")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Eicosanoids and Eosinophilic Inflammation of Airways in Stable COPD
Authors Celejewska-Wójcik N, Kania A , Górka K , Nastałek P, Wójcik K , Gielicz A, Mastalerz L , Sanak M, Sładek K
Received 30 December 2020
Accepted for publication 22 March 2021
Published 25 May 2021 Volume 2021:16 Pages 1415—1424
DOI https://doi.org/10.2147/COPD.S298678
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Natalia Celejewska-Wójcik,1 Aleksander Kania,1 Karolina Górka,1 Paweł Nastałek,1 Krzysztof Wójcik,2 Anna Gielicz,3 Lucyna Mastalerz,1 Marek Sanak,3 Krzysztof Sładek1
1Department of Pulmonology, 2nd Department of Internal Medicine, Faculty of Medicine, Jagiellonian University Medical College, Kraków, Poland; 2 2nd Department of Internal Medicine, Faculty of Medicine, Jagiellonian University Medical College, Kraków, Poland; 3Department of Molecular Biology and Clinical Genetics, Faculty of Medicine, Jagiellonian University Medical College, Kraków, Poland
Correspondence: Natalia Celejewska-Wójcik
2nd Department of Internal Medicine, Faculty of Medicine, Jagiellonian University Medical College, ul. Jakubowskiego 2, Krakow, 30-688, Poland
Tel +48 12 400 30 53
Fax +48 12 400 30 67
Email [email protected]
Purpose: Lipid mediators, particularly eicosanoids, are associated with airway inflammation, especially with the eosinophilic influx. This study aimed to measure lipid mediators and cells in induced sputum, that could possibly reflect the inflammatory process in the bronchial tree of COPD subjects.
Patients and Methods: Eighty patients diagnosed with COPD and 37 healthy controls participated in the study. Induced sputum samples were ascertained for differential cell count and induced sputum supernatant concentrations of selected eicosanoids by the means of gas chromatography/mass spectrometry and high-performance liquid chromatography/tandem mass spectrometry.
Results: Increased sputum eosinophilia was associated with higher concentrations of selected proinflammatory eicosanoids. In COPD subjects prostaglandin D2 and 11-dehydro-thromboxane B2 correlated negatively with airway obstruction measured by FEV1 and FEV1/FVC values. COPD subjects with disease exacerbations during past 12 months had significantly higher concentrations of prostaglandin D2, 12-oxo-eicosatetraenoic acid and 5-oxo-eicosatetraenoic acid.
Conclusion: Stable COPD is often associated with eosinophil influx in the lower airways and elevated concentrations of eicosanoids that is reflected by some disease characteristics.
Keywords: chronic obstructive pulmonary disease, eosinophils, eicosanoids
Introduction
As stated in the definition proposed by The Global Initiative for Obstructive Lung Disease (GOLD), chronic obstructive pulmonary disease (COPD) is characterized by a progressive airflow limitation triggered by the response in the airways and lung to noxious particles or fumes. Airway inflammation is one of the processes that drives airway limitation.1
Eosinophilic airway inflammation which is most commonly thought to play a vital role in asthma has also been described in patients with COPD. It has been demonstrated in up to 40% of stable COPD patients.2,3 Moreover, approximately 20% disease exacerbations are thought to be associated with eosinophil influx in the airways.4,5 Two previous studies: Genetic Epidemiology of COPD (COPDGene) study and the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) study, proved that the blood eosinophil count of 300 cells/µL or greater was associated with increased exacerbation risk.6,7
Both innate and adaptive immune response can start eosinophilic inflammation. Epithelium cells damaged by exposure to air pollution, irritants, microbes and allergens release cytokines: interleukin 33 (IL-33), IL-25 and thymic stromal lymphopoietin.8–10 In case of allergic triggers, dendritic cells stimulate naive T lymphocytes to differentiate to type 2 T helper cells (Th2) producing IL-4, IL-5 and IL-13, while non-allergen triggers initiate innate immune response via stimulation of type 2 innate lymphoid cells (ILC-2), which also leads to the production of Th2 type cytokines (IL-5 and IL-13).8,11 Eosinophils release mediators like eosinophilic cationic protein and eosinophil peroxidase that cause damage to bronchial epithelium and cytokines which sustain inflammation.
5-Lipoxygenase (5-LO) is the key enzyme in leukotriene synthesis transforming arachidonic acid (AA) into 5-hydroperoxyeicosatetraenoic acid (5-HpETE), a precursor of leukotriene A4 (LTA4). This intermediate can generate bioactive eicosanoids by hydrolysis into LTB4 or by conjugation with glutathione into LTC4. Next steps of extracellular metabolism produce LTD4 and LTE4. Cysteinyl leukotrienes (cys-LTs) are mediators of allergic inflammation potently contracting smooth muscles, causing vascular leak and mucus production, released mainly from eosinophils and mast cells.12 The intermediate product of 5-LO activity 55-HpETE is reduced and peroxidated to 5-hydroxyeicosatetraenoic acid (5-HETE). The activity of 12 and 15-LO leads to the generation of 12 and 15-HETE. HETEs play an important role in physiological functions such as regulating inflammation.13 Cyclooxygenase (COX) activity is at the beginning of the pathway leading to prostanoid generation, transforming AA into prostaglandins (PG) PGG2 and PGH2. Specific synthases convert cyclic endoperoxides of AA to prostaglandins: PGE2, PGD2, PGF2a, and prostanoids: thromboxane A2 (TXA2) and prostacyclin. Due to their short half-life, they act through auto and paracrine signaling and their biological effect is determined by density and type of receptors. Both PGD2 and PGF2a are potent bronchoconstrictors, while PGE2, via EP2 and EP4 receptors, is involved in bronchodilation, antifibrotic and anti-inflammatory effect.12,14 In asthma, overproduction of cys-LTs and PGD2 was linked with aspirin hypersensitivity but also with disease severity.15,16 Increased LTE4 to PGE2 ratio correlated with worse clinical course of aspirin exacerbated respiratory disease.
