Back to Journals » International Journal of Nanomedicine » Volume 16
Endoglin-Aptamer-Functionalized Liposome-Equipped PD-1-Silenced T Cells Enhance Antitumoral Immunotherapeutic Effects
Authors Xie S, Hou X, Yang W, Shi W ![]() , Yang X, Duan S, Mo F, Liu A, Wang W, Lu X
, Yang X, Duan S, Mo F, Liu A, Wang W, Lu X
Received 28 April 2021
Accepted for publication 11 August 2021
Published 31 August 2021 Volume 2021:16 Pages 6017—6034
DOI https://doi.org/10.2147/IJN.S317220
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Shenxia Xie,1,2,* Xiaoqiong Hou,1,3,* Wei Yang,1,3,* Wei Shi,1,3 Xiaomei Yang,1,3 Siliang Duan,3 Fengzhen Mo,3 Aiqun Liu,3 Wu Wang,1,4 Xiaoling Lu1,3,5
1School of Preclinical Medicine, Guangxi Medical University, Nanning, Guangxi, People’s Republic of China; 2Pharmaceutical College, Guangxi Medical University, Nanning, Guangxi, People’s Republic of China; 3International Nanobody Research Center of Guangxi, Guangxi Medical University, Nanning, Guangxi, People’s Republic of China; 4Laboratory of Tropical Biomedicine and Biotechnology, School of Tropical Medicine and Laboratory Medicine, Hainan Medical University, Haikou, Hainan, 571101, People’s Republic of China; 5College of Stomatology, Guangxi Medical University, Nanning, Guangxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaoling Lu
College of Stomatology, Guangxi Medical University, Nanning, Guangxi, 530021, People’s Republic of China
Email [email protected]
Wu Wang
Laboratory of Tropical Biomedicine and Biotechnology, School of Tropical Medicine and Laboratory Medicine, Hainan Medical University, Haikou, Hainan, 571101, People’s Republic of China
Email [email protected]
Background: The broader application of adoptive cell therapy (ACT) in cancer immunotherapies (particularly for solid tumors) has always been limited by the immunosuppressive tumor microenvironment (TME) and the insufficient targetability of effector T cells, resulting in unsatisfied therapeutic outcome. Here, we designed a new strategy by using aptamer-based immunoliposomes to modify PD-1-silencing T cells, which were activated by dendritic cell (DC)/tumor fusion cells (FCs) to improve the antitumor potency of cytotoxic T lymphocytes (CTLs/CD8+ T cells).
Methods: PD-1 gene was knocked out from CD8+ T cells using CRISPR/Cas9 system to liberate T cell activity from immunosuppression. The PD-1− T cells were stimulated with DC/tumor FCs, followed by further functional modification of tumor-specific nanoliposomes (hEnd-Apt/CD3-Lipo) to generate FC/PD-1− CTLs. The activation and proliferation and specificity of the modified FC/PD-1− CTLs were measured. The antitumor activity of these CTLs against HepG2-tumors was evaluated in xenograft NOD/SCID mice, and the antitumor mechanism was investigated based on tissue immunohistochemistry and serum ELISA.
Results: Our results indicated that the modification of hEnd-Apt/CD3-Lipo nanocomposites on the FC/PD-1− CTLs had a more substantial synergetic effect in inhibiting tumor growth and prolonging animal survival, rather than other control liposomes. Furthermore, the hEnd-Apt/CD3-Lipo-modified FC/PD-1− CTLs showed a stronger antitumor outcome in the tumor-bearing mouse model, through the mechanisms of suppressing tumor cell proliferation, promoting tumor apoptosis, reducing angiogenesis but increasing the infiltration of the FC/PD-1− CTLs in the tumor tissue, as well as upregulating the systemic levels of IFN-γ, IL-2, TNF-α and IL-6 cytokines, by comparison of the control settings.
Conclusion: In sum, our investigation suggests an enhancement of antitumor effect by the surface modification of endoglin-targeting nanoliposomes upon DC/tumor FC-activated PD-1− CTLs, therefore, provides a new tumoral endoglin-targeted approach as a promising strategy to reduce immunosuppression of tumor microenvironment and improve the immunotherapeutic outcome of anticancer ACT.
Keywords: nanoliposome, PD-1, CRISPR/Cas9, endoglin, aptamer, antitumor immunotherapy
Graphical Abstract:

Introduction
Today, malignant tumors (cancer) still severely imperil human health and cause millions of global mortality rates.1,2 Deep-seated solid tumors are challenging to cure by most therapeutic tools, mainly blamed on the complex tumor microenvironment (TME).3,4 Adoptive cell therapy (ACT), as one of the effective immunotherapeutic means for cancer treatment, aims to rebuild the “normal” immune system to fight cancer cells via intravenously transferring either tumor-resident or peripheral blood modified immune cells into the cancer patients that mediates efficient antitumor effects.5,6 More recently, two strategies are applied for ACT to improve the antitumor specificity and activity of the cultured T cells: 1) recurrently stimulating the T cells with tumor antigens and 2) genetically engineering T cells with tumor antigen-specific T cell receptors (TCRs) or chimeric antigen receptors (CARs).7,8 However, these ACT approaches always fail to efficiently exert and sustain the antitumor effect of the adoptive allogeneic or autogenous T cells against most solid tumors in the clinic, despite that some successes have been achieved in T cell-based ACTs for leukemia and melanoma.9,10 The dense physical and immunological barriers of solid tumors, as well as the complex TME (which has not yet been fully understood), may result in poor persistence and limited targetability of the adoptive T cells and make these effector cells quite tricky to fully infiltrate into the deeper tissue of solid tumor with sufficient activity after being transfused into patients.11–13 Besides, the engagement with tumor cells causes the interaction of immune checkpoints to exert a negative regulation and impairs the normal functions of T cells, which may also be another critical influence factor that is involved. Hence, there is an urgent need for a more efficient ACT technique to push forward the innovation and development of new technologies that can increase T cell potency as a research priority in the field of cancer immunotherapy.
As a potent professional antigen-presenting cell in the immune system, DC activates and endows the antigenic information to primary T cells.14 Tumor cells fuse with DC to form DC/tumor fusion cell (FC) vaccines,15 which can not only load known or unknown tumor antigens but can also act DC to present and process antigens to initial tumor-specific T cell responses, thus more effectively activate T cells to produce a more efficient antitumor effect. DC/tumor FC vaccine is known to be a promising immunotherapy approach.16,17 A series of our preliminary studies have also validated the improved antitumor effects of T cells mediating by our developed DC/tumor FC vaccine, which were prepared with high fusion efficiency of DCs and various tumor cells.18,19 In this point, by virtue of the properties of tumor-specific DC, it follows then that DC/tumor FC vaccine would utilize as an ideal T cell enhancer in two ways: promoting the activation and proliferation of T cells and improving the specificity and response of T cells against tumor cells.20
Another strategy for boosting T cell activity is to target immune checkpoints.21 It is known that the negative regulation of immune checkpoints greatly strangles T cells’ activation and cytotoxic activity.22 Programmed death-1 (PD-1), a membrane-bound molecule, is highly expressed on the T cell surface, and its specific ligand PD-L1 is known as a pair of the most important immune checkpoint molecules.23,24 Upon the engagement of the cognate ligands PD-L1 and PD-L2 that are expressed on many types of tumor cells, PD-1 signaling inhibits T cell activation and induces T cell anergy, thus significantly contributes to tumor evasion within the tumor microenvironment.25 Blocking PD-1 signaling has become a promising strategy that targets the immunosuppressive milieu and obtained some gratifying outcomes in cancer patients.26,27 The recent advent of readily programmable methods for clustered regularly interspersed short palindromic repeats/associated protein 9 (CRISPR/Cas9) based gene editing of primary human T cells raises the prospect of enhanced antitumor efficacy of ACT via PD-1 gene disruption.28,29 In this current study, we utilized the CRISPR/Cas9 gene editing technology as an advanced gene knockout tool to silence PD-1 in the T cells for antitumor treatment.
