Back to Journals » Cancer Management and Research » Volume 11
DNMT3a promotes proliferation by activating the STAT3 signaling pathway and depressing apoptosis in pancreatic cancer
Authors Jing W ![]() , Song N, Liu YP, Qu XJ, Qi YF, Li C, Hou KZ, Che XF, Yang XH
, Song N, Liu YP, Qu XJ, Qi YF, Li C, Hou KZ, Che XF, Yang XH
Received 15 January 2019
Accepted for publication 9 May 2019
Published 10 July 2019 Volume 2019:11 Pages 6379—6396
DOI https://doi.org/10.2147/CMAR.S201610
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Harikrishna Nakshatri
Wei Jing,1 Na Song,2,3 Yun-Peng Liu,2,3 Xiu-Juan Qu,2,3 Ya-Fei Qi,4 Ce Li,2,3 Ke-Zuo Hou,2,3 Xiao-Fang Che,2,3 Xiang-Hong Yang4
1The First Department of Oncology, Shengjing Hospital of China Medical University, Shenyang, People’s Republic of China; 2Department of Medical Oncology, The First Hospital of China Medical University, Shenyang, People’s Republic of China; 3Key Laboratory of Anticancer Drugs and Biotherapy of Liaoning Province, The First Hospital of China Medical University, Shenyang, People’s Republic of China; 4Department of Pathology, Shengjing Hospital of China Medical University, Shenyang, People’s Republic of China
Background: Although aberrant DNA methyltransferase 3a (DNMT3a) expression is important to the tumorigenesis of pancreatic ductal adenocarcinoma (PDAC), the role of DNMT3a in PDAC prognosis is not clarified yet due to the limited studies and lacking of underlying molecular mechanism.
Methods: The expression of DNMT3a was examined by immunohistochemistry in PDAC tissues. Gene expression profiles assays were conducted to explore the impact of DNMT3a on biological processes and signal pathways. Cell cycle and apoptosis were measured by flow cytometry. Western blotting and real-time qPCR assays were used to explore the impact of DNMT3a on expression of protein and mRNA related to cell cycle, STAT3 signaling pathway and apoptosis.
Results: DNMT3a was overexpressed and closely associated with poor outcomes of PDAC. DNMT3a knockdown restrained PDAC cell proliferation, induced cell cycle arrest and promoted apoptosis in vitro. Affymetrix GeneChip Human Transcriptome Array identified that the cell cycle-related process was most significantly associated with DNMT3a. DNMT3a knockdown induced G1-S phase transition arrest by decreasing the expression of cyclin D1, which was mediated by the reduction of IL8 and the subsequent inactivation of STAT3 signaling pathway. Furthermore, exogenous apoptosis was also promoted after DNMT3a knockdown, probably via up-regulation of DNA transcription and expression in CASP8.
Conclusion: These findings indicate that DNMT3a plays an important role in PDAC progression. DNMT3a may serve as a prognostic biomarker and a therapeutic strategy candidate in PDAC.
Keywords: DNA methyltransferase 3a, pancreatic ductal adenocarcinoma, prognosis, proliferation
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a malignant tumor with high mortality, with nearly 80% of PDAC patients diagnosed as advanced stage.1,2 Despite many advances achieved in comprehensive treatment, the overall 5-year survival rate for PDAC is still approximately 5%.3 Due to the rapid distal metastasis and local recurrence after surgical resection, the 5-year survival rate is <20% even in the patients with early stage.4 The prognosis of PDAC is still far from optimistic.
The genetic background of pancreatic cancer is intricate and complicated.5,6 Although KRAS, CDKN2A, SMAD4, and TP53 are known as driver genes in PDAC, these genes could neither predict the potential prognosis, nor influence the treatment strategy in PDAC. Therefore, there is still an urgent need to find new candidates of driver gene in PDAC. A variety of genetic changes exist during the tumorigenesis of pancreatic cancer, including epigenetic changes and driver gene mutations.7 Aberrant DNA methylation is the most common epigenetic alteration in human cancers.8 DNA methylation is a covalent chemical modification, which adds a methyl group to carbon-5 of the cytosine ring in the CpG islands. DNA promoter hypermethylation, especially in tumor suppressor genes, results in transcription silence and facilitating the occurrence and development of tumors.9,10 Current investigations have demonstrated the important role of DNA hypermethylation in pancreatic tumorigenesis, whereas the reversals of DNA methylation lead to the reduction of pancreatic tumor burdens.11,12 Thus, DNA methylation alteration may be a valuable target for PDAC therapy.
DNA methyltransferases (DNMTs), including DNMT1, DNMT3a and DNMT3b, are known as the key cellular enzymes transferring the methyl group to 5ʹcytosine. DNMT1 keeps methylation patterns in newly-biosynthesized DNA during replication.13,14 However, DNMT3a is referred to as de novo methyltransferase for its ability to maintain the methylation in hemi-methylated and unmethylated CpG sites.14,15 Recent clinical pathological analyses have confirmed that the expression of three DNMTs increased in PDAC when compared with adjacent tissues, and the high expression of DNMTs are relevant to poor overall survival (OS) in PDAC patients.16,17 Zhang et al have proposed that DNMT1, DNMT3a, and DNMT3b were significant poor prognostic factors via analysis of 57 patients with PDAC. Multivariate analyses indicated that high expression of DNMT3b was a significant independent poor prognostic factor.17 Another multivariate analysis, from the research of 66 PDAC patients, reported that high expression of DNMT1, but not DNMT3a or DNMT3b, was a statistically significant independent poor prognostic factor. Further investigations have revealed that DNMT1 promotes the cell proliferation of pancreatic cancer by accelerating cell cycle progression and inhibiting apoptosis caused by BAX gene methylation.18 However, the underlying mechanisms of DNMT3a have not been revealed due to the limited participants and the lack of basic experimental research. It is still a great significance to clarify the pathological characteristics and the functions of DNMT3a in PDAC.
In the present study, we investigated the expression of DNMT3a, its correlation with clinical pathological parameters and prognosis in PDAC. Furthermore, we found that DNMT3a could promote the proliferation of PDAC cells by accelerating cell cycle progression and suppressing apoptosis, partially through the regulation of caspase-8 expression. The results of our study clarified the significant prognostic role of DNMT3a in PDAC and characterized the underlying molecular mechanisms, which may help to ameliorate the poor prognosis of PDAC patients.
Materials and methods
Patients and specimens
A total of 136 PDAC patients undergoing surgery between January 2009 and December 2012 were selected from Shengjing Hospital of China Medical University. Six patients were excluded because of death within 30 days after surgery. Prognostic data and pathological specimens were not available in 12 patients. The remaining 118 patients were finally included in the study. The study was approved by the Institutional Research Ethics Committee of Shengjing Hospital and conducted according to the Declaration of Helsinki. Medical records including clinical and pathological characteristics were gathered and organized. The pathological diagnoses of PDAC were confirmed by two pathologists. The clinical stage was defined according to the 7th Edition of American Joint Committee on Cancer (AJCC). The OS was defined as the interval from surgery to death from any cause. The final follow-up ended on January 20, 2016. The median follow-up time was 62.2 months.
Cell cultures
The human PDAC cells, PANC-1 and SW1990, were obtained from the Type Culture Collection of the Chinese Academy of Sciences. The cells were cultured in RPMI1640 medium (Gibco, Gaithersburg, MD, USA) supplemented with 100 U/mL penicillin–streptomycin and 10% FBS (Hao Yang Biological Technology Limited Liability, TianJin, China) at 5% CO2 and 37°C.
Immunohistochemistry
PDAC patients’ tumor sections (3 μm thick) were deparaffinized and rehydrated in a graded series of ethanol. The expressions of DNMT3a and Ki67 were detected by standard methods according to the S-P immunohistochemical kit (Fuzhou Maixin Biological Technology, Fuzhou, China). All sections incubated with Ki67 (working solution; Fuzhou Maixin Biological Technology) and anti-DNMT3a antibody (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA) were preserved overnight at 4°C. The immune complex was visualized using 3,30-diaminobenzidine tetrahydrochloride (DAB kit; Fuzhou Maixin Biological Technology). Nuclear staining was considered as a positive stain for DNMT3a and Ki67. The evaluation of Ki67 was based on the percentage of positive staining cells. The final Ki67 staining score index was graded as follows: 0–20%, weakly positive expression and >20%, strongly positive expression. The evaluation of DNMT3a was based on the intensity staining and percentage of positively stained cells. The intensity of DNMT3a staining was graded as follows: 0, no staining; 1, mild staining; 2, moderate staining; and 3, intense staining. The percentages of positive staining cells were scored as follows: 0, no staining of cells; 1, 1–25%; 2, 26–50%; 3, 51–75%; and 4, 76–100%. The final DNMT3a staining score index was graded as follows: 0–5, weakly positive expression and >=6, strong positive expression. Final scores were evaluated by two pathologists blinded to the clinical pathological data.