Induced sputum (IS) is obtained by simple noninvasive procedure and has been validated for studies on inflammatory cells and mediators in the assessment of respiratory diseases.17 High-performance liquid or gas chromatography/mass spectrometry is a highly sensitive and specific method of quantification of eicosanoids in biological samples. We assumed that the lipid mediators and cells measured in the induced sputum would reflect the inflammation process in the bronchial tree that determines clinical characteristics of COPD patients. To address this goal, we analyzed a group of 80 COPD patients and 37 healthy individuals who underwent sputum induction.
Patients and Methods
Studied Subjects
In total, 80 patients diagnosed with COPD and 37 sex and age-matched healthy controls were enrolled to the study. Patients remained without any COPD exacerbation or respiratory tract infections during the 6 weeks preceding the study and received medications as currently prescribed. Clinical characteristics of study subjects is presented in Table 1.
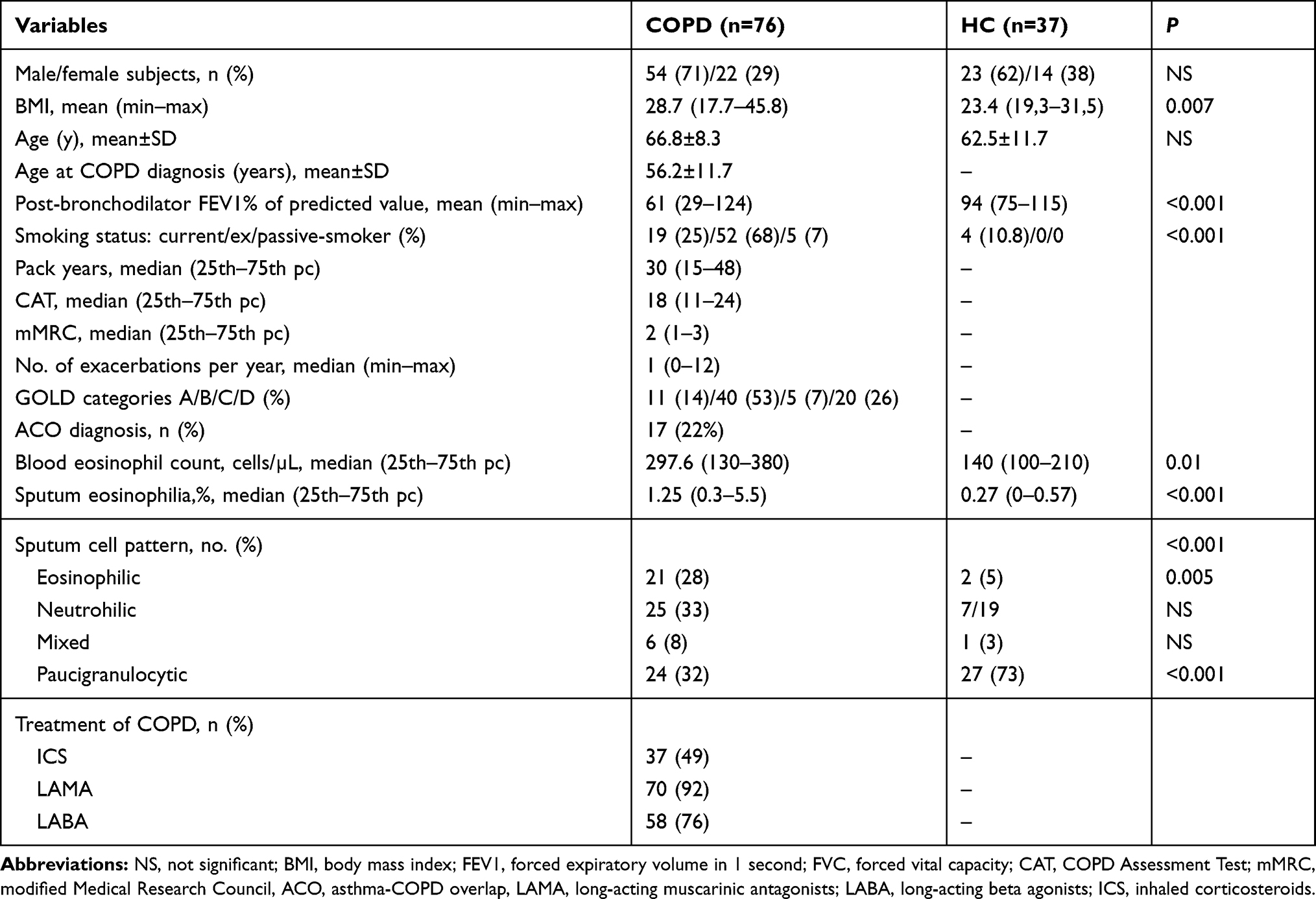 |
Table 1 Clinical Characteristics of the COPD Cohort |
Data Collection
A structured questionnaire was used to collect data including demography, smoking status, symptoms, number of COPD exacerbations and pharmacological treatment. COPD patients were classified into one of four GOLD 2019 categories, based on history of COPD exacerbations and intensity of respiratory symptoms using the modified Medical Research Council (mMRC) dyspnea scale and the COPD Assessment Test (CAT).1 Pre- and post-bronchodilator spirometry was performed using a flow-integrating computerized pneumotachograph (Pneumoscreen, Jaeger, Germany). Basic laboratory tests included peripheral blood eosinophils count.
Induced Sputum
All subjects underwent sputum induction. In COPD patients incapable to complete this procedure due to poor lung function spontaneous sputum samples were obtained. Induced sputum samples were investigated for sputum differential cell count and induced sputum supernatant concentrations of selected eicosanoids.
