Back to Journals » International Journal of Nanomedicine » Volume 19
Efficient Sequential Co-Delivery Nanosystem for Inhibition of Tumor and Tumor-Associated Fibroblast-Induced Resistance and Metastasis
Authors Li C, Wang Z, Zhang Y, Zhu Y, Xu M, Lei H, Zhang D ![]()
Received 21 July 2023
Accepted for publication 20 December 2023
Published 23 February 2024 Volume 2024:19 Pages 1749—1766
DOI https://doi.org/10.2147/IJN.S427783
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Chunhong Li,1,* Zhen Wang,1,* Yifeng Zhang,1 Yuqing Zhu,1 Maochang Xu,1 Hui Lei,1 Dan Zhang1,2
1Department of Pharmaceutical Sciences, School of Pharmacy, Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China; 2Green Pharmaceutical Technology Key Laboratory of Luzhou, School of Pharmacy, Southwest Medical University, Luzhou, 646000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hui Lei; Dan Zhang, Department of Pharmaceutical Sciences, School of Pharmacy, Southwest Medical University, 1-1 Xianglin Road, Luzhou, Sichuan, 646000, People’s Republic of China, Tel +86 17360593759 ; +86 18982700067, Email [email protected]; [email protected]
Purpose: Triple-negative breast cancer (TNBC) is the most lethal subtype of breast cancer. However, the effect of current treatment strategies by inducing tumor cell apoptosis alone is not satisfactory. The growth, metastasis and treatment sensitivity of tumors can be strongly influenced by cancer-associated fibroblasts (CAFs) in the microenvironment. Effective cancer therapies may need to target not only the tumor cells directly but also the CAFs that protect them.
Methods: Celastrol and small-sized micelles containing betulinic acid were co-encapsulated into liposomes using the thin-film hydration method (CL@BM). Folic acid was further introduced to modify liposomes as the targeting moiety (F/CL@BM). We established a novel NIH3T3+4T1 co-culture model to mimic the tumor microenvironment and assessed the nanocarrier’s inhibitory effects on CAFs-induced drug resistance and migration in the co-culture model. The in vivo biological distribution, fluorescence imaging, biological safety evaluation, and combined therapeutic effect evaluation of the nanocarrier were carried out based on a triple-negative breast cancer model.
Results: In the present study, a novel multifunctional nano-formulation was designed by combining the advantages of sequential release, co-loading of tretinoin and betulinic acid, and folic acid-mediated active targeting. As expected, the nano-formulation exhibited enhanced cytotoxicity in different cellular models and effectively increased drug accumulation at the tumor site by disrupting the cellular barrier composed of CAFs by tretinoin. Notably, the co-loaded nano-formulations proved to be more potent in inhibiting tumor growth in mice and also showed better anti-metastatic effects in lung metastasis models compared to the formulations with either drug alone. This novel drug delivery system has the potential to be used to develop more effective cancer therapies.
Conclusion: Targeting CAFs with celastrol sensitizes tumor cells to chemotherapy, increasing the efficacy of betulinic acid. The combination of drugs targeting tumor cells and CAFs may lead to more effective therapies against various cancers.
Keywords: liposomes, targeted delivery, triple-negative breast cancer, anti-tumor, combination therapy, sequential drug delivery
Introduction
Breast cancer, the most frequent cancer among women and the second most frequent newly diagnosed cancer worldwide,1,2 remains a challenge to treat because of its heterogeneity and often aggressive behavior.3 While efforts to develop treatments have targeted the tumor cells themselves,4,5 recent research has discovered that the tumor microenvironment can strongly influence tumor growth, metastasis and response to treatment.6–8 In particular, cancer-associated fibroblasts (CAFs), the most abundant cells in the stroma around the tumor, regulate tumor development by sending signals to the tumor and by altering the extracellular matrix around it.9–11 Overexpression of α-smooth muscle actin (α-SMA) in CAFs in breast cancer can produce dense collagen secretion around blood vessels, forming a physical blockade preventing drug delivery to tumor cells.12,13 This can lead to resistance and metastasis. Traditional therapeutic approaches that target cancer cells alone are insufficient in controlling CAF activation, resulting in recurrence and metastasis of breast cancer.14,15 In order to improve antitumor efficacy, there is an increasing emphasis on combination therapy, ie, the use of two or more drugs with different therapeutic mechanisms to enhance antitumor efficacy through synergistic effects.16,17 The efficacy of free drug combinations is usually compromised by insufficient bioavailability, and nanocarrier co-delivery systems are potentially advantageous in combination therapy. To date, many nanosystems have been proposed to deliver two or more molecules with synergistic pharmacological activity to different cellular targets in the tumor microenvironment.16,17 For example, small dendritic macromolecules loaded with adriamycin (PAMAM-ss-DOX) (DP) were synthesized and encapsulated into pH-responsive nanoliposomes along with adjuvants toll-like receptor 7/8 (TLR7/8) agonist resiquimod (R848) and losartan (LOS).18 Among them, LOS effectively reduced the activity of CAF. To ensure that each drug was precisely released and targeted to the site of action, Li et al prepared novel liposomes modified with hyaluronic acid and glycyrrhetinic acid for co-delivery of aprepitant (APR) and curcumin (CUR), where APR inhibited the activation of CAFs by blocking the SP/neurokinin-1 receptor, and CUR induced apoptosis in tumor cells.19 However, removal of the stromal barrier is a prerequisite for more effective cancer treatment. When the targets of co-delivered drugs are the tumor extracellular mechanism barrier and the tumor cell intracellularly, respectively, to maximize the anti-tumor effect, the development of a strategy that can deliver active molecules in a sequential manner would be the best choice. That is, one drug is released to act on the extracellular target of the tumor cell, and then the other drug is internalized into the tumor cell along with the nanocarrier for intracellular targeting.
In our efforts to develop such a therapy, we have focused on celastrol20 from the root bark of Tripterygium wilfordii, which induces apoptosis of CAFs while reversing the drug resistance of tumors.21,22 We hypothesized that administering celastrol followed by a chemotherapeutic drug might potentiate the efficacy of the latter, leading to synergistic effects against CAFs and tumor. To test this idea, we used the pentacyclic triterpene betulinic acid [3b-hydroxy-20(29)-lupaene-28-oic acid], which shows excellent anti-cancer activity23 but the experimental results are often unsatisfactory because tumors develop resistance to it.24
Given that both celastrol (CEL) and betulinic acid (BA) show relatively low solubility, permeability, and bioavailability and can cause off-target toxicity,25,26 we decided to encapsulate them within nanoparticles,27,28 an approach known to improve breast cancer treatments.29,30 In addition, the co-loaded chemotherapy drug BA and the drug CEL that induces apoptosis of CAFs have different sites of action, meaning that CEL is released into the tumor microenvironment, while BA enters the tumor cells to induce their apoptosis. Our focus was on designing a suitable nano-system that has the ability to sequentially release two chemotherapy drugs. Among nanoparticle systems, we selected liposomes and micelles because they can simultaneously be loaded with hydrophobic compounds and they are generally quite stable in vivo.31–33 Our micelles to deliver betulinic acid (BM)were based on the amphiphilic vitamin E derivative D-α-tocopherol polyethylene glycol 1000 succinate (TPGS), which the US Food and Drug Administration has approved as a drug adjuvant. TPGS contains a hydrophilic polar head and a lipophilic alkyl tail, allowing the encapsulation of betulinic acid within the hydrophobic core of micelles with a hydrophilic surface. CEL was inserted into the phospholipid bilayer of the liposomes (CL). Correspondingly, by making full use of the ascendancies of liposomes co-loading hydrophobic and hydrophilic compounds at the same time, a new strategy of encapsulating the BA-loaded polymeric micelles and CEL into one liposome (CL@BM) is proposed in our study. Meanwhile, liposomes around 100 nm are more likely to avoid the reticuloendothelial system and accumulate at the tumor site than polymer micelles of small particle size.34 Moreover, tumor-targeting delivery can be improved using multifunctional Lip modified by special ligands. We coated the liposomes with folic acid (F/CL@BM) to target them to tumors, many of which overexpress folate receptors on their surface.35
In this way, we hoped to deliver CEL to CAFs in the tumor microenvironment in order to induce their apoptosis, which should reduce the cellular density and collagen content of the matrix, in turn promoting the entry of BA into tumor cells, where it can induce apoptosis (Scheme 1). In this study, we describe the development and validation of our micelle-in-liposome nanosystem for dual drug delivery. Our work demonstrates the potential of cancer therapies that simultaneously target the tumor and its supportive microenvironment.
