Back to Journals » OncoTargets and Therapy » Volume 14
Durable Response to Crizotinib in a Patient with Pulmonary Adenocarcinoma Harboring MET Intron 14 Mutation: A Case Report
Authors Leyrat B, Durando X ![]() , Veyssiere H
, Veyssiere H ![]() , Bernadach M
, Bernadach M
Received 26 March 2021
Accepted for publication 29 May 2021
Published 29 June 2021 Volume 2021:14 Pages 3949—3958
DOI https://doi.org/10.2147/OTT.S312889
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Arseniy Yuzhalin
Brice Leyrat,1,2 Xavier Durando,1– 5 Hugo Veyssiere,3– 5 Maureen Bernadach1,3– 5
1Département d’Oncologie Médicale, Centre Jean Perrin, Clermont-Ferrand, 63011, France; 2Université Clermont Auvergne, UFR Médecine, Clermont-Ferrand, 63000, France; 3Université Clermont Auvergne, INSERM, U1240 Imagerie Moléculaire et Stratégies Théranostiques, Centre Jean Perrin, Clermont-Ferrand, 63011, France; 4Division de Recherche Clinique, Délégation Recherche Clinique et Innovation, Centre de Lutte contre le Cancer, Centre Jean Perrin, Clermont-Ferrand Cedex 1, 63011, France; 5Centre d’Investigation Clinique, UMR501, Clermont-Ferrand, 63011, France
Correspondence: Hugo Veyssiere
Division de Recherche Clinique, Centre de Lutte contre le Cancer, Centre Jean Perrin, 58, Rue Montalembert, Clermont-Ferrand, 63011, France
Email [email protected]
Background: For patients with non-epidermal non–small-cell lung cancer (NSCLC), molecular alterations should always be investigated, especially in non-smokers, who have a very high frequency of targetable alterations (EGFR 52%; ALK 8% in particular). MET exon 14 alterations are identified in 3– 4% of NSCLCs and MET gene amplification and high protein expression are associated with a poor prognosis. The French recommendations only authorize the use of capmatinib and crizotinib if the mutation concerns exon 14. However, several different types of mutation in exon 14 of MET and its flanking introns can induce a jump in exon 14, activate the MET gene and thus be sensitive to anti-MET tyrosine kinase inhibitors.
Case Summary: This case concerns a 76-year-old Caucasian male with a medical history including idiopathic thrombocytopenic purpura, chronic myelomonocytic leukemia (CMML), atrial fibrillation, arterial hypertension, obesity (BMI 36kg/m2), and a 5– 10 pack-per-year smoking history. A left upper lobe pulmonary nodule of 12.4 mm was discovered in March 2019. The patient received adjuvant chemotherapy with carboplatin AUC 5 and vinorelbine 25.00 mg/m2. At the end of the adjuvant treatment, the patient was in complete remission for 5 months. In February 2020, the CT scan revealed a mediastinal lymph node progression. A complementary molecular analysis was realized on the initial surgical specimen. A c.3082+3A>T mutation in the MET gene was identified. This mutation confers susceptibility to anti-MET tyrosine kinase inhibitors. Treatment with crizotinib was initiated with an initial dose of 250 mg/day for 15 days and then increased to 250 mg twice a day. After 7 months of treatment with crizotinib, the disease was still stable according to RECIST 1.1.
Conclusion: We report here the original case of a patient presenting a lung adenocarcinoma with an intron 14 mutation and having a durable TKI response.
Keywords: crizotinib, next-generation sequencing, MET intron 14 mutation, non-small-cell lung cancer
Introduction
In patients with metastatic non–small-cell lung cancer (NSCLC) onset or metastatic relapse in first-line treatment, chemotherapy based on platinum salts in combination with anti-PD1 immunotherapy is recommended for PS 0–1 patients under 70 years of age.1,2 For patients with PDL1 ≥ 50%, pembrolizumab could be used alone instead of chemotherapy.3
For patients with non-epidermal NSCLC, molecular alterations should always be investigated, especially in non-smokers (<100 total cigarettes smoked), who have a very high frequency of targetable alterations (EGFR 52%; ALK 8% in particular).4 Driver mutations for which a specific inhibitor is available that were sought in first intention are epidermal growth factor receptor [EGFR], anaplastic lymphoma kinase [ALK], c-ROS oncogene 1 [ROS1], and BRAF V600E. Different tyrosine kinase inhibitors (TKI) are used in first-line treatment for EGFR activating mutations (afatinib, erlotinib, gefitinib, osimertinib), ALK‐rearrangement (alectinib), and ROS1 rearrangement (crizotinib). Patients with a BRAF V600E mutation benefit from a therapeutic combination of dabrafenib and trametinib.5 Other clinically relevant mutations (HER2 mutations, MET alterations, RET and other fusions) are less common, while MET exon 14 alterations are identified in 3–4% of NSCLCs.
MET exon 14 mutations are more common in adenocarcinomas than in squamous cell carcinomas and large-cell carcinomas.6 Patients with MET exon 14 mutations are generally elderly, predominantly female and include a significant proportion of non-smokers.7
MET gene amplification and high protein expression are associated with a poor prognosis.8,9 MET copy gain was initially recognized in association with secondary resistance to EGFR inhibitors,10 prompting the development of targeted therapies that showed disappointing results.11 More recently, interest in targeting MET has been rekindled by the discovery of activating mutations that may respond to targeted inhibition. Indeed, recent studies report that patients with MET amplification or MET exon 14 mutation can be sensitive to crizotinib6,12–15 and according to NCCN recommendations, capmatinib and crizotinib can be used in first line therapy or subsequent therapy for patients with MET exon 14 skipping mutation.