Sputum induction was performed by inhalation of hypertonic saline solution at concentrations increasing from 3% to 5%, using ultrasonic nebulizer (Ultraneb 2000; DeVilibiss, Somerset, PA, USA). Expectorated onto an ice-cooled Petri dish, sputum was transferred to the laboratory. After manual separation from saliva, mucus plugs were processed to obtain cytospin slides for differential cell count and supernatant for eicosanoid measurements. In short words, mucus plugs were incubated with Sputolysin reagent (EMD Millipore Merck, Darmstadt, Germany) at 1:4 weight/volume dilution for 1 hour at 37°C. The solubilized material was diluted 1:1 vol/vol in the phosphate-buffered saline (PBS), filtered through the filter (48 lm mesh), and separated by centrifugation at 700 g for 10 minutes. Cell counts were ascertained using May-Grunwald-stained cytospin preparations, while supernatant was aliquoted and stored in −70°C until analysis. Due to poor quality of sputum specimens collected from four COPD subjects, they were excluded from further analysis.
Differential count of sputum cells allowed for classification into the following inflammatory patterns: eosinophilic – defined as induced sputum eosinophilia ≥3%; neutrophilic – that is induced sputum neutrophilia ≥50%, mixed cellular with sputum eosinophilia ≥3% and neutrophilia ≥50%; and paucigranulocytic – when no predominant inflammatory cells were present.17,18
Sputum supernatant concentrations of eicosanoids were measured by the means of gas chromatography/mass spectrometry (GC-MS) for PGE2, PGD2, 8-iso-PGE2 and 8-iso-PGF2a and by high-performance liquid chromatography/tandem mass spectrometry (HPLC-MS/MS) for 5-HETE, 12-HETE, 15-HETE, 5-oxo-ETE, 12-oxo-ETE, 15-oxo-ETE, LTD4, LTE4, LTB4, PGA2, tetranor-PGE-M, tetranor-PGD-M and 11-dehydro-TXB2 as previously described.19 Briefly, organic-phase extraction was performed on samples acidified to pH 3.5 with acetic acid and spiked with chemically identical deuterated internal standards (500 pg each; Cayman Chemical Co., Ann Arbor, MI, USA). Concentrations of eicosanoids were calculated using a stable isotope dilution method from the area under the peak. Limits of quantification for eicosanoid measurements are presented in Table 2.
Statistical Analysis
Summary statistics were presented as the mean, standard deviation, median, 25th and 75th percentiles, and number in each category with the percentage of total. Normality was checked using the Shapiro–Wilk W-test. The nonparametric U-Mann–Whitney test was applied to analyze the differences between groups in quantitative data. Categorical data was analyzed using the x2 and Fisher exact tests. A P-value<0.05 was considered statistically significant. All the statistical analyses were performed with the use of Dell Statistica (v.13) and GraphPad Prism (v.8).
Results
Clinical Characteristics of the COPD Cohort
Mean age of COPD subjects was 66.8±8.3 years and mean age of COPD diagnosis was 56.2±11.7 years. Patients were predominantly males (71%) and ex-smokers (68%) with a median smoking burden of 30 pack years. COPD severity as described by GOLD categories was: 11 (14%) subjects A, 40 (53)% as B, five (7%) as C, and 20 (26%) as category D. Forty-two (56%) subjects presented disease exacerbations in the past 12 months, while 18 (23.7%) had severe exacerbations requiring hospitalization. Severe or very severe airway obstruction (FEV1<50% pred.) was observed in 21 (28%) subjects. Asthma-COPD overlap (ACO) was diagnosed in 17 (22%) patients. In almost half of the patients (49%), COPD treatment included inhaled corticosteroids. Clinical characteristics for the COPD cohort are presented in Table 1.
Induced Sputum Cells and Eicosanoids
In COPD subjects, increased sputum eosinophilia was found in 27 (35%) subjects, while among healthy controls in three (8%) subjects. Median blood eosinophilia was higher in COPD compared to healthy control subjects. More detailed characteristics of inflammatory patterns can be found in Table 1.
Induced sputum eosinophilia was strongly positively correlated with blood eosinophilia (ρ=0.84; P<0.001). There was a significant positive correlation between sputum eosinophilia and induced sputum concentrations of LTE4 (ρ=0.47; P<0.001), LTD4 (ρ=0.29; P=0.002), PGE2 (ρ=0.26; P=0.004) 8-iso-PGE2 (ρ=0.41; P<0.001), 5-HETE (ρ=0.34; P<0.001), 5-oxo-ETE (ρ=0.2; P=0.01), PGD2 (ρ=0.36; P<0.001), and 11-dehydro-TBX2 (ρ=0.24; P=0.01), while for prostaglandin metabolites tetranor-PGE-M and tetranor-PGD-M, the correlation was negative (ρ=−0.28; P=0.003 and ρ =−0.37; P<0.001, respectively).
Induced sputum concentrations of LTE4, LTD4, PGD2, PGE2, 8-iso-PGE2, 8-iso-PGF2a, 5-oxo-ETE, 12-oxo-ETE, and 11-dehydro-TBX2 were significantly higher in COPD subjects compared to healthy controls whereas concentrations of tetranor-PGE-M and tetranor-PGD-M were lower (Table 2). Figure 1 presents differences in concentrations of selected eicosanoids between healthy controls, COPD subjects without and with sputum eosinophilia.
