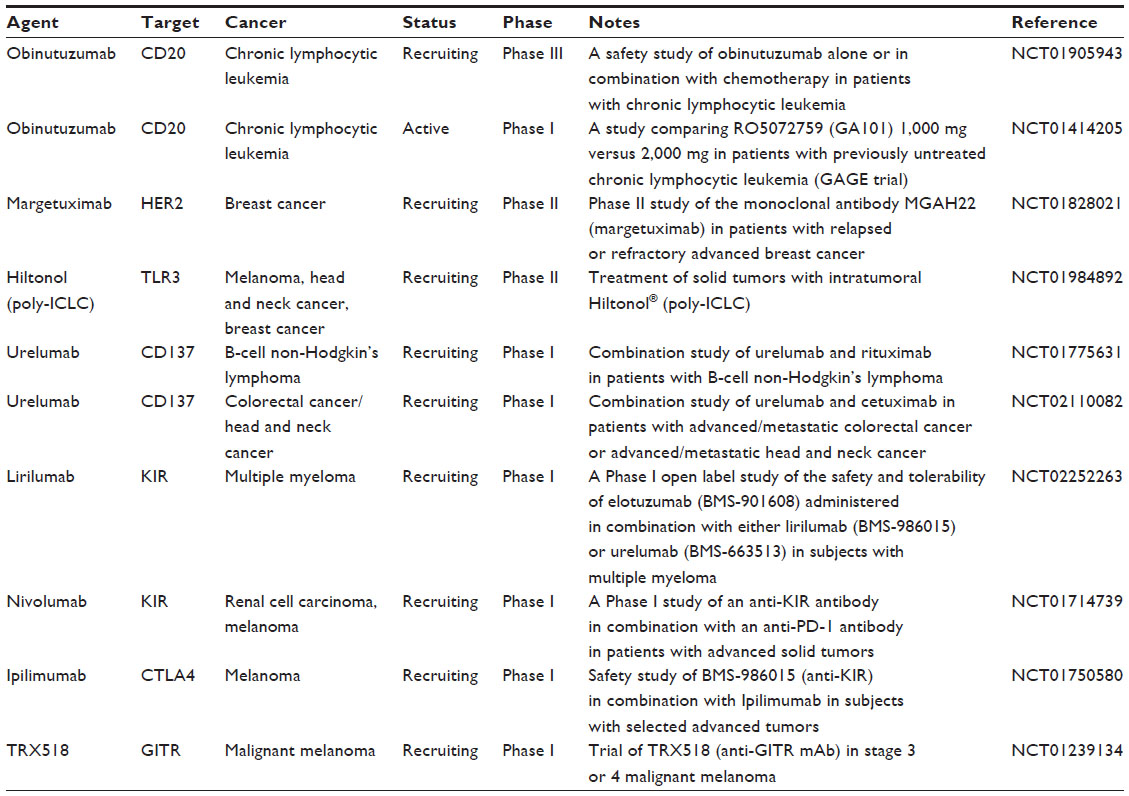 | Table 3 A list of therapeutic reagents mentioned in the article in clinical trials |
Disclosure
The authors report no conflicts of interest in this work.