Aptamers are a class of synthetic single-stranded DNA or RNA molecules that bind to a wide range of targets with high affinity and specificity, which could be usually utilized as a probe to target specific antigens for disease diagnosis and therapeutic purpose.30,31 Aptamers are generally isolated in vitro through an integrated approach named Systematic Evolution of Ligands by Exponential Enrichment (SELEX). Aptamers with higher binding affinity to tumor antigens can screen after rounds of selection-amplification cycles.32 Because of the high specificity, rapid plasma clearance, and extremely low immunogenicity, aptamers offer a tremendous advantage in terms of tumor antigen targeting therapy. Conjugating aptamers with nanocarriers, such as liposomes, facilitates the more efficient delivery of specific aptamers to the tumor sites and achieves therapeutic effects.33 For example, aptamer-functionalized hybrid nanoparticles have been used to treat breast cancer, melanoma, and osteosarcoma.34–36 Aptamer-conjugated liposomes as common pharmaceutical nanocarriers will bring more opportunities for drug delivery to target tumor sites more precisely.37
The selection of therapeutic targets directly determines the targeting specificity of adoptive T cells and the fate of antitumor therapy outcomes. Endoglin, a transmembrane glycoprotein of the transforming growth factor (TGF)-β co-receptors, is highly expressed on activated endothelial cells and is involved in vascular development and remodeling.37 Remarkably, endoglin is recently identified to be significantly involved in the development and progression of hepatocellular carcinoma and is widely used to target antiangiogenic therapy in many solid cancers.38,39 Our previous study generated an aptamer hEnd-Apt that specifically binds human endoglin based on a SELEX approach.40 Here, we developed a new therapeutic liposomal biomaterial by conjugating nanoliposomes with hEnd-Apt and an anti-CD3 antibody to allow faster encounter between T cells and tumors, with the purpose to render efficient tumor elimination. Because PD-1− negative (PD-1−) T cells show more superior tumor clearance than PD-1-positive (PD-1+) T cells, and have the ability to reduce negative impacts from the PD-1/PD-L1 axis.28 We used PD-1− CTLs as the adoptive antitumor effectors, in which the PD-1 expression was erased on human blood-sourced CD8+ T cells through CRISPR-Cas9 technology. By equipping the PD-1− CTLs with the hEnd-Apt/CD3-Lipo nanocomposites prepared in this study, we found that PD-1− CTLs modified with hEnd-Apt/CD3-Lipo had more robust capability to inhibit HepG2 tumor growth than other control nanoliposomes, and extend the survival of the tumor-bearing mice. Our study suggests that joint application of the Apt/CD3-Lipo nanocomposite coupling and the PD-1 silencing on CD8+ T cells (CTLs), together with the pre-stimulation of DC/tumor fused cell vaccine, would be a promising strategy for efficient solid tumor ACT, which may have significant research value and application prospects.
Materials and Methods
Animals and Cell Lines
Non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice (SPF-grade, female, 4–6 weeks old, 16–20 g average body weight) and BALB/c mice (SPF-grade, female, 4–6 weeks old, 18–22 g average body weight) purchased from Charles River Laboratories (Beijing, China). All mice were housed in an SPF-grade facility at the Animal Center of Guangxi Medical University at room temperature (22 ± 1 °C) with a 12/12 hours light/dark cycle and supplied with food and water ad libitum. All operations on experimental animals including animal handling, welfare and medical treatments were approved by the Institutional Animal Care and Use Committee of Guangxi Medical University, and conducted by following the Guiding Opinions on the Ethical Treatment of Laboratory Animals issued by the Science and Technology Ministry of People’s Republic of China, and the National Standard Guidelines for Laboratory Animal Welfare and Ethical Review of the People’s Republic of China (GB/T35892-2018).
Human hepatocellular carcinoma cell line HepG2, human lung adenocarcinoma cell line A549, and human embryonic kidney cell line 293T were initially obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). These cell lines were authenticated by STR (Short Tandem Repeat) DNA profiling analysis and tested as mycoplasma contamination-free by the vendor. Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT, USA), 100 units/mL of penicillin, 100 µg/mL of streptomycin (Gibco), and grown in a humidified atmosphere with 5% CO2 at 37 °C.
Isolation of Dendritic Cells and T Cells
Peripheral blood collected from three healthy donors (male, aging between 25 and 30 years) were subjected to density gradient centrifugation using the lymphocyte separation medium (Haoyang Biotech, Tianjin, China) according to manufacturer’s instructions. As a common set of techniques, the preparation of DC and T cells was following our previously published work procedure.41 In brief, peripheral blood mononuclear cells (PBMCs) were plated and cultured in Roswell Park Memorial Institute (RPMI) 1640 complete medium (Gibco, USA) supplemented with 10% FBS, 100 U/mL of penicillin and 100 µg/mL of streptomycin, at 37 °C for 2 h. In addition, the adherent cells were treated with 1000 U/mL granulocyte-macrophage colony-stimulating factor (GM-CSF; PeproTech, USA) and 500 U/mL rhIL-4 (PeproTech, USA) to induce the differentiation of DCs. The suspended PBMCs were further purified through magnetic beads-based method using human CD8 Dynabeads® Flow CompTM (Invitrogen, Carlsbad, CA, USA) and cultured in RPMI 1640 complete medium containing 100 U/mL recombinant human interleukin 2 (rhIL-2; PeproTech, Rocky Hill, NJ, USA) to obtain the CD8+ T lymphocytes. All experiments involving human samples were ethically approved and conducted in accordance with the Declaration of Helsinki and the guidelines established by the Institutional Review Board of Guangxi Medical University. All volunteers provided written informed consent to participate in the study.
PD-1 Silencing in T Cells
The PD-1 gene was knocked out in the sorted CD8+ T cells using a CRISPR-Cas9 gene-editing system. The 20-base-pair guide RNA (gRNA) sequence specifically targeting the human PD-1 gene was designed by Sangon Biotech (Shanghai) Co., Ltd. (Shanghai, China). The gRNA sequence inserted into an existing px330 plasmid (Addgene; Cambridge, MA, USA) that co-expresses with human codon-optimized Cas9 to yield px330-PD-1-gRNA plasmid. After DNA sequencing, the px330-PD-1-gRNA plasmid was transfected into human CD8+ T cells to generate PD-1− CD8+ T cells by electroporation using a Neon electroporation system (Life Technologies, Carlsbad, CA, USA). PD-1-targeting magnetic beads (Invitrogen/Thermo Fisher Scientific, Carlsbad, CA, USA) were used to separate the CD8+ T cells that were successfully transfected with the px330-PD-1-gRNA plasmid to lose PD-1 expression (PD-1− CTLs) by negative selection from the non-transfected T cell population that retained the PD-1 expression (PD-1+ CTLs), collected and washed separately. Isolation of genomic DNA and plasmid were carried out using the kits from QIAGEN Ltd. (Valencia, CA, USA). The silencing of human PD-1 was verified by T7 endonuclease 1 (T7E1) digestion on genomic DNA from the PD-1− T cells. In brief, the genomic DNA from PD-1− T cells amplified by PCR after CRISPR/Cas9 gene editing. The riboEDIT™ T7E1 Enzyme was added into the obtained DNA, and incubated at 37 °C for 30 min for Enzyme cutting. The digestion product was analyzed by 2% AGAR gel electrophoresis for the enzyme-cutting bands and the editing efficiency.
 |
Figure 1 Schematic illustration of the hEnd-Apt/CD3-Lipo-equipped PD-1- CD8+ T lymphocytes with enhanced antitumor activity of adoptive T cell treatment. |
DC/Tumor Fusion Cell Preparation and T Cell Stimulation
DC/tumor fusion cells (FCs) were prepared as previously described.40 Briefly, human PBMC-original DCs were mixed with HepG2 hepatoma cells at a ratio of 2:1 and co-incubated with pre-warmed serum-free RPMI 1640 medium supplementary with polyethylene glycol (PEG; Sigma, St Louis, MO, USA) at 40 °C for 5 min and then stopped reaction by diluting PEG density with fresh pre-warmed serum-free RPMI 1640 medium. After centrifugation, the cell pellets were resuspended in pre-warmed serum-free RPMI 1640 medium supplementary with 50 μg/mL type I collagen and incubated at 37 °C for 30 min. After three washings with PBS, FCs were pelleted by centrifugation, resuspended in RPMI 1640 complete medium containing 10% (v/v) FBS, 1000 U/mL GM-CSF and 500 U/mL rhIL-4, in 5% CO2 at 37 °C for 5 days. The successful establishment of DC/tumor (herein refer to DC/HepG2) FCs was judged by integrating the mixed cells into a single entity that was loosely adhesive to the bottom of the culture. Tumor-specific T cell populations were then generated by stimulating naïve CD8+ T cells (FC/T CTLs), or PD-1+ CD8+ T cells (FC/PD-1+ CTLs), or PD-1-silencing CD8+ T cells (FC/PD-1− CTLs), with the prepared DC/HepG2 FCs in the presence of 20 U/mL of IL-2.
Preparation and Characterization of hEnd-Apt/CD3-Lipo Nanocomposites
The maleimide (MAL)-modified liposome (Lipo) was prepared by the film hydration method,42 and then the hEnd-Apt/CD3-Lipo was further generated by jointly ligating hEnd-Apt and the commercial CD3 mAb (OKT3; eBioscience, USA) to the Lipo using the post-insertion method according to our previous work,43 with some slight alterations. Briefly, chloroform-dissolved POPC, cholesterol, PEG2000-DSPE, DDAB and MAL-PEG2000-DSPE were mixed under a molar ratio of 51.8:40:5:3:0.3 in a round-bottom bottle and incubated at 4 °C under nitrogen protection for 24 h in the dark. The chloroform in the bottle was then drained in the rotary evaporator for 10–15 min under vacuum conditions to form a uniform lipid film at the bottle bottom. After sufficient drying in a vacuum dryer, the crude Lipo suspension was formed by adding 1 mL HEPES buffer (pH 6.5, 10 mM) following 5 min ultrasonication in the water bath. The white suspension was then passed through 200-nm and 100-nm polycarbonate membranes for 20 times, respectively, to obtain MAL-modified Lipo. To obtain the hEnd-Apt/CD3-Lipo nanocomposites, the prepared hEnd-Apt and CD3 mAb were mixed in a ratio of 2:1, then thoroughly mixed with 10 times the volume of MAL-PEG2000-DSPE. After reacting for 24 h under nitrogen protection, the suspension dialyzed (50 mM Tris-HCl, 140 mM NaCl, pH = 7) to remove the unbounded aptamer or antibody. The preparation scheme for hEnd-Apt/CD3-Lipo is shown in Figure 1. Lipo nanocomposites conjugating with the hEnd-Apt alone (hEnd-Apt-Lipo), the CD3 mAb alone (CD3-Lipo), or an irrelevant aptamer and the CD3 mAb (S2.2/CD3-Lipo), or naked liposomes (Lipo), was prepared similarly.