Transient transfection
Small interfering RNA (siRNA) sequences specifically targeting DNMT3a (1# siRNA: 5ʹ-GCGUCACACAGAAGCAUAUTT-3ʹ, 5ʹ-AU AUGCUUCUGUGUGACGCTT-3ʹ; 2# siRNA: 5ʹ-CGGCUCUUCUUUGAGUUCUTT-3ʹ, 5ʹ-AGAACUCAAAGAAGAGCCGTT-3ʹ), siRNA CASP8: 5ʹ-GAUCAGAAUUGAGGUCUUUtt-3ʹ, 5ʹ-AAAGACCUCAAUUCUGAUCtg-3ʹ and negative control siRNA (siRNA NC: 5ʹ-AATTCTCCGAACGTGTCACGT-3ʹ) were constructed by Gemma Pharmaceutical Technology (Shanghai, China). For transient transfection, PDAC cells were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and siRNA. Methods were performed according to the manufacturer’s instructions.
Gene expression profiles and data analysis
PANC-1 cells transfected with siRNA NC or 1# siRNA DNMT3a for 48 hrs were collected and washed in 4°C PBS three times, and dissolved in TRIzol for analysis. Affymetrix GeneChip Human Transcriptome Array analysis (Affymetrix, Santa Clara, CA, USA) was performed by Shanghai Oebiotech (Shanghai, China). Raw data were acquired using AGCC 4.0 (Affymetrix GeneChip Command Console, version 4.0; Affymetrix).
Western blot analysis
PANC-1 cells and SW1990 cells were collected and lysed in lysis buffer. The supernatant concentration was measured by the Coomassie Brilliant Blue G-250 method. The proteins were diluted in 3× SDS loading buffer, then boiled for 5 mins at 95°C. Equal amounts of protein were separated with 10% or 8% SDS-PAGE, then transferred to PVDF membranes. The PVDF membranes were blocked with 5% skim milk in TTBS buffer for 1 hr at room temperature, and then incubated overnight with the indicated antibodies at 4°C. The following day, after washing with TTBS buffer, the PVDF membranes were incubated with secondary antibodies for 30 mins at room temperature. The MicroChemi 4.2 Electrophoresis Gel Imaging Analysis System (DNR Bio-Imaging Systems, Jerusalem, Israel) was used to visualize the protein expression bands.
MTT assay
PANC-1 cells and SW1990 cells were trypsinized; then, 6×103 cells were plated into each well in 96-well plates with three replicate wells for each condition. After transfection, cells were cultured in 96-well plates for 48 or 72 hrs. Twenty μL MTT (5 mg/mL) was then added to each well, and incubated at 37°C for 4 hrs. After removal of the MTT mixture, 200 μL dimethyl sulfoxide was added to each well to dissolve the formazan crystals. A Model 550 microplate reader (Bio-Rad Laboratories, Hercules, CA, USA) was used to measure the optical density at 570 nm. Independent experiments were repeated three times.
Colony formation assay
The transfected PANC-1 cells and SW1990 cells were plated into 12-well plates (5×102 cells/well) and cultured in RPMI 1640 medium containing 10% FBS with three replicate wells for each condition. After incubation for 2 weeks, the supernatants were carefully removed. The colonies were fixed with 4% formaldehyde for 1 min; then, stained with 0.1% Giemsa stain for 40 mins. The colonies containing >50 cells were counted and photographed. Independent experiments were repeated three times.
Cell cycle analysis
After transfection with siRNA for 48 hrs, PANC-1 cells were collected and washed with cold PBS twice, then fixed with 4°C 70% ethanol overnight at 4°C. RNase A dissolved in PBS (100 μg/mL) was incubated with the cells for 30 mins at 37°C. Propidium iodide (PI; 5 mg/mL) was used for DNA staining. Each group of cells was incubated with 5 μL of PI for another 30 mins at 37°C in the absence of light. Cell cycle distribution was analyzed using a Becton Dickinson flow cytometer (BD Accuri C6 FACScan; BD Biosciences, San Jose, CA, USA) and ModFit LT software (http://www.vsh.com/products/mflt/index.asp), version 3.3. All experiments were repeated three times.
RNA extraction, reverse transcription (RT), and real-time quantitative PCR (qPCR)
According to the manufacturer’s instructions, total RNA from cell cultures was extracted using TRIzol reagent (Invitrogen). The cDNA was then synthesized from total RNA using the PrimeScript RT reagent kit with the gDNA Eraser (Takara, Shiga, Japan) method. Real-time qPCR was performed using an Applied Biosystems 7500 Real-Time PCR Systems (Thermo Fisher Scientific, Waltham, MA, USA) using the SYBR Premix EX TaqTM II (Tli RNaseH Plus; Takara). The 18S was treated as an internal control. Data were analyzed using the Applied Biosystems 7500 software program, version 2.3 (Applied Biosystems, Foster City, CA, USA) with the automatic Ct setting for adapting baseline and a threshold for Ct determinations. The relative expression levels of mRNA were calculated by the comparative cycle threshold (Ct) and 2−ΔΔCT method.
The sequences of the primers used were the following:
IL6-forward: 5ʹ-TCTCCACAAGCGCCTTCG-3ʹ;
IL6-reverse: 5ʹ-CTCAGGGCTGAGATGCCG-3ʹ;
IL8-forward: 5ʹ-CACCACTTTCTGGAGCAT-3ʹ;
IL8-reverse: 5ʹ-CATTGAGGAATAGGAGGG-3ʹ;
IL11-forward: 5ʹ-TCTCTCCTGGCGGACACG-3ʹ;
IL11-reverse: 5ʹ-AATCCAGGTTGTGGTCCCC-3ʹ;
18S-forward: 5ʹ-CCCGGGGAGGTAGTGACGAAAAAT-3ʹ;
18S-reverse: 5ʹ-CGCCCGCCCGCTCCCAAGAT-3ʹ.
The reaction was performed for 30 s at 95°C, followed by 45 cycles at 95°C for 5 s, 59°C for 34 s, 72°C for 30 s, with a final extension at 72°C for 30 s. Each reaction was performed in triplicate.
ELISA
The cells were trypsinized, and 3×105 cells were plated in each well in 6-well plates. After transfection with the negative-control siRNA, IL8 siRNA, or DNMT3a siRNA for 48 hrs, the supernatant was collected for the ELISA reaction. According to the manufacturer’s instructions, the ELISA reaction was conducted using the Human CXCL8/IL-8 DouSet ELISA kit (15 Plate, DY208) from R&D Systems (Minneapolis, MN, USA).
Apoptosis analysis
A total of 3×105 cells per well were plated into 6-well plates. After transfection with negative control siRNA, DNMT3a siRNA and CASP8 siRNA using Lipofectamine 2000 for 72 hrs, the cells were harvested and resuspended in 1× binding buffer containing Annexin V-FITC and PI according to the AnnexinV-FITC/PI Apoptosis Detection Kit instructions. The apoptotic cells were analyzed using a BD flow cytometry Accuri C6 (BD Biosciences). Independent experiments were repeated three times.
Co-immunoprecipitation
Ten μL of rabbit anti-FADD antibody or control IgG mixed with Protein A agarose beads (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) was used with 200 μg protein of the cell lysate for co-immunoprecipitation. A final volume of 400 μL of intermixture was gently rocked overnight at 4°C. The beads bound to target proteins and antibodies were centrifuged at 14,000 rpm for 1 min, then washed with lysis buffer four times. Sixty μL of sampling buffer was added, and boiled for 5 mins at 95°C. All samples were then subjected to Western blotting.
Statistical analysis
Data statistical analysis was conducted using SPSS statistical software for Windows, version 17.0 (SPSS, Chicago, IL, USA). All values are expressed as the mean ± standard error. Data of PDAC patients were compared with Fisher’s exact test or the chi-square test, as appropriate. The Kaplan–Meier method was used for survival data analysis, and the differences were tested using the log-rank test. The Cox proportional hazard model was used for multivariate analysis, and a forward stepwise method was used to include variables in the model. Student’s t-test (two-tailed) was used to evaluate the differences between two groups. Differences between the multiple groups were evaluated by one-way analysis of variance with a post-hoc LSD test. A value of P<0.05 was considered statistically significant.
Results
Overexpression of DNMT3a is associated with poor prognosis of PDAC patients
To identify the exact role of DNMT3a in the progression of PDAC, immunohistochemistry was performed to examine the expression levels of DNMT3a in human PDAC tissues. It was found that the positive rate of DNMT3a was 86.4% and the positive rate of Ki67 was 99.1%. DNMT3a significantly overexpressed in PDAC tissues compared with the adjacent normal tissues (Figure 1A). Furthermore, the relationship between the overexpression of DNMT3a and the clinicopathological status of PDAC patients was analyzed. As shown in Table 1, higher DNMT3a expression was significantly associated with TNM (P=0.019), worse differentiation (P=0.012), and a higher expression of Ki67 (P=0.001). However, there was no significant correlation between DNMT3a expression and other clinicopathological features, such as patient age, tumor size, vascular invasion, and perineural invasion.
