Back to Journals » Drug Design, Development and Therapy » Volume 15
Effectiveness and Anticancer Activity of a Novel Phenolic Compound from Garcinia porrecta Against the MCF-7 Breast Cancer Cell Line in vitro and in silico
Authors Darwati D, Safitri AN, Ambardhani N, Mayanti T, Nurlelasari N, Kurnia D ![]()
Received 9 June 2021
Accepted for publication 15 July 2021
Published 12 August 2021 Volume 2021:15 Pages 3523—3533
DOI https://doi.org/10.2147/DDDT.S321824
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Darwati Darwati, Ayu Nadila Safitri, Nurul Ambardhani, Tri Mayanti, Nurlelasari Nurlelasari, Dikdik Kurnia
Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Padjadjaran, Jatinangor, 45363, West Java, Indonesia
Correspondence: Darwati Darwati
Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Padjadjaran, Jatinangor, 45363, West Java, Indonesia
Email [email protected]
Background: Cancer is a leading cause of death worldwide, with breast cancer being the most common invasive cancer type in women. Several therapeutic strategies have been explored to reduce the mortality rates of breast cancer. Chemotherapy is the most commonly used systemic treatment, but associated with numerous side-effects. Development of anticancer agents with high efficacy and minimal negative effects is therefore an important focus of research. Natural materials provide an excellent source of bioactive compounds. For instance, Garcinia porrecta from the Clusiaceae family has multiple pharmacological activities, including antioxidant, anti-inflammatory, antibacterial, antiviral, anti-HIV, antidepressant, and anticancer properties.
Purpose: The main objective of this study was to investigate the potential anticancer effects of compounds extracted from the bark of G. porrecta.
Materials and Methods: Our experiments were divided into three steps: (1) chromatographic isolation of compounds using various separation techniques, such as extraction, separation and purification, (2) characterization via infrared (IR), nuclear magnetic resonance (NMR) and mass spectroscopy, and (3) evaluation of anticancer activity in vitro (MTT assay) and in silico (via analysis of molecular docking against caspase-9, tumor necrosis factor alpha (TNF-α), estrogen receptor alpha (ER-α), and human epidermal growth factor receptor 2 (HER-2)).
Results: Depsidone ( 1) and benzophenone ( 2) from the ethyl acetate extract of bark of G. porrecta were identified as bioactive components. Examination of the activities of these compounds against MCF-7 cells revealed an IC50 value of 119.3 μg/mL for benzophenone, whereas IC50 for depsidone could not be estimated. Benzophenone activity was lower than that of the positive control doxorubicin (6.9 μg/mL). Depsidone showed the highest binding affinity for HER-2 (− 9.2 kcal.mol-1) and benzophenone for ER-α (− 8.0 kcal.mol-1).
Conclusion: Benzophenone displays potency as an anticancer agent through blocking ER-α.
Keywords: Garcinia porrecta, Clusiaceae, phenolic, MCF-7 cells, in vitro, in silico
Introduction
Breast cancer is the most commonly diagnosed cancer type in Indonesia.1 Several therapeutic strategies have been explored over recent years in addition to surgery, radiotherapy and chemotherapy.2 Chemotherapy is the standard treatment regimen for multiple tumor types, including breast cancer.3 However, one major limitation is the bystander effect whereby normal cells are eliminated along with cancer cells.4 Moreover, a number of chemotherapeutic drugs induce negative side-effects and chronic toxicities that may be irreversible, particularly in the heart, lung, and kidney.5
The MCF-7 cancer cell line has complex morphological and molecular characteristics. An important marker for prognosis is the presence of estrogen receptor (ER).6 Around 60% breast cancer cells overexpress estrogen receptor-α (ER-α),7 which plays an important role in transcription of nuclear DNA essential for mammary gland development8 and is necessary for the breast cancer signaling network.9 ER-α additionally regulates cell proliferation and differentiation through paracrine mechanisms.10 These findings support the utility of ER-α as a potential molecular target for inhibiting the progression of malignant cells. The development of molecular targeted drugs with anticancer efficacy is a predominant focus of current research efforts.
The search for novel bioactive compounds as therapeutic agents for deadly diseases, such as breast cancer, is an ongoing a hot topic and numerous potential drug designs have been explored as pharmacological treatments.11 Active components of many herbal remedies are under extensive investigation in view of their affordability, with the aim of improving therapeutic efficacy and minimizing side-effects.12 In vitro and in vivo approaches and studies on molecular mechanisms are commonly employed to evaluate the active components of natural products. Additionally, computational methods have been widely used for the prediction and design of anticancer drugs.13
Selection of drug candidates and isolation of novel bioactive compounds from plants are necessary preliminary steps for identification of effective anticancer agents. Structure–activity relationship (SAR) analyses are frequently utilized to determine candidates that are specific, effective, and selective for target receptors. To establish the molecular mechanisms of action of novel bioactive compounds, in silico clinical trials that effectively simulate and model the interactions between chemical molecules and binding targets are conducted,14 generating data that support in vitro and in vivo findings and accelerating the drug discovery process.