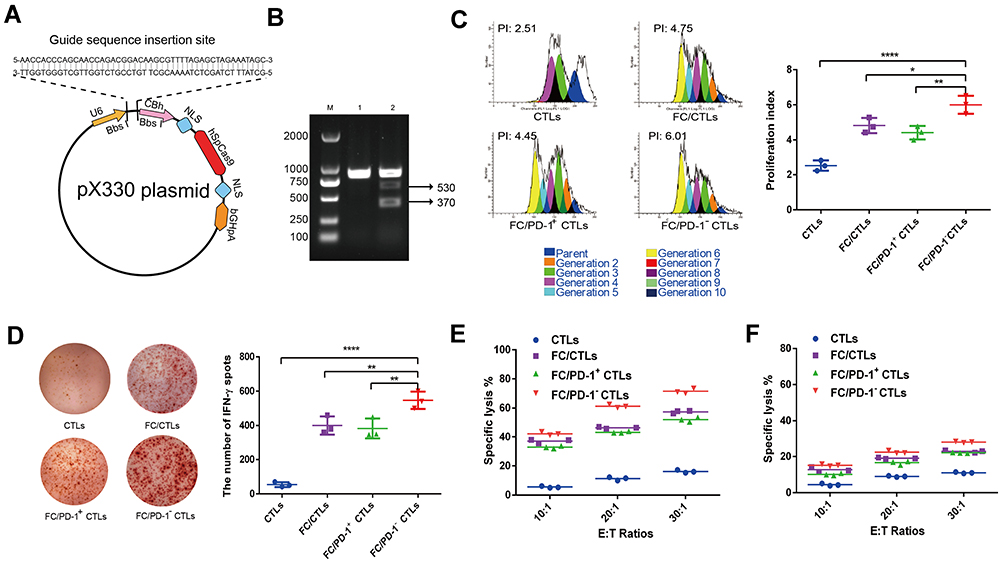 |
Figure 2 CRISPR/Cas9-mediated PD-1 silencing significantly enhanced T cell proliferation, IFN-γ production and the cytotoxicity of T cells against HepG2 cells. (A) Illustration of the DNA sequence structure design of the recombinant plasmid px330-PD-1-gRNA for CRISPR/Cas9 gene knockout of PD-1. (B)The effective CRISPR/Cas9-mediated gene silencing of the human PD-1 gene in human PBMC-derived CD8+ T cells (CTLs) was verified by T7E1 restriction enzyme digestion and agarose gel electrophoresis. Lane M: DNA markers; Lane 1: control genomic DNA from untransfected CTLs; Lane 2: genomic DNA from PD-1− CTLs. (C) The indicated cells were labeled with CFSE and co-cultured with DC/HepG2 FCs at a ratio of 10:1 for 7 days, and cell proliferation was determined by flow cytometric assay of CFSE dilution. Representative flow cytometry images from each group are presented, and the quantification of the proliferation index was summarized from three independent experiments. (D) The frequency of IFN-γ-secreted CTLs was determined by ELISPOT assay. Representative image of cells producing IFN-γ from each group is presented, and the density of IFN-γ+ cells was summarized from three independent experiments with similar results. (E) DC/HepG2 FCs-stimulated PD-1− T cells exhibited increased and endoglin-specific cytotoxicity against HepG2 cells. After stimulation with DC/HepG2 FCs for 7 days, indicated T cells (effector cells, E) were co-cultured with PKH26-labeled HepG2 (E) or A549 cells (F) (target cells, T) at the indicated E:T ratios for 6 hours. Data represent one of three independent experiments with similar results. *p < 0.05, **p < 0.01, ****p < 0.0001, compared between indicated groups. |
The hEnd-Apt was selected from large-scale library screening that demonstrated the most prominent binding ability to HepG2 cells. Aptamers (hEnd-Apt and S2.2) were synthesized by Shanghai Sangon Biotech Co., Ltd. (Shanghai, China). Anti-human CD3 antibody (monoclonal, OKT3) was purchased from eBioscience/Thermo Fisher Scientific (Carlsbad, CA, USA). The microscopic images of Lipo nanocomposites were obtained with a transmission electron microscope H-7650 (Hitachi, Ltd., Tokyo, Japan). The particle size and potential of the samples were measured by a Zetasizer analyzer with the Dynamic Light Scattering (DLS).
Cytotoxicity Evaluation
To determine the cytotoxicity of the nanocomposites against normal or cancer cells in vitro, 293T, A549 and HepG2 cells were incubated with 1 nM, 10 nM, 100 nM, 500 nM or 1000 nM of hEnd-Apt/CD3-Lipo nanocomposites in RPMI 1640 complete medium (containing 10% FBS) for 24 hours or 48 hours. The concentration of hEnd-Apt/CD3-Lipo was quantitated by the concentration of the total protein modifying on the surface of Lipo using a BCA assay kit (Beyotime, Beijing, China). The cell viability of the hEnd-Apt/CD3-Lipo nanocomposite to these cell lines was detected by Cell Counting Kit-8 (CCK-8; Sigma, USA) following the specifications of the manufacturer.
Histology was conducted using hematoxylin and eosin (H&E) staining method to examine possible infiltration of inflammatory cells in the heart, liver, spleen, lung and kidney of the mice administered with the hEnd-Apt/CD3-Lipo nanocomposites to assess the in vivo toxicity. Briefly, six BALB/c mice were randomly assigned to the experimental group (intraperitoneal injection of 1000 mM hEnd-Apt/CD3-Lipo nanocomposites suspending with 200 μL PBS) and the control group (intraperitoneal injection of an equal volume of PBS). At 7 days after treatments, mice were sacrificed, and the tissues of heart, liver, spleen, lung, and kidney were respectively sampled, fixed and sectioned for H&E staining following the standard procedures.
Binding Specificity of Functionalized Nanocomposites
The fluorescence intensity of FITC was detected by flow cytometry to determine the binding rate of lipid nanocomposite to human T cells and tumor cells. The FITC-conjugated lipo-nanocomposites including hEnd-Apt/CD3-Lipo, hEnd-Apt-Lipo, CD3-Lipo and S2.2/CD3-Lipo, were incubated with PD-1− CTLs or HepG2/A549 tumor cells (50 nM per 106 cells) in PBS on ice for 30 min. After washing 3 times, T cells or tumor cells resuspended in 200 µL washing buffer were measured for FITC signal. The cells without incubation were used for control. Flow cytometry was performed with a flow cytometer (FACSCalibur, BD Biosciences; Franklin Lakes, NJ, USA), and the exported data were analyzed by the FlowJo 10 software (FlowJo, LLC; Ashland, OR, USA).
T Cell Proliferation Assay
The indicated human DC/HepG2 FCs stimulated T cells were labeled with CFSE (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s instructions. Briefly, after washing with RPMI 1640 medium (without FBS), T cells were incubated with CFSE (1:1000 dilution, 5 µM) in PBS at 37 °C in the dark for 20 min. Then, T cells were resuspended in RPMI 1640 complete medium (containing 10% FBS) and incubated at 37 °C for another 5 min. CFSE-labeled T cells were then seeded into 12-well plates at a density of 1×106 cells/well and incubated with DC/HepG2 FCs at a ratio of 10:1 at 37 °C in the dark for 7 days. T cell proliferation was analyzed by flow cytometry, and the data were calculated with a ModFit LT program (version 5.0, Verity Software House, Topsham, ME, USA).
Cytokine Assay and T Cell Activation Assay
The secretion of IFN-γ from T cells co-cultured with DC/HepG2 FCs was measured by enzyme-linked immune absorbent spot (ELISPOT). After the co-culturing of DC/HepG2 FCs and T cells at a 1:10 ratio at 37 °C for 20 hours, the IFN-γ+ T cells were detected using an ELISPOT kit (Dakewe Biotech Co., Ltd, Shenzhen, China) following the manufacturer’s instructions.
The levels of IFN-γ and IL-2 in the culture supernatant of liposome/Lipo nanocomposites-equipped FC/PD-1− CTLs were determined by enzyme-linked immunosorbent assay (ELISA). The FC/PD-1− CTLs were incubated with nanocomposites at a density of 105 cells in 100 µL culture medium/well in 48-well plates at 37 °C for 1 hour. Then, 100 µL of HepG2 or A549 tumor cells (106 cells/mL) were added to each well co-cultured for another 3 hours. Cells were collected and stained with FITC-conjugated anti-human CD25 or anti-human CD69 antibodies (BD Biosciences, San Jose, CA, USA) and then subjected to flow cytometric analysis to detect levels of T cell activation. The contents of IFN-γ and IL-2 in the co-culture supernatants of T cells and the tumor cells were detected by ELISA kits (R&D Systems, Minneapolis, MN, USA) by following the manufacturer’s instructions.