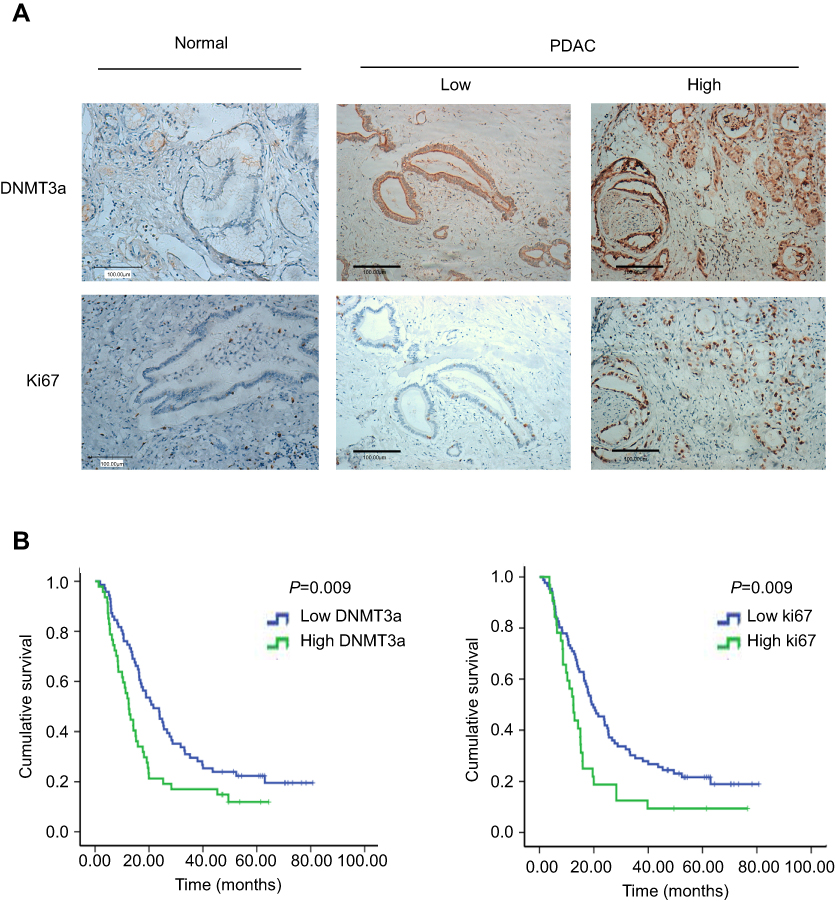 |
Figure 1 DNMT3a is elevated in PDAC patients and predicts poor prognosis. (A) Representative images of DNMT3a and Ki67 protein staining in normal pancreatic ductal epithelium and PDAC. DNMT3a was localized within the cytoplasm and nucleus, while Ki67 was localized within the nucleus only. Both DNMT3a and Ki67 were elevated in PDAC tumor cells. Scale bar: 100 μm. (B) Univariate Cox regression survival analysis and the log-rank test indicated high DNMT3a (left) and high Ki67 (right) expressions were associated with poor overall survival. Abbreviations: DNMT, DNA methyltransferase; PDAC, pancreatic ductal adenocarcinoma. |
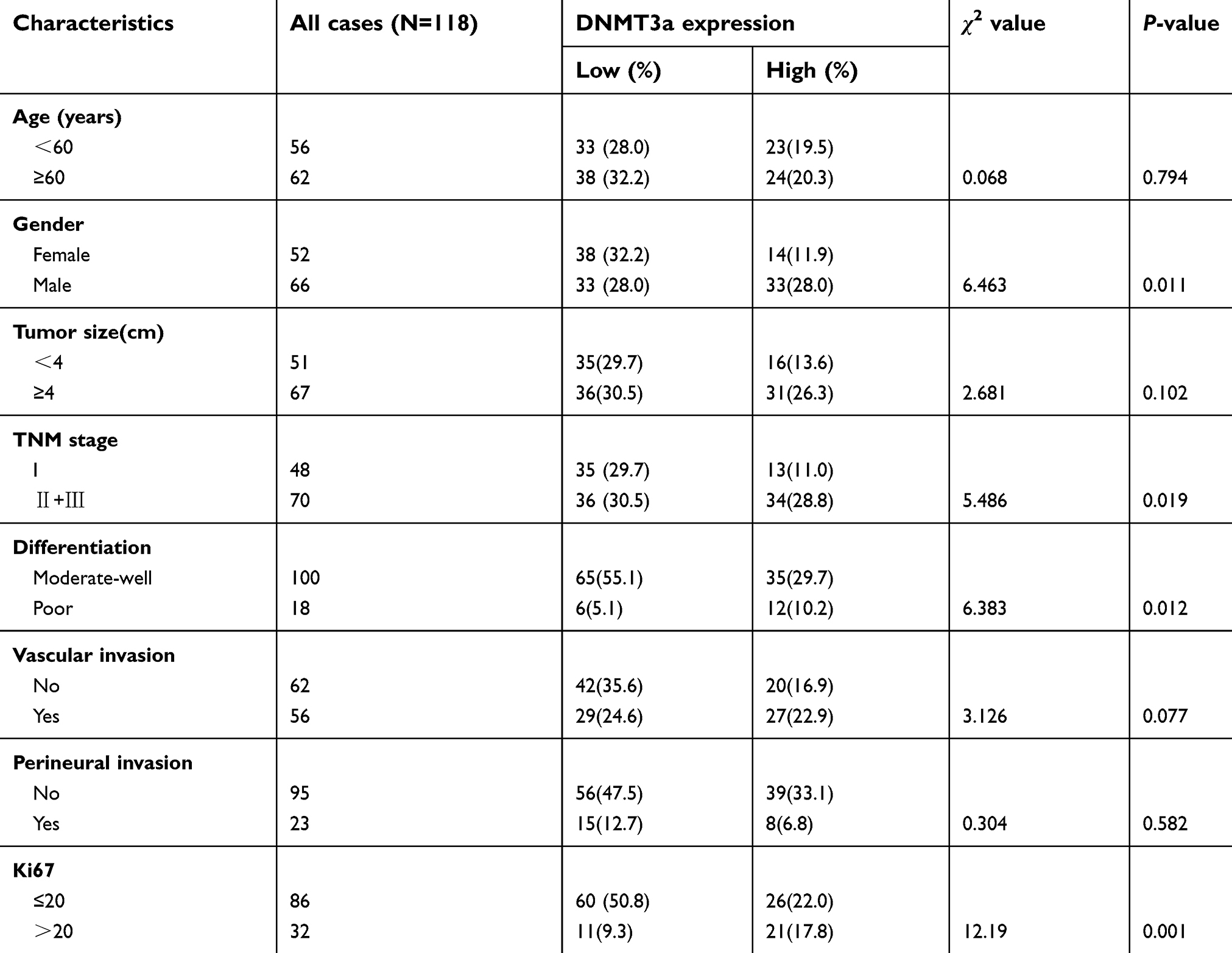 |
Table 1 Correlation between DNMT3a expression and clinicopathological characteristics in 118 cases of PDAC |
Kaplan-Meier analysis showed that PDAC patients with higher expression of DNMT3a had poor OS. The median survival time of high DNMT3a expression PDAC patients was 12.49 months, while the median survival time of PDAC patients with low DNMT3a expression was 21.60 months (P=0.009) (Figure 1B). As shown in Table 2, high expression of DNMT3a, larger tumor size, lymph node metastasis, poor tumor differentiation, high TNM staging, high serum CA199, and high expression of Ki67 were associated with poor prognosis. We fitted the seven factors above into multivariate Cox proportional hazard model analyses. The multivariate analyses identified excessive DNMT3a expression (P=0.005 HR=1.887; 95% CI=0.214–2.933) and high CA199 levels (P=0.004; HR=2.826; 95% CI=1.401–5.701) as independent prognostic factors for poor OS (Table 3). Taken together, these results suggested that excessive DNMT3a expression was an independent and promising prognostic factor in PDAC.