The Clusiaceae or Guttiferae family belongs to the Malpighiales order comprising 40 to 50 genera with 1000 species of herbs, shrubs, and trees that thrive throughout tropical and subtropical areas. One of the genera belonging to this family is Garcinia, distributed in India, Indonesia, West and Central Africa, and Brazil.15 Various genera of Garcinia have been used as folk medicine for treating cancer, oxidative stress, inflammation, and infectious disorders.16 Several chemical classes of Garcinia have been identified to date, including xanthones,17–22 benzophenones,23 anthocyanins and bioflavonoids.24,25 These phytochemical compounds have multiple pharmacological activities, including antioxidant, anti-inflammatory, antibacterial, antiviral, anti-HIV, antidepressant, and anticancer properties.16,26
Garcinia porrecta is a tropical-flowering tree that grows abundantly in Indonesia. Plant parts such as fruit, pericarp, leaf, bark, and stem, are traditionally used as medication for fever. Three xanthone (dulxanthone E-G) isolates from bark of G. porrecta with strong cytotoxic activity against murine leukemia L1210 cells have been identified.26 In the current study, the anticancer activity of phytochemical extracts of G. porrecta bark was investigated against breast cancer cells. Previously, we isolated a novel polyoxygenated dimer-type xanthone, 5.5ʹ-oxybis(1,3,7-trihydroxy-9H-xanthen-9-one),27 from ethyl acetate extract of G. porrecta. Here, we present the isolation and structural determination of two compounds, depsidone (1) and benzophenone (2) derivatives, from G. porrecta (Figure 1). The cytotoxicity of these newly isolated compounds against the human breast cancer cell line MCF-7, were examined with the MTT assay and molecular docking simulations conducted to validate our in vitro findings.
 |
Figure 1 Chemical structures of compounds 1 (depsidone derivative) and 2 (benzophenone derivative). |
Materials and Methods
Materials
G. porrecta bark was collected from Bogor Botanical Garden (Bogor, Indonesia) in April 2019. The plant was identified and deposited in the Herbarium Bogoriense (No. IV.K.78a; Center of Biological Research and Development, National Institute of Science, Bogor, Indonesia). Distilled organic solvents used for extraction and purification included methanol, n-hexane, ethyl acetate, acetone, ethanol, and distilled water, while the chemicals for spectroscopic analysis were used pro-analyzed (p.a) grades. Chromatographic separations were conducted on Silica G 60 (0.063–0.200 mm and 0.200–0.500 mm) (Merck, Darmstadt, Germany) and ODS RP-18 (0.040–0.063 mm) (Merck) columns. Silica G 60 F254 plates (0.25 mm, Merck) and ODS RP-18 F254S plates were used for thin-layer chromatography (TLC) and detection achieved by spraying with 5% AlCl3 in ethanol (v/v), followed by heating.
Human breast cancer cell line (MCF-7 ATCC-HBT-22TM) was used in this study. This cell line was a generous gift from Prof. Ahmad Faried, dr., PhD., SpBS(K), FICS (Faculty of Medicine, Universitas Padjadjaran) and the use of these cells was approved by the Research Ethics Committee Universitas Padjadjaran. MCF-7 cells were suspended in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum solution (Gibco), 2% penicillin-streptomycin, and 0.5% Fungizone (Gibco) and added by RPMI 1640 media 100% and incubated for 2–3 days at 37°C in a 5% CO2 incubator. The number of viable cells was counted using a hemocytometer.
The three-dimensional structure of caspase 9 used in the computational study was obtained from the Protein Data Bank (PDB) ID: 1NW9. TNF-α (PDB ID: 1TNF), ER-α (PDB ID: 1A52), and HER-2 (PDB ID: 3PP0) were used for screening of compounds with activity against breast cancer cells. 3D structures were retrieved from the RSCB Protein Data Bank (https://www.rcsb.org/) in PDB format. Doxorubicin (CID 31703) was used as a positive control ligand as for compounds 1 and 2 ligands retrieved from the PubChem compound database (https://pubchem.ncbi.nlm.nih.gov/) in SDF format and converted to PDB format with Open Babel 2.4.2 program.
Instruments
The chemical structures of isolated compounds were determined using a Fourier transform infra-red spectrophotometer (FTIR) Shimadzu 8400 instrument. 1D and 2D NMR spectra were recorded on a Bruker Avance-600 spectrometer (at 600 MHz for 1H and 150 MHz for 13C) using tetramethylsilane (TMS) as an internal standard (Billerica, MA, USA). Mass spectrometry (MS) was performed with a Waters Xevo QTOFMS instrument (Waters, Milford, MA, USA). TLC plates were visualized with UV detector lamps at 254 and 365 nm. For the cytotoxicity assay, 96-well microplates, micropipettes, microtubes, incubators, and Biochrom microplate readers were used.
Isolation of Components of G. porrecta Bark
Air-dried bark of G. porrecta (2 kg) was ground into powder and extracted with n-hexane (5×2 L), ethyl acetate (5×2 L) and methanol (5×2 L) at room temperature. Solvents were concentrated with a rotary evaporator under pressure to yield crude n-hexane (21 g), ethyl acetate (12.5 g), and methanol (25 g) extracts. The ethyl acetate extract (12.5 g) was fractionated via vacuum liquid chromatography on Silica G 60 using a gradient of n-hexane-ethyl acetate-methanol (0:100; v/v, each 1 L) to obtain eight fractions (A-H).
Fraction F (2.07 g) was further fractionated via Silica G 60 (0.063–0.200 mm) column chromatography using a combination of n-hexane-ethyl acetate-methanol solvents (5.5:4:0.5) for elution, resulting in nine subfractions (F1-F9). The F8 (287 mg) subfraction was purified via column chromatography on an ODS RP-18 column and eluted using H2O-MeOH at a gradient of 10% (v/v) to yield compounds 1 (15.7 mg) and 2 (5.5 mg).
Cytotoxicity Assay of Compounds 1 and 2
The cytotoxicity of isolated compounds against human breast cancer MCF-7 cells was examined using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) method. The stock culture was grown in flasks containing RPMI supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin-streptomycin and incubated at 37°C for 24 h. After the medium was changed, MCF-7 cells were detached and seeded in 96-well microtiter plates. After 24 h, the compounds were added to the wells. Cell viability was determined after 48 h by measuring the metabolic conversion of the yellow salt of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide to insoluble formazan, a purple colored product resulting from its reduction in viable cells. Insoluble formazan was diluted with DMSO.28
Data from the MTT assay were read using a microplate reader at 450 nm. Eight concentrations of all the compounds were evaluated (7.81, 15.63, 31.25, 62.50, 125.00, 250.00, 500.00, 1000.00 µg/mL) in 100% DMSO, with a final concentration of 2.5% DMSO in each well.