In vitro Killing Assay
To test the cytotoxicity of T cells irritated by DC/HepG2 FCs, human peripheral blood T cells were incubated with DC/HepG2 FCs at a ratio of 10:1 in the presence of rhIL-2 (10 U/mL) at 37 °C for 7 days. HepG2 or A549 cells were labeled with the PKH26 red fluorescent cell linker kit (Sigma, USA). Total CTLs, FC/CTLs, FC/PD-1+ CTLs and FC/PD-1− CTLs (effector cells) were then co-cultured with PKH26-labeled tumor cells (target cells) at different effector cell versus target cell (E:T) ratios at 37 °C for 6 hours. The apoptosis rates of HepG2 and A549 cells were measured by flow cytometry after staining of the cell mixture with 7-aminoactinomycin D (7-AAD) at room temperature for 10 min.
The tumor-killing ability of modified or unmodified liposomes bound FC/PD-1− CTLs were evaluated. FC/PD-1− CTLs were incubated with 50 nM hEnd-Apt-Lipo, CD3-Lipo, S2.2/CD3-Lipo, and hEnd-Apt/CD3-Lipo, respectively, at a density of 2×105 cells/100 µL/well in 48-well plates at 37 °C for 1 hour. PKH26-labeled HepG2 or A549 cells were incubated with liposome-modified FC/PD-1− CTLs at the E:T ratios of 2.5:1, 5:1, and 10:1 at 37 °C for 6 hours. Then, cells were stained with propidium iodide (PI) in the dark at room temperature for 15 min. The apoptosis rates of HepG2 and A549 cells were measured with flow cytometry, and the specific lysis rates were calculated with the following formula: T cell-specific lysis rate = (apoptosis rate in the experimental group – naturally occurring apoptosis rate in the control group)/(1 – naturally occurring apoptosis rate in the control group) × 100%.
Human Tumor Xenograft Model and Treatments
The HCC tumor-bearing mouse model was built by inoculating HepG2 cells on the inguinal region of female NOD/SCID mice subcutaneously. HepG2 cells growing in the logarithmic phase were washed and resuspended in PBS at 2×106 cells/mL, and 100 µL of the cell suspension was injected into the right inguinal region on day 0. From then on, the tumor length (L) and width (W) were measured from day 5 after the inoculation of HepG2 cells and every 5 days after that. For evaluating the in vivo tumor-elimination ability of liposome-imposed FC/PD-1− CTLs, mice were randomly divided into seven groups (6 mice per group), and treated with 5×104 unimposed CTLs (FC/PD-1− CTLs), pure liposome-equipped CTLs (Lipo + FC/PD-1− CTLs), hEnd-Apt-Lipo-equipped CTLs (hEnd-Apt-Lipo + FC/PD-1− CTLs), CD3-Lipo-equipped CTLs (CD3-Lipo + FC/PD-1− CTLs), S2.2/CD3-Lipo-equipped CTLs (S2.2/CD3-Lipo + FC/PD-1− CTLs), and hEnd-Apt/CD3-Lipo-equipped CTLs (hEnd-Apt/CD3-Lipo + FC/PD-1− CTLs), or 200 µL PBS through continuous tail vein injection from day 5 to day 9, once per day.
Immunohistochemistry (IHC)
Mice from the above therapeutic experiment were sacrificed. The tumor tissues were separated, fixed in 4% paraformaldehyde, processed, and embedded in paraffin. To evaluate the intratumoral cell proliferation and apoptosis, the ultrathin tissue sections were either incubated with an anti-Ki-67 antibody (1:200 dilution; Maixin Biotechnology, China)-based IHC for cell proliferation, or subjected to a TUNEL assay (Click-iT Plus TUNEL assay, Invitrogen, Carlsbad, CA) for cell apoptosis, by following the manufacturer’s instructions. Similarly, the density of tumoral microvessels (CD31+) and the infiltration of T cells (CD3+) were quantified by IHC based on an anti-CD31 mAb (1:500; Abcam, Britain) and an anti-CD3 mAb (1:500; Abcam, Britain) as primary antibodies, following an HRP-labeled IgG monoclonal antibody as a secondary antibody staining, and the sections were counterstained with DAB. The number of positive cells was counted and analyzed to evaluate the density of intratumoral proliferated cells, apoptotic cells, microvessels or the T cell infiltration among different treatments.
Statistical Analysis
Statistical analysis was performed using SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA). Data in each figure represent the mean ± standard deviation (SD) from at least three independent experiments. Differences between the two groups were compared using the two-tailed Student’s t post hoc test and one-way analysis of variance (ANOVA). The survival rate was analyzed by the Kaplan–Meier method, and the difference between the groups was calculated by weighted Log rank test. Data analyses and graphing were performed using GraphPad Prism statistical software version 6.0. p < 0.05 was considered to be statistically significant.
Results
PD-1 Silence Significantly Enhanced the Proliferation and IFN-γ Production of T Cell, and Promoted Cytotoxic T Cell Response Against HepG2 Cells in vitro
Construction of the recombinant plasmid (px330-PD-1-gRNA) by CRISPR/Cas9 technology is shown schematically in Figure 2A. To validate PD-1 silencing in T cells, we amplified the genomic DNA from PD-1− T cells and performed restriction digestion using T7 endonuclease 1. As expected, genomic DNA from PD-1− T cells presented two bands with the molecular weight of 370 bps and 530 bps after double-enzyme digestion (Figure 2B). In response to the stimulation with DC/HepG2 FCs, all CD8+ T cell populations (including FC/CTLs, FC/PD-1+ and FC/PD-1− CTLs) demonstrated significantly boosted proliferation, compared with the naïve CD8+ T cells without DC/HepG2 FC stimulation. Remarkably, FC/PD-1− CTLs had the most enhanced proliferation and displayed the highest proliferation index (Figure 2C). Consistently, a much similar trend of IFN-γ production was identified from different T cell groups in response to DC/HepG2 FCs stimulation, and PD-1− CTLs demonstrated the most IFN-γ+ spots (Figure 2D). These results suggest that CRISPR/Cas9-mediated PD-1 silencing promotes T cell proliferation and activation following DC-mediated tumor antigen presentation.
Antitumor ability of T cells was examined by co-culturing with HepG2 cells at different E:T ratios (10:1, 20:1, and 30:1). Flow cytometric results showed that DC/HepG2 FCs stimulation significantly enhanced the cytotoxicity of CTLs, compared with un-stimulated CTLs (CTLs alone). At the E:T ratio of 20:1, the cytotoxicity of FC/PD-1− CTLs was significantly higher than that of the other three groups (Figure 2E). Furthermore, tumor lysis mediated by the DC/HepG2 FCs stimulated CTLs was specific to HepG2 cells, while all groups of CTLs presented much minimal toxicity against A549 cells (Figure 2F).
The hEnd-Apt/CD3-Lipo Nanocomposites Demonstrated Good Physical Properties and Biocompatibility
To improve the antitumor effect of adoptive PD-1− T cell therapy, which was developed in our previous study, we modified these T cells with tumor-targeted Liponanocomposites that conjugated with a hEnd-Apt and an anti-CD3 mAb, naming “hEnd-Apt/CD3-Lipo”. The physical properties of these nanocomposites were characterized. TEM graph and DLS analyses showed all nanocomposites generated in our study (including hEnd-Apt-Lipo and the control nanocomposites) were all well assembled and exhibited significant dispersibility, as well as size uniformity at a diameter of ~85 nm (Figure 3A, B and D). The zeta potential of hEnd-Apt-Lipo nanocomposites was approximately −30 mV, the absolute value of which was higher than that of any control particular (Figure 3C and E).
 |
Figure 3 Characterization of hEnd-Apt/CD3-Lipo nanocomposite. (A) Representative transmission electron micrograph of various nanocomposites. (B) The particle size distribution of the indicated Lipo nanocomposites. The result was summarized from three independent experiments (D). (C and E) The hEnd-Apt/CD3-Lipo nanocomposite demonstrated a suitable zeta potential. The zeta potential distributions of indicated lipo nanocomposites are shown in (C), and the summarized data from three independent experiments are shown in (E). |
Figure 4 demonstrates the biocompatibility of the hEnd-Apt/CD3-Lipo nanocomposites via assessing the in vitro toxicity to living cells (293T, A549 and HepG2), and in vivo toxicity to normal BALB/c mice. CCK8 assay indicated no statistical difference in cell viability between the hEnd-Apt/CD3-Lipo group and PBS control group was identified in 293T (Figure 4A), A549 (Figure 4B), and HepG2 (Figure 4C), regardless of the dosage (1–1000 nM) and the duration (24 h or 48 h) of the incubation. Histology based on H&E staining demonstrated that intravenously injecting the hEnd-Apt/CD3-Lipo did not cause visible damage to the tissue of heart, liver, spleen, lung or kidney of the mice, while no significant inflammatory cell infiltration was observed when compared with the controls (Figure 4D). Our data suggest the hEnd-Apt/CD3-Lipo nanocomposites demonstrate good biocompatibility and nontoxicity.