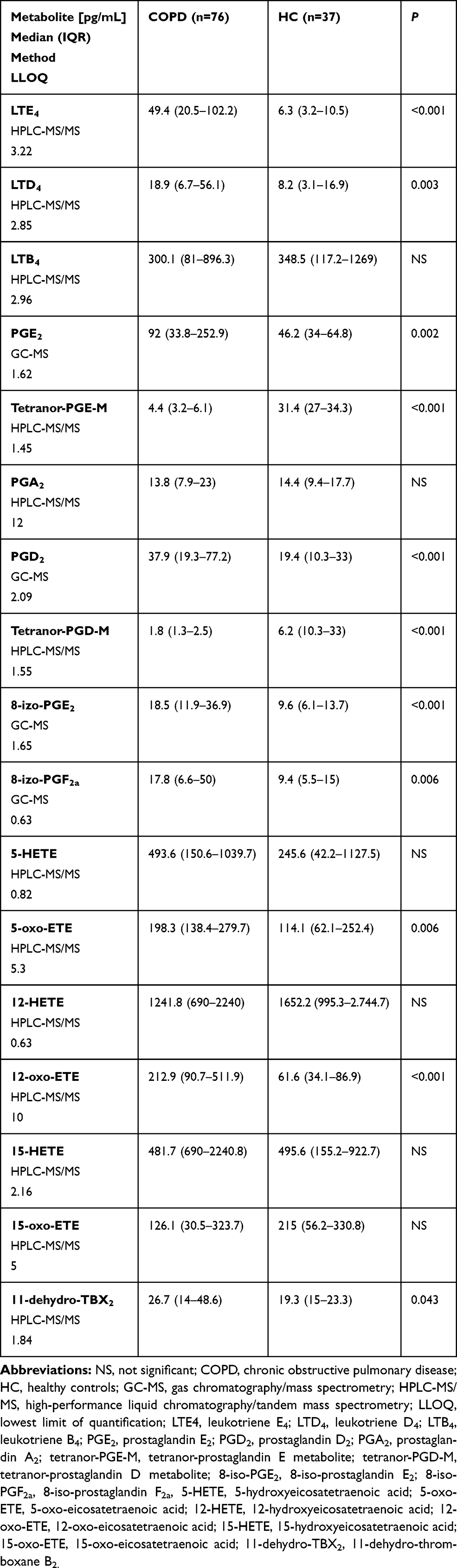 |
Table 2 Concentrations of Eicosanoids in Induced Sputum in COPD and Healthy Controls |
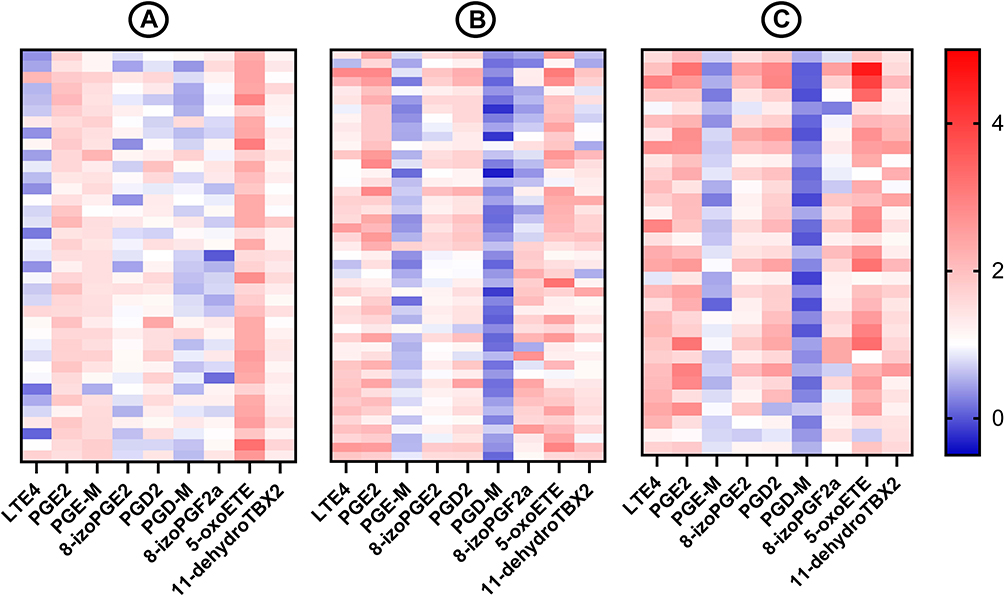 |
Figure 1 Heat map presenting concentrations of selected eicosanoids. Notes: (A) Healthy controls, (B) COPD subjects with noneosinophilic airway inflammation, (C) COPD subjects with eosinophilic airway inflammation. |
Induced Sputum Inflammatory Markers and COPD Course
Among induced sputum eicosanoids in COPD subjects, PGD2 and 11-dehydro-TBX2 correlated negatively with airway obstruction measured by FEV1 (ρ=−0.25; P=0.04 and ρ=−0.3; P=0.015, respectively) and FEV1/FVC (ρ=−0.28; P=0.028 and ρ=−0.33; P=0.006) values. COPD subjects that experienced any disease exacerbations during the past 12 months had significantly lower FEV1 (mean 58.1±27 vs 75.5+23.5, P=0.02) values, higher blood and sputum eosinophilia, yet statistical significance was reached only for blood eosinophilia (median=310, IQR=170–410 cells/µL vs no exacerbations: median=185, IQR=110–300, P=0.02). In this group, significantly higher concentrations of PGD2 (median=51, IQR=25.9–120 pg/mL vs median=25.2, IQR=19.1–55.8, P=0.025) and 12-oxo-ETE (median=320.9, IQR=126.1–557.1 pg/mL vs median=117.1, IQR=78.7–238.7, P=0.022) and 5-oxo-ETE (median=160.8 IQR=51–361 pg/mL vs median=75.8, IQR=20.2–163.1, P=0.042) were observed (see Supplementary Table 1). Figure 2 presents correlations between PGD2 in induced sputum and both FEV1 and sputum eosinophilia. When ACO subjects were compared to the rest of the COPD cohort no differences in eicosanoid profile were observed (see Supplementary Table 2). ACO subjects had significantly higher blood (310 cells/µL median, 220–460 cells/µL IQR vs 210 cells/µL median, 110–370 cells/µL IQR, P=0.039) but not sputum eosinophilia. No differences in induced sputum eicosanoids nor eosinophilia were observed when COPD subjects treated with ICS were compared with subjects receiving only bronchodilators.