 |
Scheme 1 Assembly and mechanism of folate-coated micelles-in-liposomes loaded with BA and CEL (F/CL@BM) to accumulate at tumor sites and induce apoptosis in, respectively, tumor-associated fibroblasts (TAFs) and tumor cells. Abbreviations: TPGS, Vitamin E derivative D-α-tocopherol polyethylene glycol 1000 succinate; DSPE-PEG-FA, 1.2-distearoyl-sn-glycero-3-phosphoethanolamine-N- [methoxy (polyethylene glycol)-2000] -folic acid; SPC, soybean phospholipid; CHOL, cholesterol. |
Materials and Methods
Reagents
Celastrol was bought from Chengdu Zhibiaohuachun Biotechnology (Chengdu, China); betulinic acid, cholesterol and coumarin-6, from J&K Scientific (Beijing, China); 1.2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] -folic acid and soybean phospholipid (SPC), from Xi’an Ruixi Biotechnology (Xi’an, China); fetal bovine serum (FBS), calf serum, RPMI-1640 medium, and Dulbecco’s modified Eagle medium (DMEM), from GIBCO (Thermo Fisher Scientific, Carlsbad, California, USA); 3.3′-dioctadecyloxacarbocyanine perchlorate (DiO) and 1.1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI), from Beyotime Biotechnology; and DAPI, from Solarbio Life Sciences (Beijing, China).
The Cell Counting Kit-8 (CCK-8) was obtained from APExBIO Technology (Houston, TX, USA). 2’,7’-Dichlorodihydrofluorescein diacetate and JC-1 mitochondrial membrane potential fluorescent probe was obtained from Solarbio (Beijing, China). Antibodies were obtained from Abcam (Cambridge, UK) against α-SMA (catalog no. ab124964), Bcl-2 (ab196495) or collagen I (ab260043). Antibody against P-glycoprotein 1 (catalog no. AF5185) was obtained from Affinity Biosciences (Jiangsu, China).
Cell Lines
Mouse mammary breast tumor cell line 4T1 and mouse embryonic fibroblast cell line NIH 3T3 were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cultures were maintained in RPMI-1640 medium supplemented with 10% FBS or DMEM supplemented with 10% FBS in a standard incubator at 37 °C in an atmosphere of 5% CO2.
Animals
All procedures and experiments were carried out in accordance with the guidelines of the Laboratory Animal Ethics Committee at Southwest Medical University. Six-week-old female BALB/c mice (16 ± 2 g) were purchased from Chengdu Yaokang Biotechnology (Chengdu, China) and housed individually in cages with ad libitum access to food and water. Animals were co-injected subcutaneously into the fourth mammary pad with 4T1 cells (1 × 105) and NIH 3T3 cells (5 × 104).
tumor volumes were regularly determined according to the formula [length × (width)2] / 2.
Preparation of BM and F/CL@BM
To prepare BM, we mixed TPGS and betulinic acid in a weight ratio of 10:1 in ethanol (5 mL), The mixed solution was evaporated on a rotary evaporator at 50 °C, forming a lipid film, which was rehydrated in 10 min in PBS (5 mL).
To prepare CL@BM, we mixed SPC and cholesterol in a weight ratio of 10:2:1 in methylene dichloride (5 mL), celastrol (1mg) was added, and the mixture was evaporated on a rotary evaporator at 35 °C to form a lipid film. The film was rehydrated for 30 min with the above-prepared BM (10 mL), followed by probe sonication at 65 W for 10 min in ice water.
To prepare F/CL@BM, we incubated CL@BM and DSPE-PEG2000-folic acid (2 mg) at 37 °C for 60 min.
Before use in experiments, all these preparations were passed through a 220-nm filter to remove unencapsulated drugs.
Characterization of Formulations
Particle size and zeta potential were assessed using dynamic light scattering (Nano ZS 90, Malvern, Malvern city, UK). Morphology was assessed using transmission electron microscopy (JEM-200CX, JEOL, Tokyo, Japan) after diluting the samples to 2 mg/mL with PBS, dispersing the dilution onto copper grids, staining with 10 μL of 1% (v/v) phosphotungstic acid in PBS, and drying in air.
Loading capacity and encapsulation efficiency for betulinic acid and celastrol were measured using high-performance liquid chromatography at, respectively, 220 and 425 nm. Encapsulation efficiency (EE) was calculated as follows:

Co-loading of betulinic acid and celastrol was confirmed using FRET experiments, in which celastrol was replaced with DiO to serve as energy donor and betulinic acid was replaced with DiI to serve as energy acceptor. For comparison, liposomes containing DiO (L-DiO) and micelles containing DiI (M-DiI) were combined in a mass ratio of 1:1 to form M@DiI@L-DiO. Fluorescence emission spectra were recorded after excitation at 470 nm using a Synergy H1 Microplate Reader (BioTek Instruments, Winooski, VT, USA). The FRET ratio was quantified as the ratio of fluorescence emission at 568 nm to the sum of the emission at 568 and 503 nm.
A dialysis method was used to measure the release of betulinic acid and celastrol from BM@CL in vitro. BM@CL in PBS (1 mL, 1 mg/mL for both drugs) was placed in a dialysis bag with a molecular weight cut-off of 10 kDa (Shanghai Titan Technology), which was immersed in 20 mL of PBS (pH adjusted to 6.5) containing 0.5% Tween 80 with stirring (100 rpm). At the indicated times, an aliquot (1 mL) was withdrawn and replaced with the same volume of fresh PBS. The aliquot was assayed for betulinic acid and celastrol using high-performance liquid chromatography.
The stability of F/CL@BM in vitro was assessed in PBS or DMEM by measuring size and polydispersity index every other day for two weeks by dynamic light scattering.
Effect of Formulations on Cell Viability in vitro
4T1 cells alone or together with NIH 3T3 fibroblasts (1 × 104 cells per well, 1:1 ratio of NIH 3T3:4T1 cells) were seeded into 96-well plates and incubated for 48 h. Then cells were treated for 24 h with F/CL@BM at concentrations (μg/mL) of CEL and BA of 0, 0.025, 0.125, 0.25, 0.5 and 1. CCK-8 reagent (10 μL) was added to each well, and the plates were incubated at 37 °C for 30 min. Optical density was measured at 450 nm.
As a complementary assessment of cell viability, 4T1 cells were seeded into 6-well plates (5 × 105 cells per well), incubated for 24 h, then exposed for 4 h to F/CL@BM containing CEL and BA at concentrations of 0.1 µg/L. Cells were washed with PBS, stained with calcein-AM and propidium iodide, and analyzed under a fluorescence microscope (Leica, Germany).
