Back to Journals » Journal of Pain Research » Volume 16
DNA Methylation Changes in Blood Cells of Fibromyalgia and Chronic Fatigue Syndrome Patients
Authors Przybylowicz PK, Sokolowska KE, Rola H ![]() , Wojdacz TK
, Wojdacz TK
Received 8 September 2023
Accepted for publication 13 November 2023
Published 30 November 2023 Volume 2023:16 Pages 4025—4036
DOI https://doi.org/10.2147/JPR.S439412
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Alaa Abd-Elsayed
Patrycja Kamila Przybylowicz, Katarzyna Ewa Sokolowska, Hubert Rola, Tomasz Kazimierz Wojdacz
Independent Clinical Epigenetics Laboratory, Pomeranian Medical University, Szczecin, Poland
Correspondence: Tomasz Kazimierz Wojdacz, Independent Clinical Epigenetics Laboratory, Pomeranian Medical University, Unii Lubelskiej 1, Szczecin, 71-252, Poland, Tel +48 91 44 17 201, Email [email protected]
Purpose: Fibromyalgia (FM) and Chronic Fatigue Syndrome (CFS) affect 0.4% and 1% of society, respectively, and the prevalence of these pain syndromes is increasing. To date, no strong association between these syndromes and the genetic background of affected individuals has been shown. Therefore, it is plausible that epigenetic changes might play a role in the development of these syndromes.
Patients and Methods: Three previous studies have attempted to elaborate the involvement of genome-wide methylation changes in blood cells in the development of fibromyalgia and chronic fatigue syndrome. These studies included 22 patients with fibromyalgia and 127 patients with CFS, and the results of the studies were largely discrepant. Contradicting results of those studies may be attributed to differences in the omics data analysis approaches used in each study. We reanalyzed the data collected in these studies using an updated and coherent data-analysis framework.
Results: Overall, the methylation changes that we observed overlapped with previous results only to some extent. However, the gene set enrichment analyses based on genes annotated to methylation changes identified in each of the analyzed datasets were surprisingly coherent and uniformly associated with the physiological processes that, when affected, may result in symptoms characteristic of fibromyalgia and chronic fatigue syndrome.
Conclusion: Methylomes of the blood cells of patients with FM and CFS in three independent studies have shown methylation changes that appear to be implicated in the pathogenesis of these syndromes.
Keywords: epigenetics, chronic pain, microarray studies, cytosine methylation
Introduction