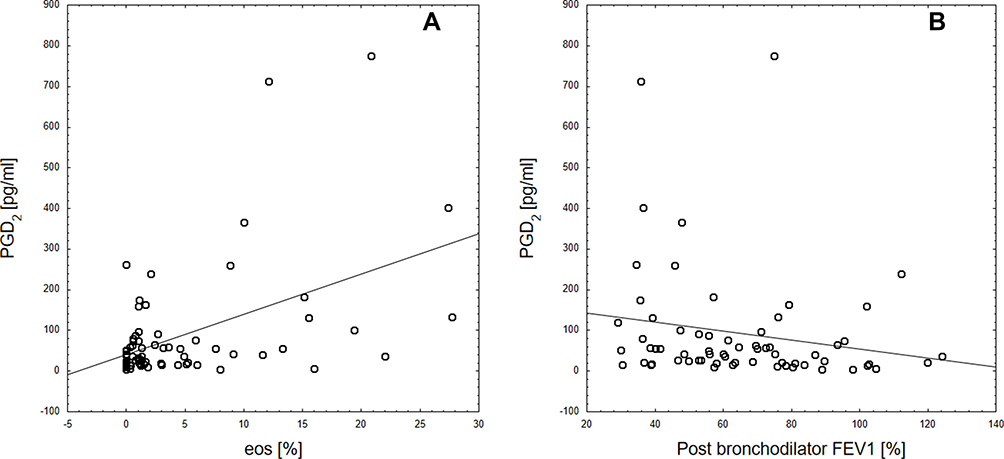 |
Figure 2 Correlations between PGD2 and sputum eosinophilia (A) and post-bronchodilator FEV1 (B). |
Discussion
In this study, we investigated concentrations of selected eicosanoids in bronchial secretions and inflammatory cell patterns of COPD patients. A highly sensitive and specific high-performance liquid or gas chromatography/mass spectrometry method of quantification of eicosanoids was used.19
The eosinophil count and percentage vary among different respiratory compartments, with sputum eosinophilia usually defined as 3% or more eosinophils of all non-epithelial sputum cells.20,21 Eosinophilic airway inflammation, perceived as a typical feature of asthma, has also been found in COPD patients. Frequency of eosinophilic pattern in COPD in our sample was comparable to other studies reporting numbers from 32–40%.2,3,22–25 Median sputum eosinophila was comparable with a study analyzing pure COPD patients by Zahraei et al,26 while blood eosinophilia was higher, possibly due to the fact that our study included ACO. In our COPD cohort sputum eosinophilia correlated positively with blood eosinophilia. This observation is well documented in several studies on asthma.18,27,28 In COPD, on the other hand, the existing data on that correlation are unclear. In studies that included patients with pure COPD, the presence and strength of correlation seem to be dependent on the severity of the disease. In stable mild-to-moderate COPD, no such correlation was observed, while in moderate-to-severe disease a weak correlation was found, and peripheral eosinophils could not predict sputum eosinophilia.24,27,29,30 Despite these rather poor correlations in stable COPD, blood eosinophil counts have been proposed as a marker to guide ICS treatment.1 The eosinophilic pattern in COPD patients results either from co-existing asthma or poorly understood mechanisms that may lead to airway eosinophilia in COPD patients without any asthmatic features. It is thought either Th2 cells or ILC2 produce cytokines that promote recruitment and activation of eosinophils and lead to tissue eosinophilia.30 Studies in asthmatics have shown that cys-LTs and PGD2 released by mast cells recruit and activate the ILC2 cells.31 By measurement of ISS, we confirmed that eosinophilic inflammation in COPD patients was associated with overproduction of cys-LTs: LTD4 and LTE4, lipoxygenation products HETEs: 5-HETE and 5-oxo-ETE and prostanoids: PGD2, PGE2, 8-izo-PGE2, 11-dehydro-TBX2 in the bronchial compartment. A study by Sanak et al32 has proved that asthmatics compared to healthy controls present increased concentrations of 5-HETE, 12-HETE, and 15-HETE, LTC4, LTD4, and LTE4, metabolites of PGD2, PGE2, PGF2a, 8-iso-PGF2a, and 11-dehydro-TXB2 in exhaled breath condensate (EBC). Imbalance between pro- and anti-inflammatory compounds is important in the pathogenesis of airway inflammation in asthma. Decreased synthesis of lipoxines and PGE2 accompanied by overproduction of proinflammatory cys-LTs and PGD2 was linked with nonsteroidal antiinflammatory drug exacerbated respiratory disease and disease severity.32–36 Studies investigating eicosanoids in induced sputum have shown increased concentrations of cys-LTs and PGD2 to result from eosinophil influx of the bronchial tree.15,18 Concentrations of tetranor-PGE-M and tetranor-PGD-M in ISS were lower along with higher concentrations of initial compounds PGE2 and PGD2 in COPD compared to healthy controls. Higher PGE2 with lower metabolite concentrations in ISS were previously observed in a subtype of severe asthma with impaired lung function characterized by neutrophil and mixed cellular patterns in a study by Mastalerz et al. A possible explanation for that may lay in the altered metabolism of prostaglandins or imbalance between different PGE2 receptor types.
There are conflicting data on the potential differences in clinical characteristics of COPD patients with eosinophilic and non-eosinophilic airway inflammation. In some studies, there were no differences in the severity of airway obstruction between eosinophilic and non-eosinophilic patients, while other authors reported higher post-bronchodilator FEV1 in non-eosinophilic patients.23,24,29,37 In our study there was no relationship between blood or sputum eosinophilia and basal lung function measured by FEV1. Similar findings concerning airway and blood eosinophilia support the hypothesis that there might be some dissociation between the lung function in terms and eosinophils in COPD.24,38 Yet in our sample eicosanoids PGD2 and 11-dehydro-TBX2 correlated negatively with airway obstruction measured by FEV1. The negative correlation between eicosanoid metabolites HETEs and HpETEs measured in serum and FEV1/FVC ratio was observed by Cai et al.39 Prospective studies have shown that COPD patients with increased baseline blood eosinophil levels (≥300 cells/µL) were at a greater risk of disease exacerbations.7,40 COPD subjects in our study that experienced any disease exacerbations during the past 12 months had significantly higher blood eosinophilia and concentrations of PGD2, 12-oxo-ETE and 5-oxo-ETE measured in ISS. The percentage of our patients who experienced COPD exacerbation seems to be relatively high compared to the rates observed in published studies.41,42 A possible explanation might be the fact that our study subjects were recruited in an academic center where patients with more severe clinical course of COPD are referred. Moreover, in some cases, patients were referred to our center after an exacerbation requiring hospitalization in the previous year.