As recommended by the European Society for Medical Oncology (ESMO), as none of the respective targeted agents has regulatory approval, routine MET molecular testing is not advised.16 In France, the search for mutations in the exon 14 of MET is recommended as an option at the initial diagnosis and always before second-line treatment. Several studies have shown that MET exon 14 mutations represent a clinically unique molecular subtype of NSCLC, therefore making it necessary to include MET as part of larger testing panels performed either initially or when routine EGFR, ALK, and ROS1 testing is negative.8,17–22
Anti-MET TKIs (crizotinib, capmatinib, tepotinib, savolitinib) do not currently have marketing authorization in France. In the case of MET exon 14 mutations, a request for a temporary recommendation of crizotinib use is pending at the time of writing. This request does not apply to other MET alterations. It concerns treatment of adult patients with non-emergent locally advanced or metastatic small-cell bronchial cancers with a mutation of exon 14 of c-MET, after at least one treatment line based on a platinum dipole with or without immunotherapy. Capmatinib is available in nominative temporary use authorization for patients with advanced or metastatic non–small-cell lung cancer with a c-MET mutation affecting exon 14 who have already received first-line treatment and are not eligible for a clinical trial currently underway in France.
Thus, contrary to the American recommendations which authorize the use of capmatinib and crizotinib in the case of a mutation skipping exon 14 of METs, the French recommendations only authorize the use of these treatments if the mutation concerns exon 14. However, several different types of mutation in exon 14 of MET and its flanking introns can induce a jump in exon 14, activate the MET gene and thus be sensitive to anti-MET tyrosine kinase inhibitors.12
We report here the original case of a patient presenting a NSCLC with an intron 14 mutation and having a durable TKI response.
Case Report Presentation
This case concerns a 76-year-old Caucasian male with a medical history including idiopathic thrombocytopenic purpura, chronic myelomonocytic leukemia (CMML), atrial fibrillation, arterial hypertension, obesity (BMI 36kg/m2), and a 5–10 pack-per-year smoking history.
A left upper lobe pulmonary nodule of 12.4 mm was discovered in March 2019 following symptoms of coughing (Figure 1). A PET scan showed an hypermetabolism of the left upper lobe pulmonary nodule (SUVmax: 3.29) and an intense hypermetabolic focus in the left interlobular lymph node (SUVmax: 5.55) (Figure 2). The rest of the spreading assessment was negative.
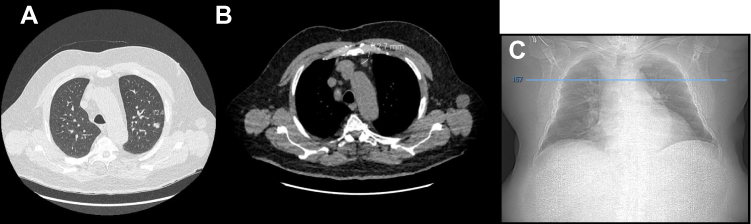 |
Figure 1 Chest CT scan of February 19, 2019 representing a left upper lobe pulmonary nodule of 12.4 mm in lung window (A) and a prevascular adenopathy in mediastinal window of 12.7 mm (B). Chest X-Ray with the cross-sectional height at which pulmonary nodule is located (C). |
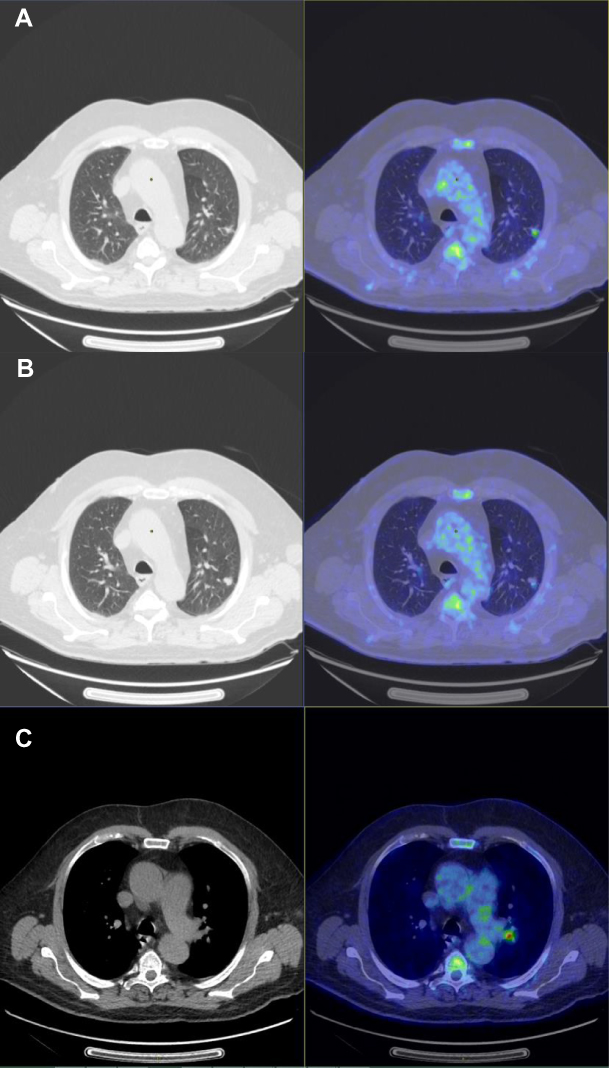 |
Figure 2 Pre-therapeutic TEP scan of March 19, 2019 representing a hypermetabolism of the left upper lobe pulmonary nodule (SUVmax: 3.29) (A and B); and an intense hypermetabolism focus in the left interlobular lymph node (SUVmax: 5.55) (C). |
A left upper lobectomy with mediastinal lymph node dissection by lateral thoracotomy was performed on April 3, 2019.23 Anatomopathological examination of the surgical specimen prompted the diagnosis of primary invasive pulmonary adenocarcinoma, associated with a sarcomatoid contingent with spindle cells (10%) of 1 cm, not infiltrating the visceral pleura, and complete exeresis, but with the presence of endolymphatic tumor emboli. PDL1 was 70%. Six metastatic lymph nodes and 9 lobar and peribronchial lymph nodes were present. No mediastinal lymph nodes were involved. The tumor was classified pT1aN1M0, stage IIB (TNM 8th edition).