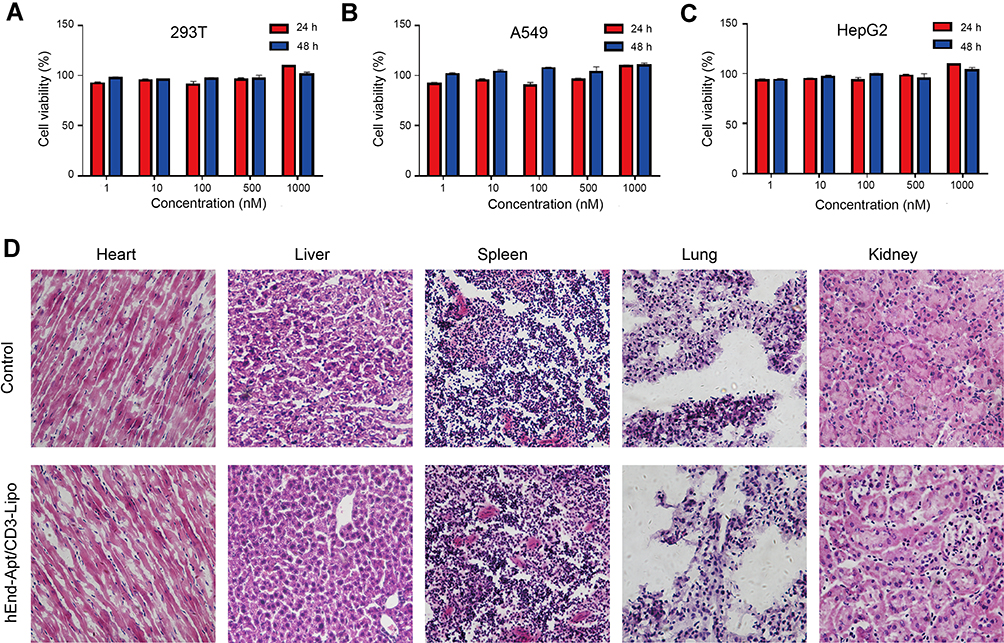 |
Figure 4 The biocompatibility of hEnd-Apt/CD3-Lipo nanocomposite in vitro and in vivo. (A–C) 293T cells (A), A549 cells (B), and HepG2 cells (C) were treated with the hEnd-Apt/CD3-Lipo nanocomposite at five different concentrations for 24 and 48 hours, respectively. The CCK-8 assay was performed to determine the percentages of cell viability, showing no significant difference in cell viability between designed groups. Data were representative of three independent experiments with three replicates in each group. (D) The BALB/c mice were treated with PBS or the hEnd-Apt/CD3-Lipo nanocomposite. Mice were sacrificed at 7 days after treatment. H&E staining of heart, liver, spleen, lung, and kidney sections demonstrated no noticeable histology change between the hEnd-Apt/CD3-Lipo treatment and control, indicating no organ toxicity. Representative H&E images were selected from three mice per group with similar results. |
The hEnd-Apt/CD3-Lipo Nanocomposites Specifically Bound to FC/PD-1− CTLs and HepG2 Hepatocellular Carcinoma Cells
The binding of the lipo-nanocomposites to both T cells and tumor cells is the basis for the more efficient elimination of tumors by the CTLs equipped with the hEnd-Apt/CD3-Lipo nanocomposite. Flow cytometry results showed that the FITC-prelabeled hEnd-Apt/CD3-Lipo nanocomposites specifically bound FC/PD-1− CTLs at a positive ratio of 76.4% after co-incubation with the FC/PD-1− CTLs for 0.5 h (Figure 5A and D). Similarly, to test the binding specificity and potency against the endoglin-expressing targets, the FITC-prelabeled hEnd-Apt/CD3-Lipo was incubated with HepG2 (hepatoma line, endoglinhigh or A549 (lung cancer line, endoglinlow), following the fluorescence detection by flow cytometry. As shown in Figure 5B, and E, the percentage and MFI of FITC+ HepG2 cells were significantly higher than those of any of the control. On the contrary, FITC+ A549 cells were hardly detected to bind the hEnd-Apt/CD3-Lipo (Figure 5C and F), suggesting these nanocomposites barely bind with the target that lacks the expression of endoglin.
 |
Figure 5 The hEnd-Apt/CD3-Lipo nanocomposites were specifically bound to FC/PD-1− CTLs and HepG2 cells. (A-C) The hEnd-Apt/CD3-Lipo nanocomposites were incubated with effector cells (FC/PD-1− CTLs) or target cells (HepG2 cells or A549 cells) for 30 min. Flow cytometric analyses showed that the hEnd-Apt/CD3-Lipo nanocomposites were specifically bound to FC/PD-1− CTLs (A) and HepG2 cells (B), but hardly bound to A549 cells (C). “F” in the bracket means FITC-fluorescence labeling on the protein or the nanocomposite. The mean fluorescence intensity (MFI) of FITC for the above three cell types was summarized from three replicates (D-F for A-C, respectively). n=3 for each group; ****p < 0.0001 for comparison of hEnd-Apt/CD3-Lipo group to control group. |
The hEnd-Apt/CD3-Lipo Nanocomposites Reinforced the Activation of FC/PD-1− CTLs in Response to HepG2 Cells
The activation of FC/PD-1− CTLs mediated by the hEnd-Apt/CD3-Lipo in response to tumor targets (HepG2 here) was analyzed based on the expression of lymphocyte activation markers CD25 and CD69, and the production of IFN-γ and IL-2 cytokines. According to the flow cytometric results, the percentages of CD25+ cells (Figure 6A and C) and CD69+ cells (Figure 6B and D) within total FC/PD-1− CTLs were both upregulated after incubation with hEnd-Apt/CD3-Lipo, CD3-Lipo or S2.2/CD3-Lipo for 3 hours, comparing those of hEnd-Apt-Lipo, naked Lipo or the PBS control, and the hEnd-Apt/CD3-Lipo induced significantly much higher expression of surface CD25 and CD69 than any other group. In addition, the enhanced activation of PD-1− CTLs induced by hEnd-Apt/CD3-Lipo, was also indirectly supported by the significant increase of IFN-γ (Figure 6E) and IL-2 (Figure 6F) productions, detecting from the culture supernatant of HepG2 cell in the presence of hEnd-Apt/CD3-Lipo and FC/PD-1− CTLs after 5-day’s co-incubation. Evidently, hEnd-Apt/CD3-Lipo can promote the activation of FC/PD-1− CTLs in response to the stimulation of endoglin-expressing target cells.
 |
Figure 6 The hEnd-Apt/CD3-Lipo nanocomposite reinforced the activation of FC/PD-1− CTLs in response to HepG2 cells. (A-D) The hEnd-Apt/CD3-Lipo nanocomposite up-regulated surface expression of CD25 and CD69 on FC/PD-1− CTLs exposed to HepG2 cells. The representative histogram plots with percentages of CD25+ (A) or CD69+ (B) cells are shown, and the percentages of CD25+ (C) or CD69+ (D) cells were summarized from 3 replicates. (E-F) The hEnd-Apt/CD3-Lipo nanocomposite enhanced the secretion of IFN-γ and IL-2 from FC/PD-1− CTLs exposed to HepG2 cells. PD-1− CTLs pre-incubated with various lipo nanocomposites were co-cultured with HepG2 cells for 5 days, and the supernatant was subjected to ELISA to determine the levels of IFN-γ (E) and IL-2 (F). n=3 for each group; ns, not significant, **p < 0.01, ****p < 0.0001, for comparison of hEnd-Apt/CD3-Lipo+ PD-1− CTLs group to control group. |
The hEnd-Apt/CD3-Lipo Nanocomposites Promoted the Cytotoxicity of FC/PD-1− CTLs Against HepG2 Cells in vitro and in vivo
Our flow cytometric data (Figure 7A) demonstrated that after co-culturing the FC/PD-1− CTLs and HepG2 target cells at various E:T ratios in the presence of appropriate dose hEnd-Apt/CD3-Lipo nanocomposites, the lytic rates of HepG2 cells were increased over any of the control treatment. The specific lysis of HepG2 cells induced by the synergy of hEnd-Apt/CD3-Lipo and FC/PD-1− CTLs was over 50% under the E:T ratio of 10:1. In contrast, equal amount of hEnd-Apt/CD3-Lipo showed no significant lysis of A549 target cells by FC/PD-1− CTLs and at any E:T ratio (Figure 7B), which was similar to the effects by the control nanocomposites, suggesting minimal cytotoxicity of FC/PD-1− CTLs on target cells expressing low or none endoglin epitope.
 |
Figure 7 The hEnd-Apt/CD3-Lipo nanocomposite significantly augmented the killing ability of FC/PD-1− CTLs against HepG2 cells in vitro and in vivo. (A and B) FC/PD-1− CTLs modified with the hEnd-Apt/CD3-Lipo nanocomposite exhibited significantly improved and specific in vitro cytotoxicity against HepG2 cells. Indicated effector cells (FC/PD-1− CTLs modified with various nanocomposite) were co-cultured with PKH26-labeled HepG2 (A) or A549 cells (B) at the indicated E:T ratios for 6 hours. Apoptosis of tumor cells was examined by PI staining, and the specific lysis is presented as the percentage of PKH26+ PI+ cells as analyzed by flow cytometry. Data represent one of three independent experiments with similar results. (C and D) The hEnd-Apt/CD3-Lipo nanocomposite significantly augmented the in vivo killing ability of FC/PD-1− CTLs against HepG2 cells. The tumor sizes were measured (C), and the survival of mice over time was examined by Kaplan-Meier analysis (D), n=6 for each group. (E-H) The concentrations of IFN-γ, IL-2, TNF-α and IL-6 in the serum were detected by ELISA. **p < 0.01, between indicated groups. |
Therapeutic potency of the hEnd-Apt/CD3-Lipo nanocomposites in vivo was also evaluated in our study. The human-original FC/PD-1− CTLs and different lipo nanocomposites generated in our study were jointly given to treat HepG2 tumor xenografic NOD/SCID mice through intravenous tail injection. The size of the subcutaneous tumors was monitored and measured for 30 days. We found that the tumor volumes in the mice of all treatment groups were significantly smaller than those who received PBS sham treatment, to a greater or lesser degree (Figure 7C). Among these groups, the treatment of hEnd-Apt/CD3-Lipo displayed the highest suppression of tumor growth than any other nanocomposites. Comparison of the survival curve for HepG2 tumor-bearing mice treated with hEnd-Apt/CD3-Lipo or the designed controls upon adoptive treatment of FC/PD-1− CTLs indicated that hEnd-Apt/CD3-Lipo could coordinate better with FC/PD-1− CTLs to the extent the survival period of mice against HepG2 tumors (Figure 7D). Additionally, the combination of hEnd-Apt/CD3-Lipo and FC/PD-1− CTLs mediated increased serum IFN-γ, IL-2, TNF-α and IL-6 in the tumor-bearing mice, when comparing with the treatment of FC/PD-1− CTLs together with the other nanocomposite (Figure 7E–H). Collectively, our data indicated that the hEnd-Apt/CD3-Lipo nanocomposites generated in our study augmented the killing ability of FC/PD-1− CTLs against HepG2 tumor cells in vitro and in vivo.