Numerous studies have investigated eosinophilic airway inflammation in COPD as a promising target for inhaled corticosteroid (ICS) therapy. It has been demonstrated that in patients with COPD, ICS titration to normalize sputum eosinophil count resulted in a reduction of a severe exacerbation rate.4 In our sample we did not document any differences in eicosanoid concentrations or sputum eosinophilia between patients treated and not treated with ICS. None of the study subjects had an ICS dose titrated based on sputum eosinophilia. Studies on asthma patients have shown no correlation between glucocorticosteroid treatment and eicosanoids in EBC.32 Also high concentrations of proinflammatory eicosanoids in ISS were observed in eosinophilic asthma subjects treated with high glucocorticosteroid regimens.15
A study by Cai et al39 that investigated serum eicosanoids has shown that certain HETEs differed between ACO and pure COPD. In our study, no differences in eicosanoid profile in bronchial compartment were observed. ACO subjects had significantly higher blood but not sputum eosinophilia compared to other COPD subjects. There are many reports in the literature where, compared to COPD patients, ACO patients are characterized not only by higher eosinophilia in peripheral blood but also in sputum.43–46 In this study we used the diagnostic criteria presented in the GOLD and GINA reports, in which ACO is defined as persistent airflow limitation with several features usually associated with asthma and several usually associated with COPD.1,47 However, many other criteria (Soler-Cataluna or Rhee criteria) include sputum eosinophilia as one of the ACO diagnostic criteria.48,49 Therefore, it is not surprising that many publications have shown higher sputum eosinophilia in ACO subjects compared to COPD patients.
We are aware of the limitations of our study. The study sample is limited and some analyses, e.g., comparison between GOLD A-D groups, could not be performed. In some patients, due to poor lung function, a spontaneous sputum sample was taken. What is more, a large number of patients with severe COPD were included in this study, as it was performed in a tertiary center which could possibly bias the results. It might be questioned if a measurement of local production of eicosanoids fully reflects the complexity of inflammation and its relationship with clinical course of COPD. It also is known that inflammatory patterns of influx cells in the airways may change over time.
Conclusion
This study provides information on patterns of inflammatory cells and profiles of eicosanoids measured in induced sputum in stable COPD compared to healthy controls. Stable COPD is often associated with eosinophil influx in the lower airways and elevated concentrations of eicosanoids that are reflected by some disease characteristics.
Abbreviations
COPD, chronic obstructive pulmonary disease; BMI, body mass index; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; CAT, COPD Assessment Test; mMRC, modified Medical Research Council; ACOS, asthma-COPD overlap syndrome; LAMA, long-acting muscarinic antagonists; LABA, long-acting beta agonists; GC-MS, gas chromatography/mass spectrometry; HPLC-MS/MS, high-performance liquid chromatography/tandem mass spectrometry; GOLD, The Global Initiative for Obstructive Lung Disease; COPDGene, Genetic Epidemiology of COPD; ECLIPSE, Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints; IL-33, interleukin 33; Th2, type 2 T helper cells; ILC-2, type 2 innate lymphoid cells; 5-LO, 5-Lipoxygenase; AA, arachidonic acid, 5-HpETE, 5-hydroperoxyeicosatetraenoic acid; LTA4, leukotriene A4; cys-LTs, Cysteinyl leukotrienes; 5-HETE, 5-hydroxyeicosatetraenoic acid; COX, Cyclooxygenase; TXA2 thromboxane A2; IS, induced sputum; ACO, asthma-COPD overlap; EBC, exhaled breath condensate; ICS inhaled corticosteroid; HC, healthy controls; LTE4, leukotriene E4; LTD4, leukotriene D4; LTB4, leukotriene B4; PGE2, prostaglandin E2; PGD2, prostaglandin D2; PGA2, prostaglandin A2; tetranor-PGE-M, tetranor-prostaglandin E metabolite; tetranor-PGD-M, tetranor-prostaglandin D metabolite; 8-iso-PGE2, 8-iso-prostaglandin E2; 8-iso-PGF2a, 8-iso-prostaglandin F2a, 5-HETE, 5-hydroxyeicosatetraenoic acid; 5-oxo-ETE, 5-oxo-eicosatetraenoic acid; 12-HETE, 12-hydroxyeicosatetraenoic acid; 12-oxo-ETE, 12-oxo-eicosatetraenoic acid; 15-HETE, 15-hydroxyeicosatetraenoic acid; 15-oxo-ETE, 15-oxo-eicosatetraenoic acid; 11-dehydro-TBX2, 11-dehydro-thromboxane B2.
Ethics Approval and Informed Consent
Subjects signed informed consent to participate in the study. Study protocol complied with Helsinki Declaration and was approved by the Jagiellonian University Ethics Committee (KBET 122.6120.40.2015).
Acknowledgments
This study was supported by the National Science Centre (NCN) grant (2014/15/N/NZ5/036/32).
Preliminary results from this study were presented as a poster presentation at the ERS International Congress. The poster’s abstract was published in European Respiratory Journal 2018 52: Suppl. 62, PA4235: https://erj.ersjournals.com/content/52/suppl_62/PA4235
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest.
References
1. Gold Reports - Global Initiative for Chronic Obstructive Lung Disease - GOLD. Available from: https://goldcopd.org/gold-reports/.