The patient received adjuvant chemotherapy with carboplatin AUC 5 and Vinorelbine 25.00 mg/m2, for which he received four courses from June 6 to August 30, 2019, instead of taxol-carboplatin because he did not want an alopecia-causing treatment.24 At the end of the adjuvant treatment (September 2019), the patient was in complete remission for 5 months. In February 2020, the CT scan revealed a mediastinal lymph node progression (RECIST 1.1) with a 21 mm adenopathy in front of the aortic arch and a second one of 18 mm in the pretracheal space (Figure 3). At that time, a complementary molecular analysis by next-generation sequencing (NGS) was realized on the initial surgical specimen. A c.3082+3A>T mutation in the MET gene was identified with no EGFR mutation, ALK or Ros 1 translocation. This substitution is in the splice donor site of intron 14 and most likely causes a jump in exon 14. This mutation confers susceptibility to anti-MET tyrosine kinase inhibitors.25
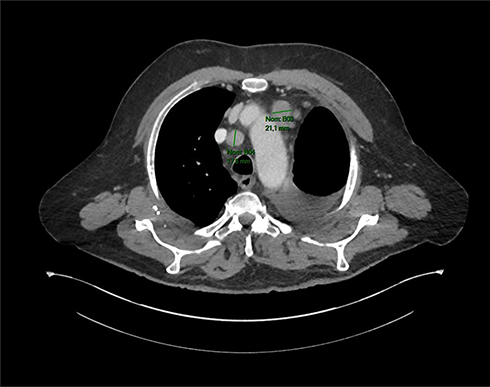 |
Figure 3 CT scan of February 5, 2020 revealing a mediastinal lymph node progression (RECIST 1.1) with a 21 mm adenopathy in front of the aortic arch and a second one of 18 mm in the pretracheal space. |
Based on his history of idiopathic thrombocytopenic purpura, his age, the COVID-19 pandemic and the presence of a mutation in MET intron 14, treatment with crizotinib was initiated with an initial dose of 250 mg/day for 15 days and then increased to 250 mg twice a day, in accordance with recommendations for use.
An early CT scan at six weeks (April 9) showed an excellent and almost complete response to mediastinal lymph node involvement, according to RECIST 1.1 (adenopathy of the aortic arch at 5 mm and of the pretracheal space at 5 mm) with a good clinical and hematological tolerance (Figure 4).
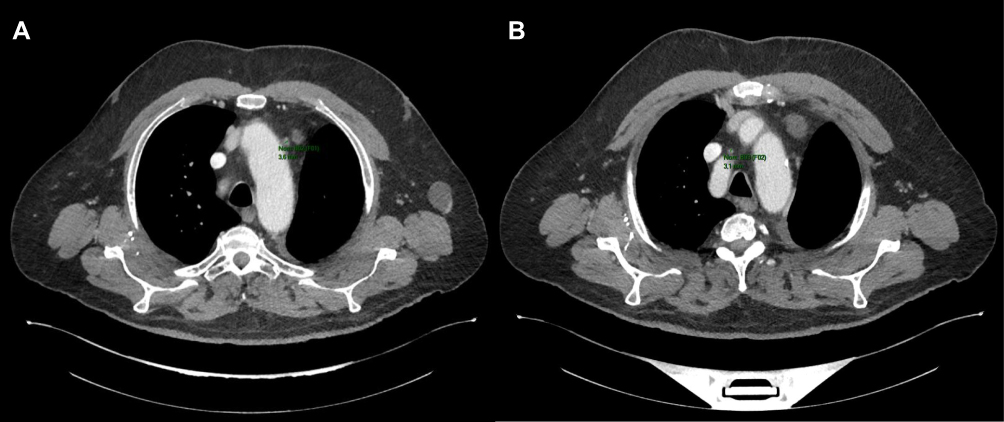 |
Figure 4 CT scan of June 11, 2020 showing a partial response aspect (RECIST 1.1) of two mediastinal lymph node involvements, the first in front of the aortic arch (A) and a second one of 18 mm in the pretracheal space (B). |
After an additional two months of treatment, the re-evaluation CT scan (June 2020) found a continuation of the partial response aspect (RECIST 1.1), while the initial target mediastinal adenopathies were no longer measurable and appeared partly calcified (3 mm in front of the aortic arch or prevascular upper mediastinal). In September 2020, after 7 months of treatment with crizotinib, the disease was still stable according to RECIST 1.1. However, at the end of September 2020, the patient developed anemia and deep thrombocytopenia, which led to the discontinuation of crizotinib. The myelogram revealed 70% of medullary blasts in favor of acutization of the CMML in acute myeloid leukemia (AML), excluding a toxicity of crizotinib. In December 2020, 3 months after discontinuation of crizotinib, the patient presented a lymph node and pleural progression (Figure 5).