References
Jarboe J, Gupta A, Saif W. Therapeutic human monoclonal antibodies against cancer. Methods Mol Biol. 2014;1060:61–77. | |
Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9(10):729–740. | |
Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000; 6(4):443–446. | |
Manches O, Lui G, Chaperot L, et al. In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood. 2003;101(3):949–954. | |
Arnould L, Gelly M, Penault-Llorca F, et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br J Cancer. 2006;94(2):259–267. | |
Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10(5):317–327. | |
Topham NJ, Hewitt EW. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology. 2009;128(1):7–15. | |
Cassatella MA, Anegón I, Cuturi MC, Griskey P, Trinchieri G, Perussia B. Fc gamma R(CD16) interaction with ligand induces Ca2+ mobilization and phosphoinositide turnover in human natural killer cells. Role of Ca2+ in Fc gamma R(CD16)-induced transcription and expression of lymphokine genes. J Exp Med. 1989;169(2):549–567. | |
Sica A, Dorman L, Viggiano V, et al. Interaction of NF-kappaB and NFAT with the interferon-gamma promoter. J Biol Chem. 1997;272(48):30412–30420. | |
Alderson KL, Sondel PM. Clinical cancer therapy by NK cells via antibody-dependent cell-mediated cytotoxicity. J Biomed Biotechnol. 2011;2011:379123. | |
Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19: 275–290. | |
Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol. 2002;2(8):580–592. | |
Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8(1):34–47. | |
Robertson MJ, Caligiuri MA, Manley TJ, Levine H, Ritz J. Human natural killer cell adhesion molecules. Differential expression after activation and participation in cytolysis. J Immunol. 1990;145(10):3194–3201. | |
Walzer T, Jaeger S, Chaix J, Vivier E. Natural killer cells: from CD3(−)NKp46(+) to post-genomics meta-analyses. Curr Opin Immunol. 2007;19(3):365–372. | |
Penack O, Gentilini C, Fischer L, et al. CD56dimCD16neg cells are responsible for natural cytotoxicity against tumor targets. Leukemia. 2005;19(5):835–840. | |
Stabile H, Nisti P, Morrone S, et al. Multifunctional human CD56low CD16low natural killer cells are the prominent subset in bone marrow of both pediatric healthy donors and leukemic patients. Haematologica. Epub January 16, 2015. | |
Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol. 2011;89(2):216–224. | |
Cartron G, Dacheux L, Salles G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99(3):754–758. | |
Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21(21):3940–3947. | |
Addeo R, Caraglia M, Cerbone D, et al. Panitumumab: a new frontier of target therapy for the treatment of metastatic colorectal cancer. Expert Rev Anticancer Ther. 2010;10(4):499–505. | |
Kennedy B, Hillmen P. Immunological effects and safe administration of alemtuzumab (MabCampath) in advanced B-cLL. Med Oncol. 2002;19 Suppl:S49–S55. | |
Hudis CA. Trastuzumab–mechanism of action and use in clinical practice. N Engl J Med. 2007;357(1):39–51. | |
Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood. 1997;90(3):1109–1114. | |
Wu J, Edberg JC, Redecha PB, et al. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest. 1997;100(5):1059–1070. | |
Persky DO, Dornan D, Goldman BH, et al. Fc gamma receptor 3a genotype predicts overall survival in follicular lymphoma patients treated on SWOG trials with combined monoclonal antibody plus chemotherapy but not chemotherapy alone. Haematologica. 2012;97(6):937–942. | |
Rodríguez J, Zarate R, Bandres E, et al. Fc gamma receptor polymorphisms as predictive markers of cetuximab efficacy in epidermal growth factor receptor downstream-mutated metastatic colorectal cancer. Eur J Cancer. 2012;48(12):1774–1780. | |
Walsh G, Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol. 2006;24(10):1241–1252. | |
Wright A, Morrison SL. Effect of glycosylation on antibody function: implications for genetic engineering. Trends Biotechnol. 1997;15(1):26–32. | |
Peipp M, Lammerts van Bueren JJ, Schneider-Merck T, et al. Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood. 2008;112(6):2390–2399. | |