2. Singh D, Kolsum U, Brightling CE, et al. Eosinophilic inflammation in COPD: prevalence and clinical characteristics. Eur Respir J. 2014;44(6):1697–1700. doi:10.1183/09031936.00162414
3. Saha S, Brightling CE. Eosinophilic airway inflammation in COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(1):39–47. doi:10.2147/copd.2006.1.1.39
4. Siva R, Green RH, Brightling CE, et al. Eosinophilic airway inflammation and exacerbations of COPD: a randomised controlled trial. Eur Respir J. 2007;29(5):906–913. doi:10.1183/09031936.00146306
5. Saetta M, Di Stefano A, Maestrelli P, et al. Airway eosinophilia in chronic bronchitis during exacerbations. Am J Respir Crit Care Med. 1994;150(6 Pt 1):1646–1652. doi:10.1164/ajrccm.150.6.7952628
6. Keene JD, Jacobson S, Kechris K, et al. Biomarkers predictive of exacerbations in the SPIROMICS and COPD Gene cohorts. Am J Respir Crit Care Med. 2017;195(4):473–481. doi:10.1164/rccm.201607-1330OC
7. Yun JH, Lamb A, Chase R, et al. Blood eosinophil count thresholds and exacerbations in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2018;141(6):2037–2047.e10. doi:10.1016/j.jaci.2018.04.010
8. Narendra DK, Hanania NA. Targeting IL-5 in COPD. Int J Chron Obstruct Pulmon Dis. 2019;14:1045–1051. doi:10.2147/COPD.S155306
9. Ying S, O’Connor B, Ratoff J, et al. Expression and cellular provenance of thymic stromal lymphopoietin and chemokines in patients with severe asthma and chronic obstructive pulmonary disease. J Immunol. 2008;181(4):2790–2798. doi:10.4049/jimmunol.181.4.2790
10. Gorska K, Nejman-Gryz P, Paplinska-Goryca M, Korczynski P, Prochorec-Sobieszek M, Krenke R. Comparative Study of IL-33 and IL-6 levels in different respiratory samples in mild-to-moderate asthma and COPD. COPD. 2018;15(1):36–45. doi:10.1080/15412555.2017.1416074
11. Jacobsen E, Doyle A, Colbert D, et al. Differential activation of airway eosinophils induces IL-13 mediated allergic Th2 pulmonary responses in mice. Allergy. 2015;70(9):1148–1159. doi:10.1111/all.12655
12. Fanning LB, Boyce JA. Lipid mediators and allergic diseases. Ann Allergy Asthma Immunol. 2013;111(3):155–162. doi:10.1016/j.anai.2013.06.031
13. Romano M, Cianci E, Simiele F, Recchiuti A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur J Pharmacol. 2015;760:49–63. doi:10.1016/j.ejphar.2015.03.083
14. Woodward DF, Jones RL, Narumiya S. International union of basic and clinical pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev. 2011;63(3):471–538. doi:10.1124/pr.110.003517
15. Mastalerz L, Celejewska‐Wójcik N, Wójcik K, et al. Induced sputum supernatant bioactive lipid mediators can identify subtypes of asthma. Clin Exp Allergy. 2015;45(12):1779–1789. doi:10.1111/cea.12654
16. Celejewska‐Wójcik N, Wójcik K, Ignacak‐Popiel M, et al. Subphenotypes of nonsteroidal antiinflammatory disease-exacerbated respiratory disease identified by latent class analysis. Allergy. 2020;75(4):831–840. doi:10.1111/all.14141
17. Djukanović R, Sterk PJ, Fahy JV, Hargreave§ FE. Standardised methodology of sputum induction and processing. Eur Respir J. 2002;20(37suppl):1s–2s. doi:10.1183/09031936.02.00000102
18. Mastalerz L, Celejewska-Wójcik N, Wójcik K, et al. Induced sputum eicosanoids during aspirin bronchial challenge of asthmatic patients with aspirin hypersensitivity. Allergy. 2014;69(11):1550–1559. doi:10.1111/all.12512
19. Sanak M, Gielicz A, Nagraba K, Kaszuba M, Kumik J, Szczeklik A. Targeted eicosanoids lipidomics of exhaled breath condensate in healthy subjects. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878(21):1796–1800. doi:10.1016/j.jchromb.2010.05.012
20. Rutgers SR, Timens W, Kaufmann HF, van der Mark TW, Koëter GH, Postma DS. Comparison of induced sputum with bronchial wash, bronchoalveolar lavage and bronchial biopsies in COPD. Eur Respir J. 2000;15(1):109–115. doi:10.1183/09031936.00.15110900
21. Weiszhar Z, Horvath I. Induced sputum analysis: step by step. Breathe. 2013;9(4):300–306. doi:10.1183/20734735.042912
22. Kim VL, Coombs NA, Staples KJ, et al. Impact and associations of eosinophilic inflammation in COPD: analysis of the AERIS cohort. Eur Respir J. 2017;50(4):1700853. doi:10.1183/13993003.00853-2017
23. Leigh R, Pizzichini MMM, Morris MM, Maltais F, Hargreave FE, Pizzichini E. Stable COPD: predicting benefit from high-dose inhaled corticosteroid treatment. Eur Respir J. 2006;27(5):964–971. doi:10.1183/09031936.06.00072105
24. Proboszcz M, Mycroft K, Paplinska-Goryca M, et al. Relationship between blood and induced sputum eosinophils, bronchial hyperresponsiveness and reversibility of airway obstruction in mild-to-moderate Chronic Obstructive Pulmonary Disease. COPD. 2019;16(5–6):354–361. doi:10.1080/15412555.2019.1675150
25. Negewo NA, McDonald VM, Baines KJ, et al. Peripheral blood eosinophils: a surrogate marker for airway eosinophilia in stable COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:1495–1504. doi:10.2147/COPD.S100338