Fibromyalgia (FM) and Chronic Fatigue Syndrome (CFS) are characterized by chronic pain, fatigue, and weakness. Patients with these symptoms also suffer from sleep abnormalities and report affected cognitive processes such as memory. The diagnosis of these two syndromes is challenging and is based on questionnaires that make the diagnosis rather difficult and prone to be subjective. Currently, the American College of Rheumatology (ACR) criteria are the most widely used in the diagnosis of FM1–4 and for CFS diagnosis the National Academy of Medicine (NAM)5 criteria, which recently replaced 1999 Fukuda and 2003 Canadian criteria,5,6 are the most widely adopted. Morbidity statistics show that FM affects 0.4–9.3% of people in different geographical regions7,8 and even 1% of the worldwide population may suffer from CFS.9 Women are three times more affected by each disease than men.4,9 The most frequent age of onset for FM is between 50 and 60 years,4 while CFS is most often diagnosed in two age groups: 10–19 and 30–39 years old.5 The treatment of both syndromes is challenging and depends on patient-specific symptoms such as post-exertional malaise, orthostatic intolerance, sleep issues, cognitive dysfunction, fatigue, immune dysfunction, pain, and gastrointestinal issues.5 As both diseases affect young and middle-aged people, and a large proportion of FM and CFS patients require continuous medical attention and are frequently unable to work, the management of FM and CFS presents a significant challenge for both healthcare systems and the labor market. To date, only weak evidence of the genetic background of FM and CFS has been reported in the literature. In the case of FM, the results of an observational study indicate a familiar aggregation of FM and odds ratio of FM in relatives of FM probands was found to be 8.5 (95% CI: 2.8–26).10 A more recent GWAS-based study estimated the heritability of FM in different age groups; however, the estimates differed significantly between the groups.11 Other studies on the genetic background of FM or chronic widespread musculoskeletal pain (CWP), which is a symptom of fibromyalgia, also suggested a genetic predisposition to this condition, but the results of these studies are largely discrepant.12,13
Similar to FM, a genetic contribution to CFS has been suggested by familial aggregation.14–16 Here, disease-associated genetic variants GRIK2 and NPAS217 have been reported, but these findings were not corroborated by large-scale GWASes.18,19
Considering rather weak evidence for a genetic predisposition to FM and CFS, it is plausible that aberrations in epigenetic mechanisms of gene expression regulation are involved in the development of these syndromes. Here, we reanalyzed available methylomics data for patients with FM and CFS from three independent studies using an updated and uniform bioinformatics data analysis framework. We found remarkable coherence of the physiological processes potentially affected by the identified methylation changes between those studies. Our results add to the body of evidence that epigenetic changes play a key role in the development of fibromyalgia and chronic fatigue syndromes.
Materials and Methods
Patients’ and Samples Characteristics