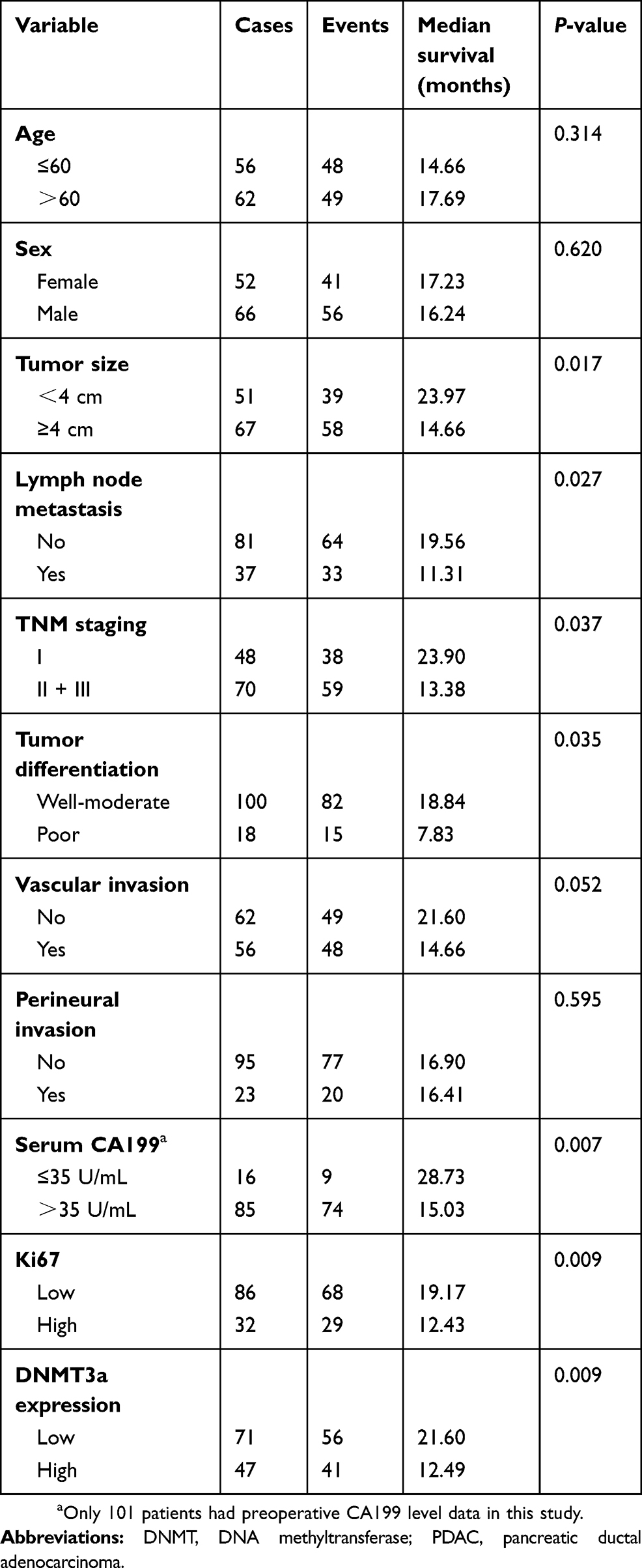 |
Table 2 Univariate survival analysis (Log-Rank) of the clinicopathological characteristics, DNMT3a and Ki67 in 118 cases of PDAC |
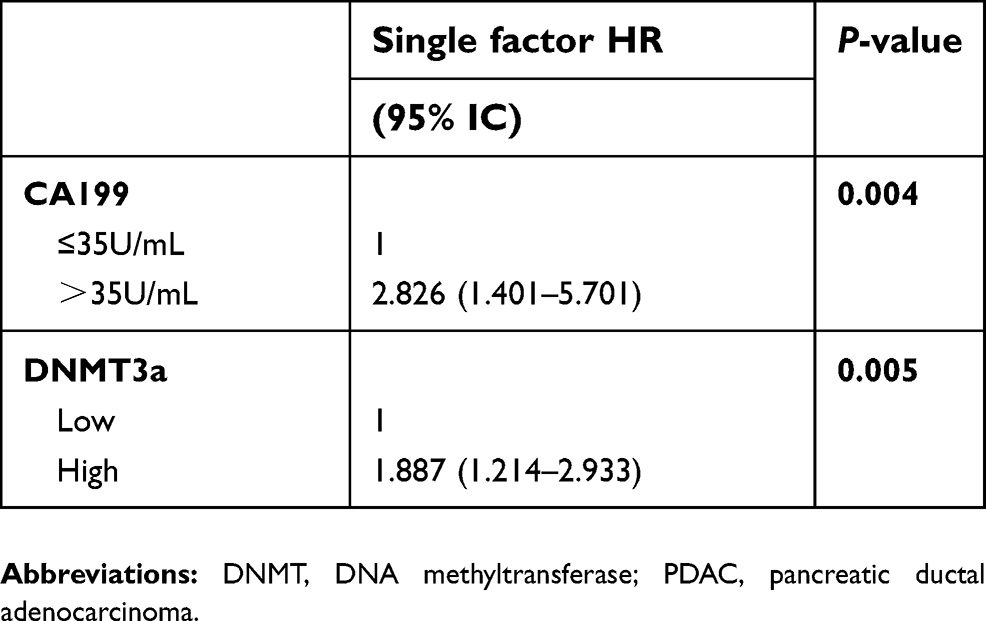 |
Table 3 Multivariate survival analysis (Cox regression) of the clinicopathological characteristics in 118 cases of PDAC |
DNMT3a promotes cell proliferation in PDAC cells
From the result that excessive DNMT3a expression was associated with the high expression of Ki67, which suggesting that DNMT3a could promote the proliferation in PDAC, we then investigated the potential role of DNMT3a in PDAC cells proliferation. The efficacy of DNMT3a knockdown was confirmed by Western blotting after 48 and 72 hrs transiently transfection with DNMT3a siRNA in PANC-1 cells and SW1990 cells (Figure 2A). Then, MTT and colony formation assays were performed to assess the effect of DNMT3a on cell proliferation. As shown in Figure 2B, the cell viability of 1# siRNA DNMT3a or 2# siRNA DNMT3a groups at 48 and 72 hrs was significantly declined compared with the siRNA NC group. Similarly, the colony formation abilities were also impaired in DNMT3a knockdown PANC-1 cells and SW1990 cells when compared with control groups (Figure 2C). These results indicated that DNMT3a played a key role in cell growth in PDAC.
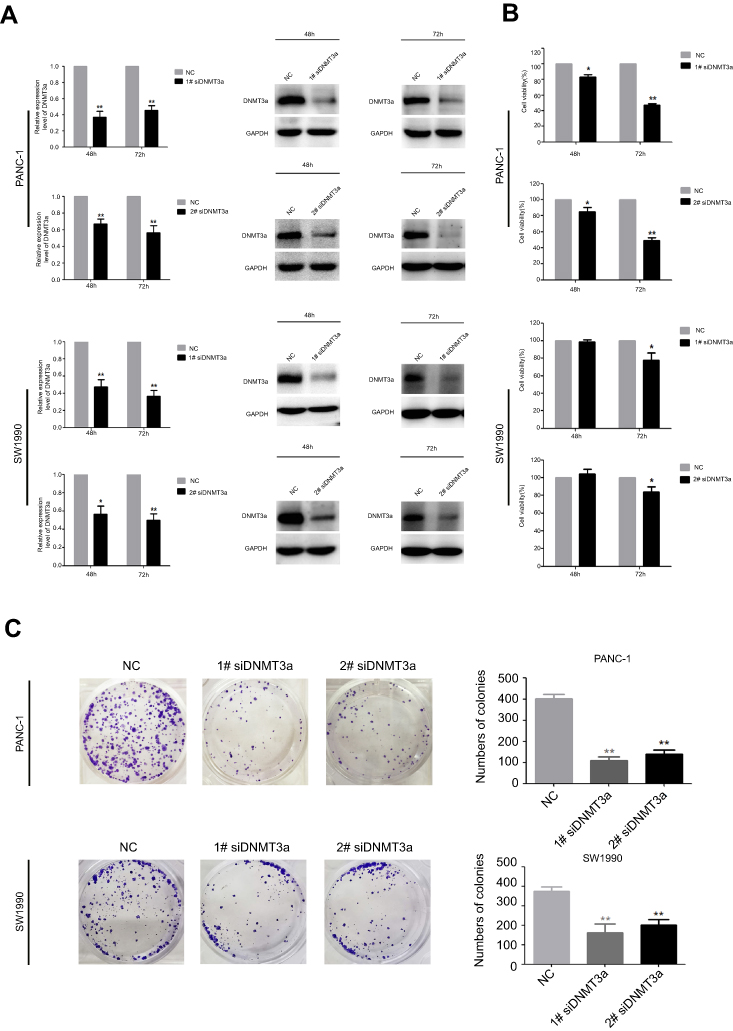 |
Figure 2 DNMT3a promotes the proliferation in PDAC cells. (A) PANC-1 and SW1990 cells were transfected with siRNA DNMT3a for 48 and 72 hrs. The expression levels of DNMT3a protein were confirmed by Western blotting. (B) The MTT assays showed that DNMT3a knockdown restrained proliferation of PDAC cells. (C) The colony formation ability of PANC-1 and SW1990 cells was significantly depressed after DNMT3a knockdown in vitro. *P<0.05 vs parental cells with siRNA NC treatment; **P<0.01 vs parental cells with siRNA NC treatment.Abbreviations: DNMT, DNA methyltransferase; PDAC, pancreatic ductal adenocarcinoma. |
DNMT3a down-regulation induces multiple biological processes inhibited in PANC-1 cells
To further insight into the effect of DNMT3a on biological processes and signal pathways in PDAC, Affymetrix GeneChip Human Transcriptome Array analysis was performed in DNMT3a knockdown PANC-1 cells. Among the affected biological processes, the cell cycle-related process was identified as most relevant to DNMT3a expression (Figure 3A). Other depressed biological processes also included DNA replication, the cytokine-mediated signaling pathway, the interferon-gamma-mediated signaling pathway, chromosome segregation, positive regulation of protein autophosphorylation, and negative regulation of the apoptotic process. As shown in Figure 3B, cell cycle-related signal pathways of KEGG pathway analysis were suppressed after DNMT3a knockdown. Overall, the results suggested that the cell cycle played a major role in DNMT3a-induced proliferation in PDAC cells.
