")
Back to Journals » OncoTargets and Therapy » Volume 14
Endometrial Tumors with MSI-H and dMMR Share a Similar Tumor Immune Microenvironment
Authors Song Y, Gu Y, Hu X, Wang M, He Q , Li Y
Received 23 June 2021
Accepted for publication 30 July 2021
Published 16 August 2021 Volume 2021:14 Pages 4485—4497
DOI https://doi.org/10.2147/OTT.S324641
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Yunfeng Song,1,* Ye Gu,1,* Xiang Hu,1 Mengfei Wang,1 Qizhi He,2 Yiran Li1
1Department of Gynecology, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai, People’s Republic of China; 2Department of Pathology, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yiran Li
Department of Gynecology, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, No. 2699, West GaoKe Road, Shanghai, 201204, People’s Republic of China
Tel +86 21-20261000
Fax +86-21-20261234
Email [email protected]
Qizhi He
Department of Pathology, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, No. 2699, West GaoKe Road, Shanghai, 201204, People’s Republic of China
Tel +86 21-20261000
Fax +86-21 20261234
Email [email protected]
Purpose: Microsatellite instability (MSI) and mismatch repair deficiency (dMMR) are important biomarkers for predicting responses to immune checkpoint inhibitor (ICI) therapies. Although PCR-based tests for high MSI (MSI-H) and dMMR yield highly concordant results in endometrial cancer (EC), it is unclear whether this is true for MSI-H and MMR detected by next-generation sequencing (NGS) and immunohistochemistry (IHC), respectively. This study investigated whether EC with MSI-H identified by NGS and dMMR identified by IHC have similar tumor immune microenvironments.
Patients and Methods: EC tissue and corresponding peripheral blood lymphocyte samples were collected from 99 randomly selected patients. MSI status and tumor mutation burden (TMB) were examined by NGS. MMR protein and programmed death ligand (PD-L)1 expression and tumor-infiltrating lymphocyte (TIL) abundance were evaluated by IHC.
Results: Of the 99 EC samples, 29 (29%) had dMMR by IHC, while 18 (18%) had MSI-H by NGS. MSI and MMR status identified by the two methods were discordant in the 99 EC patients, and 2/18 NGS-identified MSI-H patients (11%) retained MMR protein expression. MSI-H and dMMR endometrial tumors had similar numbers of cluster of differentiation (CD)3+ TILs (T cells) and CD8+ TILs (cytotoxic T cells) in the tumor center and periphery, which differed from those in microsatellite stable (MSS) and mismatch repair-proficient (pMMR) EC; they also showed similar TMB, PD-L1 expression, and TIL counts with higher TMB and PD-L1 expression than MSS and pMMR ECs. The abundance of CD3+ and CD8+ TILs was increased in PD-L1-positive EC.
Conclusion: NGS-identified MSI status and IHC-identified MMR status were inconsistent in EC, and 11% of NGS-identified MSI-H tumors retained MMR protein expression. Conversely, MSI and MMR status determined by the two methods provided similar data on TMB, PD-L1 expression, and TIL abundance, which can guide treatment decisions with ICIs.
Keywords: TMB, PD-L1, TIL, checkpoint therapy, endometrial cancer
Introduction
The incidence of endometrial cancer (EC) is increasing yearly, and EC is one of the three most prevalent gynecologic tumors affecting women.1,2 Most patients with EC have a low risk of recurrence and can be managed by surgery alone.3 For metastatic and relapsed EC, chemotherapy and hormone therapy are the only available treatment options, but these are toxic and do not significantly improve 5-year overall survival, progression-free survival, or relapse rate.4,5 Many new targeted therapies have recently been evaluated in clinical trials, but only a few have improved survival and therapeutic responses. Therefore, it is important to identify molecular subgroups of EC that are likely to benefit from targeted therapies including immunotherapy.
EC is a heterogeneous disease comprising multiple histotypes with different risk factors, clinical behaviors, and outcomes. In the 1970s, a traditional classification of EC into types 1 and 2 was proposed.6 The classification applied in clinical practice to guide therapeutic decision-making is based on histopathologic assessment of cell type and grade;7,8 however, this can be highly subjective, difficult to validate, and has limited utility for predicting responses to therapy.9,10 Thus, there is a need for a more reproducible, objective, and biologically informative classification system for EC. The Cancer Genome Atlas (TCGA) research network described four distinct prognostic EC subtypes in 2013 based on genomic abnormalities that reflect EC tumor biology, which can provide more precise guidance for surgery, adjuvant therapy, and disease surveillance.11 One of these subtypes is high microsatellite instability (MSI-H) or a hypermutated phenotype resulting from a defective mismatch repair (MMR) mechanism (known as MMR-deficient [dMMR]), which is observed in approximately 30% of endometrial tumors.3,12
The algorithm for EC screening may include an immunohistochemistry (IHC)-based test for MMR, PCR-based test for MSI, or both, as they are equally valid initial screening tools in EC. However, IHC is the preferred method because of the high false negative rates of MSI tests in EC.13–15 Next-generation sequencing (NGS) platforms use different technologies to decode the identity of nucleotides in DNA or detect covalent modifications such as methylated nucleotides; a distinctive genomic signature for the high MSI (MSI-H) phenotype of EC was identified using a targeted NGS gene panel. Although PCR-based tests for MSI and MMR in EC have high concordance, there is little or no agreement between NGS gene panel results for MSI and IHC results for MMR.11,16 This is clinically significant, because tumor-infiltrating lymphocytes (TILs) are more abundant in the immune microenvironment of MSI-H EC compared to microsatellite-stable (MSS) EC.17 Additionally, programmed death ligand (PD-L)1 expression was elevated in dMMR EC compared to MMR-proficient (pMMR) cases. It is therefore important to compare the tumor immune microenvironment in EC with NGS-identified MSI-H and IHC-identified dMMR to determine whether there are any discrepancies that could influence decisions concerning immune checkpoint inhibitor (ICI) therapies.
In the present study we analyzed the degree of concordance between NGS-identified MSI-H and IHC-identified dMMR ECs—specifically with respect to the tumor immune environment (eg, neoantigens, PD-L1 expression, and TIL levels)—with the aim of identifying EC subgroups that could potentially benefit from targeted immunotherapy regimens.
Materials and Methods
Patients and Tissues
We obtained EC tissues and corresponding peripheral blood lymphocyte samples from 99 randomly selected (in terms of age and personal or family history of cancer) patients who were treated at our institution between 2015 and 2018. Clinical information obtained from hospital records included age at diagnosis, surgical stage, adjuvant treatment, and disease status. The patients were staged according to the International Federation of Gynecology and Obstetrics criteria. Tumor grade, histologic subtype, depth of myometrial invasion, and lymphovascular invasion were reviewed by a gynecologic pathologist based on the 2014 World Health Organization criteria. This study was approved by the Human Investigation Ethics Committee of the Shanghai First Maternity and Infant Hospital. Samples were collected from patients after they had provided written, informed consent.
IHC
Deparaffinized tissue sections were rehydrated in dewaxing solution (Maixin, Fuzhou, China) for 10 min, and antigens were retrieved by heating the sections in EDTA (pH 9.0) for 20 min at 99°C. IHC was performed using a Titan S autostainer (Lumatas Biosystems, Fremont, CA, USA) as follows. Endogenous peroxidase activity was blocked with H2O2 (Maixin, Fuzhou, China) and the sections were incubated with primary antibodies diluted 1:100 or 1:200 (Table S1) at room temperature for 30 min, followed by incubation with secondary antibodies. The sections were stained with 3,3′-diaminobenzidine (DAB) as the chromogen (Maixin, Fuzhou, China) using the Elivision Super kit (Maixin, Fuzhou, China), followed by staining with prediluted anti–PD-L1 antibody (clone SP263; Ventana Medical Systems, Tucson, AZ, USA). The negative control was a matched rabbit IgG. The sections were assessed using an OptiView DAB IHC Detection Kit (Thermo Fisher Scientific, Waltham, MA, USA) on a BenchMark ULTRA automated staining platform (Roche, Basel, Switzerland). The specificity and sensitivity of the assay were evaluated using control placental tissue samples with known PD-L1 expression levels.