Cytotoxic activity was expressed as IC50 and analyzed using a linear regression equation. The percentage of cell viability was calculated as follows:
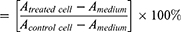
A = Absorbance
Molecular Docking of Caspase 9/TNF-α/ER-α/HER-2 with Compounds 1 and 2
Autodock Vina is open-source software in PyRx 0.8 used for ligand-protein docking and virtual screening for anticancer activity of compounds 1 and 2. Binding of compounds 1 and 2 and doxorubicin to caspase 9, TNF-α, ER-α, and HER-2 protein targets was examined and the ligand was free for bind docking. Conformations were selected based on binding energy, specifically, that with the lowest binding affinity score with a root-mean-square deviation value less than 1.0Å.
Docking results were visualized with PYMOL and analyzed using the Discovery Studio 2020 Client program. Ligand-residue interactions and docking poses in the 3-dimensional molecular images are shown in PYMOL program. Next, these interactions were observed in three-dimensional molecular images for optimal visualization using the Discovery Studio 2020 Client program. The docking pose of each protein-ligand complex was compared to the three-dimensional structures of caspase 9, TNF-α, ER-α, and HER-2 bound to ligands on fatty acid sites. The similarities with ligation pose of compounds bound to fatty acid sites were determined and the relation of the docking pose of the ligands and protein targets analyzed.
Results
Structural Characterization of Compounds 1 and 2 from Bark of G. porrecta
Detailed structural characterization of compounds 1 and 2 are provided below based on the data interpretation spectroscopic results, including FTIR, HR-TOFMS 1H-NMR, and 13C-NMR.
Spectral data of compound 1 are as follows: IR: 3200, 2653, 1677, and 1600 cm−1 (Supplementary Material 1). HR-TOFMS (positive ion mode) (m/z): 273.0395. 1H-NMR (500 MHz, acetone-d6): δH 6.8 (1H, d, 8.0 Hz, H-1), 7.4 (1H, dd, 8.0; 2.0 Hz, H-2), 7.5 (1H, d, 2.0 Hz, H-4), 6.8 (1H, d, 8.0 Hz, H-6), 7.4 (1H, dd, 8.0; 2.0 Hz, H-7), 7.5 ppm (1H, d, 2 Hz, H-9) (Supplementary Material 3). 13C-NMR (125 MHz, acetone-d6): δC 115.6 (C-1), 123.6 (C-2), 150.6 (C-3), 117.4 (C-4), 145.5 (C-4a), 167.5 (C-5), 123.1 (C-5a), 115.6 (C-6), 123.6 (C-7), 150.6 (C-8), 117.4 (C-9), 145.5 (C-9a), 167.5 (C-10), 123.1 ppm (C-10a) (Supplementary Material 4).
Spectral data of compound 2 are as follows: IR: 3381, 2969, 1597, 1475 cm−1 (Supplementary Material 2). HR-TOFMS (positive ion mode) (m/z): 291.0859. 1H-NMR (500 MHz, acetone-d6): δH 6.2 (1H, s, H-3), 6.2 (1H, s, H-5), 6.7 (1H, d, 2.0 Hz, H-2ʹ), 6.5 (1H, t, 2.0 Hz, H-4ʹ), 6.7 (1H, d, 2.0 Hz, H-6ʹ), 3.6 (3H, s, 2-OMe), 3.6 ppm (3H, s, 6-OMe) (Supplementary Material 5). 13C-NMR (125 MHz, acetone-d6): δC 111 (C-1), 161 (C-2), 92.6 (C-3), 159.3 (C-4), 92.6 (C-5), 161 (C-6), 195 (C-7), 141.7 (C-1ʹ), 108.3 (C-2ʹ), 159.4 (C-3ʹ), 108.1 (C-4ʹ), 159.4 (C-5ʹ), 108.3 (C-6ʹ), 55.9 (2-OMe), 55.9 ppm (6-OMe) (Supplementary Material 6).
Compound 1 was obtained as a pale yellow crystal and HR-TOFMS revealed an ion peak m/z 273.0395 [M+H]+ corresponding to the molecular formula C14H8O6 with 11 degrees. The IR spectrum contained absorption bands at νmax 3200 (hydroxyl), 2653 (C-H stretching of aliphatic groups), 1677 (carbonyl), and 1600 cm−1 (aromatic C=C), respectively.
The 1H-NMR spectrum displayed signals corresponding to two doublet aromatic protons at δH 6.8 (1H, d, J = 8.5 Hz, H-1,6) and 7.5 (1H, d, J = 2 Hz, H-2,7) as well as one double doublet aromatic proton at δH 7.4 (1H, dd, J = 8.5 and 2 Hz, H-4,9). The 13C-NMR spectrum contained 14 signals attributable to two benzene rings and two carbonyl carbons. These signals were classified by their chemical shifts on distortionless enhancement by polarisation transfer (DEPT) and heteronuclear single-quantum correlation (HSQC) spectra as six sp2 methine carbons, four sp2 oxygenated carbons, two sp2 quaternary carbons, and two carbonyl carbons at δC 167.5 ppm.
The heteronuclear multiple-bond correlation (HMBC) spectrum showed correlations from H-4,9 (δH 7.5) to C-4a,9a (δC 145.5) and C-3,8 (δC 150.6), confirming the location of a hydroxyl group at the 3,8-position (Figure 2). The carbonyl position was determined on the basis of correlations from H-4,9 (δH 7.5) to C-5,10 (δC 167.5). Another aromatic proton was identified at the 2,7-position based on correlations from H-2,7 (δH 7.4) to C-4,9 (δC 117.4), C-3,8 (δC 150.6) and at the 1,6-position based on correlations from H-1,6 (δH 6.8) to C-3,8 (δC 150.6), C-4a,9a (δC 145.5), C-2,7 (δC 123.6), and C-5a,10a (δC 123.1). 1H-1H COSY and further HMBC correlation data were in agreement with this finding, leading to the identification of the compound as a depsidone derivative (1), as shown in Figure 1.
 |
Figure 2 Selected HMBC correlations for compounds 1 and 2. |
Compound 2 was obtained as a yellow-brown gum with a molecular weight (m/z) of 291.0859 [M+H]+ corresponding to a molecular formula of C15H14O6 with nine degrees. The IR spectrum exhibited absorption bands at Vmax 3381 (hydroxyl), 2969 (C-H stretching of aliphatic bond), 1597 (carbonyl), and 1475 cm−1 (aromatic C=C), respectively.