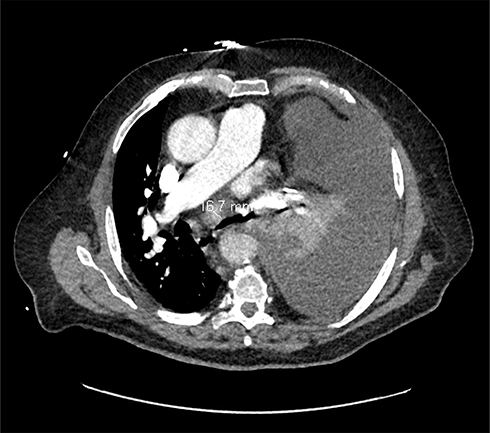 |
Figure 5 CT scan of December 28, 2020 representing a lymph node and pleural progression. |
To date, no case of transformation of CMML in acute leukemia on crizotinib has been described in the literature. Studies have shown the efficacy of crizotinib in cases of AML with ALK rearrangement/ALK expression, which in some cases resulted in a reduction in cell load until normal hematopoiesis was restored.26,27
Discussion
The new-generation sequencing used in our center to search for the recommended mutations in first-line metastatic recurrent pulmonary adenocarcinomas has allowed us to identify a molecular alteration of MET that is not accessible to targeted therapy according to the recommendations in force in our country (mutation c.3082+3A>T of intron 14) (Figure 6). However, the analysis of the literature shows that this substitution is in the splice donor site of intron 14 and most likely causes a jump in exon 14 and that this mutation confers susceptibility to anti-MET tyrosine kinase inhibitors.25 In a multidisciplinary consultation meeting, we therefore decided to initiate treatment with crizotinib with early re-evaluation in order to limit hematological toxicity in this elderly, fragile patient also presenting chronic myelomonocytic leukemia, particularly in this COVID-19 pandemic period when oral treatments were widely recommended.
 |
Figure 6 Schematic representation of a part of MET gene, including exon 14, its splicing site, and the site of the mutation. |
The MET gene encodes the receptor for hepatocyte growth factor (HGFR), a tyrosine kinase receptor for hepatocyte growth factor (HGF), present in epithelial cells. The binding of active HGF to the MET receptor induces activation of MET tyrosine kinase by dimerization and trans-phosphorylation of carboxy-terminal tail-clustered tyrosine residues of tyrosine kinase. Its activation results in downstream signaling pathways activation, including mitogen-activated protein kinase (MAPK), phosphoinositide-3 kinase (PI3K/AKT), and signal transducer and activator of transcription 3 (STAT3) pathway, which promote cell migration, proliferation, and survival.28–31 MET protein receptor stability and degradation are regulated by the intracellular juxtamembrane domain which is encoded in part by MET exon 14 and contains the tyrosine Y1003 residue that, when phosphorylated, serves as the binding site for the casitas B-lineage lymphoma (CBL) E3 ubiquitin ligase (Figure 7). CBL-mediated ubiquitination results in receptor internalization from the cell membrane to endocytic vesicles and subsequent proteasomal degradation (Figure 8).32
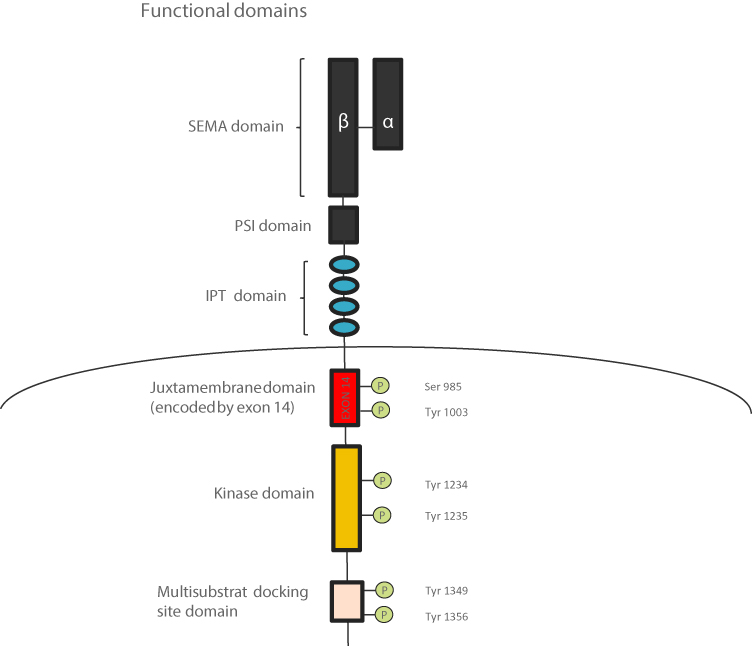 |