Evaluation of IHC Staining
Three pathologists who were blinded to the MSI status of patient samples independently reviewed the stained sections. Nuclear staining of adjacent normal endometrial lymphocytes served as the internal positive control. We judged the complete absence of nuclear staining in the tumor cells as a loss of MMR protein expression, and manually counted TILs in the five areas with the highest lymphocyte aggregation within or adjacent to the tumor (Figure 1). The sections were scanned at 20× magnification using an Aperio Scanscope AT Turbo scanner (Leica Biosystems, Nussloch, Germany) as previously described.17 Immune cells in the tumor periphery (Figure 1, green boxes) were defined as those that were +1 or −1 mm from the invasive tumor front or endometrium–myometrium interface. The center (Figure 1A, yellow boxes) was defined as the area >1 mm away from the invasive tumor front or endometrium–myometrium interface toward the luminal aspect (Figure 1B). The box with the highest signal intensity was considered as a hotspot (Figure 1C, red boxes). Intraepithelial TILs were counted in a 1-mm2 area of tumor located within the boundary of tumor cell nests or glands. Stromal TILs were those in the stroma of the endometrial tumor. The numbers of positive cells in the periphery, center, hotspot, and intraepithelial and stromal areas were summed and divided by the number of cells in the total area (mm2). The results for cluster of differentiation (CD)3 and CD8 cells are shown as positive cells/mm2.
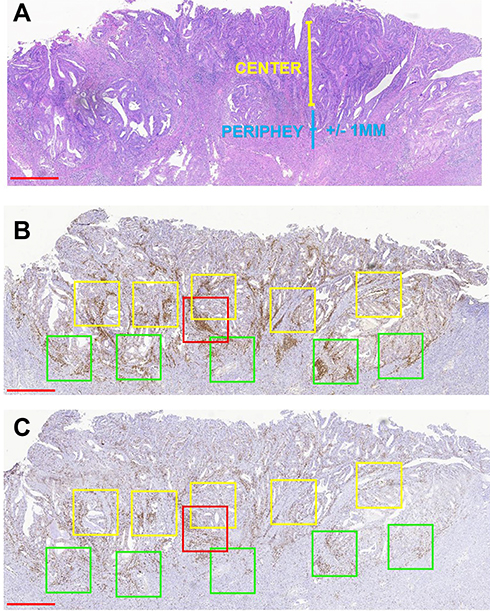 |
Figure 1 Strategy for quantifying TILs in endometrioid EC. (A) Representative H&E staining of endometrioid EC to define the tumor center and periphery. (B and C) Quantification of CD3+ cells (B) and CD8+ cells (C) in the tumor center and periphery. Red box, hotspot (single 1-mm2 box); green boxes, tumor periphery (five 1-mm2 boxes); yellow boxes, central areas (five 1-mm2 boxes). Scale bars: 1 mm. Abbreviation: H&E, hematoxylin and eosin. |
PD-L1 was predominantly expressed at the margin or the infiltrating edge of the interface between the tumor and immune stroma (Supplementary Figure S1). PD-L1 expression was evaluated in tumor cells and tumor-infiltrating immune cells. The proportion of PD-L1–positive tumor cells was estimated as the ratio (%) of total tumor cells, and the ratio of PD-L1–positive tumor-infiltrating immune cells in tumors was also calculated. We chose 1% and 5% cutoffs as the PD-L1–positive thresholds for tumor and immune cells, respectively (Supplementary Figure S2).
Detection of MSI-H by NGS
MSI status was determined by analyzing total genomic DNA extracted from formalin-fixed paraffin-embedded (FFPE) tumor tissues by NGS. Areas of interest with the highest tumor cellularity and viability (minimum 10% for both parameters) in sections stained with hematoxylin and eosin were selected. Tumor cellularity was enriched by macrodissection using a sterile 1-mm tissue punch. Total genomic DNA was extracted from tissue cores or unstained slides using the QIAamp DNA FFPE tissue kit (Qiagen, Valencia, CA, USA), and DNA concentrations were measured using Qubit dsDNA assays (Thermo Fisher Scientific). Fragments of DNA obtained using a Covaris M220 focused ultrasonicator (Covaris, Woburn, MA, USA), were selected using AMPure beads and the Agencourt AMPure XP kit (Beckman Coulter, Brea, CA, USA). The genetic profile of all tissue samples was assessed by capture-based targeted deep sequencing using the NGS genetic testing panel OncoScreen Plus (Burning Rock Dx, Guangzhou, China) with a modified Roche/Nimblegen SeqCap EZ method (Roche). This panel selects 520 genes that are associated with cancer pathogenesis and targeted therapies via probe hybridization and high-throughput sequencing, including whole exon regions of 312 genes and hotspot mutation regions (exons, introns, and promoter regions) of 208 genes. The panel can comprehensively and accurately detect variations in gene mutations, amplifications, and fusions with clinical relevance to tumors as well as biomarkers for response to ICIs such as the tumor mutation burden (TMB) and MSI. DNA quality and size were assessed using a high-sensitivity DNA assay and bioanalyzer. All indexed samples were sequenced using a NovaSeq 6000 system (Illumina, San Diego, CA, USA) with paired-end reads.
TMB
TMB is reported as mutations per megabases of sequenced DNA and was calculated based on the number of somatic mutations identified by NGS to exclude any known single nucleotide polymorphisms in the database.18
Statistical Analysis
Statistical analyses were performed using SPSS v20.0 (SPSS Inc, Chicago, IL, USA) and Prism v8.1.2 (GraphPad, La Jolla, CA, USA). Categorical data are described as frequency and percentage, and quantitative variables are expressed as mean±SEM. Fisher’s exact test or the chi-squared test was used to compare categorical variables. The Student’s t test was used to compare parametric data with a normal distribution, and the Mann–Whitney U-test was used for nonparametric data. Differences with P<0.05 were considered significant.
Results
MSI Status of EC Determined by NGS and dMMR Detected by IHC
The clinical and pathologic characteristics of the 99 patients included in this study are summarized in Table S2. Most patients were diagnosed with grade 1 EC (Table 1). The MSI status of the patients as determined by NGS and IHC was compared (Table S4). The levels of the MMR proteins MutL homolog (MLH)1, MSH2, MSH6, and PMS1 homolog (PMS)2 in 99 endometrial tumors were assessed by IHC. Staining of MMR proteins was normal in 70 tumors with pMMR (78%), indicating MSS; 29 (29%) showed no immunoreactivity against one or two MMR proteins and were thus identified as dMMR, indicating MSI-H. Most of these tumors lacked expression of both MLH1 and PMS2 (Figure 2). The NGS results showed that 18/99 tumors (18%) had MSI; 2 of these (11%) retained MMR protein expression, and 16 (89%) were confirmed as dMMR by IHC. Additionally, 10 tumors (10%) had germline mutations in MMR-related genes, of which 6 (60%) had NGS-identified MSI-H (Table S3). A total of 26 tumors (26%) had somatic mutations in MMR-related genes and 11 of these had NGS-identified MSI-H (Table S4). Compared to the detection of MSI status by NGS, IHC-identified dMMR had a sensitivity of 55.2% (95% confidence interval [CI]: 37.07%, 73.27%), specificity of 97.1% (95% CI: 93.24%, 101.05%), positive predictive value of 88.9% (95% CI: 74.37%, 103.41%), and negative predictive value of 84.0% (95% CI: 75.96%, 91.94%) for detecting MSI status (Table 2).