The 1H-NMR spectrum contained signals corresponding to a singlet aromatic proton at δH 6.2 (1H, s, H-3,5), two aromatic methoxy groups at δH 3.6 (3H, s, OCH3-2,6), two doublet aromatic protons at δH 6.7 (1H, d, J = 2 Hz, H-2ʹ,6ʹ) and a triplet aromatic proton at δH 6.5 (1H, t, J = 2 Hz, H-4ʹ). The 13C-NMR spectrum exhibited 15 signals attributable to two benzene rings and one carbonyl carbon. These signals were classified by their chemical shifts on DEPT and HSQC spectra as five sp2 methine carbons, five sp2 oxygenated carbons, two sp3 oxygenated carbons (methoxy), and a carbonyl carbon at δC 195 ppm.
The HMBC spectrum showed correlations from OCH3-2,6 (δH 3.6) to C-2,6 (δC 161), confirming the presence of a methoxy group at the 2,6-position (Figure 2). The carbonyl position was determined on the basis of the correlation from H-2ʹ,6ʹ (δH 6.7) to C-7 (δC 195). Another aromatic proton was determined at the 3,5-position based on correlations from H-3,5 (δH 6.2) to C-1 (δC 111) and C-2,6 (δC 161). 1H-1H COSY and further HMBC correlations were in agreement with this finding, leading to the identification of the compound as a benzophenone derivative (2), as shown in Figure 1.
Cytotoxic Bioassay of Compounds 1 and 2
The cytotoxic activities of compounds 1 and 2 were evaluated against the MCF-7 breast cancer cell line according to a previously published method.28 The isolated compounds were considered active inhibitors of cell growth at IC50 ≤ 20 µg/mL, moderately cytotoxic at 21–200 µg/mL, weakly cytotoxic at 201–500 µg/mL, and not cytotoxic at IC50 >501 µg/mL.29,30 According to these thresholds, the cytotoxicity of depsidone (1) could not be estimated (NE) since cell viability was >80% while benzophenone (2) exerted a cytotoxic effect with an IC50 value of 119.3 µg/mL. The anticancer activity of doxorubicin against MCF-7 was additionally compared as a positive control. The IC50 value of doxorubicin was determined as ~6.9 µg/mL using the linear regression equation. The plot of the MTT assay of these compounds are shown in the Supplementary Materials 7–9.
Molecular Interactions of Compounds 1 and 2 with Caspase 9/TNF-α/ER-α/HER-2
Following the in vitro cytotoxicity assay of depsidone (1) and benzophenone (2), the molecular mechanisms of action were predicted via computational simulation. Estrogen receptors (ER) are a group of receptor proteins activated by estrogen hormones (17β-estradiol).31 The MCF-7 cell line is ER-α-positive and proliferates in response to estradiol.32 Here, we examined interactions of the novel isolated compounds with ligand binding domains (LBD) of caspase 9/TNF-α/ER-α/HER-2. The strength of interactions between each compound and ER-α was determined from the relative binding affinity (RBA).
Tables 1 and 2 present interpretations of the docking results (binding affinity and hydrophilic interactions) for compound 1, compound 2, doxorubicin, tamoxifen and herceptin against caspase-9, TNF-α, ER-α, and HER-2, respectively.
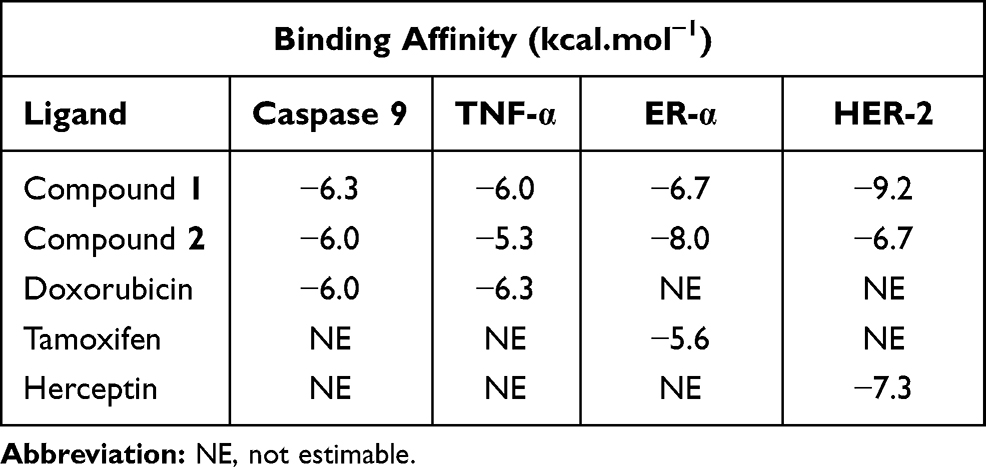 |
Table 1 Binding Affinities of Ligand-Protein Complexes |
 |
Table 2 Hydrophilic Interactions of Ligand-Protein Complexes |
Binding affinity of the compound 1–HER-2 complex showed the greatest value (−9.2 Kcal/mol) relative to the other complexes while the highest value was obtained for the compound 2-ER-α (−8.0 Kcal/mol).