Figure 7 MET structure, consists of several domains. The extracellular part is composed by SEMA domain shared between the two chains, a PSI domain, and four IPT domains. The intracellular part is composed by the juxtamembrane domain encoded by exon 14, the kinase domain, and the multisubstrate docking site. Abbreviations: PSI, plexin-semaphorin-integrin; IPT, an immunoglobin-like fold shared by plexins and transcription factors. |
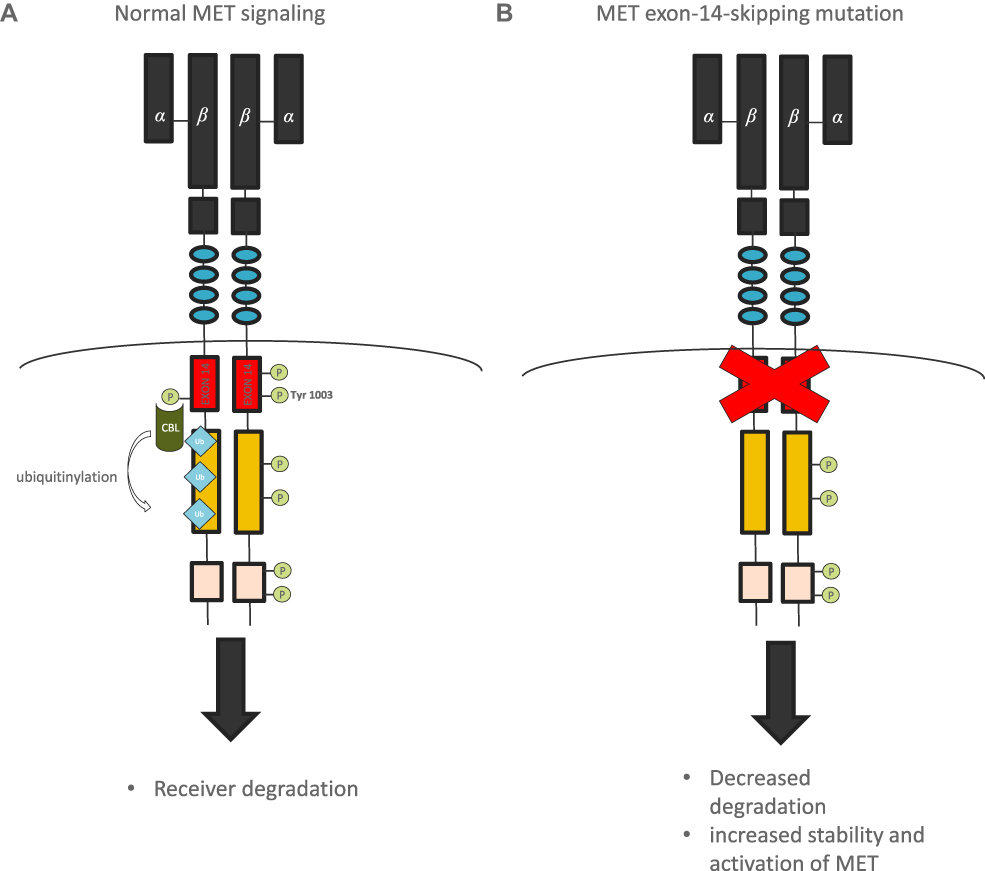 |
Figure 8 Mechanism of MET signaling regulation. Normal MET signaling (A) and abnormal signaling with MET-exon-14 skipping mutation (B). |
Different mechanisms of the MET gene activation leading to oncogenesis are as follows: an amplification of the MET gene resulting in high expression of the receptor, tyrosine kinase domain mutations resulting in constitutive activation of the receptor,33 and splicing mutations resulting in the skipping of exon 14 and loss of Y1003 during pre-mRNA splicing, resulting in loss of the casitas B-lineage lymphoma (CBL)-binding site and increased half-life of the MET receptor. In the absence of Y1003, CBL is unable to regulate ubiquitin-mediated degradation of the MET receptor by the lysosomes, leading to decreased turnover of MET and increased MET signaling that drives oncogenesis.32,34
Many somatic splice site alterations resulting in MET exon 14 skipping have been reported (more than 100). Mutations at the splice sites of MET exon 14 have been found in 2–3% of lung tumors, with a higher frequency for adenocarcinoma and sarcomatoid tumors. These mutations are very diverse, with numerous variants, including deletions, insertions, or point mutations.35 Although most of the exon-skipping mutations involve canonical splice sites, some are located further into the intronic sequence, up to 25 base pairs, adjacent to the splice acceptor sites. These mutations in exon splicing, especially the intronic noncoding region, can be difficult to interpret or may be missed by assays examining only exons and the immediately adjacent 50 and 30 splice acceptor and donor sites. Among the population of patients with a mutated MET exon 14, the prevalence of each type of mutation has been listed. Deletion of the acceptor splice site (polypyrimidine sequence or branching point) is involved in approximately 41% of mutations, deletion of donor splice site in approximately 11%, and point mutation of the donor splicing site in approximately 48%.6,35 This large degree of variation will have to be taken into account when designing clinical diagnostic sequencing assays to capture all possible activating MET mutations.12 Many MET inhibitors could be used in patients with a MET exon-14-skipping mutation.