Antitumor Mechanism of the FC/PD-1− CTLs Coupled with hEnd-Apt/CD3-Lipo Nanocomposite Combination Treatment
Given the improvement of the antitumor effect boosted by hEnd-Apt/CD3-Lipo and FC/PD-1− CTLs, we then sought to explore the in vivo mechanisms for this therapy. IHC of PCNA staining showed nearly 4 times lower intratumoral PCNA-positive cell number of the treatment with FC/PD-1− CTLs coupled with hEnd-Apt/CD3-Lipo than that of the PBS sham-treatment, or the FC/PD-1− CTLs coupled with either CD3-Lipo or S2.2/CD3-Lipo. In contrast, no significant difference in the number of PCNA-positive cells was observed between the latter two control treatments (Figure 8A and E). Upon joint treatment of HepG2-bearing mice with hEnd-Apt/CD3-Lipo and FC/PD-1− CTLs, it was found that the level of tumor cell apoptosis in tumor tissues detected by TUNEL assay was notably higher than that of the other control groups (Figure 8B and F). Anti-CD31 IHC staining showed significantly fewer dense micro-vessels expressing in tumor tissues of mice treated with hEnd-Apt/CD3-Lipo nanocomposite and FC/PD-1− CTLs (Figure 8C and G). IHC staining of tumor tissues for CD3 expression illustrated that increased the infiltration of CD3+ T cells in the tumor after hEnd-Apt/CD3-Lipo nanocomposite and FC/PD-1− CTLs treatment (Figure 8D and H).
 |
Figure 8 Combination treatment with hEnd-Apt/CD3-Lipo and FC/PD-1− CTLs inhibits tumor cell proliferation, promotes tumor cell apoptosis, suppresses tumor angiogenesis and improves the T cells infiltration. (A) Detection for proliferating cell nuclear antigen expression (PCNA) by IHC staining. (B) Results of tumor apoptosis detection by TUNEL assay; nuclei stained blue, positive cells stained green. (C) Tumor microvascular densities were detected by CD31 monoclonal antibodies. (D) The infiltrating of CD3+ T cell in the tumor tissue was detected by CD3 monoclonal antibodies (E-H) Statistical quantification of the numbers of PCNA+ cells, TUNEL+ cells, CD31+ cells and CD3+ T cell in various groups. Scale bar: 100 µm. The mean ± SD of three independent experiments is shown. **p < 0.01 for comparison of hEnd-Apt/CD3-Lipo+FC/PD-1− CTLs group to other controls. |
Discussion
Immunotherapy, which has attracted much attention in recent years, has brought a new dawn to the treatment of cancers. Using ex vivo cultured and engineered T cells with tumor specificity for ACT has recently become an important method to treat cancer patients.44,45 In this study, we use CRISPR/Cas9 gene engineering technique and DC/tumor fusion cells vaccine to prepare the FC/PD-1− CTLs, and assemble a new type of nanoliposomal preparation (hEnd-Apt/CD3-Lipo), which has excellent physical properties and biocompatibility and bridges tumor cells and T cells as a highly specific “guide” for the FC/PD-1− CTLs toward tumor local to ruin tumor cells.
Immune checkpoints, like the CTLA-4/B7 axis and PD-1/PD-L1 axis, are known to be important “braking systems” in the body that prevent over-reaction of the immune system and maintain the homeostasis of immune responses and immune cell activations.23,46 However, cancer cells can also take advantage of such a unique “braking” mechanism of immune checkpoint molecules to escape from the normal surveillance and attack of the host immune system to survive.47 Thus, blocking the function of immune checkpoints could theoretically restore the recognition and cytotoxicity of the T cells against the tumor.48 It has been reported that blocking the signal transduction of PD-1/PD-L1 in patients alone or combination with ACT improves the clinical antitumor outcome and prognosis in cancer patients.49,50 Here, our data clearly showed that DC/tumor FC-primed PD-1− CTLs effectively improved the antitumor outcome in the tumor-bearing xenograft mice in the comparison of the result in mice that infused with normal human T cells alone, suggesting the enhancement of ACT by PD-1 repression in the preclinical model. Moreover, by using non-viral vector-mediated CRISPR/Cas9 genomic editing technology to knock out PD-1 in T cells in this study, unpredictable risks caused by integrating exogenous genetic material into host cells could be largely reduced,51,52 while the generated PD-1− CTLs can survive longer and produce more consistently, more efficient and safer antitumor effects.29,53 This part of data has again provided direct evidence for the benefit of PD-1/PD-L1 blockade in T cell-based tumor immunotherapy.
Sufficient expansion and activation of the T cells ex vivo, are essential for the outcome of ACT in clinical application. DC/tumor FCs, which are the hybrid cells of DCs and tumor cells, express MHC-I and -II molecules, as well as costimulatory molecules, and have a better potency in stimulating the proliferation and activation of T cells.54,55 Our study therefore utilized the well-established DC/tumor FCs to induce a more efficient tumor-specific cytotoxicity of these PD-1− CTLs, and the data showed that these modified CTLs displayed a more significant proliferation and activation comparing the normal T cells, or T cells with PD-1 silencing or FC stimulation alone, as well as a higher proportion of IFN-γ-secreting PD-1− CTLs. The DC/HepG2 FC-mediated PD-1− CTLs were found to target and kill HepG2 cells effectively, significantly decrease tumor growth, and prolong the survival of SCID mice. These results strongly support our theory and design, and suggest a remarkable superposition of PD-1-silencing and DC/tumor FC vaccine pretreatment upon T cells for ACT against the solid tumor.
Improving the in vivo targeting recruitment and accumulation of adoptive effector cells into the tumor local is considered another significant technical challenge and concern that must be solved. Today, nanoliposomal materials are widely applied as drug carriers in the field of medical bioengineering, because of the unique properties of small size, good histocompatibility, and sustained drug release, which can passively target the carried drugs to the reticuloendothelial system to reduce their toxicity.56–58 Here, in the purpose of transporting tumor-specific PD-1− CTLs toward the tumor more accurately, we designed and generated a new nanoliposomal carrier that assembled with a commercial CD3 mAb and our previous selected hEnd-Apt, which could bridge T cells (via CD3 mAb arm) and hEnd-expressing tumor cells (via hEnd-Apt arm) via nanoliposome as the carrier, at a nanoscale. Compared to antibody modalities, nucleic-acid aptamers are easier produced by in vitro selection and synthesized by a cell-free chemical system, have very low immunogenicity and superior tissue penetration ability due to the smaller size.59 Besides, aptamers possess high efficiency for target accumulation, as well as a shorter retention time in circulation and non-targeting tissues.60 These features make aptamer a more attractive agent for targeted cancer therapy. To take these advantages of aptamer, in the current study, we design an aptamer to target endoglin antigen, which is known as an important marker for tumor angiogenesis thus an ideal target for antitumor immunotherapy.61,62 The hEnd-Apt generated by SELEX technology has been validated to bond endoglin antigen with high specificity and affinity, evidenced by a 57.9% binding rate of liposomes modified with the selected hEnd-Apt against HepG2 cells (high-End expression), but much lower binding rate (only 2.37%) against A549 cells (low-End expression). The highly specific and stable binding of the immunoliposome nanocomposites against tumor target cells is the most important basis for the effective targeted killing of the tumor. Accordingly, the hEnd-Apt/CD3-Lipo generated in our study have fully highlighted the characteristic and potential to specific target and kill the target cells with endoglin over-expression in vitro and in vivo.