In terms of the ligand-caspase 9 complex, binding affinity of compound 1–caspase 9 was greater than doxorubicin-caspase 9 and compound 2–caspase 9 complexes, which had similar binding affinities. However, the binding pockets of compound 1–caspase 9 and doxorubicin–caspase 9 complexes were in the same position, as characterized by interactions with the same residue (Tyr329). Compound 1–Caspase 9 displayed more hydrophilic interactions relative to the doxorubicin–caspase 9 complex (Figure 3).
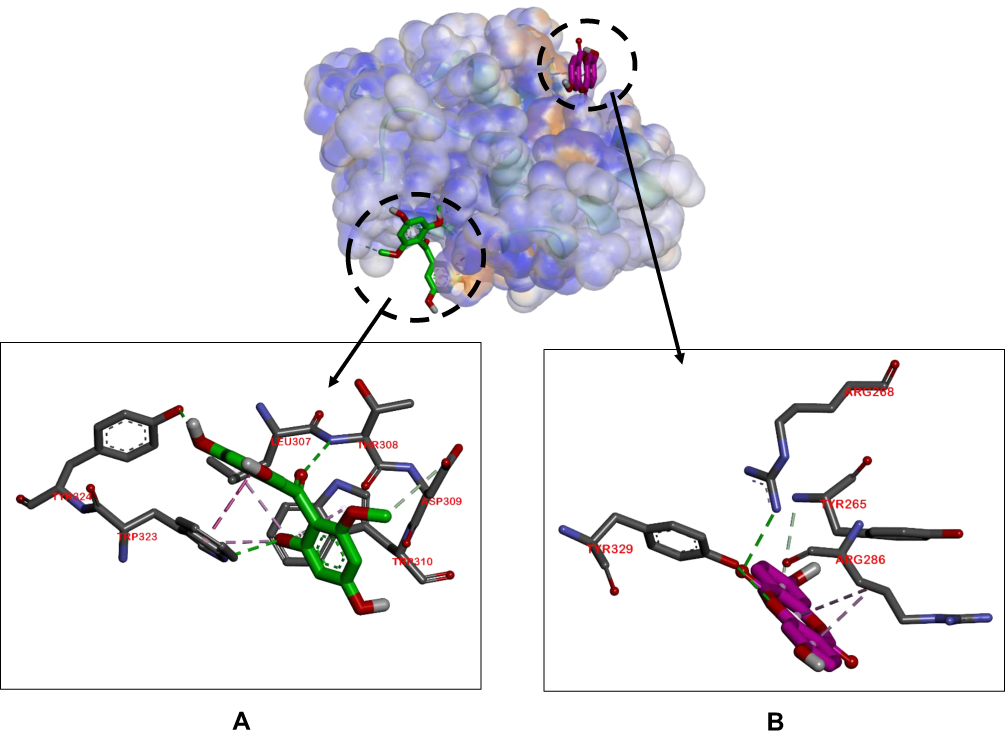 |
Figure 3 Binding sites on caspase 9 for 2 (A) and 1 (B). |
The binding affinities of compounds 1 and 2 for TNF-α were lower than that of doxorubicin, but to a non-significant extent, with values of −6.0, −5.3, and −6.3 Kcal/mol, respectively. The ligand–TNF-α complexes utilized the same binding pocket, as shown in Figure 4.
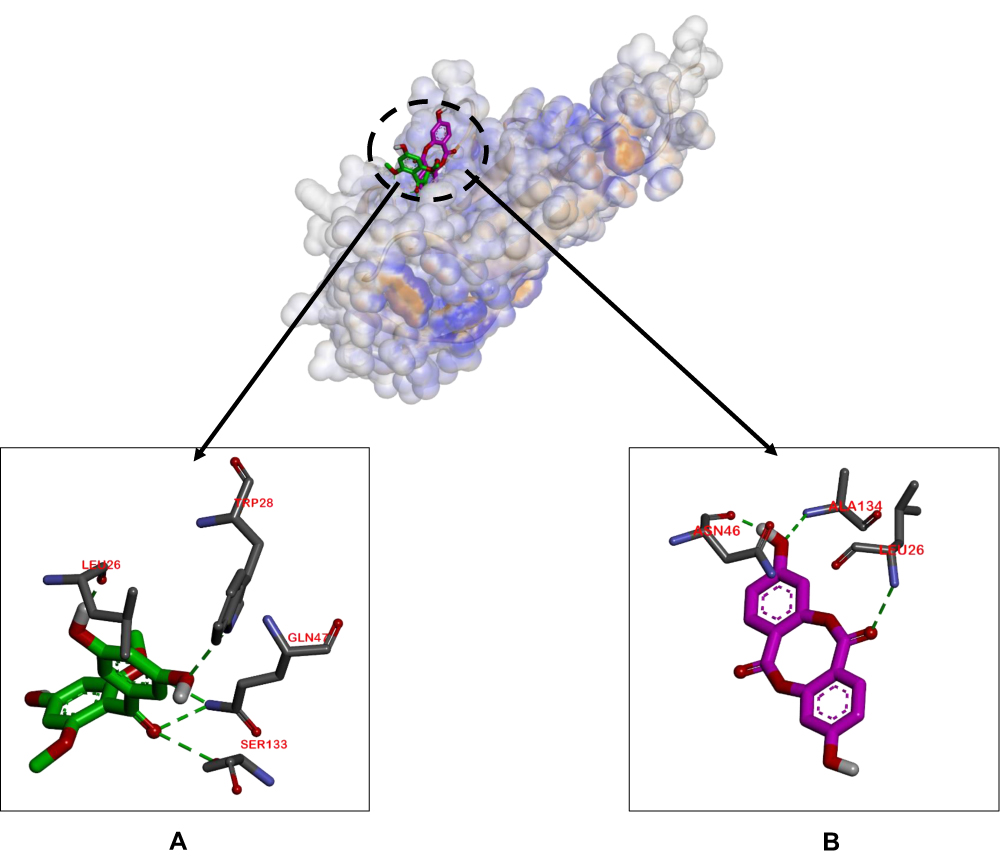 |
Figure 4 Binding sites on TNF-α for 2 (A) and 1 (B). |
For the ligand–ER-α complex, the binding affinity of compound 2–ER-α was higher compared to compound 1–ER-α and tamoxifen–ER-α complexes (−8.0, −6.7, and −5.6 Kcal/mol, respectively). Figure 5 shows the similarity in binding pockets between complexes of compounds 1 and 2 with ER-α although the residual bonds were different.