Schuster M, Umana P, Ferrara C, et al. Improved effector functions of a therapeutic monoclonal Lewis Y-specific antibody by glycoform engineering. Cancer Res. 2005;65(17):7934–7941. | |
Stavenhagen JB, Gorlatov S, Tuaillon N, et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res. 2007;67(18):8882–8890. | |
Rafiq S, Siadak A, Butchar JP, et al. Glycovariant anti-CD37 monospecific protein therapeutic exhibits enhanced effector cell-mediated cytotoxicity against chronic and acute B cell malignancies. MAbs. 2013;5(5):723–735. | |
Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014; 370(12):1101–1110. | |
Mössner E, Brünker P, Moser S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010;115(22):4393–4402. | |
Patz M, Isaeva P, Forcob N, et al. Comparison of the in vitro effects of the anti-CD20 antibodies rituximab and GA101 on chronic lymphocytic leukaemia cells. Br J Haematol. 2011;152(3):295–306. | |
Lazar GA, Dang W, Karki S, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A. 2006;103(11):4005–4010. | |
Benencia F, Courrèges MC, Conejo-García JR, et al. HSV oncolytic therapy upregulates interferon-inducible chemokines and recruits immune effector cells in ovarian cancer. Mol Ther. 2005;12(5):789–802. | |
Toda M, Rabkin SD, Kojima H, Martuza RL. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum Gene Ther. 1999;10(3):385–393. | |
Li H, Dutuor A, Fu X, Zhang X. Induction of strong antitumor immunity by an HSV-2-based oncolytic virus in a murine mammary tumor model. J Gene Med. 2007;9(3):161–169. | |
Li H, Dutuor A, Tao L, Fu X, Zhang X. Virotherapy with a type 2 herpes simplex virus-derived oncolytic virus induces potent antitumor immunity against neuroblastoma. Clin Cancer Res. 2007;13(1):316–322. | |
Miller CG, Fraser NW. Requirement of an integrated immune response for successful neuroattenuated HSV-1 therapy in an intracranial metastatic melanoma model. Mol Ther. 2003;7(6):741–747. | |
Prestwich RJ, Errington F, Ilett EJ, et al. Tumor infection by oncolytic reovirus primes adaptive antitumor immunity. Clin Cancer Res. 2008;14(22):7358–7366. | |
Aghi M, Martuza RL. Oncolytic viral therapies – the clinical experience. Oncogene. 2005;24(52):7802–7816. | |
Patel MR, Kratzke RA. Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Transl Res. 2013;161(4):355–364. | |
Seymour LW, Cawood R, Fisher KD. Cellular carriers or free viruses? Curr Opin Mol Ther. 2008;10(4):321–322. | |
Fields BN, Knipe DM, Howley PM. Fields Virology. 3rd ed. Philadelphia, PA: Lippincott-Raven Publishers; 1996. | |
Coffey MC, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282(5392):1332–1334. | |
Marcato P, Shmulevitz M, Lee PW. Connecting reovirus oncolysis and Ras signaling. Cell Cycle. 2005;4(4):556–559. | |
Marcato P, Shmulevitz M, Pan D, Stoltz D, Lee PW. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis-dependent release. Mol Ther. 2007;15(8):1522–1530. | |
Shmulevitz M, Marcato P, Lee PW. Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer. Oncogene. 2005;24(52):7720–7728. | |
Shmulevitz M, Marcato P, Lee PW. Activated Ras signaling significantly enhances reovirus replication and spread. Cancer Gene Ther. 2010;17(1):69–70. | |
Shmulevitz M, Pan LZ, Garant K, Pan D, Lee PW. Oncogenic Ras promotes reovirus spread by suppressing IFN-beta production through negative regulation of RIG-I signaling. Cancer Res. 2010;70(12):4912–4921. | |
Prestwich RJ, Harrington KJ, Pandha HS, Vile RG, Melcher AA, Errington F. Oncolytic viruses: a novel form of immunotherapy. Expert Rev Anticancer Ther. 2008;8(10):1581–1588. | |
Gujar SA, Marcato P, Pan D, Lee PW. Reovirus virotherapy overrides tumor antigen presentation evasion and promotes protective antitumor immunity. Mol Cancer Ther. 2010;9(11):2924–2933. | |
Prestwich RJ, Ilett EJ, Errington F, et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin Cancer Res. 2009;15(13):4374–4381. | |
Gujar SA, Pan DA, Marcato P, Garant KA, Lee PW. Oncolytic virus-initiated protective immunity against prostate cancer. Mol Ther. 2011;19(4):797–804. | |
Errington F, White CL, Twigger KR, et al. Inflammatory tumour cell killing by oncolytic reovirus for the treatment of melanoma. Gene Ther. 2008;15(18):1257–1270. | |