Cellular Uptake of Formulations
4T1 and NIH 3T3 cells were seeded into 12-well plates (4×104 cells per well, 1:1 ratio of NIH 3T3:4T1 cells), incubated overnight, then the medium was replaced with 1 mL of fresh medium containing free coumarin-6 (C6), micelles containing C6 (MC), liposomes containing C6 (LC), micelles-in-liposomes containing C6 (MC@LC) or folate-coated micelles-in-liposomes with C6 (F/MC@LC). In all cases, the C6 concentration was 1 μg/mL. After 2 h, the cells were fixed, stained with DAPI, and examined under a confocal laser scanning microscope (TCS SP2, Leica). In other experiments, the cells were harvested, resuspended in PBS, and analyzed by flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA) to quantitate cellular uptake.
In some experiments, cultures were pretreated for 30 min with 1 mg/mL free folic acid to saturate folate receptors on the surface of tumor cells. Then cultures were exposed to the different formulations as described above.
Effect of Formulations on Cell Migration
4T1 cells alone or together with NIH 3T3 cells were seeded into six-well plates (1×105 cells per well, 1:1 ratio of NIH 3T3:4T1 cells) and incubated for 24 h, then the monolayer was scratched with a sterile pipette tip. Scratched cells were washed away using PBS, and the adherent cells were treated with free betulinic acid, free celastrol, a physical mixture of the two drugs, CL, CL@BM or F/CL@BM. Wound width was measured using an Eclipse Ti-S microscope (Nikon) immediately after the scratch and at 24 h later.
Effect of Formulations on Intracellular Production of Reactive Oxygen Species
4T1 cells were seeded into 6-well plates (1 × 105 cells per well) or 96-well plates (1 × 104 cells per well), incubated for 24 h, washed three times with PBS, then treated for 24 h with free BA, free CEL, a physical mixture of the two drugs, BM, CL, CL@BM, or F/CL@BM. The cells were washed three times with PBS, incubated for 30 min with 2’,7’-Dichlorodihydrofluorescein diacetate (DCFH-DA), and washed free DCFH-DA. Six-well plates were then examined for the presence of the fluorescent product ROS under a fluorescence microscope, while 96-well plates were analyzed on a microplate reader to quantitate levels of the fluorescent product ROS (excitation 488 nm, emission 525 nm).
Effect of Formulations on Mitochondrial Membrane Potential
4T1 cells were seeded into six-well plates (5 × 104), incubated for 24 h, then treated for 30 min with free betulinic acid, free celastrol, a physical mixture of the two drugs, CL, CL@BM, or F/CL@BM. The cultures were incubated for 20 min with 5 μg/mL JC-1 in RPMI 1640 medium, washed with PBS, then observed under a fluorescence microscope.
Western Blotting
4T1 cells alone or together with NIH 3T3 cells were seeded into 6-well plates (1 × 105 cells per well, 1:1 ratio of NIH 3T3:4T1 cells), incubated for 24 h, then treated for 24h with various drug formulations. As a control, 4T1 cells were cultured alone and treated the same way. Cells were harvested, lysed with radio-immunoprecipitation assay (RIPA) buffer, microcentrifuged to pellet debris, and the supernatant was fractionated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were incubated 12h with primary rabbit antibodies against α-SMA, Bcl-2, P-gp or GAPDH, followed by incubation for 1 h at 25 °C with goat anti-rabbit secondary antibody conjugated to horseradish peroxidase. Antibody binding was detected using enhanced chemiluminescence (Image Quant LAS 4000 Mini; Fuji, Tokyo, Japan) and quantitated using Gel Image System 4.00 (Tanon, Shanghai, China).
Biodistribution of Formulations in vivo
Mice bearing 4T1/CAF desmoplastic tumors with a volume of approximately 200 mm3 were injected through the tail vein with DiD-loaded micelles (BM), DiD-loaded micelles-in-liposomes (CL@BM), and DiD-loaded, folate-modified micelles-in-liposomes (F/CL@BM) (three animals per condition). In all cases, the dose of DiD was 0.1 mg/kg. At the indicated time points, mice were anesthetized and imaged (Caliper Life Sciences, Mountain View, CA, USA). At 24 h after injection, tumors and major organs were excised and imaged.
Antitumor Efficacy of Formulations in vivo
Mice bearing 4T1/CAF desmoplastic tumors with a volume of approximately 80 mm3 were injected through the tail vein with PBS (Control), free betulinic acid, free celastrol, a physical mixture of the two drugs, BM, CL, CL@BM or F/CL@BM). The doses of betulinic acid and celastrol were 2 mg/ kg. tumor volume and body weight were measured throughout the experiment.
On day 14 after injection, mice were sacrificed, tumors and organs were excised, and blood was sampled. The excised tumors were weighed, photographed, and sectioned for TUNEL staining. In parallel, tumor samples were fixed in 10% formalin, embedded in paraffin, cut into sections, deparaffinized and subjected to antigen recovery. Sections were immunostained overnight at 4 °C against α-SMA or collagen I, followed by incubation with a secondary antibody conjugated to horseradish peroxidase. Antibody binding was detected by incubating the sections. Sections were also stained with hematoxylin.
Organs were sectioned for hematoxylin-eosin staining. Blood was analyzed for levels of creatinine, AST and ALT.
Statistical Analysis
Data were reported as mean ± standard deviation (SD) and analyzed using GraphPadPrism 9.4.0. Pairwise differences were assessed for significance using the two-tailed Student’s t-test, while differences among three or more groups were assessed using one-way analysis of variance.
Results and Discussion
Characterization of F/CL@BM
Transmission electron microscopy showed BMs and F/BM@CL to be uniformly spherical, and the complete micelles-in-liposomes showed the expected bilayer structure (Figure 1A). The respective zeta potentials were −3.05 and −15.33 mV (Figure 1B) and respective diameters of the two particles were 25.6 and 121.3 nm (Figure 1C) implying that mutual repulsion would prevent them from self-aggregating in solution or in the body. The polydispersity index was substantially lower for F/CL@BM (0.257) than BM (0.360) (Figure 1D), suggesting that BM are stabilized by encapsulation within liposomes. Their encapsulation efficiency (EE), and loading capacity (LC), are shown in Table 1. The LC of F/CL@BM was 8.46 ± 0.20% (BA) and 8.17 ± 0.05% (CEL), and the EE was 82.77 ± 0.06% (BA) and 88.34 ± 0.05% (CEL), respectively. Indeed, the size and polydispersity of F/CL@BM did not alter during standing at 4 °C in phosphate-buffered saline (PBS) or culture medium (Figure 1E). The diameter of F/CL@BM is slightly larger when in the presence of 10% fetal bovine serum (FBS) than in a PBS solution. This size discrepancy is attributed to the formation of a protein corona on the surface of nanoparticles in the presence of FBS.