According to the different combination of MET kinase domain structure, MET-TKIs can be divided into three types (type I, II, III), with type I inhibitors which can be subdivided into Ia and Ib.29,36 Type I inhibitors bind to the active conformation of the kinase in the ATP pocket; type II inhibitors bind to the inactive conformation of the kinase in the ATP pocket; and type III inhibitors are non-ATP-competitive allosteric inhibitors and bind outside the ATP pocket.
Recent reports demonstrated that patients with MET amplification or MET exon 14 mutation are sensitive to crizotinib.13,15 Crizotinib is a multi-tyrosine kinase inhibitor discovered through pharmacological screening. Among 120 human kinases, 13 kinases are inhibited by crizotinib, including c-MET and ALK, which shares 36% kinase domain sequence identity with c-MET.37
Crizotinib is used for the treatment of ALK- or ROS1-rearranged advanced NSCLCs.38,39 A type Ia inhibitor, it was the first MET tyrosine kinase inhibitor to be explored in MET-exon-14-altered NSCLCs and has shown its effectiveness in patients with MET amplification and MET exon 14 mutation.25 Drilon et al showed that among 65 patients with MET exon-14-altered NSCLCs treated with crizotinib, an objective response rate (ORR) was 32%, with a median progression-free survival (PFS) of 7.3 months, independent of the molecular heterogeneity or concurrent increased MET copy number.40
Recently, highly selective type Ib MET-TKI, more selective than crizotinib and potentially more potent in preclinical models, have been created, such as capmatinib,41 tepotinib,42 and savolitinib,43 which are under study in clinical trials.
Wolf et al showed in a recent Phase II GEOMETRY mono-1 study that the ORR with capmatinib was 67.9% and median PFS was 9.69 months among 28 patients with treatment-naïve disease; the overall response rate was 40.6% and median PFS was 5.42 months among 69 previously treated patients.41 Capmatinib has been approved by the Food and Drug Administration (FDA) for adult patients with a MET exon-14-skipping mutation. Paik et al showed in a single-arm phase II trial of 152 patients with MET exon-14 skipping mutations, the overall response rate with tepotinib was 46%, with a median response duration of 11.1 months.44 For these molecules, toxicity was acceptable, with essentially peripheral edema and nausea. While these are promising results, studies are still ongoing, and a longer follow-up is needed to know if these molecules are more effective than crizotinib. Other drugs such as cabozantinib45 and glesatinib,46 which are multi-targeted inhibitors (type II MET-TKI), are also being studied.
Nevertheless, patients showing initial benefits from these treatments invariably develop acquired resistance, leading to a disease progression. Drug resistance mechanisms have already been explored in diverse cases reports of patients with MET exon 14-altered lung cancer treated with MET-TKIs. The main mechanisms described were the decrease of mutation abundance and the change of MET mutation site after treatment with crizotinib.36 Guo et al (ASCO) tried to set out to identify potential resistance mechanisms among 74 lung cancer patients with stage IV MET exon 14 mutation treated with MET-TKI, of whom 91% received crizotinib at first-line treatment. Tumor samples underwent targeted NGS. This research showed that a lack of MET expression or RAS pathway activation is associated with poor MET-TKI outcomes in MET exon 14-altered lung cancers. On-target acquired resistance was found in less than 25% of patients and HGF amplification was found as a novel mechanism. Potential off-target acquired drug resistance mechanisms could be mediated by RAS/MDM2/EGFR pathway activation.47 Many studies suggest that switching from a type I MET-TKI (essentially crizotinib) to type II MET-TKI such as glesatinib or cabozantinib may overcome resistance mutations.36,48,49
Conclusion
In this case of pulmonary adenocarcinoma harboring intron 14 MET mutation, crizotinib was responsible for an effective therapeutic response, despite the transformation of LMMC in AML which is probably not linked with crizotinib based on the literature data. Several alterations in the exon or in the intron sequence including splicing sites can be involved in MET exon-14-skipping mutation, and thus leading to sensitivity to anti-MET tyrosine kinase inhibitors. Given their wide variability and complexity, mutations in MET splicing sites require the optimisation of molecular biology techniques. Targeted NGS-based assays interrogating MET as part of a larger gene panel should be preferred for screening purposes. The detection of mutations in MET exon 14 splice sites requires the development of specific techniques capable of identifying intronic alterations. For DNA-based tests, the test design must allow accurate and complete sequencing of exon 14 and its adjacent introns. The quantity of material (DNA and cells) can be limiting factors in detecting them. The search for an exon 14 jump by mRNA sequencing could be a solution, but currently its use in current practice is not possible given its cost and liability.
Abbreviations
ALK, anaplastic lymphoma kinase; CMML, chronic myelomonocytic leukemia; EGFR, epidermal growth factor receptor; NGS, Next-Generation Sequencing; MAPK, mitogen-activated protein kinase; NSCLC, non–small-cell lung cancer; PI3K, phosphoinositide-3 kinase; ROS1, c-ROS oncogene 1; STAT3, Signal transducer and activator of transcription 3.
Ethical Approval
Institutional approval was not required to publish the case details.
Consent for Publication
Written informed consent was obtained from the patient himself for publication of this case report.
Acknowledgments
The authors thank the patient for his participation and agreement to publication of the report.