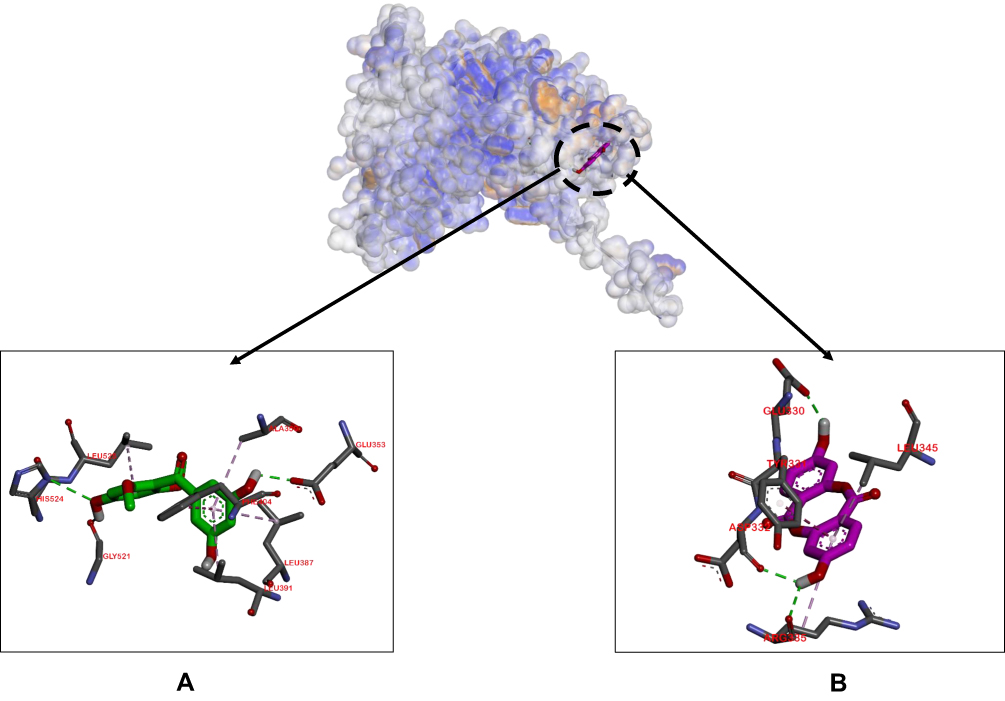 |
Figure 5 Binding sites on ER-α for 2 (A) and 1 (B). |
The binding affinities of compound 1, compound 2 and herceptin to HER-2 were −9.2, −6.7, and −7.3 Kcal/mol, respectively, with compound 1 displaying the highest affinity. We observed binding of Asp808, Arg849, and Cys805 residues to all the ligands tested (Figure 6).
 |
Figure 6 Binding sites on HER-2 for 2 (A) and 1 (B). |
Discussion
The cytotoxic activities of compounds 1 and 2 were evaluated via the MTT assay, a classic, simple, and economical colorimetric method for evaluating anticancer properties.
The viability of MCF-7 cell lines after exposure to a range of concentrations of each compound (7.81 to 1000 µg/mL) for 24 h was assessed using DMSO as a negative control. At a final concentration of 2.5% DMSO and lower (same percentage of DMSO at all compound concentrations), cell viability was not affected. Untreated cells represented the control group and were considered 100% viable.
According to the results, compound 1 induced no obvious inhibition of MCF-7 cell viability at all tested concentrations. The IC50 value could not be extrapolated and >80% cell viability was observed with all the experimental concentrations. Meanwhile, our novel compound 2 clearly exerted a cytotoxic effect, inducing inhibition of cell viability with an IC50 value of 119.3 µg/mL. However, the cytotoxic effect of compound 2 was significantly lower than that of the available anticancer agent, doxorubicin (IC50 of 6.99 µg/mL). The IC50 value of doxorubicin against MCF-7 cell lines has been previously reported as 0.417 µg/mL.33
Docking experiments showed that compound 2 was more effective in inhibiting ER-α activity while compound 1 was more effective against HER-2, as indicated by the greater binding affinity values compared to other compounds. Binding affinity depicts the strength of the ligand-protein bond, whereby greater negative affinity is correlated with a more stable and stronger bond. Hydrophilic interactions, such as hydrogen bonds, play an important role in determining binding affinity value and affect the stability of ligand-protein complexes owing to stronger interactions.34,35
In experiments on ligand–ER-α complex formation, compound 2 had the highest binding affinity, suggesting efficient occupation of the active ER-α pocket. Compound 2 displayed more hydrophilic bonds (Glu353, Gly521, His524, Phe404, Ala350, Leu387, Leu391, Leu525) relative to compound 1 and tamoxifen. Glu353A, Ala350, His524, Phe404, Leu387, Leu391 and Leu525 residues are located at the active pocket of ER-α.36 No residual similarity was evident between the complexes of compound 1–ER-α and compound 2–ER-α. However, binding sides of the residues are sufficiently close to allow competition between the two.
Compound 1 showed the greatest binding affinity (−9.2 Kcal/mol) to HER-2, a growth factor receptor with implications in cancer, compared to compound 2 and herceptin. Heterodimerization of cancer is mediated by HER-2.37 In our experiments, Asp808, Arg849, and Cys805 residues bound to all test ligands within similar binding pockets, which could potentially lead to competition, especially between compounds 1 and 2 displaying the same number of hydrophilic interactions.