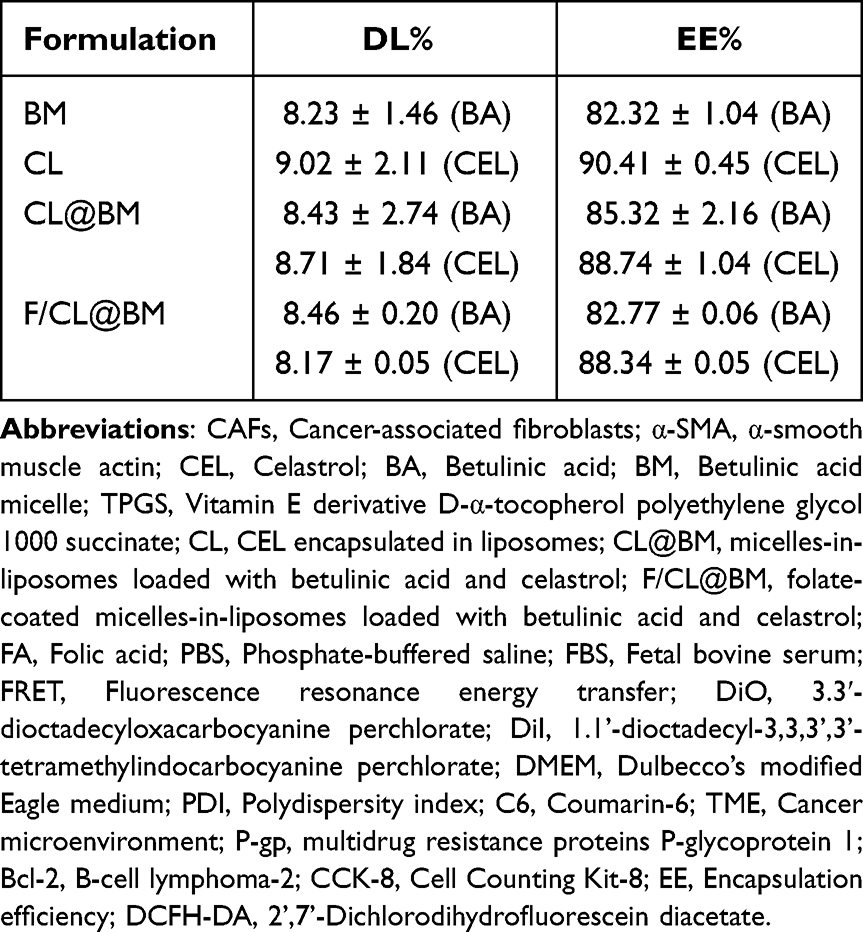 |
Table 1 DL% and EE% of Different Liposomal Formulations |
 |
Figure 1 Characterization of Betulinic acid micelles (BM) and folate-coated liposomes co-loaded with BM and celastrol (F/CL@BM). For reference, Celastrol-loaded liposomes (CL) and liposomes co-loaded with BM and celastrol (CL@BM) were also examined in many cases. (A) Transmission electron micrographs. (B) Zeta potential. (C) Particle size distribution. (D) Polydispersity index. (E) Stability of F/CL@BM at 37°Cin phosphate-buffered saline (PBS) or Dulbecco’s modified Eagle medium (DMEM), based on average particle diameter (“particle size”) or polydispersity index (PDI). (F) Fluorescence emission spectra of fluorophore-containing BM or CL alone, physically mixed together (“+”) or assembled into liposomes (“@”). DiI, 1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate; DiO, 3.3′-dioctadecyloxacarbocyanine perchlorate. (G) Ratios of I568/ (I568 + I503) in BM and CL in a physical mixture or constituted as liposomes, following excitation at 470 nm. (H) Release of betulinic acid (BA) from BMs or CL@BM (left plot), and comparison of the release of celastrol (CEL) or BA from CL@BM (right plot). Experiments were conducted in PBS (pH6.5). Data are mean ± SD (n = 3). ***p < 0.001. |
Co-loading of celastrol and betulinic acid into F/CL@BM was confirmed using fluorescence resonance energy transfer (FRET).36 In this assay, excitation of 3.3′-dioctadecyloxacarbocyanine perchlorate (DiO, representing celastrol) should induce FRET to 1.1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI, representing betulinic acid) only if the two lie within 1–10 nm of each other. Fluorescence spectrum (Figure 1G) showed significantly decreased emission at 503 nm for the donor DiO dye while increased emission at 568 nm for the acceptor DiI dye compared with that of CL-DiO and BM-DiI. The FRET ratio for the two drugs in F/CL@BM was significantly higher for the ratio when they were in a simple physical mixture (56.53% vs 28.40%, p < 0.01; Figure 1F), confirming co-loading into F/CL@BM. The absorption spectrum of F/CL@BM showed a peak at 283 nm similar to the peak for a solution of folic acid (Supplementary Figure 1), indicating successful loading of folic acid onto the liposome surface.
The release of celastrol and betulinic acid from F/CL@BM was examined in vitro by dialyzing them into PBS (pH6.5) containing 0.5% Tween 80. Neither drug was released to an appreciable extent during the first 4 h, after which release increased, leading to 72 h cumulative release of 94.60% for celastrol but 64.00% for betulinic acid (Figure 1H). Under the same dialysis conditions, betulinic acid was released more slowly from BMs than from F/CL@BM. Therefore the micelle-in-liposome structure37 of F/CL@BM leads to sequential release of CEL, followed by BA. The same trend of release was shown for the in vitro release of tretinoin and betulinic acid in PBS (pH 7.0) containing 0.5% Tween 80 (Supplementary Figure 2).
Cytotoxicity of F/CL@BM Against Tumor Cells Alone or Co-Cultured with Fibroblasts
Cytotoxicity of F/CL@BM against tumor cells alone or co-cultured with fibroblasts
The CCK-8 assay was used to assess the cell viability of 4T1 cells or co-cultured cells, and Supplementary Figure 3 showed that the cell viability of both groups of blank liposomes was above 90% at concentrations ranging from 5 to 500 μg/mL, indicating their biocompatibility and potential as safe carriers for anti- tumor drugs. To explore the therapeutic efficacy of F/CL@BM, we first examined their ability to kill 4T1 mouse mammary breast cancer cells cultured on their own or together with NIH 3T3 mouse embryonic fibroblasts. All drug formulations killed the cells in a concentration-dependent manner (Figure 2A and B, Supplementary Figure 4). F/CL@BM performed the best, even better than the same liposomes without the surface coating of folic acid (CL@BM), consistent with the ability of this coating to target tumor cells. Furthermore, the live/dead cell viability assay provided similar results (Figure 2C). This suggested that co-culturing with NIH3T3 cells may have increased the drug resistance of the tumor cells. Conversely, BA+CEL exhibited stronger cytotoxicity than BA alone. These findings suggested that CEL might help restore the sensitivity of 4T1 cells to BA.
 |
Figure 2 Cytotoxicity of different drug formulations against 4T1 tumor cells cultured alone or together with NIH 3T3 fibroblasts. The formulations described in Figure 1 were used, as well as free betulinic acid (BA) and free celastrol (CEL). (A and B) Cytotoxicity was assessed using the Cell Counting Kit-8 assay with 4T1 tumor cells cultured (A) alone or (B) together with NIH 3T3 fibroblasts. Data are mean ± SD (n = 5). ****p < 0.0001, ***p < 0.001, *p < 0.05. (C) Cytotoxicity against 4T1 monocultures was assessed through staining with calcein-AM and propidium iodide. Live cells appear green; dead cells, red. Data are mean ± SD (n = 3). Scale bar, 50 μm. |
Cellular Uptake of F/CL@BM by Tumor Cells Alone or Co-Cultured with Fibroblasts
Next, we examined the internalization of the various drug formulations into co-cultured cells after labeling the formulations with the green dye coumarin-6. F/CL@BM was internalized to the greatest extent, even more than CL@BM (Figure 3A). This likely reflects internalization mediated by the folate receptor, because the internationalization of F/CL@BM was inhibited in the presence of excess free folic acid. Similar results were observed in 4T1 monocultures or co-cultures of 4T1 tumor r cells and NIH3T3 fibroblasts (Supplementary Figure 5A). These experiments with confocal microscopy were confirmed using flow cytometry (Figure 3B). Consistent with the qualitative cell uptake experiment by CLSM, the results of flow cytometry shown in Figure 3C demonstrated that folate-modified nanocarriers were detected with more C6 than other groups, with increases of 2.3-fold and 1.4-fold compared to the C6 and CL@BM groups, respectively (p < 0.0001). In addition, the quantitative results of FCM could demonstrate that tumor cells alone have a stronger ability to take up F/CL@BM than cells co-cultured with fibroblasts. This may be due to the stronger uptake of folic acid by tumor cells (Supplementary Figure 5).