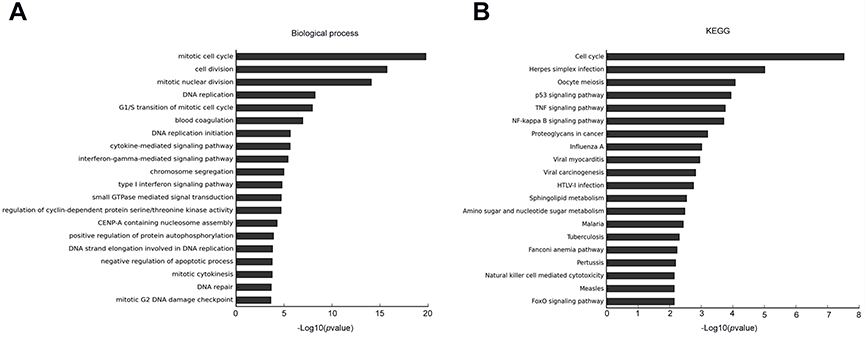 |
Figure 3 Knockdown of DNMT3a inhibits the key biological processes and signal pathway in PANC-1 cells. (A) DNMT3a down-regulation induced a mitotic cell cycle, G1/S transition of the mitotic cell cycle, and other multiple biological processes inhibited in PANC-1 cells. (B) DNMT3a down-regulation remarkably inhibited the cell cycle in PANC-1 cells according to KEGG analysis. PANC-1 cells transfected with 1# siRNA DNMT3a were chosen as an appropriate cellular model to test the effects of DNMT3a knockdown.Abbreviations: DNMT, DNA methyltransferase; PDAC, pancreatic ductal adenocarcinoma. |
DNMT3a accelerates cell cycle G1–S phase transition progression via IL8 by activating the STAT3 signaling pathway
To further explore the possible mechanism of DNMT3a in the regulation of PDAC cells proliferation, the distribution of cell cycle phases of PANC-1 cells was detected by flow cytometric analysis after DNMT3a transient knockdown of for 48 hrs. The percentage of G0/G1 phase cells increased and S phase cells decreased significantly in the PANC-1 cells after DNMT3a down-regulation compared with control cells (G0/G1 phase: 73.17%±0.90% vs 61.13%±1.25%, P=0.005; S phase: 14.68%±0.86% vs 24.33%±1.20%, P=0.0008, respectively) (Figure 4A and B). The expression of G1–S phase transition related-proteins, cyclin D1, p21 and p16, was decreased in DNMT3a knockdown PANC-1 cells, whereas no change observed in CDK4 (Figures 4C and S1). Moreover, the similar result was also obtained in SW1990 cells (Figure S2). According to the previous results of Affymetrix GeneChip Human Transcriptome Array analysis, the level of CCND1 mRNA down-regulated more than twofold in DNMT3a knockdown PANC-1 cells compared with control group. We speculated that down-regulation of transcription factors led to transcriptional inhibition in the CCND1 gene after DNMT3a transient knockdown of in PANC-1 cells. p-STAT3, as a transcription factor of CCND1, is widely activated in PDAC. The phosphorylated STAT3 (Y705) was decreased in PANC-1 cells with DNMT3a down-regulation (Figure 4D). The changes in mRNA levels of different cytokines (IL6, IL8, and IL11) were confirmed by PCR according to Affymetrix GeneChip Human Transcriptome Array analysis (Figure 4E). Significant reduction occurred in the transcription of the IL8 gene and the level of IL8 in the supernatant of PANC-1 cells treated with siRNA DNMT3a, while no changes were obtained in IL6 and IL11 (Figure 4F and G). The siRNA IL8 was used to transiently knockdown the expression of IL8. We found a notable decline of cyclin D1 and p-STAT3 (Y705) in IL8 knockdown PANC-1 cells (Figure 4H). STAT3 (Y705) phosphorylation elevated in DNMT3a knockdown PANC-1 cells after exogenous IL8 treatment (Figure S3).
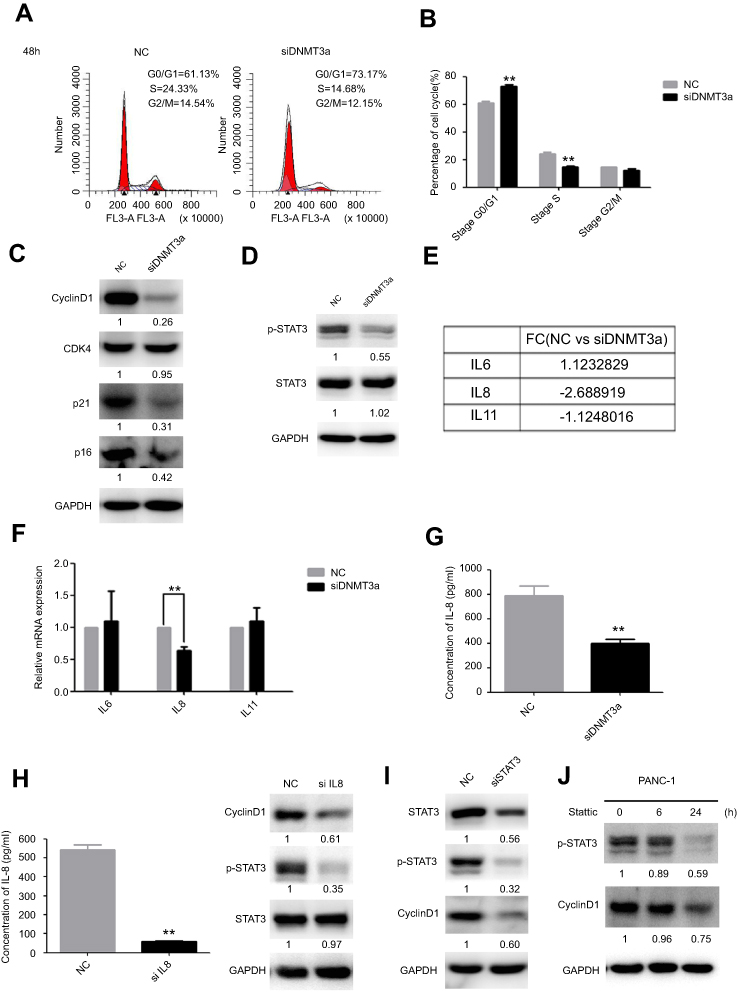 |
Figure 4 DNMT3a down-regulation inhibits cell cycle progression in PDAC cells. (A) The G1/S cell cycle block was detected by flow cytometry in PANC-1 cells after siRNA DNMT3a treatment for 48 hrs. (B) The related percentages of cells in G0/G1, S, and G2/M phase were depicted by a histogram, **P<0.01 vs parental cells with siRNA NC treatment. (C) Western blotting showed that the expression of cyclin D1, p16 and p21 were decreased after siRNA DNMT3a transfection, while there was no significant alteration in CDK4. (D) Western blotting also showed that there were significantly reduced expressions of p-STAT3 in DNMT3a knockdown PDAC cells. (E) The changes in mRNA levels of different cytokines (IL6, IL8, and IL11) according to Affymetrix GeneChip Human Transcriptome Array analysis. (F) Real-time qPCR measured the changes in mRNA levels of different cytokines (IL6, IL8, and IL11) in DNMT3a knockdown PANC-1 cells, while there was significant decrease in mRNA levels of IL8. (G) IL8 level decreased in DNMT3a knockdown PANC-1 cells supernatant. (H) The down-regulation of IL8 was confirmed by an ELISA. Western blotting showed that p-STAT3and cyclin D1were significantly decreased in PANC-1 cells treated with siRNA IL8, while there was no significant alteration in STAT3. (I) STAT3 knockdown was observed in PANC-1 cells treated with siRNA STAT3 for 48 hrs. Western blotting analysis showed that the expression of p-STAT3 and cyclin D1 decreased in PANC-1 cells treated with siRNA STAT3. (J) The expressions of p-STAT3 and cyclin D1 declined in PANC-1 cells after treatment with stattic, especially after stattic treatment for 24 hrs. **P<0.01 vs parental cells with siRNA NC treatment.Abbreviations: DNMT, DNA methyltransferase; PDAC, pancreatic ductal adenocarcinoma. |
Accompanied by the significant decrease of STAT3 and phosphorylation of STAT3 (Y705), the expression of cyclin D1 was also significantly reduced in STAT3 transiently knockdown PANC-1 cells (Figure 4I). Similar, expression of cyclin D1 was also significantly down-regulated after treatment of inactivator of STAT3 (stattic, 10 μM) for 6 and 24 hrs (Figure 4J).
Together, these results indicated that DNMT3a regulated proliferation of PDAC cells, partly through accelerating cell cycle G1–S phase transition progression by regulation of extracellular IL8 levels, which activating the STAT3 signal pathway and eventually inducing the expression of cyclin D1.
DNMT3a restrains cell apoptosis in PDAC cells
Next, the changes of apoptosis proportion were detected by flow cytometry in DNMT3a knockdown PANC-1 cells. As shown in Figure 5A, DNMT3a knockdown increased the cell apoptosis from 7.73%±0.64% to 20.60%±1.06% (P=0.0056) in PANC-1 cells (Figure 5B). In addition, the increase of cleavage of caspase-3, caspase-8, and PARP was also confirmed by Western blotting in siRNA DNMT3a treated PANC-1 cells (Figure 5C). These results indicated that DNMT3a could also inhibit apoptosis in PDAC cells.