Doxorubicin is often used in the treatment of breast cancer based on induction of higher activities of caspase 9 protein and TNF-α.38,39 The docking results of ligand–caspase 9 complexes showed that all ligands had similar binding affinity and activity against caspase 9. In particular, the Tyr329 residue was involved in hydrophilic interactions of compound 1–caspase 9 and doxorubicin–caspase 9 complexes. In terms of ligand–TNF-α complexes, compound 1 was predicted to have equivalent activity as doxorubicin, but with different binding pocket positions, therefore leading to no competitive reactions.
Conclusion
Novel depsidone (1) and benzophenone (2) derivatives were isolated from stem bark of G. porrecta belonging to the Clusiaceae family for the first time. Compound 1 showed no inhibitory activity against the human breast cancer cell line MCF-7 while compound 2 induced inhibition with an IC50 value of 119.3 µg/mL. Docking simulation and binding affinity experiments revealed that compound 1 was more effective in inhibiting HER-2 while compound 2 was more effective against ER-α. Our data support the utility of G. porrecta as a natural plant source of potentially therapeutic bioactive compounds. In view of the collective findings, further research on drug discovery from Garcinia genus, including in vitro, in vivo and clinical studies, is warranted.
Acknowledgments
This research was funded by the Directorate General of Scientific Resources, Technology, and Higher Education, Ministry of Research, Technology, and Higher Education, Indonesia (PDUPT, number, 2788/UN6.D/LT/2019, by D.S.). The authors are grateful to Dr. Mohamad Nurul Azmi (M.Sc.) from Universiti Sains Malaysia for assistance with NMR measurements, Mr. Kansi Haikal at the Central Laboratory (Universitas Padjadjaran) for QTOFMS Measurements, and Mr. Suharto from Bogor Botanical Garden (Bogor, Indonesia) for plant samples.
Disclosure
The authors have no conflicts of interest for this work to declare.
References
1. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Prayogo AA, Wijaya AY, Hendrata WM, et al. Dedifferentiation of MCF-7 breast cancer continuous cell line, development of breast cancer stem cells (BCSCs) enriched culture and biomarker analysis. Indones Biomed J. 2020;12(2):115–123. doi:10.18585/inabj.v12i2.977
3. Fikroh RA, Matsjeh S, Anwar C. Synthesis and anticancer activity of (E)-2ʹ-hydroxy-2-bromo-4,5-dimethoxychalcone against breast cancer (MCF-7) cell line. Molekul. 2020;15(1):34–39. doi:10.20884/1.jm.2020.15.1.558
4. Ketabforoosh SHME, Kheirollahi A, Safavi M, et al. Synthesis and anti-cancer activity evaluation of new dimethoxylated chalcone and flavanone analogs. Arch Pharm Chem Life Sci. 2014;347(11):853–860. doi:10.1002/ardp.201400215
5. Roche VF. Cancer and chemotherapy. In: Lemke TL, Williams DA, Roche VF, Zito SW, editors. Foye’s Principles of Medicinal Chemistry.
6. Giacinti L, Claudio PP, Lopez M, et al. Epigenetic information and estrogen receptor alpha expression in breast cancer. Oncologist. 2006;11(1):1–8. doi:10.1634/theoncologist.11-1-1
7. Hanstein B, Djahansouzi S, Dall P, et al. Insight into molecular biology of the estrogen receptor define novel therapeutic targets for breast cancer. Eur J Endocrinol. 2004;150(3):243–255. doi:10.1530/eje.0.1500243
8. Hayashi SI, Eguchi H, Tanimoto K, et al. The expression and function of estrogen receptor α and β in human breast cancer and its clinical application. Endocr Relat Cancer. 2003;10(2):193–202. doi:10.1677/erc.0.0100193
9. Suganya J, Radha M, Naorem DL, et al. In silico docking studies of selected flavonoids - natural healing agents against breast cancer. Asian Pac J Cancer Prev. 2014;15(19):8155–8159. doi:10.7314/APJCP.2014.15.19.8155
10. Tan H, Zhong Y, Pan Z. Autocrine regulation of cell proliferation by estrogen receptor-alpha in estrogen receptor-alpha-positive breast cancer cell lines. BMC Cancer. 2009;9(31):1–12. doi:10.1186/1471-2407-9-31
11. Yu Y, Shen MY, Song QQ, et al. Biological activities and pharmaceutical applications of polysaccharide from natural resources: a review. Carbohydr Polym. 2018;183:91–101. doi:10.1016/j.carbpol.2017.12.009
12. Sudha A, Srinivasan P, Kanimozhi V, et al. Antiproliferative and apoptosis-induction studies of 5-hydroxy 3ʹ,4ʹ,7-trimethoxyflavone in human breast cancer cells MCF-7: an in vitro and in silico approach. J Recept Signal Transduct. 2018;38:1–13. doi:10.1080/10799893.2018.1468780
13. Tabassum S, Zaki M, Afzal M, et al. Synthesis and characterization of Cu(II)-based anticancer chemotherapeutic agent targeting topoisomerase Ia: in vitro DNA binding, pBR322 cleavage, molecular docking studies and cytotoxicity against human cancer cell lines. Eur J Med Chem. 2014;74(2014):509–523. doi:10.1016/j.ejmech.2013.12.046
14. Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3(11):935–949. doi:10.1038/nrd1549
15. Brito LC, Berenger ALR, Figueiredo MR. An overview of anticancer activity of Garcinia and Hypericum. Food Chem Toxicol. 2017;109:847–862.