 |
Figure 3 Uptake of drug formulations by 4T1 tumor cells. The formulations described in Figure 1 were used. In each experiment, some cultures were treated with F/CL@BM in the presence of excess free folic acid (“+FA”) or with free fluorescent dye coumarin-6 (C6). (A) Confocal micrographs showing uptake of drug formulations by 4T1 cells cultured alone. Scale bar, 50 μm. (B) FCM image of C6 fluorescence intensity. (C) Quantitation of uptake using flow cytometry. Data are mean ± SD (n = 3). ****p < 0.0001. Scale bar, 50 μm. |
Effects of F/CL@BM on Migration of Tumor Cells Alone or Co-Cultured with Fibroblasts
To evaluate the anti-migration activity of different drug formulations, we performed a scratch assay on 4T1 cells alone (Figure 4A). The results showed that all drug formulations inhibited wound healing compared to the control group. The group treated with both BA and CEL, and the group treated with CL@BM had slightly stronger inhibitory effects than the free CEL group, while the group treated with ligand-modified Lip showed significantly stronger anti-migration activity. As shown in Figure 4C, the cell migration rate of the group treated with F/CL@BM was 15.92%, lower than that of the other groups, indicating that it had better anti-migration activity. In the tumor microenvironment (TME), the interaction between tumor and stromal cells plays a role in tumor metastasis. In stroma cells, CAFs can secrete pro-migratory molecules to promote tumor migration and metastasis.38,39 To examine the anti-migratory activity of different drug formulations in the presence of CAFs, the cell migration efficiency of different drug formulations was measured in the co-culture scratch assay model. As shown in Figure 4B and D, regardless of the treatment conditions, 4T1 migration was greater in monocultures than in co-cultures, reflecting that CAFs secrete molecules promoting tumor migration and metastasis.
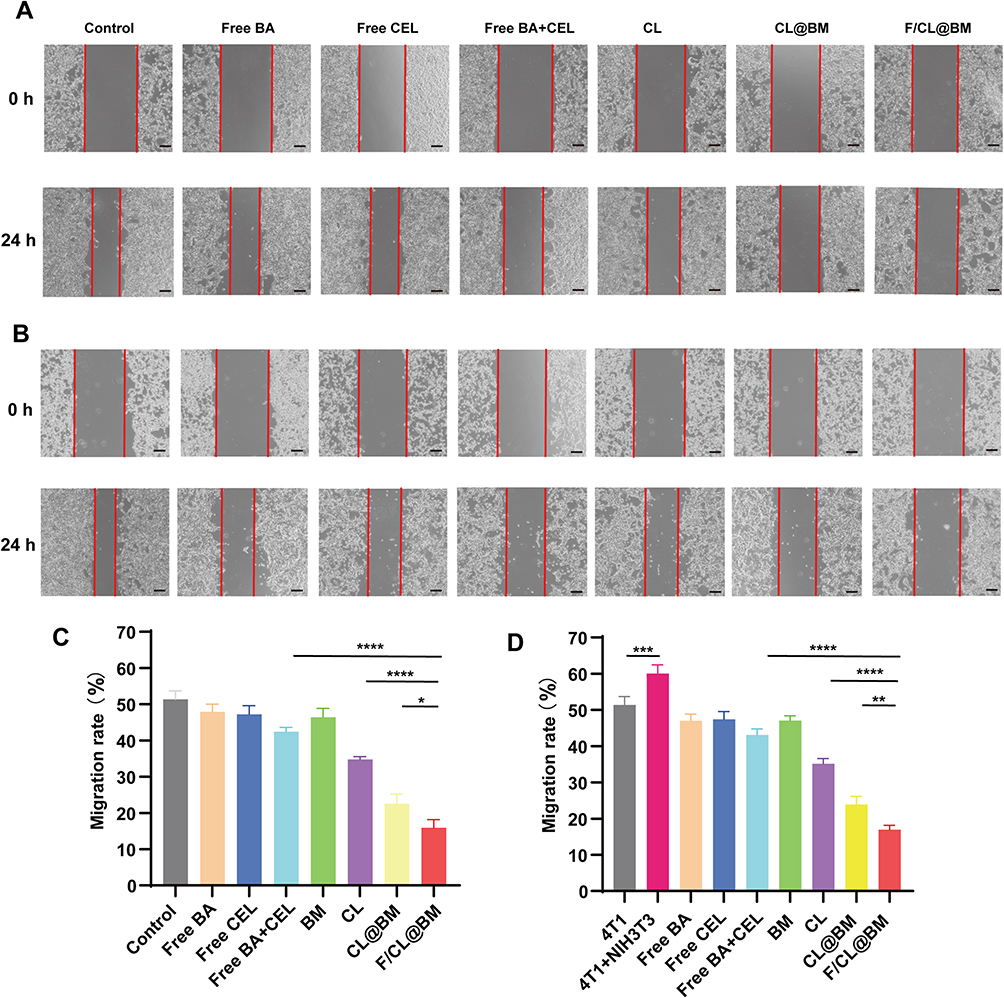 |
Figure 4 Effects of drug formulations on tumor cell migration in a wound healing assay. 4T1 tumor cells were cultured alone or together with NIH 3T3 fibroblasts, the monolayer was scratched, and migration by tumor cells into the wound area was quantitated. Cultures were exposed to the formulations described in Figure 2. “Control” cultures were not exposed to any formulation. (A and B) Representative photomicrographs of (A) monocultures and (B) co-cultures immediately after the scratch and at 24 h later. (C and D) Quantitation of migration assays in (C) monocultures and (D) co-cultures. Data are mean ± SD (n = 3). *p< 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, Scale bar, 100 μm. |
Effects of F/CL@BM on Mitochondrial Membrane Potential in Tumor Cell Monocultures
BA can penetrate the mitochondrial membrane, disrupting its ability to function as a physical barrier and to support oxidative phosphorylation, as well as inducing the production of cytotoxic reactive oxygen species (ROS).40 We confirmed that F/CL@BM induced ROS production in 4T1 tumor cells cultured alone or together with NIH 3T3 fibroblasts, and the production was greater than with either drug on its own (Figure 5A and B). We further confirmed that this ROS production depolarized the mitochondrial membrane, and the effect was stronger with F/CL@BM than with either drug on its own (Figure 5C).