In purpose to further clarify the mechanisms involving the enhanced antitumor response for the therapeutic protocol of DC/tumor FC-induced and hEnd-Apt/CD3-Lipo-modified PD-1− CTLs, we evaluated alterations in aspects of neoangiogenesis, tumor cell proliferation and apoptosis, and PD-1− CTLs proliferation and functions before and after the treatment in human tumor-bearing xenograft mice, in addition to the measurement of tumor growth and animal survival length. It is clear that this new protocol, the hEnd-Apt/CD3-Lipo equipment upon FC-induced PD-1− CTLs more significantly activated and expanded these adoptive T cells in the mice, and raised antitumor cytokine production, comparing any control protocol. The synergy between the FC-induced PD-1− CTLs and the hEnd-Apt/CD3-Lipo not only reduced the proliferation but also promoted the apoptosis of the HepG2 tumor cells, whereas the intratumoral vasculature density of the mice was appeared to decrease more than the control sets. These factors eventually led to the suppression of tumor growth, thus extending the survival period of the mice, demonstrating an enhanced antitumor potency of this well-designed therapeutic protocol. Therefore, it appears to be necessary to block the PD-1/PD-L1 axis to reduce the possible exhaustion and deactivation of ex vivo cultured T cells before antitumor ACT treatment (especially of solid tumors), just as important as to select better targets for the improvement of tumor-specific targetability of these T cells, if a maximum therapeutic outcome is expected.
It is also worthy of highlighting that the new type hEnd-Apt/CD3-Lipo nanomaterial generated in the current study has appropriate molecular potential, good dispersibility and stability, as well as nanoscale particular size. Such unique characteristics maintain the biocompatibility, biodegradability and passive targetability of nanoliposomal carrier.63 As a medicine using on patients, the request for the safety of any biomaterial must not be less strict than the requirement of the antitumor efficiency. According to some clinical investigations, currently, most routine ACT approaches for cancers by using adoptive T cells exist the shortages of the loss of tumor antigen expression and on-target off-tumor toxicity.64 Besides, these treatments often inevitably lead to side effects like cytokine release syndrome (“cytokine storm”) and unexpected neurotoxicity,65,66 which will definitely adverse outcome and tumor recurrence in cancer patients, or even cause death. It serves to show that developing new antitumor agents requires both antitumor efficacy and the safety. As shown in our preclinical data, the hEnd-Apt/CD3-Lipo showed no toxic effect on the cultured cells and complete-immunity mice, in addition of the enhanced antitumor outcome upon tumor-specific FC-induced PD-1− human T cells in human tumor xenograft mice. Both virtues showcase the values and advantages of hEnd-Apt/CD3-Lipo nanocomposite as a promising and excellent candidate of nanomedicine or nanoparticulate drug carrier for tumor immunotherapy. Anyway, for further application, the antitumor efficacy and safety of hEnd-Apt/CD3-Lipo–based ACT protocol still require further validation in more preclinical and clinical trials.
Conclusion
Our study provides the first experimental evidence on the enhanced antitumor potential of FC/PD-1− CTLs, as well as the synergetic enhancement by equipping with the tumor-targeted functionalized nanoliposome (hEnd-Apt/CD3-Lipo). Disrupting the PD-1/PD-L1 axis by CRISPR/Cas9 system-mediated PD-1 silence in T cells significantly rescued and improved the functions and antitumor cytotoxicity of the ex vivo cultured T cells, which greatly promoted the tumor cell clearance and the tumor destruction in the tumor-bearing mice. On the other hand, through simultaneously linking tumor and T cells, the hEnd-Apt/CD3-Lipo nanocomposites were able to prime more FC/PD-1− CTLs to eliminate the endoglin-expressing HepG2 tumor cells, either in vitro or in vivo. Our findings suggest a synergy between targeted silence of PD-1 and the specific modification with the tumor/T cells bispecific aptamer-based nanoliposomes upon the DC/tumor FC-primed CD8+ T cells, could be a novel and promising therapeutic modality for immunotherapy in cancer patients. Besides, such a strategy of multi-functionalization on nanoliposomal biomaterial provides a new idea to optimize therapeutic efficacy, and sheds new light on advancing individualized and precise treatments, with the significant meaning of further clinical application.
Abbreviations
ACT, adoptive cell therapy; DC, dendritic cell; CTLs, cytotoxic T lymphocytes; FC, DC/tumor fusion cells; TME, tumor microenvironment; TCRs, T cell receptors; CARs, chimeric antigen receptors; SELEX, systematic evolution of ligands by exponential enrichment; CRISPR/Cas9 clustered regularly interspersed short palindromic repeats/associated protein 9; CCK-8, Cell Counting Kit-8; FBS, fetal bovine serum; ELISPOT, enzyme-linked immune absorbent spot; ELISA, enzyme-linked immunosorbent assay.
Acknowledgments
This work was supported, in part, by grants from the Project of National Natural Scientific Foundation of China (No. 81773254); National Key Research and Development Plan “Inter-governmental Cooperation in International Scientific and Technological Innovation” (No. 2019YFE0117300); Guangxi Science and Technology Base and Talents Project (No. GuiKe-AD20238062) and Guangxi Bagui Honor Scholars.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Romero Y, Trapani D, Johnson S, et al. National cancer control plans: a global analysis. Lancet Oncol. 2018;19(10):e546–e555. doi:10.1016/S1470-2045(18)30681-8
2. Given LS, Hohman K, Kostelecky B, Vinson C. Cancer control planning: self-assessment for pre-planning, development, implementation and evaluation of national cancer control plans. Cancer Causes Control. 2018;29(12):1297–1303. doi:10.1007/s10552-018-1123-z
3. Hirata E, Sahai E. Tumor Microenvironment and differential responses to therapy. Cold Spring Harb Perspect Med. 2017;7(7):a026781. doi:10.1101/cshperspect.a026781
4. Fane M, Weeraratna AT. How the ageing microenvironment influences tumour progression. Nat Rev Cancer. 2020;20(2):89–106. doi:10.1038/s41568-019-0222-9
5. Lee DA. Cellular therapy: adoptive immunotherapy with expanded natural killer cells. Immunol Rev. 2019;290(1):85–99. doi:10.1111/imr.12793
6. Waidmann O. Recent developments with immunotherapy for hepatocellular carcinoma. Expert Opin Biol Ther. 2018;18(8):905–910. doi:10.1080/14712598.2018.1499722
7. McHayleh W, Bedi P, Sehgal R, Solh M. Chimeric antigen receptor T-cells: the future is now. J Clin Med. 2019;8(2):207. doi:10.3390/jcm8020207
8. Roselli E, Frieling JS, Thorner K, Ramello MC, Lynch CC, Abate-Daga D. CAR-T engineering: optimizing signal transduction and effector mechanisms. BioDrugs. 2019;33(6):647–659. doi:10.1007/s40259-019-00384-z
9. Jiang X, Xu J, Liu M, et al. Adoptive CD8(+) T cell therapy against cancer: challenges and opportunities. Cancer Lett. 2019;462:23–32. doi:10.1016/j.canlet.2019.07.017
10. Zhang E, Gu J, Xu H. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol Cancer. 2018;17(1):7. doi:10.1186/s12943-018-0759-3
11. Adusumilli PS, Cherkassky L, Villena-Vargas J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6(261):261ra151. doi:10.1126/scitranslmed.3010162
12. Thistlethwaite FC, Gilham DE, Guest RD, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. 2017;66(11):1425–1436. doi:10.1007/s00262-017-2034-7
13. Sasawatari S, Okamoto Y, Kumanogoh A, Toyofuku T. Blockade of N-glycosylation promotes antitumor immune response of t cells. J Immunol. 2020;204(5):1373–1385. doi:10.4049/jimmunol.1900937