16. Hemshekhar M, Sunitha K, Santhosh MS, et al. An overview on genus garcinia: phytochemical and therapeutical aspects. Phytochem Rev. 2011;10(3):325–351. doi:10.1007/s11101-011-9207-3
17. Chin YW, Kinghorn AD. Structural characterization, biological effects, and synthetic studies on xanthones from mangosteen (Garcinia mangostana), a popular botanical dietary supplement. Mini Rev Org Chem. 2008;5(4):355–364. doi:10.2174/157019308786242223
18. Suksamrarn S, Komutiban O, Ratananukul P, et al. Cytotoxic prenylated xanthones from the young fruit of Garcinia mangostana. Chem Pharm Bull. 2006;54(3):301–305. PMID: 16508181. doi:10.1248/cpb.54.301
19. Ibrahim SRM, Abdallah HM, El-Halawany AM, et al. Garcixanthones b and c, new xanthones from the pericarps of Garcinia mangostana and their cytotoxic activity. Phytochem Lett. 2018;25:12–16. doi:10.1016/j.phytol.2018.03.009
20. Subarnas A, Diantinil A, Abdulah R, et al. Apoptosis-mediated antiproliferative activity of friedolanostane triterpenoid isolated from the leaves of Garcinia celebica against MCF7 human breast cancer cell lines. Biomed Rep. 2016;4(1):79–82. doi:10.3892/br.2015.532
21. Bui TQ, Bui A, Nguyen KT, et al. A depsidone and six triterpenoids from the bark of Garcinia celebica. Tetrahedron Lett. 2016;57(23):2524–2529. doi:10.1016/j.tetlet.2016.04.104
22. Ritthiwigrom T, Laphookhieo S, Pyne SG. Chemical constituents and biological activities of Garcinia cowa Roxb. J Sci Technol. 2013;7(2):212–231. doi:10.14456/mijst.2013.18
23. Sari AC, Elya B. Antioxidant activity and lipoxygenase enzyme inhibition assay with total flavonoid assay of Garcinia porrecta Laness. stem bark extracts. Pharm J. 2017;9:257–266. doi:10.5530/pj.2017.2.44
24. Obolskiy D, Pischel I, Siriwatanametanon N, Heinrich M. Garcinia mangostana L.: a phytochemical and pharmacological review. Phytother Res. 2009;23(8):1047–1065. doi:10.1002/ptr.2730
25. Al-Shagdari A, Alarcón AB, Cuesta-Rubio O, et al. Biflavonoids, main constituents from Garcinia bakeriana leaves. Nat Prod Commun. 2013;8(9):1237–1240.
26. Kardono LBS, Hanafi M, Sherley G, et al. Bioactive constituents of Garcinia porrecta and Garcinia parvifolia grown in Indonesia. Pak J Biol Sci. 2006;9(3):483–486. doi:10.3923/pjbs.2006.483.486
27. Safitri AN. 5,5-Oxybis(1,3,7-trihydroxy-9H-xanthen-9-one): a new xanthone from the stem bark of Garcinia porrecta (Clusiaceae). Molbank. 2020;3:1–5. doi:10.3390/M1153w
28. Heah KG, Shobri NRBM, Khoruddin NAB, et al. A review on dimethyl thiazodiphenyl-tetrazoliumbromide (MTT) assay in cell viability. Res J App Sci. 2017;12(7–9):372–378.
29. Grever MR, Schepartz SA, Chabner BA. The national cancer institute: cancer drug discovery and development program. Semin Oncol. 1992;19(6):622–638. PMID: 1462164.
30. Nguyen NH, Ta QTH, Pham QT, et al. Anticancer activity of novel plant extracts and compounds from Adenosma bracteosum (Bonati) in human lung and liver cancer cells. Molecules. 2020;25(2192):1–16. doi:10.3390/molecules25122912
31. Dahlman-Wright K, Cavailles V, Fuqua SA, et al. International union of pharmacology. LXIV. Estrogen receptors. Pharmacol Rev. 2006;58(4):773–781. PMID: 17132854. doi:10.1124/pr.58.4.8
32. Gross JM, Yee D. Commentary how does the estrogen receptor work. Breast Cancer Res. 2002;4(2):62–64. doi:10.1186/bcr424
33. Osman AM, Bayoumi HM, Al-Harthi SE, et al. Modulation of doxorubicin cytotoxicity by resveratrol in a human breast cancer cell line. Cancer Cell Int. 2012;12(1):47–53. doi:10.1186/1475-2867-12-47
34. Fitriah A, Holil K, Syarifah U, et al. In silico approach for revealing the anti-breast cancer and estrogen receptor alpha.
35. Herdiyati Y, Astrid Y, Shadrina AAN, et al. Potential fatty acid as antibacterial agent against oral bacteria of Streptococcus mutans and Streptococcus sanguinis from basil (Ocimum americanum): in vitro and in silico studies. Curr Drug Discov Technol. 2020;16:1–10. doi:10.2174/1570163817666200712171652
36. Mustarichie R, Levita J, Arpina J. In silico study of curcumol, curcumenol, isocurcumenol, and β-sitosterol as potential inhibitors of estrogen receptor alpha of breast cancer. Med J Indones. 2014;23(1):15–24. doi:10.13181/mji.v23i1.684
37. Satyanarayanajois S, Villalba S, Jianchao L, Lin GM. Design, synthesis, and docking studies of peptidomimetics based on HER2–Herceptin binding site with potential antiproliferative activity against breast cancer cell lines. Chem Biol Drug Des. 2009;74(3):246–257. doi:10.1111/j.1747-0285.2009.00855.x
38. Ümit Bağríaçík E, Uslu K, Yurtçu E, et al. Stobadine inhibits doxorubicin‐induced apoptosis through a caspase‐9 dependent pathway in P815 mastocytoma cells. Cell Biol Int. 2007;31(9):979–984. doi:10.1016/j.cellbi.2007.03.008
39. Cao W, Chi WH, Wang J, et al. TNF-α promotes doxorubicin-induced cell apoptosis and anti-cancer effect through downregulation of p21 in p53-deficient tumor cells. Biochem Biophys Res Commun. 2005;330(4):1034–1040. doi:10.1016/j.bbrc.200f5.02.188
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.