 |
Table 1 Clinical and Pathological Characteristics of the Study Population |
 |
Table 2 Classification of MSI by NGS Compared with Classification of MMR by IHC |
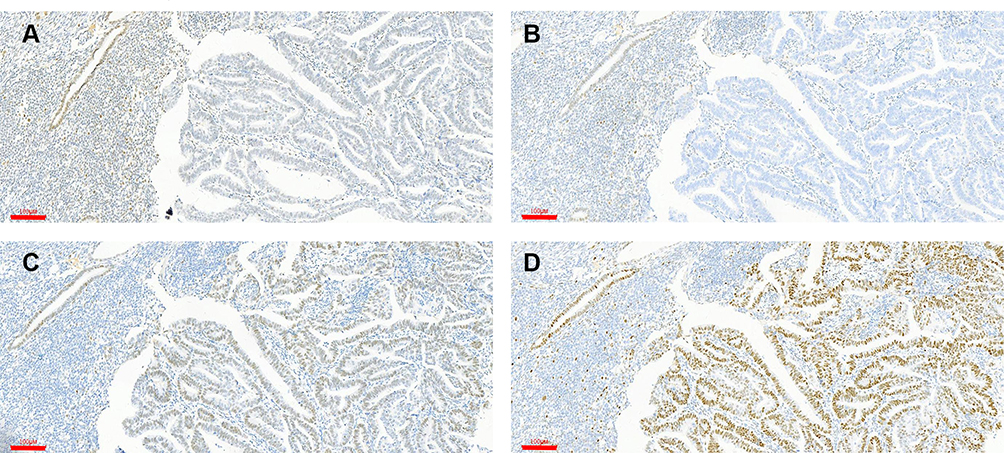 |
Figure 2 Detection of MMR protein expression by IHC. (A and B) Loss of MLH (A) and PSM2 (B) staining. (C and D) Intact MSH2 (C) and MSH6 (D) staining. Note the presence of nuclear staining in adjacent glands and benign stromal cells, validating the IHC method. Scale bars: 100 μm. |
TMB and PD-L1 Expression in MSI-H and dMMR EC
We next examined the relationship between TMB, MSI-H or dMMR, and PD-L1 expression. MSI-H tumors had high TMB (Figure 3A) and showed increased PD-L1 expression in tumor and immune cells compared to MSS tumors (Figures 3B and 4). Increased TMB (Figure 5A) and an increasing trend in PD-L1 expression in tumor and immune cells were observed in dMMR tumors compared to pMMR tumors (Figures 5B and 6). MSI-H tumors had TMB and PD-L1 expression levels similar to dMMR tumors (Figure 7A and B).
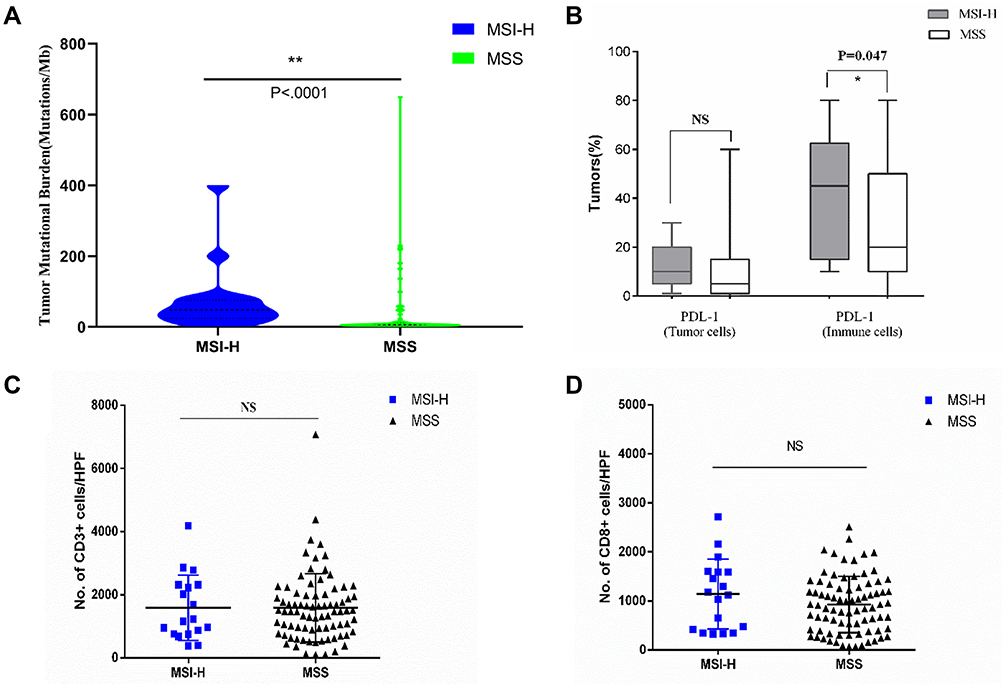 |
Figure 3 Neoantigen load and PD-L1 expression level in MSI-H and MSS EC. (A) TMB per megabase in tumor DNA exons and introns detected by ColoSeq. Blue and green represent MSI-H and MSS tumors, respectively. (B–D) Average numbers of PD-L1–expressing tumor cells and TILs (B), CD3+ immune cells (C), and CD8+ immune cells (D) identified by IHC. NS, nonsignificant at p>0.05, *p<0.05, **p<0.01 (Mann–Whitney U-test). Abbreviation: HPF, high-power field. |
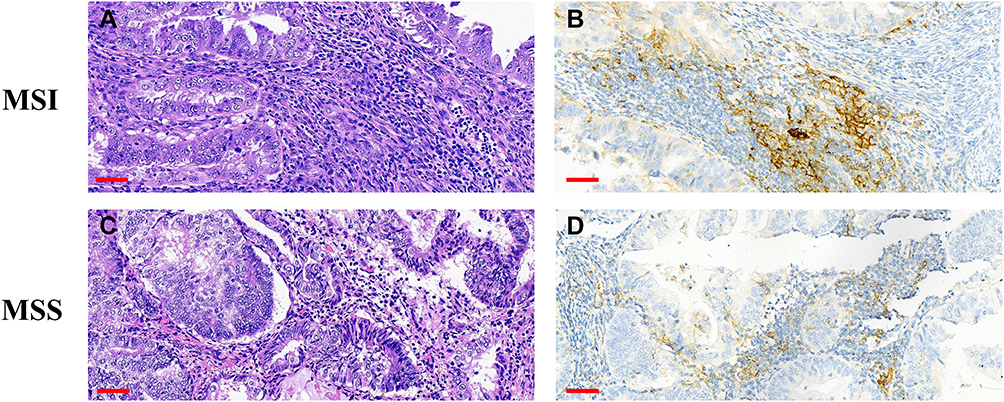 |
Figure 4 Upregulation of PD-L1 in MSI-H EC. (A–D) H&E staining and positive membrane staining for PD-L1 in MSI-H ECs. Scale bars: 60 μm. Abbreviation: H&E, hematoxylin and eosin. |
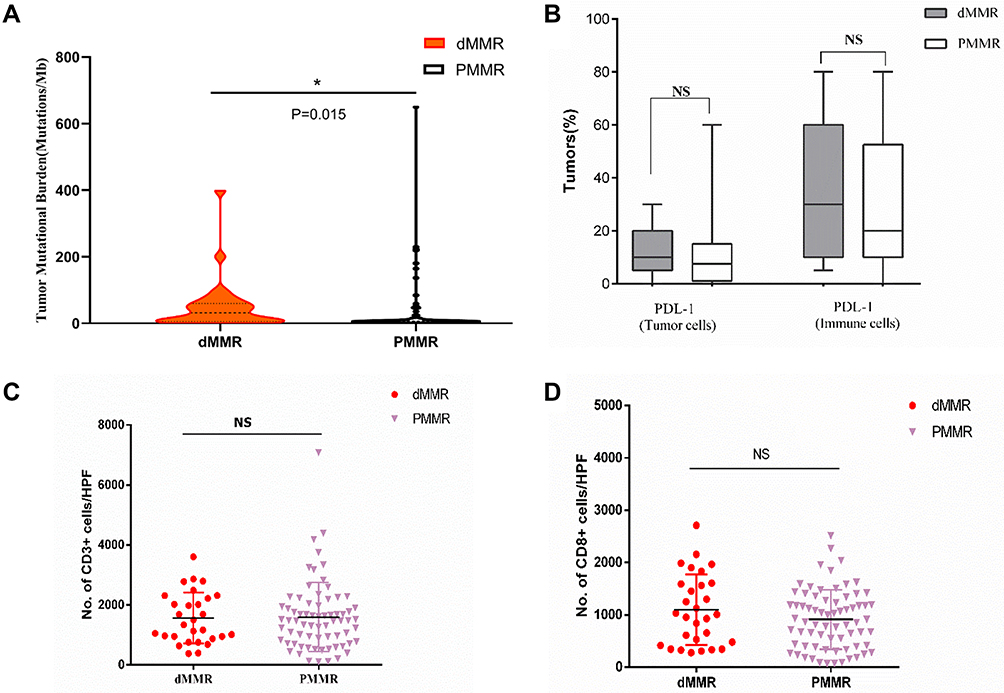 |