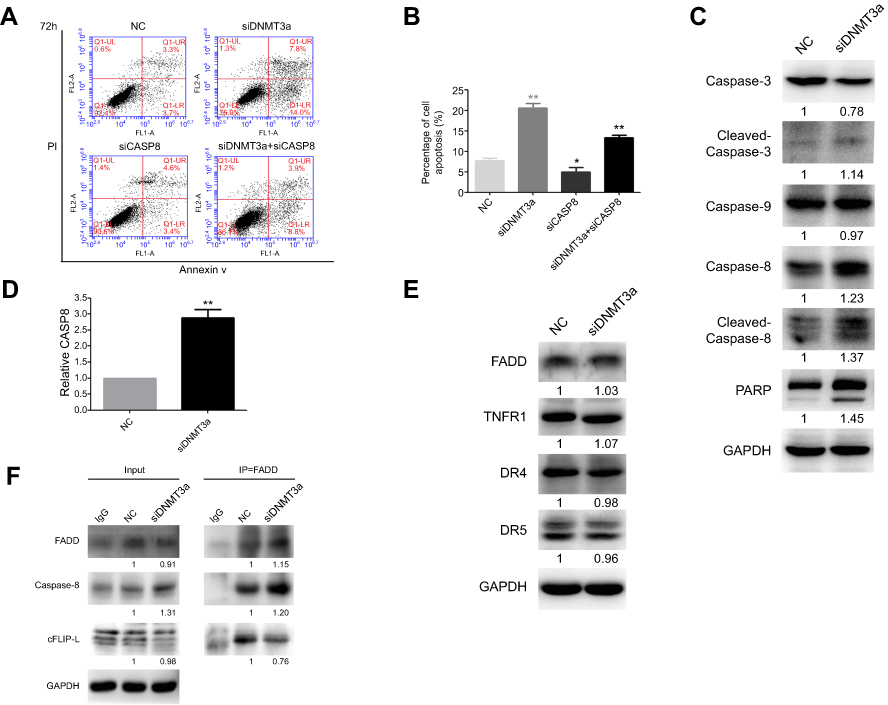 |
Figure 5 Knockdown of DNMT3a promotes apoptosis in PANC-1 cells. (A) The cell apoptosis was increased as detected by flow cytometry in PANC-1 cells after siRNA DNMT3a treatment for 72 hrs. The apoptosis was reduced in cells with both DNMT3a and caspase-8 knockdown. (B) The apoptosis rates of PANC-1 cells treated with siRNA DNMT3a and siRNA CASP8 (annexin V positive cells, %). *P<0.05 vs. parental cells with siRNA NC treatment, **P<0.01 vs cells with siRNA normal control NC treatment. (C) Western blotting analysis showed that the expression of apoptosis-related proteins (cleavage of caspase-3, caspase-8, cleavage of caspase-8, and PARP) was elevated in PANC-1 cells with siRNA DNMT3a transfection. (D) Real-time qPCR showed elevated mRNA levels of CASP8 in DNMT3a knockdown PANC-1 cells. |
According to the analysis result of above Affymetrix Gene Chip Human Transcriptome Array, the up-regulation of exogenous apoptosis-related gene, CASP8 in DNMT3a knocked-down PANC-1 cells aroused our concern, and was verified by real-time-PCR and Western blotting (Figure 5D). However, no changes of other exogenous apoptosis-related proteins and receptors, such as FADD, TNFR1, DR4 and DR5 were detected by Western blotting (Figure 5E). The apoptosis decreased after caspase-8 down-regulated in DNMT3a knockdown PANC-1 cells by siRNA CASP8 (Figures 5A, B and S4). To further reveal the mechanism of caspase-8 activation, co-immunoprecipitation was performed to identify formation of the DISC complex. The combination of FADD and caspase-8 was markedly increased, and the cFLIP-L combining with FADD decreased significantly (Figure 5F). Collectively, these results indicated that down-regulation of DNMT3a induced cell apoptosis partly via the exogenous apoptotic pathway in PDAC cells.
Discussion
DNMT3a is an active member of the DNMTs family, which is responsible for de novo methylation during embryogenesis and somatic tissue development.19 It has been demonstrated that DNMT3a is highly expressed in embryonic cells and down-regulated after differentiation in adult somatic tissues.20 Recent studies have shown that aberrant up-regulation of DNMT3a occurs in tumorigenesis, contributes to the tumor progression and refers as a poor prognostic factor in melanoma, hepatocellular carcinoma, gastric cancer, and PDAC.17,21–25 Zhang et al reported that the mRNA of DNMT3a increased in PDAC. Furthermore, the analysis indicated that although overexpression of DNMTs (DNMT1, DNMT3a, and DNMT3b) had strong value in poor prognosis, only DNMT3b was a significant independent poor prognostic factor.17 However, Gao et al reported that DNMT1 positive expression was considered as an independent poor prognostic factor, based on survival analysis in 66 PDAC patients.18 According to the limitations of previously analyzed cases, the actual role of DNMT3a in the prognosis of PDAC is still unclear.
In this study, we determined the significance and underlying mechanism of DNMT3a in PDAC through the analysis of 118 cases. Our results revealed that the overexpression of DNMT3a was significantly correlated with high TNM stages and poor differentiation, as well as an independent poor prognostic factor for OS, indicating that DNMT3a might be considered as a biomarker for PDAC prognosis. With the result that the expression of DNMT3a was positively correlated with Ki67, we speculated that DNMT3a was closely related to the proliferation in PDAC cells. Silencing DNMT3a did significantly inhibit the viability of PDAC cells, induce cell cycle arrest and apoptosis in vitro, suggesting that DNMT3a played an important role in proliferation of PDAC, and might be an important therapeutic target.
Previous studies reported that deficiency of DNMT3a remarkably inhibited the growth of melanomas in mouse melanoma models.22 DNMT3a depletion could induce reactivation of epigenetic silencing of the tumor suppressor gene PTEN, significantly inhibiting the cell proliferative ability in HCC cells.23 Recently, He et al reported that DNMT3a promoted gastric cancer cell growth by disrupting the G1/S checkpoint via methylation of the promoter of p18INK4C, which caused the down-regulation of p18INK4C.26 All these results indicated that DNMT3a facilitated tumor development. However, the relationship between DNMT3a and PDAC tumorigenesis is still uncertain. In this study, we found that the suppressed expression of DNMT3a impeded the G1/S transition by down-regulation of cyclin D1, and restrained the activation of the STAT3 pathway. According to previous studies, p-STAT3 interacted with a promoter of the cyclin D1 gene, and directly regulated transcription of cyclin D1.27,28 Therefore, cyclin D1 protein expression was positively correlated with STAT3 protein and p-STAT3 expressions.29,30 Subsequently, siRNA STAT3 and stattic were used to depress the activation of STAT3 in PDAC cells. We found that cyclin D1 declined due to decreasing p-STAT3 expression. These results suggested that one of the mechanisms of the G1–S phase arrest was the transcription inhibition of cyclin D1 via p-STAT3 (Y705) caused by DNMT3a knockdown. STAT3 is a member of the STAT family, whose activity is implicated in biological processes, including cell proliferation, differentiation, anti-apoptosis angiogenesis, and carcinogenesis.31 STAT3, as a transcription factor activated by phosphorylation of a tyrosine, translocates to the nucleus and initiates target genes transcription.32 Persistent phosphorylation of STAT3 has been reported to widely exist in multiple human cancers, including pancreatic cancer.33–35 Several proteins are known to inactivate STAT3 by dephosphorylation of STAT3 at its tyrosine or inhibition of the STAT3 DNA-binding activity, such as the protein inhibitors of activated STAT (PIAS), Src homology region 2 domain-containing phosphatase 1 (SHP-1), and the suppressors of cytokine signaling (SOCS).36–38 For the past 10 years, studies have revealed that constitutive activation of STAT3 is partly due to the decreased expression of negative regulators, which is caused by corresponding hypermethylation of CpG islands in regions in multiple tumors, including PDAC.39–41 Recently, Huang et al reported that up-regulation of DNMT1 induced SOCS3 methylation and negatively regulated SOCS3 expression.42 However, no changes of SHP-1 and SOCS3 were observed in DNMT3a knockdown PANC-1 cells (data not shown). We speculated that DNMT3a might have other activation mechanisms of the STAT3 pathway, which promoted proliferation in PDAC.
Based on our preliminary studies, DNMT3a down-regulation induced multiple biological processes inhibited in PANC-1 cells, including the cytokine-mediated signaling pathway. Our study found that the significant reduction of IL8 mRNA level and the expression level in supernatant was obtained in PANC-1 cells treated by DNMT3a siRNA. These results suggested that DNMT3a can further influence proliferation-related signal transduction by regulating the autocrine pathway of cytokines to the tumor microenvironment. O’Gorman et al reported that DNMT1 downregulation decreased IL-8 secretion in HCT116 intestinal epithelial cells via epigenetic promotion of IκBα expression, which negatively regulated the activation of NF-κB.43 However, whether DNMT3a has a similar regulatory mechanism of IL8 with DNMT1 in cancer, particularly in PDAC, is still unknown. Further investigation is needed to identify the target genes of DNMT3a related to IL8 expression to better characterize the latent mechanisms.