 |
Figure 5 Effects of drug formulations on mitochondrial membrane potential and expression of drug resistance proteins in tumor cells in vitro. Cultures were exposed to the formulations described in Figure 2. “Control” cultures were not exposed to any formulation. (A and B) 4T1 cells were cultured alone in the presence of 2′,7′-dichlorodihydrofluorescein diacetate, which is converted by reactive oxygen species into a green fluorescent product. (A) Representative micrographs. Scale bar, 50 μm. (B) Quantification of reactive oxygen species. (C) 4T1 cells were cultured alone in the presence of fluorescent dye JC-1, which appears red when it can accumulate in the mitochondrial matrix under normal conditions, but green otherwise. Thus, green color indicates abnormally permeable mitochondrial membrane. Scale bar, 50 μm. **p < 0.01, ****p < 0.0001. |
Effects of F/CL@BM on Expression of Drug Resistance Proteins in Tumor Cells Alone or Co-Cultured with Fibroblasts
To understand the proteins that might mediate the apparent resistance of 4T1 cells to BA in co-culture and that therefore may be downregulated by F/CL@BM, we focused on α-SMA as well as the multidrug resistance proteins P-glycoprotein 1(P-gp) and B-cell lymphoma-2 (Bcl-2). As expected, NIH 3T3 fibroblasts produced abundant α-SMA when co-cultured with 4T1 tumor cells, presumably reflecting CAF activation, which the drug formulations, especially F/CL@BM, downregulated (Figure 6). Meanwhile, other signature proteins of CAFs such as FAP, Fibronectin also showed significant reduction (Supplementary Figure 6). To further verify the inhibitory effect of F/CL@BM on CAFs, we measured the expression of α-SMA, FAP, Fibronectin protein-related genes. Quantitative Real-time fluorescence quantitative PCR analysis showed a significant decrease in the expression of these genes compared with the control group (Supplementary Figure 7). Our results suggested that F/CL@BM had a stronger synergistic effect on suppressing the expression of signature proteins in CAFs. These data revealed that the nanocombination of BA and CEL inhibited the activity of CAFs by rapidly reducing α-SMA, FAP, Fibronectin proteins. Analogously, co-culturing 4T1 cells with fibroblasts upregulated P-glycoprotein 1 and Bcl-2, which the drug formulations, especially F/CL@BM, downregulated. Analogously, co-culturing 4T1 cells with fibroblasts upregulated P-glycoprotein 1 and Bcl-2, which the drug formulations, especially F/CL@BM, downregulated.
 |
Figure 6 Western blotting against α-SMA, Bcl-2, and P-gp in total soluble protein fractions from (1) control 4T1 cells in monoculture, (2) control co-cultures of 4T1 cells and NIH 3T3 fibroblasts, or co-cultures treated with (3) free CEL, (4) free CEL+BA, (5) CL, (6) CL@BM or (7) F/CL@BM. (A) Western blotting assay for the expression of protein. (B) Quantitative analysis. ***p < 0.001, ****p < 0.0001. |
These findings identify three proteins that may help CAFs protect tumor cells from chemotherapy and that are downregulated by F/CL@BM, which thereby sensitives the tumor cells to chemotherapy.
Biodistribution and Tumor Accumulation of F/CL@BM in Mice
We created a mouse model of breast cancer by co-injecting 4T1 mammary gland tumour cells with NIH 3T3 fibroblasts into one of the animal’s mammary pads. Then we injected various drug formulations into these animals via the tail vein. While BM disappeared fairly quickly from tumors, substantial F/CL@BM was visible in tumors even at 24 h after injection (Figure 7A). These results were confirmed through ex vivo examination of individual tissues (Figure 7B), which showed F/CL@BM levels in tumors to be up to 3-fold higher than levels of other formulations (Figure 7C). In contrast, CL@BM accumulated significantly more in liver than in tumors (Figure 7D). These findings corroborate the tumor-targeting function of folate on the liposome surface.
 |
Figure 7 Biodistribution of drug formulations in desmoplastic orthotopic tumors in vivo. Mice were co-injected with 4T1 cells and NIH 3T3 fibroblasts in the fourth mammary pad; after tumors attained adequate size, the animals were systemically injected with indicated formulations, defined in Figure 1. (A) Time course of in vivo fluorescence images at the indicated times after injection. (B) Fluorescence of tumors was measured ex vivo at 24 h after injection. (C) Representative fluorescence images. (D) Fluorescence of major organs. Data are mean ± SD (n = 3). **p < 0.01, ***p < 0.001, ****p < 0.0001. |
Therapeutic Efficacy of F/CL@BM in Mice
Due to the good therapeutic effect in vitro and tumor distribution in vivo, we further evaluated the anti- tumor efficiency of our designed nano-system in a rich matrix orthotopic breast cancer model. Tumor volume measurements showed that different BA and/or CEL formulations significantly inhibited tumor growth (Figure 8A). However, compared with other groups, F/CL@BM had the strongest inhibitory effect on tumor growth. This may be due to the synergistic effect of BA and CEL compared with their individual use. Representative images of excised tumors (Figure 8B) roughly corresponded to the trend of tumor weight changes as shown in Figure 8C. In addition, the tumor growth inhibition effect of the F/CL@BM group was better than that of the unmodified CL@BM due to the folate-mediated endocytosis (p<0.05). Consistent with the trend of tumor volume, the tumor weight of the F/CL@BM group was the lowest, as shown in Figure 8C. Furthermore, TUNEL staining showed that there were almost no apoptotic cells in the PBS-treated group (no TUNEL, brown), whereas the F/CL@BM-treated group showed the largest apoptotic area and other groups induced varying degrees of tumor cell apoptosis consistent with the above results (Figure 8D).
 |
Figure 8 Antitumor efficacy of drug formulations in vivo. Mice were co-injected with 4T1 cells and NIH 3T3 fibroblasts in the fourth mammary pad; after tumors attained adequate size, the animals were systemically injected with indicated formulations, defined in Figure 2. “Control” animals were injected with PBS. (A) Tumor growth curves. (B) Photographs of tumors excised on day 14 after injection. (C) Tumor weight on day 14 after injection. (D) TUNEL staining of tumor tissues sectioned on day 14 after injection. Scale bar, 50μm. Data are mean ± SD (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001, ****p<0.0001. |
Lung Metastasis Inhibition
Distant metastasis is the main reason for poor prognosis and high mortality of the patients with advanced breast cancer. We performed an evaluation of the inhibitory effect of nanomaterials on pulmonary metastasis in vivo. As illustrated in Figure 9A, the H&E lung staining images depicted a substantial burden of lung tumors except for the F/CL@BM group, which indicated a significant inhibition of 4t1 tumor pulmonary metastasis due to F/CL@BM. The results from Figure 9B displayed a large number of metastatic nodules observed in the lung, indicating the successful invasion of the tumor into the lung resulting in distant metastatic foci. The combination therapy group exhibited a reduction in the number of surface nodules in the lung compared to monotherapy, indicating that combination therapy improves the anti-metastatic effect. The number of nodules was lower in the F/CL@BM group compared to other drug treatment groups, indicating inhibition of pulmonary metastasis. The probable cause of this finding is the capability of F/CL@BM to effectively deliver more drugs to the metastatic area by reducing the tumor barrier, thereby inhibiting the formation of lung nodules. In addition, the key role of CEL in this system cannot be ignored because it can significantly inhibit the expression of α-SMA, FAP, Fibronectin in activated fibroblasts, further regulating the TME and enhancing anti- tumor effects (Supplementary Figure 8). This conclusion is also supported by the results of α-SMA and collagen expression at the tumor site detected by immunohistochemistry as shown in Figure 9C. Therefore, we conclude that F/CL@BM could effectively reduce CAF activation in tumor tissues. Based on this, CAF-mediated physical barriers are broken, and the anti-cancer effect of chemotherapy drugs is enhanced.
 |
Figure 9 Effects of drug formulations on the tumor microenvironment in vivo. (A) Thin sections of lung were prepared on day 14 after injection and stained with hematoxylin-eosin. Scale bar, 50μm. (B) Representative photographs of lungs excised on day 14 after injection. Tumor nodule are circled in yellow. (C) Immunohistochemistry to detect α-SMA and collagen I in thin sections of lung like those in panel (A). Scale bar, 40μm. Data are mean ± SD (n = 5). |
Safety Evaluation
Figure 10A revealed no significant differences in the heart, liver, spleen, and kidneys of the treatment group compared to the control group, implying that the nanomaterials represent a safe delivery vehicle for anti- tumor drugs. In addition, there were no significant changes or differences in mouse body weight across all groups, as shown in Figure 10B. Furthermore, no differences were found for detected serum biochemistry values, including ALT and AST. Therefore, this finding suggested that repeated intravenous injection of prepared nanoparticles does not lead to severe liver or kidney dysfunction or complications, as presented in Figure 10C and confirmed in the hemolysis experiment Supplementary Figure 9. Consequently, F/CL@BM is a secure carrier for administering anti- tumor drugs.