14. Shang N, Figini M, Shangguan J, et al. Dendritic cells based immunotherapy. Am J Cancer Res. 2017;7(10):2091–2102.
15. Pang YB, He J, Cui BY, et al. A potential antitumor effect of dendritic cells fused with cancer stem cells in hepatocellular carcinoma. Stem Cells Int. 2019;2019:5680327. doi:10.1155/2019/5680327
16. Mayoux M, Roller A, Pulko V, et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci Transl Med. 2020;12:(534):eaav7431. doi:10.1126/scitranslmed.aav7431
17. Kajihara M, Takakura K, Ohkusa T, Koido S. The impact of dendritic cell-tumor fusion cells on cancer vaccines - past progress and future strategies. Immunotherapy. 2015;7(10):1111–1122. doi:10.2217/imt.15.73
18. Koido S. Dendritic-tumor fusion cell-based cancer vaccines. Int J Mol Sci. 2016;17(6):828. doi:10.3390/ijms17060828
19. He J, Zheng R, Zhang Z, et al. Collagen I enhances the efficiency and anti-tumor activity of dendritic-tumor fusion cells. Oncoimmunology. 2017;6(12):e1361094. doi:10.1080/2162402X.2017.1361094
20. Hegde PS, Chen DS. Top 10 Challenges in Cancer Immunotherapy. Immunity. 2020;52(1):17–35. doi:10.1016/j.immuni.2019.12.011
21. Xue S, Hu M, Iyer V, Yu J. Blocking the PD-1/PD-L1 pathway in glioma: a potential new treatment strategy. J Hematol Oncol. 2017;10(1):81. doi:10.1186/s13045-017-0455-6
22. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–1355. doi:10.1126/science.aar4060
23. He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020;30(8):660–669. doi:10.1038/s41422-020-0343-4
24. Brown CC, Wolchok JD. PD-L1 blockade therapy: location, location, location. Cancer Cell. 2020;38(5):615–617. doi:10.1016/j.ccell.2020.10.017
25. Knauss S, Preusse C, Allenbach Y, et al. PD1 pathway in immune-mediated myopathies: pathogenesis of dysfunctional T cells revisited. Neurol Neuroimmunol Neuroinflamm. 2019;6(3):e558. doi:10.1212/NXI.0000000000000558
26. Bardhan K, Anagnostou T, Boussiotis VA. The PD1: PD-L1/2 pathway from discovery to clinical implementation. Front Immunol. 2016;7:550. doi:10.3389/fimmu.2016.00550
27. Chinai JM, Janakiram M, Chen F, Chen W, Kaplan M, Zang X. New immunotherapies targeting the PD-1 pathway. Trends Pharmacol Sci. 2015;36(9):587–595. doi:10.1016/j.tips.2015.06.005
28. Hu W, Zi Z, Jin Y, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. 2019;68(3):365–377. doi:10.1007/s00262-018-2281-2
29. Rupp LJ, Schumann K, Roybal KT, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7(1):737. doi:10.1038/s41598-017-00462-8
30. Nimjee SM, White RR, Becker RC, Sullenger BA. Aptamers as therapeutics. Annu Rev Pharmacol Toxicol. 2017;57:61–79. doi:10.1146/annurev-pharmtox-010716-104558
31. Kaur H, Bruno JG, Kumar A, Sharma TK. Aptamers in the therapeutics and diagnostics pipelines. Theranostics. 2018;8(15):4016–4032. doi:10.7150/thno.25958
32. Tang Y, Liu H, Chen H, et al. Advances in aptamer screening and drug delivery. J Biomed Nanotechnol. 2020;16(6):763–788. doi:10.1166/jbn.2020.2943
33. Czechowska K, Lannigan J, Wang L, et al. Cyt-Geist: current and future challenges in cytometry: reports of the CYTO 2018 conference workshops. Cytometry A. 2019;95(6):598–644. doi:10.1002/cyto.a.23777
34. He Y, Wang M, Fu M, et al. Iron(II) phthalocyanine loaded and AS1411 aptamer targeting nanoparticles: a nanocomplex for dual modal imaging and photothermal therapy of breast cancer. Int J Nanomedicine. 2020;15:5927–5949. doi:10.2147/IJN.S254108
35. Hanafi-Bojd MY, Moosavian Kalat SA, Taghdisi SM, Ansari L, Abnous K, Malaekeh-Nikouei B. MUC1 aptamer-conjugated mesoporous silica nanoparticles effectively target breast cancer cells. Drug Dev Ind Pharm. 2018;44(1):13–18. doi:10.1080/03639045.2017.1371734
36. Zeng YB, Yu ZC, He YN, et al. Salinomycin-loaded lipid-polymer nanoparticles with anti-CD20 aptamers selectively suppress human CD20+ melanoma stem cells. Acta Pharmacol Sin. 2018;39(2):261–274. doi:10.1038/aps.2017.166
37. Dou XQ, Wang H, Zhang J, et al. Aptamer-drug conjugate: targeted delivery of doxorubicin in a HER3 aptamer-functionalized liposomal delivery system reduces cardiotoxicity. Int J Nanomedicine. 2018;13:763–776. doi:10.2147/IJN.S149887
38. Chen W, Dong W, Wang J, Wen Z, Hao X. Elevated expressions of survivin and endoglin in patients with hepatic carcinoma. Cancer Biother Radiopharm. 2019;34(1):7–12. doi:10.1089/cbr.2018.2539
39. Kasprzak A, Adamek A. Role of endoglin (CD105) in the progression of hepatocellular carcinoma and anti-angiogenic therapy. Int J Mol Sci. 2018;19:12. doi:10.3390/ijms19123887
40. Qian L, Tang Z, Yin S, et al. Fusion of dendritic cells and cancer-associated fibroblasts for activation of anti-tumor cytotoxic t lymphocytes. J Biomed Nanotechnol. 2018;14(10):1826–1835. doi:10.1166/jbn.2018.2616
41. Tang Z, Mo F, Liu A, et al. A nanobody against cytotoxic T-lymphocyte associated antigen-4 increases the anti-tumor effects of specific CD8(+) T cells. J Biomed Nanotechnol. 2019;15(11):2229–2239. doi:10.1166/jbn.2019.2859
42. Schelté P, Boeckler C, Frisch B, Schuber F. Differential reactivity of maleimide and bromoacetyl functions with thiols: application to the preparation of liposomal diepitope constructs. Bioconjug Chem. 2000;11(1):118–123. doi:10.1021/bc990122k
43. Yang X, Zhao J, Duan S, et al. Enhanced cytotoxic T lymphocytes recruitment targeting tumor vasculatures by endoglin aptamer and IP-10 plasmid presenting liposome-based nanocarriers. Theranostics. 2019;9(14):4066–4083. doi:10.7150/thno.33383
44. Tian H, Huang JJ, Golzio C, et al. Endoglin interacts with VEGFR2 to promote angiogenesis. FASEB j. 2018;32(6):2934–2949. doi:10.1096/fj.201700867RR
45. Redeker A, Arens R. Improving adoptive T cell therapy: the particular role of t cell costimulation, cytokines, and post-transfer vaccination. Front Immunol. 2016;7:345. doi:10.3389/fimmu.2016.00345
46. Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell. 2018;33(4):581–598. doi:10.1016/j.ccell.2018.03.005
47. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol. 2016;2(10):1346–1353. doi:10.1001/jamaoncol.2016.1051
48. Rotte A, Jin JY, Lemaire V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann Oncol. 2018;29(1):71–83. doi:10.1093/annonc/mdx686
49. Poggio M, Hu T, Pai CC, et al. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell. 2019;177(2):414–427.e413. doi:10.1016/j.cell.2019.02.016
50. Wu Y, Chen W, Xu ZP, Gu W. PD-L1 Distribution and perspective for cancer immunotherapy-blockade, knockdown, or inhibition. Front Immunol. 2019;10:2022. doi:10.3389/fimmu.2019.02022
51. Schumann K, Lin S, Boyer E, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A. 2015;112(33):10437–10442. doi:10.1073/pnas.1512503112
52. Li L, Hu S, Chen X. Non-viral delivery systems for CRISPR/Cas9-based genome editing: challenges and opportunities. Biomaterials. 2018;171:207–218. doi:10.1016/j.biomaterials.2018.04.031
53. Wei J, Luo C, Wang Y, et al. PD-1 silencing impairs the anti-tumor function of chimeric antigen receptor modified T cells by inhibiting proliferation activity. J Immunother Cancer. 2019;7(1):209. doi:10.1186/s40425-019-0685-y
54. Tan C, Dannull J, Nair SK, et al. Local secretion of IL-12 augments the therapeutic impact of dendritic cell-tumor cell fusion vaccination. J Surg Res. 2013;185(2):904–911. doi:10.1016/j.jss.2013.06.045
55. Wang Y, Xiang Y, Xin VW, et al. Dendritic cell biology and its role in tumor immunotherapy. J Hematol Oncol. 2020;13(1):107. doi:10.1186/s13045-020-00939-6
56. Olusanya TOB, Haj Ahmad RR, Ibegbu DM, Smith JR, Elkordy AA. Liposomal drug delivery systems and anticancer drugs. Molecules. 2018;23(4):907. doi:10.3390/molecules23040907
57. Maeki M, Kimura N, Sato Y, Harashima H, Tokeshi M. Advances in microfluidics for lipid nanoparticles and extracellular vesicles and applications in drug delivery systems. Adv Drug Deliv Rev. 2018;128:84–100. doi:10.1016/j.addr.2018.03.008
58. Gudbergsson JM, Jønsson K, Simonsen JB, Johnsen KB. Systematic review of targeted extracellular vesicles for drug delivery - Considerations on methodological and biological heterogeneity. J Control Release. 2019;306:108–120. doi:10.1016/j.jconrel.2019.06.006
59. Hori SI, Herrera A, Rossi JJ, Zhou J. Current advances in aptamers for cancer diagnosis and therapy. Cancers(Basel). 2018;10(1):9. doi:10.3390/cancers10010009
60. Delcanale P, Porciani D, Pujals S, et al. Aptamers with tunable affinity enable single-molecule tracking and localization of membrane receptors on living cancer cells. Angew Chem Int Ed Engl. 2020;59(42):18546–18555. doi:10.1002/anie.202004764
61. Rossi E, Sanz-Rodriguez F, Eleno N, et al. Endothelial endoglin is involved in inflammation: role in leukocyte adhesion and transmigration. Blood. 2013;121(2):403–415. doi:10.1182/blood-2012-06-435347
62. Gallardo-Vara E, Tual-Chalot S, Botella LM, Arthur HM, Bernabeu C. Soluble endoglin regulates expression of angiogenesis-related proteins and induction of arteriovenous malformations in a mouse model of hereditary hemorrhagic telangiectasia. Dis Model Mech. 2018;11(9):dmm034397. doi:10.1242/dmm.034397
63. Agrawal M, Ajazuddin Tripathi DK, et al. Recent advancements in liposomes targeting strategies to cross blood-brain barrier (BBB) for the treatment of Alzheimer’s disease. J Control Release. 2017;260:61–77. doi:10.1016/j.jconrel.2017.05.019
64. Xie G, Ivica NA, Jia B, et al. CAR-T cells targeting a nucleophosmin neoepitope exhibit potent specific activity in mouse models of acute myeloid leukaemia. Nat Biomed Eng. 2021;5(5):399–413. doi:10.1038/s41551-020-00625-5
65. Norelli M, Camisa B, Barbiera G, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–748. doi:10.1038/s41591-018-0036-4
66. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24(6):731–738. doi:10.1038/s41591-018-0041-7
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.