A decrease in apoptosis is considered as a mechanism of tumor progression. Aberrant methylation of apoptosis-associated genes leads to the abnormal expression of apoptosis-associated proteins and apoptosis depression. It was reported that zebularine, a DNMT inhibitor, induced apoptosis in lung cancer cells.44,45 In addition, Tan et al found that zebularine induced mitochondria-mediated apoptosis by upregulating Bax and activating caspase-3 in gastric cancer cells.46 Hypermethylation of the promoter of the CASP8 gene was found in HCC cells. Both knockdown of DNMT1 and DNMT3b-mediated demethylation of the CASP8 promoter induced up-regulation of caspase-8 and led to TRAIL-mediated apoptosis in HCC cells.47 Moreover, Gao et al reported that Bax mRNA was significantly increased in DNMT1- or DNMT3b-depleted PDAC cells due to DNA demethylation.18 Similar, another study reported that DNMT3a knockdown dramatically up-regulated 153 genes in HCC cells, including apoptosis-associated genes with CpG islands.23 These results suggested that DNMT3a might play an important role in the apoptosis of PDAC. In the present study, our results showed that DNMT3a knockdown induced cell apoptosis in PDAC cells. Caspase-8 and cleaved-caspase-8 notably increased, while there were no significant changes in FADD and exogenous apoptosis-related receptors in DNMT3a knockdown PANC-1 cells. In addition, knockdown of DNMT3a resulted in increased combination of FADD and caspase-8 in DISC, which induced the activation of caspase-8. We speculated that DNMT3a might regulate the expression of caspase-8 by DNA methylation in PDAC. Furthermore, the activation of caspase-8 was caused by the up-regulation in caspase-8 expression and the DISC complex, rather than the transmission of exogenous apoptotic signals.
Conclusion
In summary, our study showed that DNMT3a overexpressed in PDAC, and high expression of DNMT3a was closely associated with poorer outcomes of PDAC patients. Furthermore, we found that down-regulation of DNMT3a could suppress PDAC cell proliferation by inducing cell cycle arrest and apoptosis, suggesting it is a key regulator of PDAC proliferation. DNMT3a knockdown caused G1–S phase transition arrest by decreasing the expression of cyclin D1, the underlying mechanism included DNMT3a downregulation-mediated reduction of IL8 levels in the tumor microenvironment and the subsequent inactivation of STAT3 signaling pathway. Furthermore, exogenous apoptosis was induced in DNMT3a downregulation PDAC cells, probably by DNA demethylation in CASP8. However, it still needs to further elucidate that the molecular mechanisms of DNMT3a governing PDAC cell cycle progression and apoptosis. Taken together, these findings indicate the DNMT3a serve as an independent biomarker for PDAC prognosis, which may also impact the therapeutic strategy for PDAC patients.
Ethics approval and informed consent
The patients whose tissues were used for this research had provided written informed consent, and that this was conducted in accordance with the Declaration of Helsinki. Ethics approval was obtained from the Institutional Research Ethics Committee of Shengjing Hospital (Shenyang, China) for the use of pathological specimens and clinical data for research purposes (No. 2018PS162K).
Consent for publication
All authors have agreed for this publication.
Acknowledgments
This work was supported by National Science and Technology Major Project of the Ministry of Science and Technology of China (No. 2017ZX09304025), National Natural Science Foundation of China (No. 81773108), The Basic Scientific Research Projects of Liaoning Higher Education Institution (LQNK201711), Science and Technology Plan Project of Liaoning Province (No. 2015020457), The General Project of Liaoning Province Department of Education (No. LS201613), Science and Technology Plan Project of Liaoning Province (No. 2016007010), and The Key Research and Development Program of Shenyang (No. 17-230-9-01).
Author contributions
XHY, XFC and YPL contributed to the design of the study. WJ contributed to the acquisition of data, performance of experiments, analysis and interpretation of data and manuscript preparation. NS, KZH, CL and XJQ provided research materials and methods. XHY and YFQ conducted the pathological diagnoses of PDAC and the evaluation of immunohistochemistry. NS and XFC contributed to drafting the article and revising it critically for important intellectual content. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Dr Wei Jing, Prof Xiang-Hong Yang and Prof Xiao-Fang Che reports grants from the National Science and Technology Major Project of the Ministry of Science and Technology of China, National Natural Science Foundation of China, The Basic Scientific Research Projects of Liaoning Higher Education Institution, Science and Technology Plan Project of Liaoning Province, The General Project of Liaoning Province Department of Education, Science and Technology Plan Project of Liaoning Province and The Key Research and Development Program of Shenyang, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
2. Garrido-Laguna I, Hidalgo M. Pancreatic cancer: from state-of-the-art treatments to promising novel therapies. Nat Rev Clin Oncol. 2015;12(6):319–334. doi:10.1038/nrclinonc.2015.53
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
4. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. Jama. 2007;297(3):267–277. doi:10.1001/jama.297.3.267
5. Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi:10.1038/nature12477
6. Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501. doi:10.1038/nature14169
7. Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. doi:10.1038/nature11547
8. Luczak MW, Jagodzinski PP. The role of DNA methylation in cancer development. Folia Histochem Cytobiol. 2006;44(3):143–154.
9. Ma X, Wang YW, Zhang MQ, Gazdar AF. DNA methylation data analysis and its application to cancer research. Epigenomics. 2013;5(3):301–316. doi:10.2217/epi.13.26
10. Ghavifekr Fakhr M, Farshdousti Hagh M, Shanehbandi D, Baradaran B. DNA methylation pattern as important epigenetic criterion in cancer. Genet Res Int. 2013;2013:317569. doi:10.1155/2013/317569
11. Sato N, Fukushima N, Hruban RH, Goggins M. CpG island methylation profile of pancreatic intraepithelial neoplasia. Mod Pathol. 2008;21(3):238–244. doi:10.1038/modpathol.3800991
12. Oghamian S, Sodir NM, Bashir MU, et al. Reduction of pancreatic acinar cell tumor multiplicity in Dnmt1 hypomorphic mice. Carcinogenesis. 2011;32(6):829–835. doi:10.1093/carcin/bgr039
13. Miremadi A, Oestergaard MZ, Pharoah PD, Caldas C. Cancer genetics of epigenetic genes. Hum Mol Genet. 2007;16 Spec No 1:R28–49. doi:10.1093/hmg/ddm021
14. Ferguson-Smith AC, Greally JM. Epigenetics: perceptive enzymes. Nature. 2007;449(7159):148–149. doi:10.1038/449148a
15. Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19(3):219–220. doi:10.1038/890
16. Peng DF, Kanai Y, Sawada M, et al. Increased DNA methyltransferase 1 (DNMT1) protein expression in precancerous conditions and ductal carcinomas of the pancreas. Cancer Sci. 2005;96(7):403–408. doi:10.1111/j.1349-7006.2005.00071.x
17. Zhang JJ, Zhu Y, Zhu Y, et al. Association of increased DNA methyltransferase expression with carcinogenesis and poor prognosis in pancreatic ductal adenocarcinoma. Clin Transl Oncol. 2012;14(2):116–124. doi:10.1007/s12094-012-0770-x
18. Gao J, Wang L, Xu J, et al. Aberrant DNA methyltransferase expression in pancreatic ductal adenocarcinoma development and progression. J Exp Clin Canc Res. 2013;32:86. doi:10.1186/1756-9966-32-86
19. Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. 2001;20(24):3139–3155. doi:10.1038/sj.onc.1204341
20. Li H, Rauch T, Chen ZX, Szabo PE, Riggs AD, Pfeifer GP. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem. 2006;281(28):19489–19500. doi:10.1074/jbc.M513249200
21. Chen BF, Chan WY. The de novo DNA methyltransferase DNMT3A in development and cancer. Epigenetics. 2014;9(5):669–677. doi:10.4161/epi.28324
22. Deng T, Kuang Y, Wang L, Li J, Wang Z, Fei J. An essential role for DNA methyltransferase 3a in melanoma tumorigenesis. Biochem Biophys Res Commun. 2009;387(3):611–616. doi:10.1016/j.bbrc.2009.07.093
23. Zhao Z, Wu Q, Cheng J, Qiu X, Zhang J, Fan H. Depletion of DNMT3A suppressed cell proliferation and restored PTEN in hepatocellular carcinoma cell. J Biomed Biotechnol. 2010;2010:737535. doi:10.1155/2010/737535
24. Cao XY, Ma HX, Shang YH, et al. DNA methyltransferase3a expression is an independent poor prognostic indicator in gastric cancer. World J Gastroenterol. 2014;20(25):8201–8208. doi:10.3748/wjg.v20.i25.8201
25. He S, Wang F, Yang L, et al. Expression of DNMT1 and DNMT3a are regulated by GLI1 in human pancreatic cancer. PLoS One. 2011;6(11):e27684. doi:10.1371/journal.pone.0027684
26. Li Y, Nie Y, Tu S, et al. Epigenetically deregulated miR-200c is involved in a negative feedback loop with DNMT3a in gastric cancer cells. Oncol Rep. 2016;36(4):2108–2116. doi:10.3892/or.2016.4996
27. Leslie K, Lang C, Devgan G, et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 2006;66(5):2544–2552. doi:10.1158/0008-5472.CAN-05-2203
28. Liu LC, Su CH, Wang HC, et al. Contribution of personalized Cyclin D1 genotype to triple negative breast cancer risk. BioMedicine. 2014;4:3. doi:10.7603/s40681-014-0003-4
29. Zhang XM, Zhou C, Gu H, Yan L, Zhang GY. Correlation of RKIP, STAT3 and cyclin D1 expression in pathogenesis of gastric cancer. Int J Clin Exp Pathol. 2014;7(9):5902–5908.