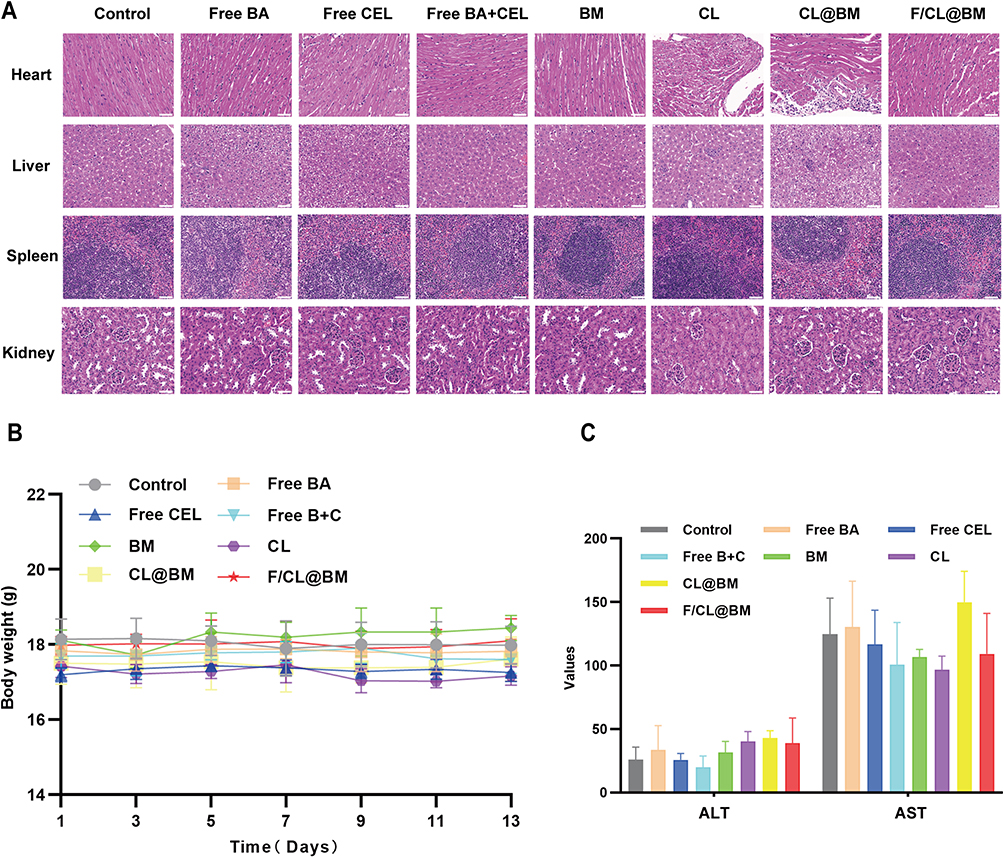 |
Figure 10 Safety evaluation. (A) H&E staining of main organs (hearts, livers, spleens, and kidneys) at the end of experiments. Scale bar=50 μm. (B) Body weight of animals during treatment. (C) Levels of serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in serum on day 14 after injection. (Compared to control group. *p<0.05, **p<0.01, ***p<0.001, n=5, mean ±SD). Data are mean ± SD (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001, ****p<0.0001. |
Overall, these results suggest that F/CL@BM effectively inhibits tumor growth in vivo, but systemic toxicity can be ignored, so co-delivery of anti- tumor drugs and anti-fibrotic agents shows great potential for breast and other CAF-rich tumors.
Limitations of the study should be taken into account while evaluating the results. One concern is with the experimental conditions; the study explored the therapeutic effect of the nano-system only on BALB/c mouse models injected with 4T1 cells. A more realistic data for clinical trials could be obtained from humanized immune system mouse models injected with human breast cancer cells or tumor tissue from patients. Due to limited experimental equipment, the study made efforts to maintain consistency of the different batch parameters (such as particle size, PDI, zeta potential, encapsulation efficiency, drug loading) of the synthesized nanoparticles. However, the differences between the batches are still outside of industrial production standards. Only the use of more precise instruments can reduce and standardize the differences between different batches of nanoparticles.
Conclusion
We have prepared a novel multifunctional nano-formulation that can target tumors in vivo with sequential release. Upon systemic administration, the formulation first specifically aggregated at mesenchymal-rich tumor sites under FA guidance, releasing CEL and BM. The released CEL was retained in the stroma to inhibit fibrosis by down-regulating the expression of proteins such as α-SMA, collegen I, etc., in CAFs, which cleared the way for the subsequently released antitumor drugs to enter into the cancer cells, where they similarly induced apoptosis. The co-loaded nano-formulations proved to be more effective in inhibiting tumor growth and significantly reduced the number of lung metastases in mice than the formulations with either drug alone. In summary, the present study was able to effectively inhibit tumor growth and metastasis in TNBC by sequential delivery of two drugs, specific removal of CAFs, and increased drug penetration, providing a new strategy for the treatment of TNBC. Given the powerful effects of CAFs in the tumor microenvironment on the growth and metastasis of many types of cancers, our approach may prove to be effective for a variety of malignancies.
Ethics Approval and Consent to Participate
All in vivo procedures have been approved by Animal Ethics Committee of the Center for Experimental Animal Research of Southern Medical University (number: swum 20220817033).
Acknowledgments
We gratefully acknowledge technical support by the Public Platform of Advanced Detecting Instruments, Public Center of Experimental Technology, Southwest Medical University.
Funding
This work was supported by the [Sichuan Science and Technology Program #1] under Grant [number 23NSFSC0666]; [Sichuan Science and Technology Program #2] under Grant [number 22YFS0614] and [the key research and development project fund of social development of [Luzhou Science and Technology Bureau #3] under Grant (number 2022-SYF-56).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Akram M, Iqbal M, Daniyal M, Khan AU. Awareness and current knowledge of breast cancer. Biol Res. 2017;2017:50.
3. Anastasiadi Z, Lianos GD, Ignatiadou E, Harissis HV, Mitsis M. Breast cancer in young women: an overview. Updates Surg. 2017;69(3):313–317. doi:10.1007/s13304-017-0424-1
4. Miller-Kleinhenz JM, Bozeman EN, Yang L. Targeted nanoparticles for image-guided treatment of triple-negative breast cancer: clinical significance and technological advances. Wiley Interd Rev Nanomed Nanobiotechnology. 2015;7(6):797–816.
5. Du ML, Ouyang Y, Meng FS, et al. Nanotargeted agents: an emerging therapeutic strategy for breast cancer. Nanomedicine. 2019;14(13):1771–1786. doi:10.2217/nnm-2018-0481
6. Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8(2):98–101.
7. Yang S, Gao HL. Nanoparticles for modulating Cancer microenvironment to improve drug delivery and Cancer therapy. Pharmacol Res. 2017;126:97–108. doi:10.1016/j.phrs.2017.05.004
8. Wagner EF. CANCER Fibroblasts for all seasons. Nature. 2016;530:7588):42–43. doi:10.1038/530042a
9. Goulet CR, Pouliot F. TGF beta Signaling in the Cancer Microenvironment. In: Birbrair A, editor. Cancer Microenvironment: Signaling Pathways, Pt B. Springer; 2021:89–105.