Figure 5 Neoantigen load and PD-L1 expression level in dMMR and PMMR endometrial tumors. (A) TMB per megabase in tumor DNA exons and introns detected by ColoSeq. Red and black represent dMMR and PMMR tumors, respectively. (B) Average numbers of PD-L1–expressing tumor cells and TILs (B), CD3+ immune cells (C), and CD8+ immune cells (D) identified by IHC. NS, nonsignificant at p>0.05, *p<0.05, (Mann–Whitney U-test). Abbreviation: HPF, high-power field. |
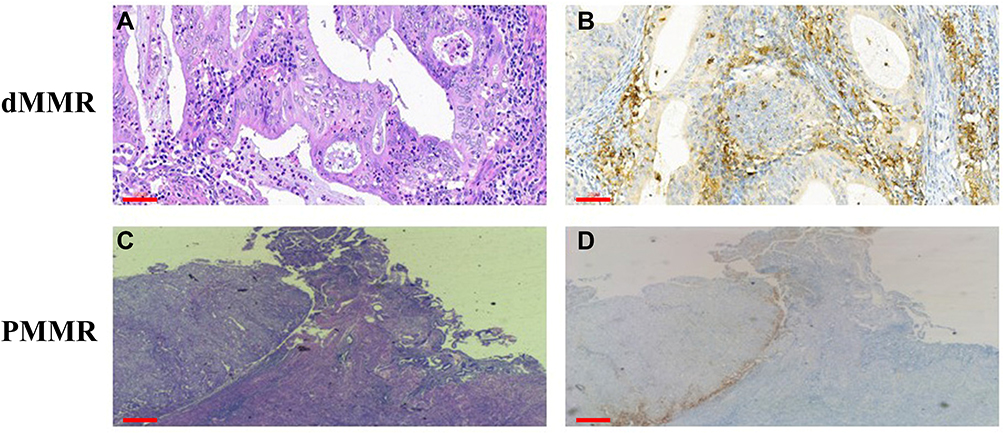 |
Figure 6 Upregulation of PD-L1 expression in dMMR EC. (A–D) H&E staining and positive membrane staining for PD-L1 in dMMR ECs. H&E, hematoxylin and eosin. Scale bars: 60 μm. |
 |
Figure 7 Neoantigen load and PD-L1 expression level in dMMR and MSI-H endometrial tumors. (A) TMB per megabase in tumor DNA exons and introns detected by ColoSeq. Red and blue represent dMMR and pMMR tumors, respectively. (B) Average numbers of PD-L1–expressing tumor cells and TILs (B), CD3+ immune cells (C), and CD8+ immune cells (D) identified by IHC. NS, nonsignificant at p>0.05 (Mann–Whitney U-test). Abbreviation: HPF, high-power field. |
TILs in MSI-H and dMMR EC
To compare the tumor immune environments in EC identified as MSI-H by NGS and dMMR by IHC, we performed IHC-based quantification of CD3+ and CD8+ TILs. CD3+ and CD8+ cells in the tumor center and periphery were counted in the tumor center (Figure 1, yellow boxes), hotspot (Figure 1, red boxes), and periphery (Figure 1, green boxes) in areas of highest staining density in five fields with an area of 1 mm2. MSI-H and MSS EC had comparable numbers of CD3+ TILs (T cells) and CD8+ TILs (cytotoxic T cells) in the tumor center (CD3+: 558.2 vs 517.3, P=0.638; CD8+: 138.6 vs 148.5, P=0.428) and periphery (CD3+: 669.9 vs 549.9, P=0.270; CD8+: 163.5 vs 199.2, P=0.953) (Table 3 and Figure 3C and D); dMMR and pMMR EC also showed similar numbers of CD3+ TILs (T cells) and CD8+ TILs (cytotoxic T cells) in the tumor center (CD3+: 571.8 vs 531.6, P=0.734; CD8+: 97.0 vs 148.5, P=0.276) and periphery (CD3+: 593.4 vs 555.5, P=0.270; CD8+: 124.1 vs 205.6, P=0.793) (Table 4 and Figure 5C and D). MSI-H tumors had similar numbers of CD3+ TILs and CD8+ TILs as dMMR tumors (Figure 7C and D).