30. Wu J, Lu WY, Cui LL. Clinical significance of STAT3 and MAPK phosphorylation, and the protein expression of cyclin D1 in skin squamous cell carcinoma tissues. Mol Med Rep. 2015;12(6):8129–8134. doi:10.3892/mmr.2015.4460
31. Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285(1–2):1–24.
32. Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282(28):20059–20063. doi:10.1074/jbc.R700016200
33. Jatiani SS, Baker SJ, Silverman LR, Reddy EP. Jak/STAT pathways in cytokine signaling and myeloproliferative disorders: approaches for targeted therapies. Genes Cancer. 2010;1(10):979–993. doi:10.1177/1947601910397187
34. Lili LN, Matyunina LV, Walker LD, Daneker GW, McDonald JF. Evidence for the importance of personalized molecular profiling in pancreatic cancer. Pancreas. 2014;43(2):198–211. doi:10.1097/MPA.0000000000000020
35. Zimmer S, Kahl P, Buhl TM, et al. Epidermal growth factor receptor mutations in non-small cell lung cancer influence downstream Akt, MAPK and Stat3 signaling. J Cancer Res Clin Oncol. 2009;135(5):723–730. doi:10.1007/s00432-008-0509-9
36. Chung CD, Liao J, Liu B, et al. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278(5344):1803–1805.
37. Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19(5):378–387. doi:10.1634/stemcells.19-5-378
38. Wang W, Liu L, Song X, et al. Crystal structure of human protein tyrosine phosphatase SHP-1 in the open conformation. J Cell Biochem. 2011;112(8):2062–2071. doi:10.1002/jcb.23125
39. Al-Jamal HA, Mat Jusoh SA, Hassan R, Johan MF. Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC Cancer. 2015;15:869. doi:10.1186/s12885-015-1584-3
40. Oshimo Y, Kuraoka K, Nakayama H, et al. Epigenetic inactivation of SOCS-1 by CpG island hypermethylation in human gastric carcinoma. Int J Cancer. 2004;112(6):1003–1009. doi:10.1002/ijc.20521
41. Wang J, Zhou H, Han Y, et al. SOCS3 methylation in synergy with Reg3A overexpression promotes cell growth in pancreatic cancer. J Mol Med. 2014;92(12):1257–1269. doi:10.1007/s00109-014-1184-8
42. Huang L, Hu B, Ni J, et al. Transcriptional repression of SOCS3 mediated by IL-6/STAT3 signaling via DNMT1 promotes pancreatic cancer growth and metastasis. J Exp Clin Canc Res. 2016;35:27. doi:10.1186/s13046-016-0301-7
43. O’Gorman A, Colleran A, Ryan A, Mann J, Egan LJ. Regulation of NF-kappaB responses by epigenetic suppression of IkappaBalpha expression in HCT116 intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2010;299(1):G96–G105. doi:10.1152/ajpgi.00460.2009
44. You BR, Park WH. Zebularine inhibits the growth of A549 lung cancer cells via cell cycle arrest and apoptosis. Mol Carcinog. 2014;53(11):847–857. doi:10.1002/mc.22042
45. You BR, Park WH. Zebularine-induced apoptosis in Calu-6 lung cancer cells is influenced by ROS and GSH level changes. Tumour Biol. 2013;34(2):1145–1153. doi:10.1007/s13277-013-0656-8
46. Tan W, Zhou W, Yu HG, Luo HS, Shen L. The DNA methyltransferase inhibitor zebularine induces mitochondria-mediated apoptosis in gastric cancer cells in vitro and in vivo. Biochem Biophys Res Commun. 2013;430(1):250–255. doi:10.1016/j.bbrc.2012.10.143
47. Kurita S, Higuchi H, Saito Y, et al. DNMT1 and DNMT3b silencing sensitizes human hepatoma cells to TRAIL-mediated apoptosis via up-regulation of TRAIL-R2/DR5 and caspase-8. Cancer Sci. 2010;101(6):1431–1439. doi:10.1111/j.1349-7006.2010.01565.x
Supplementary materials
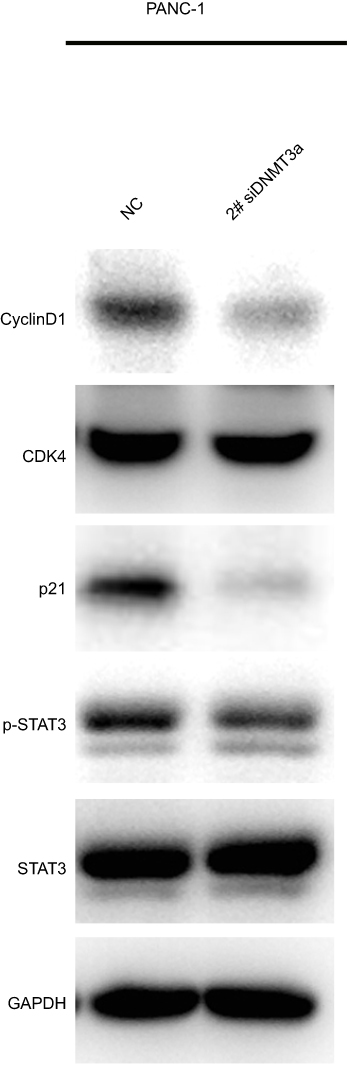 |
Figure S1 The expressions of cyclin D1 and p21 were suppressed in DNMT3a down-regulated PANC-1 cells with 2# si RNA DNMT3a. |
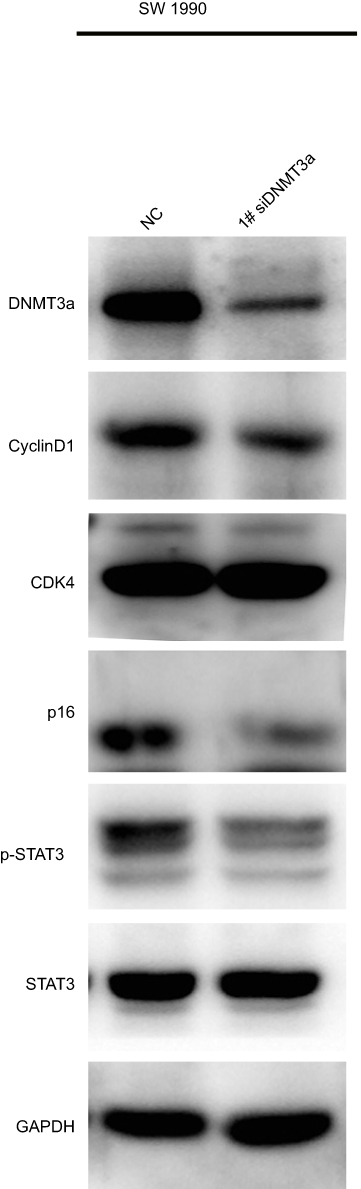 |
Figure S2 The expressions of cyclin D1 and p16 were also suppressed in DNMT3a down-regulated SW1990 cells with 1# si RNA DNMT3a. |
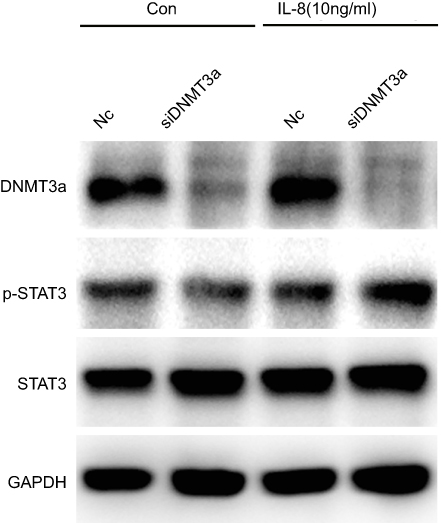 |
Figure S3 STAT3 (Y705) phosphorylation elevated in DNMT3a knockdown PANC-1 cells after IL8 (10 ng/mL) added in the media. |
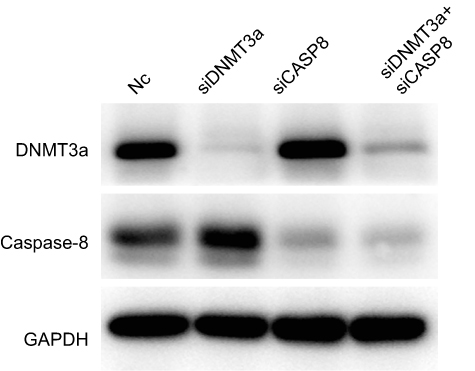 |
Figure S4 The efficacy of DNMT3a and caspase-8 knockdown was confirmed by Western blotting at 72 hrs. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.