10. Zhu Y, Yu FY, Tan Y, Yuan H, Hu FQ. Strategies of targeting pathological stroma for enhanced antiCancer therapies. Pharmacol Res. 2019;2019:148.
11. Liu J, Li M, Luo Z, Dai L, Guo X, Cai K. Design of nanocarriers based on complex biological barriers in vivo for Cancer therapy. Nano Today. 2017;15:56–90. doi:10.1016/j.nantod.2017.06.010
12. Chen XM, Song EW. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2019;18(2):99–115. doi:10.1038/s41573-018-0004-1
13. Buchsbaum RJ, Oh SY. Breast cancer-associated fibroblasts: where we are and where we need to go. Cancers. 2016;8(2):19. doi:10.3390/cancers8020019
14. Zeltz C, Primac I, Erusappan P, Alam J, Noel A, Gullberg D. Cancer-associated fibroblasts in desmoplastic Cancers: emerging role of integrins. Semi Cancer Biol. 2020;62:166–181. doi:10.1016/j.semcancer.2019.08.004
15. Salimifard S, Masjedi A, Hojjat-Farsangi M, et al. Cancer associated fibroblasts as novel promising therapeutic targets in breast cancer. Pathol Res Pract. 2020;216(5):152915. doi:10.1016/j.prp.2020.152915
16. Guo L, Zhang Y, Al-Jamal KT. Recent progress in nanotechnology-based drug carriers for celastrol delivery. Biomater Sci. 2021;9(19):6355–6380. doi:10.1039/D1BM00639H
17. Xiao YT, Li X, Mao JH, et al. Reverse anti-breast cancer drug resistance effects by a novel two-step assembled nano-celastrol medicine. Nanoscale. 2022;14(21):7856–7863. doi:10.1039/D2NR02064E
18. Liu Q, Chen FQ, Hou L, et al. Nanocarrier-mediated chemo-immunotherapy arrested cancer progression and induced cancer dormancy in desmoplastic melanoma. Acs Nano. 2018;12(8):7812–7825. doi:10.1021/acsnano.8b01890
19. Amiri S, Dastghaib S, Ahmadi M, et al. Betulin and its derivatives as novel compounds with different pharmacological effects. Biotechnol Adv. 2020;38:107409. doi:10.1016/j.biotechadv.2019.06.008
20. Yang QR, Xu J, Gu JM, et al. Extracellular vesicles in cancer drug resistance: roles, mechanisms, and implications. Adv Sci. 2022;9(34):2201609. doi:10.1002/advs.202201609
21. Qi X, Gao C, Yin C, Fan J, Wu X, Guo C. Improved anticancer activity of betulinic acid on breast cancer through a grafted copolymer-based micelles system. Drug Deliv. 2021;28(1):1962–1971. doi:10.1080/10717544.2021.1979125
22. Li Z, Guo Z, Chu D, et al. Effectively suppressed angiogenesis-mediated retinoblastoma growth using celastrol nanomicelles. Drug Deliv. 2020;27(1):358–366. doi:10.1080/10717544.2020.1730522
23. Santos AC, Pereira I, Pereira-Silva M, et al. Nanocarriers for resveratrol delivery: impact on stability and solubility concerns. Tren Food Sci Technol. 2019;91:483–497.
24. Solanki R, Jodha B, Prabina KE, Aggarwal N, Patel S. Recent advances in phytochemical based nano-drug delivery systems to combat breast cancer: a review. J Drug Del Sci Technol. 2022;2022:77.
25. Truffi M, Mazzucchelli S, Bonizzi A, et al. Nano-strategies to target breast cancer-associated fibroblasts: rearranging the cancer microenvironment to achieve anticancer efficacy. Int J Mol Sci. 2019;20(6):1263. doi:10.3390/ijms20061263
26. Singh AK, Pandey A, Tewari M, et al. Prospects of Nano-Material in Breast Cancer Management. Pathol Oncol Res. 2013;19(2):155–165. doi:10.1007/s12253-013-9609-1
27. Hwang D, Ramsey JD, Kabanov AV. Polymeric micelles for the delivery of poorly soluble drugs: from nanoformulation to clinical approval. Adv Drug Delivery Rev. 2020;156:80–118. doi:10.1016/j.addr.2020.09.009
28. Lee J, Kim J, Jeong M, et al. Liposome-based engineering of cells to package hydrophobic compounds in membrane vesicles for cancer penetration. Nano Lett. 2015;15(5):2938–2944. doi:10.1021/nl5047494
29. Li YM, Chen Z, Cui YA, Zhai GX, Li LB. The construction and characterization of hybrid paclitaxel-in-micelle-in-liposome systems for enhanced oral drug delivery. Colloids Surf B Biointerfaces. 2017;160:572–580. doi:10.1016/j.colsurfb.2017.10.016
30. Yuan F, Dellian M, Fukumura D, et al. Vascular permeability in a human Cancer xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55(17):3752–3756.
31. Wang L, Li M, Zhang N. Folate-targeted docetaxel-lipid-based-nanosuspensions for active-targeted cancer therapy. Int J Nanomed. 2012;7:3281–3294. doi:10.2147/IJN.S32520
32. Stanisavljevic M, Krizkova S, Vaculovicova M, Kizek R, Adam V. Quantum dots-fluorescence resonance energy transfer-based nanosensors and their application. Biosens Bioelectron. 2015;74:562–574. doi:10.1016/j.bios.2015.06.076
33. Zhao XZ, Yang X, Wang XD, et al. Penetration cascade of size switchable nanosystem in desmoplastic stroma for improved pancreatic cancer therapy. Acs Nano. 2021;15(9):14149–14161. doi:10.1021/acsnano.0c08860
34. Shi YF, Du LM, Lin LY, Wang Y. Tumour-associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat Rev Drug Discov. 2017;16(1):35–52. doi:10.1038/nrd.2016.193
35. Song J, Ge ZH, Yang XR, et al. Hepatic stellate cells activated by acidic Cancer microenvironment promote the metastasis of hepatocellular carcinoma via osteopontin. Cancer Lett. 2015;356(2):713–720. doi:10.1016/j.canlet.2014.10.021
36. Hsu TI, Chen YJ, Hung CY, et al. A novel derivative of betulinic acid, SYK023, suppresses lung cancer growth and malignancy. Oncotarget. 2015;6(15):13671–13687. doi:10.18632/oncotarget.3701
37. Mullauer FB, van Bloois L, Daalhuisen JB, et al. Betulinic acid delivered in liposomes reduces growth of human lung and colon cancers in mice without causing systemic toxicity. Anti-Cancer Drugs. 2011;22(3):223–233. doi:10.1097/CAD.0b013e3283421035
38. Tariq I, Ali MY, Janga H, et al. Downregulation of MDR 1 gene contributes to tyrosine kinase inhibitor induce apoptosis and reduction in Cancer metastasis: a gravity to space investigation. Int J Pharm. 2020;2020:591.
39. Liu MT, Luo Z, Li ZG, et al. Engineered celastrol and plasmid co-delivery for in situ expression and targeted mitochondrial relocation of Nur77 protein towards effective drug resistance reversion. Chem Eng J. 2023;2023:453.
40. Saw PE, Chen JN, Song EW. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer. 2022;8(7):527–555. doi:10.1016/j.trecan.2022.03.001
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.