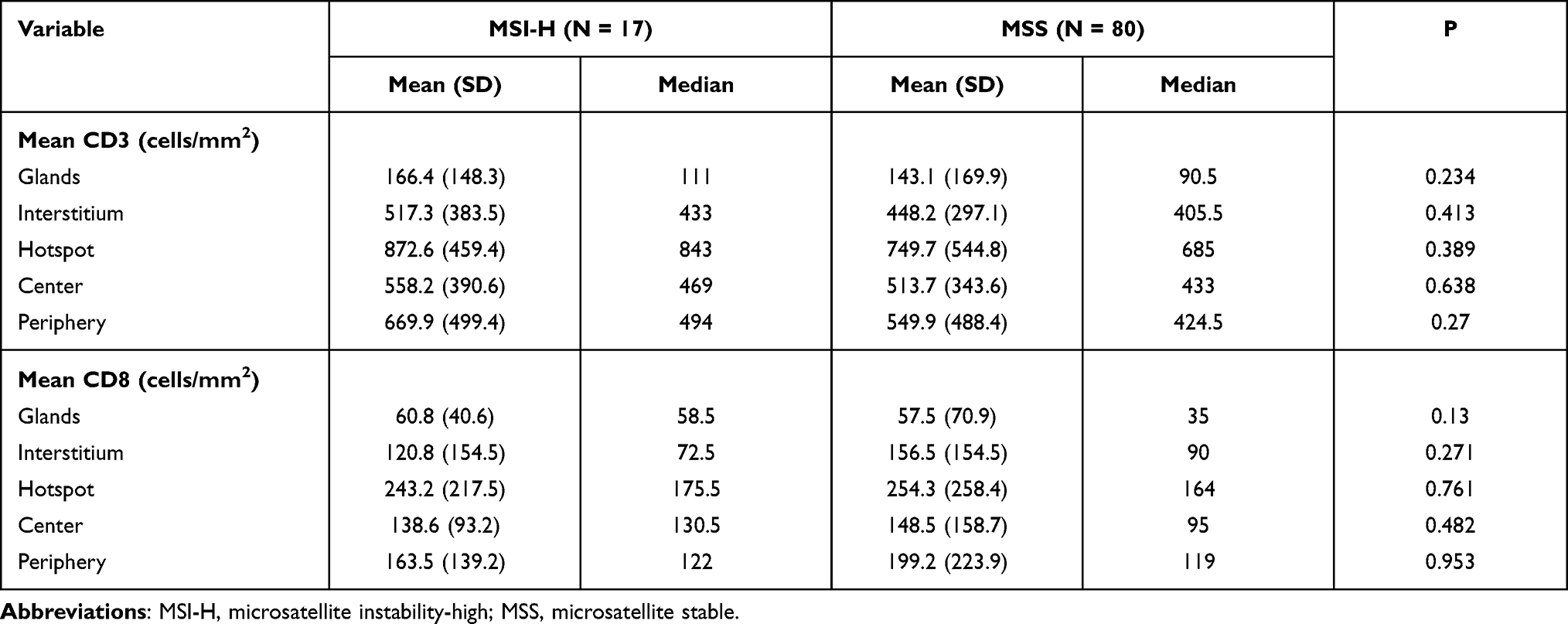 |
Table 3 TILs in MSI-H and MSS ECs |
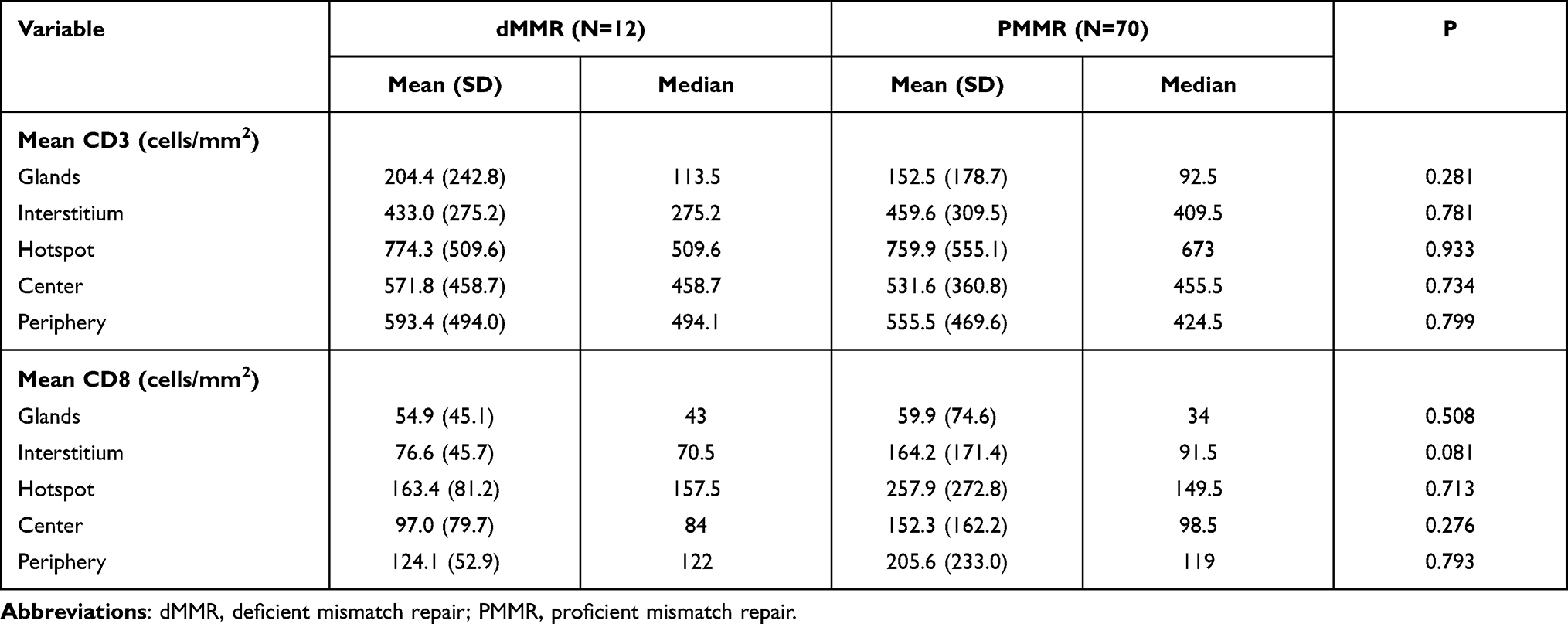 |
Table 4 TILs in dMMR and PMMR ECs |
TILs are Increased in PD-L1–Positive EC
PD-L1 expression was detected in tumor cells in 75/98 tumors (76.5%) and immune cells in 93/98 tumors (94.9%) with the 1% cutoff (Tables 5 and 6); and in tumor cells in 52/98 tumors (53.1%) and immune cells in 82/98 tumors (83.7%) with the 5% cutoff (Tables 7 and 8). PD-L1 expression increased significantly with CD3+ and CD8+ TIL numbers in the tumor microenvironment with both cutoff values (Tables 5–8). IHC analysis revealed that endometrial tumors with high PD-L1 expression had higher numbers of CD3+ and CD8+ TILs in the tumor microenvironment (Figure 8).
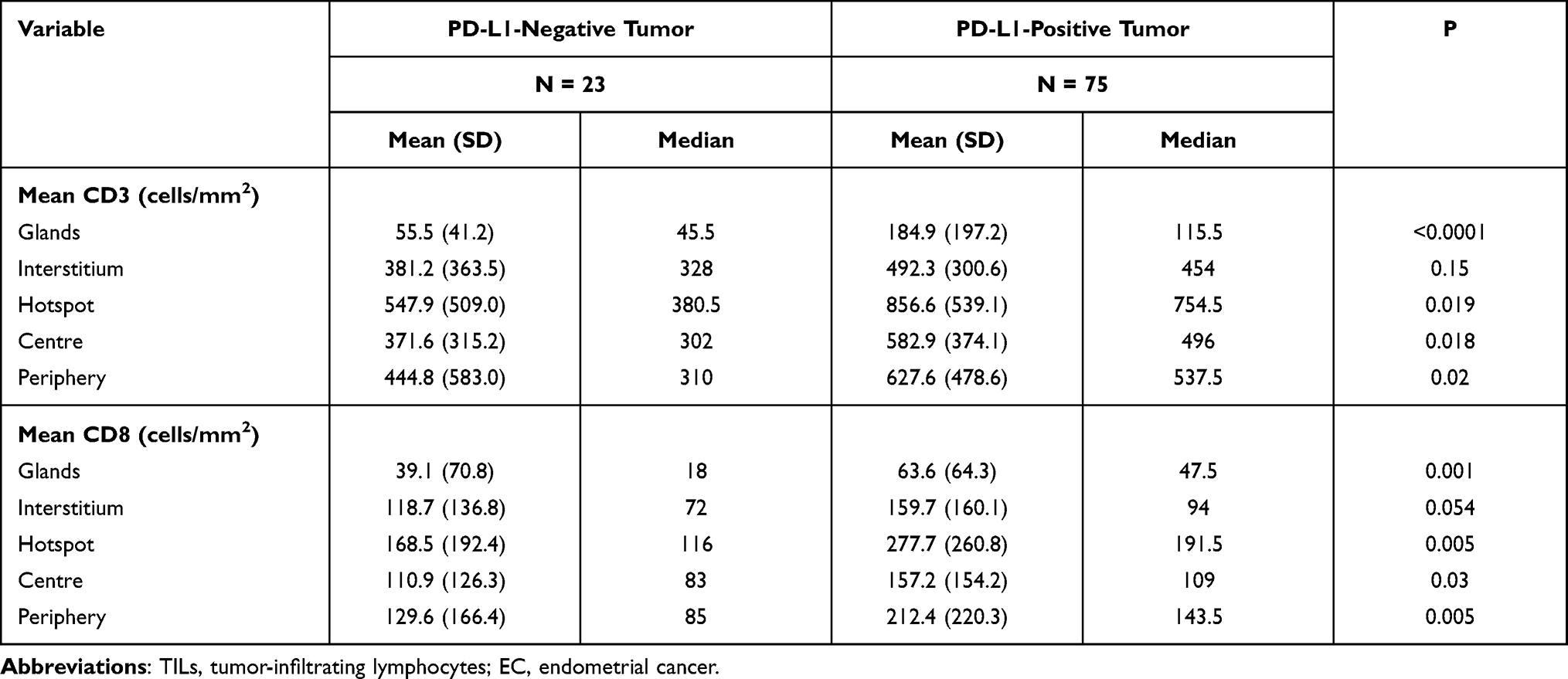 |
Table 5 TILs in ECs with or without Positive PD-L1 Expression in Tumor Cells (Cutoff Value: 1%) |
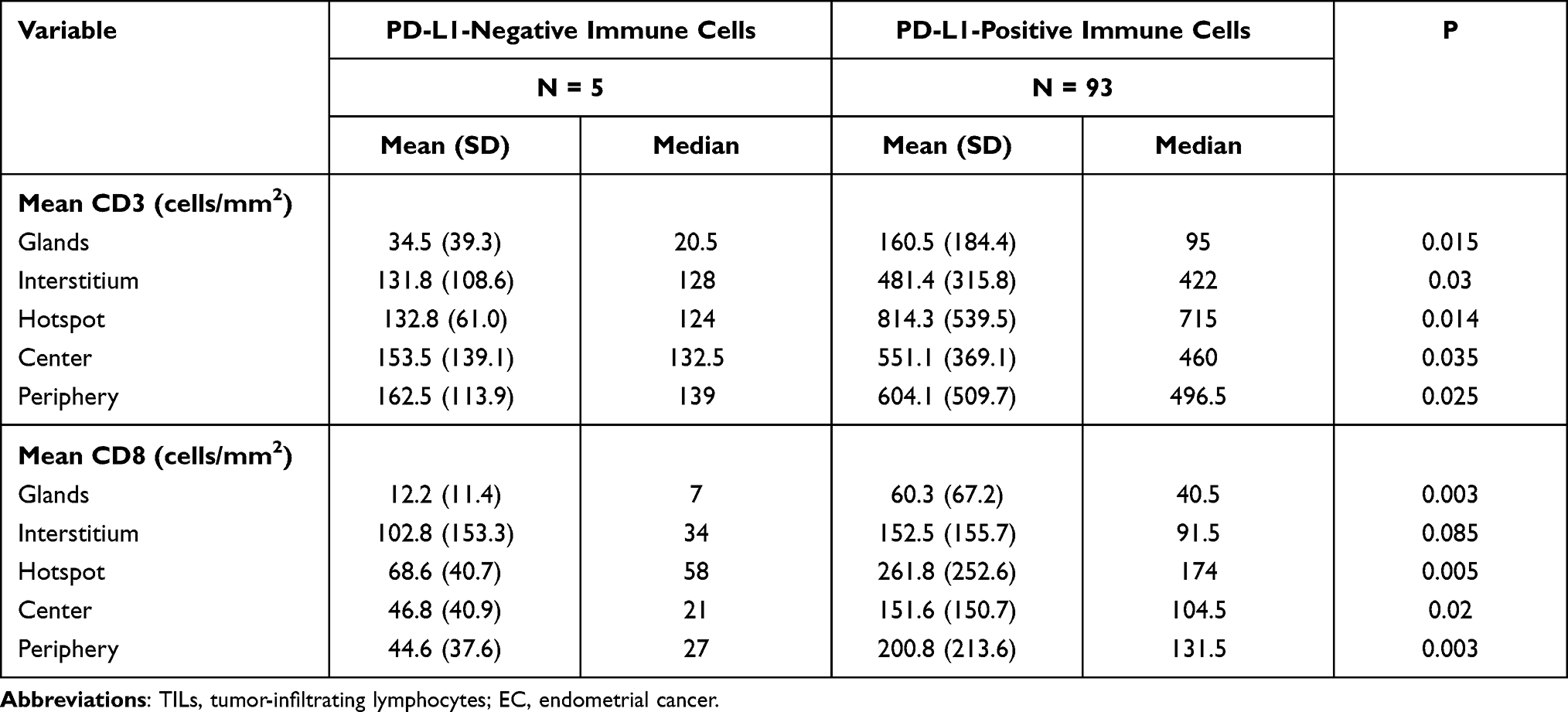 |
Table 6 TILs in ECs with or without Positive PD-L1 Expression in Immune Cells (Cutoff Value: 1%) |