Hata Y, Etoh T, Inomata M, Shiraishi N, Nishizono A, Kitano S. Efficacy of oncolytic reovirus against human breast cancer cells. Oncol Rep. 2008;19(6):1395–1398. | |
Hirasawa K, Nishikawa SG, Norman KL, Alain T, Kossakowska A, Lee PW. Oncolytic reovirus against ovarian and colon cancer. Cancer Res. 2002;62(6):1696–1701. | |
Alain T, Hirasawa K, Pon KJ, et al. Reovirus therapy of lymphoid malignancies. Blood. 2002;100(12):4146–4153. | |
Prestwich RJ, Errington F, Steele LP, et al. Reciprocal human dendritic cell-natural killer cell interactions induce antitumor activity following tumor cell infection by oncolytic reovirus. J Immunol. 2009;183(7):4312–4321. | |
Errington F, Steele L, Prestwich R, et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J Immunol. 2008;180(9):6018–6026. | |
Hamano S, Mori Y, Aoyama M, et al. Oncolytic reovirus combined with trastuzumab enhances antitumor efficacy through TRAIL signaling in human HER2-positive gastric cancer cells. Cancer Lett. 2015; 356(2 Pt B):846–854. | |
Adair RA, Roulstone V, Scott KJ, et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med. 2012;4(138):138ra77. | |
Adair RA, Scott KJ, Fraser S, et al. Cytotoxic and immune-mediated killing of human colorectal cancer by reovirus-loaded blood and liver mononuclear cells. Int J Cancer. 2013;132(10):2327–2338. | |
Kicielinski KP, Chiocca EA, Yu JS, Gill GM, Coffey M, Markert JM. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol Ther. 2014;22(5):1056–1062. | |
Forsyth P, Roldán G, George D, et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008;16(3):627–632. | |
Gollamudi R, Ghalib MH, Desai KK, et al. Intravenous administration of Reolysin, a live replication competent RNA virus is safe in patients with advanced solid tumors. Invest New Drugs. 2010;28(5):641–649. | |
Morris DG, Feng X, DiFrancesco LM, et al. REO-001: A phase I trial of percutaneous intralesional administration of reovirus type 3 dearing (Reolysin®) in patients with advanced solid tumors. Invest New Drugs. 2013;31(3):696–706. | |
Galanis E, Markovic SN, Suman VJ, et al. Phase II trial of intravenous administration of Reolysin(®) (Reovirus Serotype-3-dearing Strain) in patients with metastatic melanoma. Mol Ther. 2012;20(10):1998–2003. | |
Vidal L, Pandha HS, Yap TA, et al. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res. 2008;14(21):7127–7137. | |
Harrington KJ, Karapanagiotou EM, Roulstone V, et al. Two-stage phase I dose-escalation study of intratumoral reovirus type 3 dearing and palliative radiotherapy in patients with advanced cancers. Clin Cancer Res. 2010;16(11):3067–3077. | |
Karapanagiotou EM, Roulstone V, Twigger K, et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res. 2012;18(7):2080–2089. | |
Comins C, Spicer J, Protheroe A, et al. REO-10: a phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin Cancer Res. 2010;16(22):5564–5572. | |
Jego G, Bataille R, Geffroy-Luseau A, Descamps G, Pellat-Deceunynck C. Pathogen-associated molecular patterns are growth and survival factors for human myeloma cells through Toll-like receptors. Leukemia. 2006;20(6):1130–1137. | |
Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. | |
Lu H. TLR agonists for cancer immunotherapy: tipping the balance between the immune stimulatory and inhibitory effects. Front Immunol. 2014;5:83. | |
Kaczanowska S, Joseph AM, Davila E. TLR agonists: our best frenemy in cancer immunotherapy. J Leukoc Biol. 2013;93(6):847–863. | |
Vacchelli E, Eggermont A, Sautès-Fridman C, et al. Trial Watch: Toll-like receptor agonists for cancer therapy. Oncoimmunology. 2014;2(8):e25238. | |
Vacchelli E, Galluzzi L, Eggermont A, et al. Trial watch: FDA-approved Toll-like receptor agonists for cancer therapy. Oncoimmunology. 2012;1(6):894–907. | |
Stephenson RM, Lim CM, Matthews M, Dietsch G, Hershberg R, Ferris RL. TLR8 stimulation enhances cetuximab-mediated natural killer cell lysis of head and neck cancer cells and dendritic cell cross-priming of EGFR-specific CD8+ T cells. Cancer Immunol Immunother. 2013;62(8):1347–1357. | |
Ming Lim C, Stephenson R, Salazar AM, Ferris RL. TLR3 agonists improve the immunostimulatory potential of cetuximab against EGFR(+) head and neck cancer cells. Oncoimmunology. 2013;2(6):e24677. | |