Funding
No source of funding to declare.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gandhi L, Rodríguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378(22):2078–2092. doi:10.1056/NEJMoa1801005
2. Paz-Ares L, Luft A, Vicente D, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. 2018;379(21):2040–2051. doi:10.1056/NEJMoa1810865
3. Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi:10.1056/NEJMoa1606774
4. Couraud S, Souquet P-J, Paris C, et al. BioCAST/IFCT-1002: epidemiological and molecular features of lung cancer in never-smokers. Eur Respir J. 2015;45(5):1403–1414. doi:10.1183/09031936.00097214
5. Besse B, Adjei A, Baas P, et al. 2nd ESMO consensus conference on lung cancer: non-small-cell lung cancer first-line/second and further lines of treatment in advanced disease. Ann Oncol. 2014;25(8):1475–1484. doi:10.1093/annonc/mdu123
6. Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5(8):850–859. doi:10.1158/2159-8290.CD-15-0285
7. Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol. 2016;11(9):1493–1502. doi:10.1016/j.jtho.2016.06.004
8. Guo B, Cen H, Tan X, Liu W, Ke Q. Prognostic value of MET gene copy number and protein expression in patients with surgically resected non-small cell lung cancer: a meta-analysis of published literatures. PLoS One. 2014;9(6):e99399. doi:10.1371/journal.pone.0099399
9. Tong JH, Yeung SF, Chan AWH, et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin Cancer Res. 2016;22(12):3048–3056. doi:10.1158/1078-0432.CCR-15-2061
10. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. doi:10.1126/science.1141478
11. Spigel DR, Edelman MJ, O’Byrne K, et al. Results from the phase III randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIB or IV non-small-cell lung cancer: METlung. J Clin Oncol. 2017;35(4):412–420. doi:10.1200/JCO.2016.69.2160
12. Awad MM, Oxnard GR, Jackman DM, et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-met overexpression. J Clin Oncol. 2016;34(7):721–730. doi:10.1200/JCO.2015.63.4600
13. Paik PK, Drilon A, Fan P-D, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5(8):842–849. doi:10.1158/2159-8290.CD-14-1467
14. Heist RS, Shim HS, Gingipally S, et al. MET exon 14 skipping in non-small cell lung cancer. Oncologist. 2016;21(4):481–486. doi:10.1634/theoncologist.2015-0510
15. Liu X, Jia Y, Stoopler MB, et al. Next-generation sequencing of pulmonary sarcomatoid carcinoma reveals high frequency of actionable MET gene mutations. J Clin Oncol. 2016;34(8):794–802. doi:10.1200/JCO.2015.62.0674
16. Kerr KM, Bubendorf L, Edelman MJ, et al. Second ESMO consensus conference on lung cancer: pathology and molecular biomarkers for non-small-cell lung cancer. Ann Oncol. 2014;25(9):1681–1690. doi:10.1093/annonc/mdu145
17. Jin Y, Sun P-L, Kim H, et al. MET gene copy number gain is an independent poor prognostic marker in Korean stage I lung adenocarcinomas. Ann Surg Oncol. 2014;21(2):621–628. doi:10.1245/s10434-013-3355-1
18. Spigel DR, Ervin TJ, Ramlau RA, et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2013;31(32):4105–4114. doi:10.1200/JCO.2012.47.4189
19. Kowalczuk O, Kozlowski M, Niklinska W, Kisluk J, Niklinska BJ, Niklinski J. Increased MET gene copy number but not mRNA level predicts postoperative recurrence in patients with non-small cell lung cancer. Transl Oncol. 2014;7(5):605–612. doi:10.1016/j.tranon.2014.08.002
20. Jurmeister P, Lenze D, Berg E, et al. Parallel screening for ALK, MET and ROS1 alterations in non-small cell lung cancer with implications for daily routine testing. Lung Cancer. 2015;87(2):122–129. doi:10.1016/j.lungcan.2014.11.018
21. Noro R, Seike M, Zou F, et al. MET FISH-positive status predicts short progression-free survival and overall survival after gefitinib treatment in lung adenocarcinoma with EGFR mutation. BMC Cancer. 2015;15:31. doi:10.1186/s12885-015-1019-1
22. Weingertner N, Meyer N, Voegeli A-C, et al. Correlation between MET protein expression and MET gene copy number in a Caucasian cohort of non-small cell lung cancers according to the new IASLC/ATS/ERS classification. Pathology. 2015;47(4):320–328. doi:10.1097/PAT.0000000000000269
23. Howington JA, Blum MG, Chang AC, Balekian AA, Murthy SC. Treatment of stage I and II non-small cell lung cancer: diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(5Suppl):e278S–e313S. doi:10.1378/chest.12-2359
24. Pisters KMW, Evans WK, Azzoli CG, et al. Cancer care Ontario and American society of clinical oncology adjuvant chemotherapy and adjuvant radiation therapy for stages I-IIIA resectable non small-cell lung cancer guideline. J Clin Oncol. 2007;25(34):5506–5518. doi:10.1200/JCO.2007.14.1226