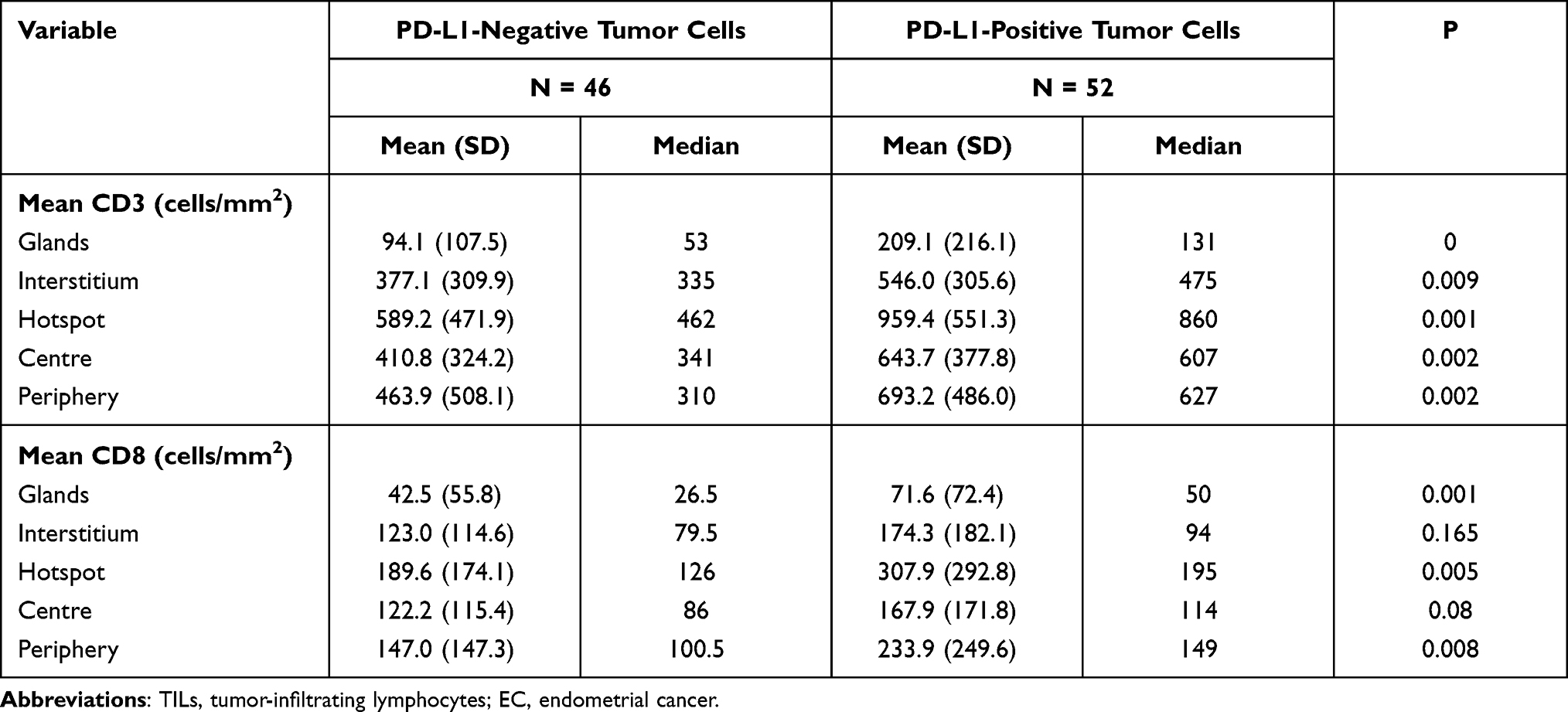 |
Table 7 TILs in ECs with or without Positive PD-L1 Expression in Tumor Cells (Cutoff Value: 5%) |
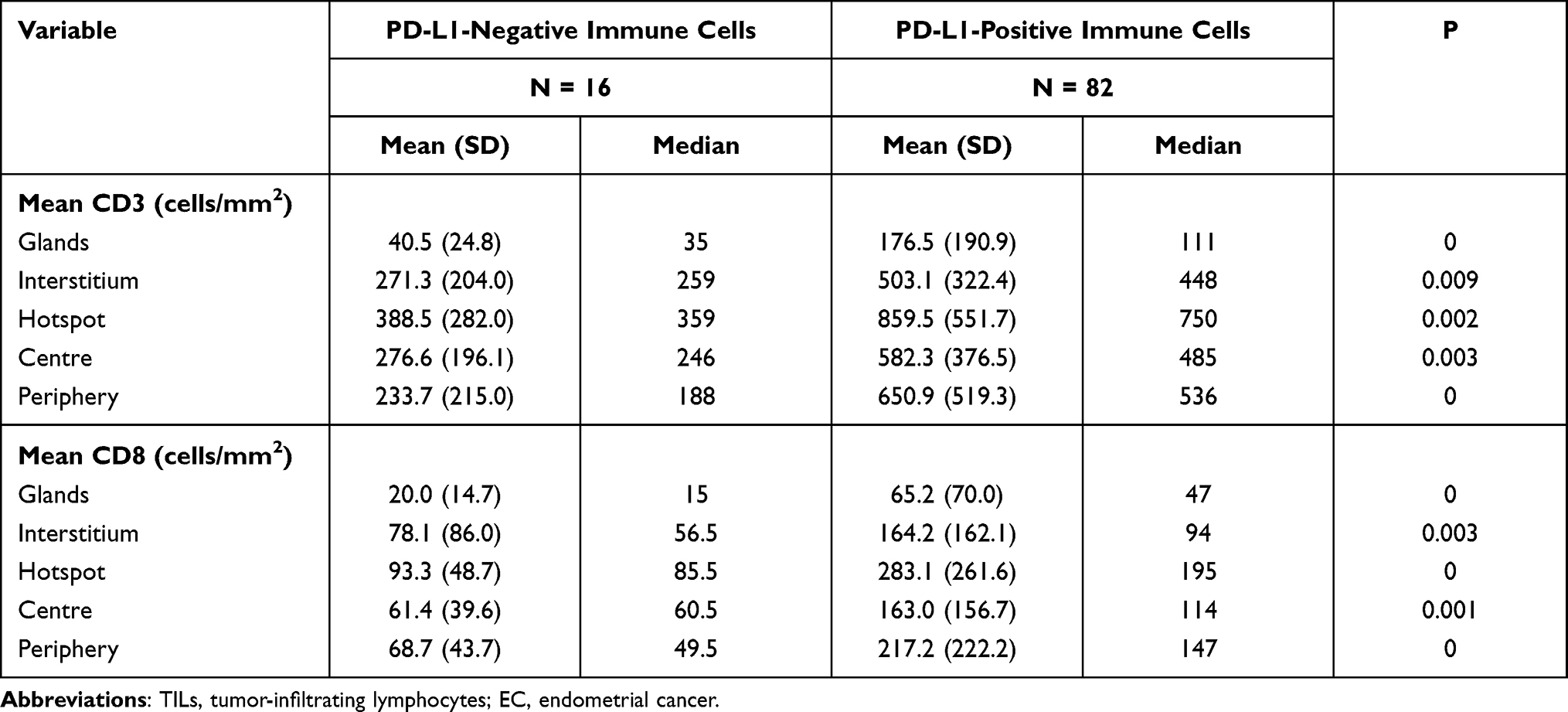 |
Table 8 TILs in ECs with or without Positive PD-L1 Expression in Immune Cells (Cutoff Value: 5%) |
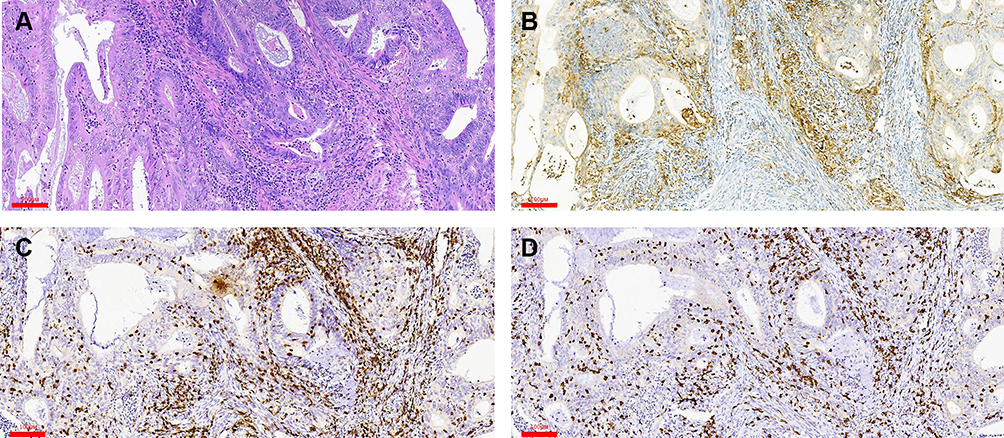 |
Figure 8 Detection of PD-L1, CD3, and CD8 expression in the tumor center by IHC. (A) H&E staining. (B–D) Positive staining for PD-L1 (B), CD3 (C), and CD8 (D). Scale bars: 60 μm. |
Discussion
MSI-H is an important biomarker for predicting response to ICI therapies, and defects in DNA MMR proteins are the primary cause of MSI.19 In our study, 100% of IHC-identified dMMR and NGS-identified MSI-H endometrial tumors belonged to different subgroups but had similar TMB, PD-L1 expression, and TIL counts, which can be useful for predicting response to ICI therapies.
MSI-H is a tumor biomarker in a variety of tumors including colorectal and gastric cancers and EC,20–22 and can be determined either by PCR with or without specific fragment sizes or by NGS depending on the individual tumor entity.23–25 DNA MMR is a critical DNA repair pathway that contributes to genomic stability,26 and tumors with dMMR—which is the primary cause of MSI-H9—have shown a remarkable response to immunotherapy in clinical trials.27 Gene mutations and slipped-strand mispairing contribute to the instability of DNA strands during the replication of repeated DNA sequences and can also give rise to a strong mutator phenotype and MSI.9,28 The expression of MMR proteins corresponds to MSI-H detection results in >90% of cases, while approximately 6% of MSI-H tumors retain MMR protein expression.29,30 In this study, 18% of tumors were identified as MSI-H by NGS and 29% as dMMR by IHC; 16% had both NGS-identified MSI-H and IHC-identified dMMR. The MMR and MSI status were discordant in all 99 endometrial tumors, and two NGS-identified MSI-H cases (11%) retained MMR protein expression. Determining the level of agreement between MSI and MMR status is critical for predicting the response to ICI therapy in EC.