26. Zahraei HN, Guissard F, Paulus V, Henket M, Donneau A-F, Louis R. Comprehensive Cluster analysis for COPD including systemic and airway inflammatory markers. J Chronic Obstr Pulm Dis. 2020;17(6):672–683. doi:10.1080/15412555.2020.1833853
27. Górska K, Paplińska-Goryca M, Nejman-Gryz P, Goryca K, Krenke R. Eosinophilic and neutrophilic airway inflammation in the phenotyping of mild-to-moderate asthma and chronic obstructive pulmonary disease. COPD. 2017;14(2):181–189. doi:10.1080/15412555.2016.1260539
28. Fowler SJ, Tavernier G, Niven R. High blood eosinophil counts predict sputum eosinophilia in patients with severe asthma. J Allergy Clin Immunol. 2015;135(3):822–824.e2. doi:10.1016/j.jaci.2014.09.034
29. Hartjes FJ, Vonk JM, Faiz A, et al. Predictive value of eosinophils and neutrophils on clinical effects of ICS in COPD. Respirol Carlton Vic. 2018;23(11):1023–1031. doi:10.1111/resp.13312
30. Mycroft K, Krenke R, Górska K. Eosinophils in COPD—Current concepts and clinical implications. J Allergy Clin Immunol Pract. 2020;8(8):2565–2574. doi:10.1016/j.jaip.2020.03.017
31. White AA, Doherty TA. Role of group 2 innate lymphocytes in aspirin-exacerbated respiratory disease pathogenesis. Am J Rhinol Allergy. 2018;32(1):7–11. doi:10.2500/ajra.2018.32.4498
32. Sanak M, Gielicz A, Bochenek G, Kaszuba M, Niżankowska-Mogilnicka E, Szczeklik A. Targeted eicosanoid lipidomics of exhaled breath condensate provide a distinct pattern in the aspirin-intolerant asthma phenotype. J Allergy Clin Immunol. 2011;127(5):1141–1147.e2. doi:10.1016/j.jaci.2010.12.1108
33. Levy BD, Bonnans C, Silverman ES, et al. Diminished lipoxin biosynthesis in severe asthma. Am J Respir Crit Care Med. 2005;172(7):824–830. doi:10.1164/rccm.200410-1413OC
34. Aggarwal S, Moodley YP, Thompson PJ, Misso NL. Prostaglandin E2 and cysteinyl leukotriene concentrations in sputum: association with asthma severity and eosinophilic inflammation. Clin Exp Allergy. 2010;40(1):85–93. doi:10.1111/j.1365-2222.2009.03386.x
35. Huynh M-LN, Malcolm KC, Kotaru C, et al. Defective apoptotic cell phagocytosis attenuates prostaglandin E2 and 15-hydroxyeicosatetraenoic acid in severe asthma alveolar macrophages. Am J Respir Crit Care Med. 2005;172(8):972–979. doi:10.1164/rccm.200501-035OC
36. Cahill KN, Bensko JC, Boyce JA, Laidlaw TM. Prostaglandin D₂: a dominant mediator of aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2015;135(1):245–252. doi:10.1016/j.jaci.2014.07.031
37. Chou K-T, Su K-C, Hsiao Y-H, et al. Post-bronchodilator reversibility of FEV1 and eosinophilic airway inflammation in COPD. Arch Bronconeumol. 2017;53(10):547–553. doi:10.1016/j.arbr.2017.01.027
38. Casanova C, Celli BR, De-torres JP, et al. Prevalence of persistent blood eosinophilia: relation to outcomes in patients with COPD. Eur Respir J. 2017;50(5):1701162. doi:10.1183/13993003.01162-2017
39. Cai C, Bian X, Xue M, et al. Eicosanoids metabolized through LOX distinguish asthma–COPD overlap from COPD by metabolomics study. Int J Chron Obstruct Pulmon Dis. 2019;14:1769–1778. doi:10.2147/COPD.S207023
40. Vedel-Krogh S, Nielsen SF, Lange P, Vestbo J, Nordestgaard BG. Blood eosinophils and exacerbations in chronic obstructive pulmonary disease. The copenhagen general population Study. Am J Respir Crit Care Med. 2016;193(9):965–974. doi:10.1164/rccm.201509-1869OC
41. Koblizek V, Milenkovic B, Barczyk A, et al. Phenotypes of COPD patients with a smoking history in Central and Eastern Europe: the POPE Study. Eur Respir J. 2017;49(5):1601446. doi:10.1183/13993003.01446-2016
42. Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363(12):1128–1138. doi:10.1056/NEJMoa0909883
43. Gao J, Iwamoto H, Koskela J, et al. Characterization of sputum biomarkers for asthma–COPD overlap syndrome. Int J Chron Obstruct Pulmon Dis. 2016;11:2457–2465. doi:10.2147/COPD.S113484
44. Inoue H, Nagase T, Morita S, Yoshida A, Jinnai T, Ichinose M. Prevalence and characteristics of asthma–COPD overlap syndrome identified by a stepwise approach. Int J Chron Obstruct Pulmon Dis. 2017;12:1803–1810. doi:10.2147/COPD.S133859
45. Kitaguchi Y, Komatsu Y, Fujimoto K, Hanaoka M, Kubo K. Sputum eosinophilia can predict responsiveness to inhaled corticosteroid treatment in patients with overlap syndrome of COPD and asthma. Int J Chron Obstruct Pulmon Dis. 2012;7:283–289. doi:10.2147/COPD.S30651
46. Kobayashi S, Hanagama M, Yamanda S, Ishida M, Yanai M. Inflammatory biomarkers in asthma-COPD overlap syndrome. Int J Chron Obstruct Pulmon Dis. 2016;11:2117–2123. doi:10.2147/COPD.S113647
47. GINA Main Report - Global Initiative for Asthma - GINA. Available from: https://ginasthma.org/gina-reports/.
48. Soler-Cataluña JJ, Cosío B, Izquierdo JL, et al. Consensus document on the overlap phenotype COPD-asthma in COPD. Arch Bronconeumol. 2012;48(9):331–337. doi:10.1016/j.arbres.2011.12.009
49. Rhee CK. Phenotype of asthma-chronic obstructive pulmonary disease overlap syndrome. Korean J Intern Med. 2015;30(4):443–449. doi:10.3904/kjim.2015.30.4.443
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.