Roda JM, Parihar R, Carson WE 3rd. CpG-containing oligodeoxynucleotides act through TLR9 to enhance the NK cell cytokine response to antibody-coated tumor cells. J Immunol. 2005;175(3):1619–1627. | |
Sommariva M, de Cesare M, Meini A, et al. High efficacy of CpG-ODN, cetuximab and cisplatin combination for very advanced ovarian xenograft tumors. J Transl Med. 2013;11:25. | |
Moga E, Alvarez E, Cantó E, et al. NK cells stimulated with IL-15 or CpG ODN enhance rituximab-dependent cellular cytotoxicity against B-cell lymphoma. Exp Hematol. 2008;36(1):69–77. | |
Wang H, Rayburn ER, Wang W, Kandimalla ER, Agrawal S, Zhang R. Immunomodulatory oligonucleotides as novel therapy for breast cancer: pharmacokinetics, in vitro and in vivo anticancer activity, and potentiation of antibody therapy. Mol Cancer Ther. 2006;5(8):2106–2114. | |
Wooldridge JE, Ballas Z, Krieg AM, Weiner GJ. Immunostimulatory oligodeoxynucleotides containing CpG motifs enhance the efficacy of monoclonal antibody therapy of lymphoma. Blood. 1997;89(8):2994–2998. | |
Betting DJ, Yamada RE, Kafi K, Said J, van Rooijen N, Timmerman JM. Intratumoral but not systemic delivery of CpG oligodeoxynucleotide augments the efficacy of anti-CD20 monoclonal antibody therapy against B cell lymphoma. J Immunother. 2009;32(6):622–631. | |
van Ojik HH, Bevaart L, Dahle CE, et al. CpG-A and B oligodeoxynucleotides enhance the efficacy of antibody therapy by activating different effector cell populations. Cancer Res. 2003;63(17):5595–5600. | |
Lin W, Voskens CJ, Zhang X, et al. Fc-dependent expression of CD137 on human NK cells: insights into “agonistic” effects of anti-CD137 monoclonal antibodies. Blood. 2008;112(3):699–707. | |
Melero I, Shuford WW, Newby SA, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3(6):682–685. | |
Wilcox RA, Flies DB, Zhu G, et al. Provision of antigen and CD137 signaling breaks immunological ignorance, promoting regression of poorly immunogenic tumors. J Clin Invest. 2002;109(5):651–659. | |
Kohrt HE, Houot R, Goldstein MJ, et al. CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood. 2011;117(8):2423–2432. | |
Kohrt HE, Houot R, Weiskopf K, et al. Stimulation of natural killer cells with a CD137-specific antibody enhances trastuzumab efficacy in xenotransplant models of breast cancer. J Clin Invest. 2012;122(3):1066–1075. | |
Kohrt HE, Colevas AD, Houot R, et al. Targeting CD137 enhances the efficacy of cetuximab. J Clin Invest. 2014;124(6):2668–2682. | |
Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol. 2002;20:217–251. | |
Koh CY, Blazar BR, George T, et al. Augmentation of antitumor effects by NK cell inhibitory receptor blockade in vitro and in vivo. Blood. 2001;97(10):3132–3137. | |
Romagné F, André P, Spee P, et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood. 2009;114(13):2667–2677. | |
Benson DM Jr, Hofmeister CC, Padmanabhan S, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood. November 22, 2012;120(22):4324–4333. | |
Vey N, Bourhis JH, Boissel N, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood. 2012;120(22):4317–4323. | |
Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. | |
Zitvogel L, Kroemer G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology. 2012;1(8):1223–1225. | |
Terme M, Ullrich E, Aymeric L, et al. Cancer-induced immunosuppression: IL-18-elicited immunoablative NK cells. Cancer Res. 2012;72(11):2757–2767. | |
Benson DM Jr, Bakan CE, Mishra A, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116(13):2286–2294. | |
Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384(9948):1109–1117. | |
Nocentini G, Riccardi C. GITR: a modulator of immune response and inflammation. Adv Exp Med Biol. 2009;647:156–173. | |
Placke T, Kopp HG, Salih HR. Glucocorticoid-induced TNFR-related (GITR) protein and its ligand in antitumor immunity: functional role and therapeutic modulation. Clin Dev Immunol. 2010;2010:239083. | |
Baltz KM, Krusch M, Bringmann A, et al. Cancer immunoediting by GITR (glucocorticoid-induced TNF-related protein) ligand in humans: NK cell/tumor cell interactions. FASEB J. 2007;21(10):2442–2454. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.