MSI-H and dMMR tumors usually harbor a large number of neoantigens that contribute to immune activation.31,32 Targeting checkpoints related to immune cell activation is the most effective way to activate the antitumor immune response.33 In advanced carcinomas, MSI-H is an established predictor of therapeutic response to checkpoint-directed immunotherapies.34,35 Additionally, dMMR was shown to enhance the efficacy of ICIs in solid tumors. We determined that NGS-identified MSI-H and IHC-identified dMMR EC has similar TMB, PD-L1 expression, and TIL counts. TMB and PD-L1 expression were higher in NGS-identified MSI-H and IHC-identified dMMR EC samples than in those with MSS and pMMR, respectively. There were no differences in CD3+ and CD8+ TIL counts between ECs with MSI-H and MSS identified by NGS or between ECs with dMMR and pMMR identified by IHC. The higher TMB and PD-L1 expression in MSI-H and dMMR endometrial tumors supports the clinical efficacy of programmed death (PD)-1 inhibitors in the treatment of solid tumors with MSI24 and dMMR.27,31,34 Furthermore, PD-L1 expression increased with TIL abundance in the endometrial tumor microenvironment, indicating that PD-1 inhibitors can elicit an effective antitumor immune response in EC with high PD-L1 expression.
Conclusion
NGS-identified MSI and IHC-identified MMR status was inconsistent in 100% of examined EC samples and 11% of NGS-identified MSI-H cases retained MMR protein expression. Furthermore, MSI-H and dMMR endometrial tumors had similar tumor immune microenvironments with comparable TMB, PD-L1 expression, and TIL abundance. A combination of MSI-H and dMMR may better reflect the pathologic status of EC patients and can facilitate clinical decision-making regarding ICI therapy.
Abbreviations
CD, cluster of differentiation; DAB, 3,3′-diaminobenzidine; dMMR, deficient mismatch repair; EC, endometrial cancer; FFPE, formalin-fixed paraffin-embedded; ICI, immune checkpoint inhibitor; IHC, immunohistochemistry; MMR, mismatch repair; MSI, microsatellite instability; MSI-H, high microsatellite instability; MSS, microsatellite-stable; NGS, next-generation sequencing; PD-1, programmed death 1; PD-L1, programmed death ligand 1; pMMR, mismatch repair-proficient; TCGA, The Cancer Genome Atlas; TIL, tumor-infiltrating lymphocyte; TMB, tumor mutation burden.
Data Sharing Statement
The datasets used and/or analyzed in the present study are available from the corresponding author Yiran Li on reasonable request.
Ethics Approval and Consent to Participate
This study was approved by the Human Investigation Ethics Committee of the Shanghai First Maternity and Infant Hospital. Written, informed consent was received from participants before their inclusion in the study. The study complies with the guidelines of the Helsinki Declaration.
Consent for Publication
The authors confirm that written, informed consent was obtained from the patients to publish their data.
Acknowledgments
The authors thank their colleagues for providing helpful suggestions concerning this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by grants from the National Natural Science Foundation of China (nos. 81472427, 32070583, 81672574, and 81702547); Shanghai Health System Outstanding Talents Program (no. 2018YQ23); Shanghai New Frontier Technology Project (no. SHDC12015110); and Shanghai Municipal Medical and Health Discipline Construction Projects (no. 2017ZZ02015).
Disclosure
The authors have declared that no conflict of interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30.
2. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
3. Kunitomi H, Banno K, Yanokura M, et al. New use of microsatellite instability analysis in endometrial cancer. Oncol Lett. 2017;14:3297–3301.
4. Wilkinson-Ryan I, Binder PS, Pourabolghasem S, et al. Concomitant chemotherapy and radiation for the treatment of advanced-stage endometrial cancer. Gynecol Oncol. 2014;134:24–28.
5. Kokka F, Brockbank E, Oram D, et al. Hormonal therapy in advanced or recurrent endometrial cancer. Cochrane Database Syst Rev. 2010;12:CD007926.
6. Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol. 1983;15:10–17.
7. Dedes KJ, Wetterskog D, Ashworth A, et al. Emerging therapeutic targets in endometrial cancer. Nat Rev Clin Oncol. 2011;8:261–271.
8. Matias-Guiu X, Prat J. Molecular pathology of endometrial carcinoma. Histopathology. 2013;62:111–123.
9. Karamurzin Y, Rutgers JK. DNA mismatch repair deficiency in endometrial carcinoma. Int J Gynecol Pathol. 2009;28:239–255.
10. Murali R, Soslow RA, Weigelt B. Classification of endometrial carcinoma: more than two types. Lancet Oncol. 2014;15:e268–e278.
11. Cancer Genome Atlas Research Network; Kandoth C, Schultz N, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73.
12. Bregar A, Deshpande A, Grange C, et al. Characterization of immune regulatory molecules B7-H4 and PD-L1 in low and high grade endometrial tumors. Gynecol Oncol. 2017;145:446–452.
13. Bruegl AS, Djordjevic B, Batte B, et al. Evaluation of clinical criteria for the identification of lynch syndrome among unselected patients with endometrial cancer. Cancer Prev Res (Phila). 2014;7:686–697.
14. Goodfellow PJ, Billingsley CC, Lankes HA, et al. Combined microsatellite instability, MLH1 methylation analysis, and immunohistochemistry for lynch syndrome screening in endometrial cancers from GOG210: an NRG Oncology and Gynecologic Oncology Group Study. J Clin Oncol. 2015;33:4301–4308.
15. Leenen CH, van Lier MG, van Doorn HC, et al. Prospective evaluation of molecular screening for lynch syndrome in patients with endometrial cancer ≤70 years. Gynecol Oncol. 2012;125:414–420.
16. Zhao S, Choi M, Overton JD, et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci USA. 2013;110:2916–2921.
17. Howitt BE, Shukla SA, Sholl LM, et al. Association of polymerase e-mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA Oncol. 2015;1:1319–1323.
18. 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74.
19. Richman S. Deficient mismatch repair: read all about it. Int J Oncol. 2015;47:1189–1202.
20. Imai K, Yamamoto H. Carcinogenesis and microsatellite instability: the interrelationship between genetics and epigenetics. Carcinogenesis. 2008;29:673–680.
21. Watson P, Lynch HT. The tumor spectrum in HNPCC. Anticancer Res. 1994;14:1635–1639.
22. Bonneville R, Krook MA, Kautto EA, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol. 2017;2017:
23. Soumerai TE, Donoghue MTA, Bandlamudi C, et al. Clinical utility of prospective molecular characterization in advanced endometrial cancer. Clin Cancer Res. 2018;24:5939–5947.
24. Vanderwalde A, Spetzler D, Xiao N, et al. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018;7:746–756.
25. Ta RM, Hecht JL, Lin DI. Discordant loss of mismatch repair proteins in advanced endometrial endometrioid carcinoma compared to paired primary uterine tumors. Gynecol Oncol. 2018;151:401–406.
26. Liu D, Keijzers G, Rasmussen LJ. DNA mismatch repair and its many roles in eukaryotic cells. Mutat Res. 2017;773:174–187.
27. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413.
28. Castillo-Lizardo M, Henneke G, Viguera E. Replication slippage of the thermophilic DNA polymerases B and D from the Euryarchaeota Pyrococcus abyssi. Front Microbiol. 2014;5:403.
29. Hechtman JF, Rana S, Middha S, et al. Retained mismatch repair protein expression occurs in approximately 6% of microsatellite instability-high cancers and is associated Latham A, Srinivasan P, Kemel Y, et al. Microsatellite instability is associated with the presence of Lynch syndrome pan-cancer. J Clin Oncol. 2019;37:286–295.
30. Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, Phase 2 study. Lancet Oncol. 2017;18:1182–1191.
31. Willis JA, Reyes-Uribe L, Chang K, et al. Immune activation in mismatch repair-deficient carcinogenesis: more than just mutational rate. Clin Cancer Res. 2020;26:11–17.
32. Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res. 2019;38:255.
33. Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520.
34. Timmermann B, Kerick M, Roehr C, et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS One. 2010;5:e15661.
35. Sahin IH, Akce M, Alese O, et al. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br J Cancer. 2019;121:809–818.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.