25. Lindeman NI, Cagle PT, Aisner DL, et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the college of American pathologists, the international association for the study of lung cancer, and the association for molecular pathology. Arch Pathol Lab Med. 2018;142(3):321–346. doi:10.5858/arpa.2017-0388-CP
26. Maesako Y, Okumura A, Takeoka K, et al. Reduction of leukemia cell burden and restoration of normal hematopoiesis at 3 months of crizotinib treatment in RAN-binding protein 2 (RANBP2)-anaplastic lymphoma kinase (ALK) acute myeloid leukemia. Leukemia. 2014;28(9):1935–1937. doi:10.1038/leu.2014.166
27. Maxson JE, Davare MA, Luty SB, et al. Therapeutically targetable ALK mutations in leukemia. Cancer Res. 2015;75(11):2146–2150. doi:10.1158/0008-5472.CAN-14-1576
28. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834–848. doi:10.1038/nrm3012
29. Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12(2):89–103. doi:10.1038/nrc3205
30. Sierra JR, Tsao M-S. c-MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol. 2011;3(1 Suppl):S21–35. doi:10.1177/1758834011422557
31. Feng Y, Thiagarajan PS, Ma PC. MET signaling: novel targeted inhibition and its clinical development in lung cancer. J Thorac Oncol. 2012;7(2):459–467. doi:10.1097/JTO.0b013e3182417e44
32. Recondo G, Che J, Jänne PA, Awad MM. Targeting MET dysregulation in cancer. Cancer Discov. 2020;10(7):922–934. doi:10.1158/2159-8290.CD-19-1446
33. Jeffers M, Schmidt L, Nakaigawa N, et al. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc Natl Acad Sci USA. 1997;94(21):11445–11450. doi:10.1073/pnas.94.21.11445
34. Ma PC. MET receptor juxtamembrane exon 14 alternative spliced variant: novel cancer genomic predictive biomarker. Cancer Discov. 2015;5(8):802–805. doi:10.1158/2159-8290.CD-15-0769
35. Baldacci S, Kherrouche Z, Descarpentries C, et al. [MET exon 14 splicing sites mutations: a new therapeutic opportunity in lung cancer]. Rev Mal Respir. 2018;35(8):796–812. French. doi:10.1016/j.rmr.2018.01.011
36. Huang C, Zou Q, Liu H, et al. Management of non-small cell lung cancer patients with MET exon 14 skipping mutations. Curr Treat Options Oncol. 2020;21(4):33. doi:10.1007/s11864-020-0723-5
37. Cui JJ, Tran-Dubé M, Shen H, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem. 2011;54(18):6342–6363. doi:10.1021/jm2007613
38. Shaw AT, Kim D-W, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. doi:10.1056/NEJMoa1214886
39. Oxnard GR, Binder A, Jänne PA. New targetable oncogenes in non–small-cell lung cancer. J Clin Oncol. 2013;31(8):1097–1104. doi:10.1200/JCO.2012.42.9829
40. Drilon A, Clark JW, Weiss J, et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat Med. 2020;26(1):47–51. doi:10.1038/s41591-019-0716-8
41. Wolf J, Seto T, Han J-Y, et al. Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): efficacy data from the phase II GEOMETRY Mono-1 Study. JCO. 2019;37(15_suppl):9004. doi:10.1200/JCO.2019.37.15_suppl.9004
42. Paik PK, Veillon R, Cortot AB, et al. Phase II study of tepotinib in NSCLC patients with METex14 mutations. J Clin Oncol. 2020. Available from: https://ascopubs.org/doi/abs/10.1200/JCO.2019.37.15_suppl.9005.
43. Lu S, Fang J, Li X, et al. Phase II study of savolitinib in patients (pts) with pulmonary sarcomatoid carcinoma (PSC) and other types of non-small cell lung cancer (NSCLC) harboring MET exon 14 skipping mutations (METex14+). JCO. 2020;38(15_suppl):9519. doi:10.1200/JCO.2020.38.15_suppl.9519
44. Paik PK, Felip E, Veillon R, et al. Tepotinib in non-small-cell lung cancer with MET exon 14 skipping mutations. N Engl J Med. 2020;383(10):931–943. doi:10.1056/NEJMoa2004407
45. Traslazionale FR. Phase II single arm study with CABozantinib in non-small cell lung cancer patients with MET deregulation. clinicaltrials.gov; 2019. Available from: https://clinicaltrials.gov/ct2/show/NCT03911193.
46. Mirati Therapeutics Inc. Phase 2, parallel-arm study of MGCD265 in patients with locally advanced or metastatic non-small cell lung cancer with activating genetic alterations in mesenchymal-epithelial transition factor. clinicaltrials.gov; 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT02544633.
47. Guo R, Offin M, Brannon AR, et al. MET inhibitor resistance in patients with MET exon 14-altered lung cancers. JCO. 2019;37(15_suppl):9006. doi:10.1200/JCO.2019.37.15_suppl.9006
48. Engstrom LD, Aranda R, Lee M, et al. Glesatinib exhibits antitumor activity in lung cancer models and patients harboring met exon 14 mutations and overcomes mutation-mediated resistance to type I MET inhibitors in nonclinical models. Clin Cancer Res. 2017;23(21):6661–6672. doi:10.1158/1078-0432.CCR-17-1192
49. Klempner SJ, Borghei A, Hakimian B, Ali SM, Ou S-HI. Intracranial activity of cabozantinib in MET exon 14-positive NSCLC with brain metastases. J Thorac Oncol. 2017;12(1):152–156. doi:10.1016/j.jtho.2016.09.127
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.