The results presented in this manuscript are based on data from published studies and were thus performed under appropriate ethical approval. The original datasets were obtained from the Gene Expression Omnibus database (Table 1). Specifically, the microarray-based genome-wide methylation profiling data included methylation profiles of blood cells from 22 women diagnosed with FM,20 profiles of T CD3+ cells from 15 male and 46 female CFS patients21 and profiles of peripheral blood mononuclear cells (PBMC) from 66 females with CFS.22,23 FM was diagnosed using the 2010 ACR criteria.2,3,20 Additionally, pain intensity and the effect of pain on daily activity patients were assessed using three questionnaires, as described in a previous study.24 CFS was diagnosed using the 1994 Fukuda and 2003 Canadian criteria, and the RAND-36 questionnaire was used to measure pain intensity and its impact on daily activity.21,22 The controls in the experiment were healthy counterparts of the cells used in specific methylation screening experiments and included methylation profiling data from 23 peripheral blood, 48 T CD3+, and 24 PBMC cells.
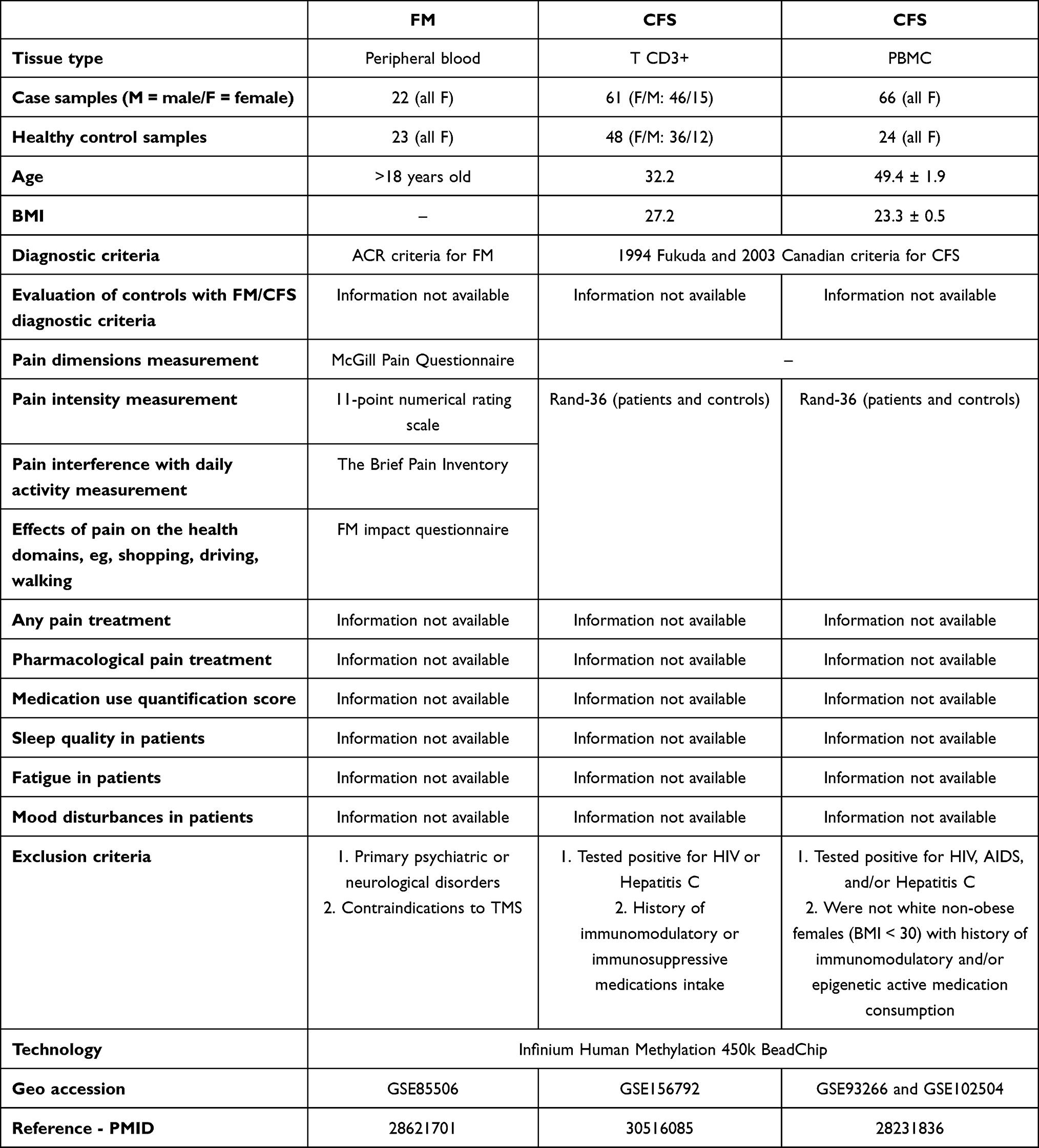 |
Table 1 Detailed Description of the Patient Cohorts Used in the Study |
Unified Genome-Wide Methylation Analysis
The main goal of our study was to analyze the Infinium HumanMethylation 450 K BeadChip (450 K, Illumina Inc.) methylation profiling data from three independent studies with one coherent and up-to-date bioinformatics data analysis framework that would allow us to compare the results across those studies. Briefly, raw data were processed using the ChAMP package25,26 and normalized using the BMIQ method. Subsequently, we used ComBat to correct for the batch effect in all datasets; this procedure was not used previously in the data analysis of data from reference.20 Correction of the cell composition of individual samples has become a standard procedure for studies based on blood samples.27,28 In two of the analyzed in our study datasets,21,22 cell-type proportion correction was not used or was used as a covariate,22 which has been shown to be less precise than cell fraction correction according to a previous study.29 We adjusted all our analyses for cell fraction differences with the EpiDISH R package modified as described by Bińkowski et al30 with reference restricted only to cell types present in individual samples. We then used linear regression (function in ChAMP) to identify differentially methylated probes (DMPs) between cases and controls. In the gene GSEA and enrichment analyses, we considered only DMPs displaying more than 0.05 absolute mean β-value difference between cases and controls, with adjusted p-value (Benjamini-Hochberg) of less than 5%. All analyses were performed using R 4.1.2.
Enrichment of DMPs in Genomic Regulatory Regions
Statistically significant enrichment or depletion of methylation changes in specific functional genomic regions is likely to suggest the function of these changes. Thus, we analyzed the distribution of DMPs identified with our data analysis frame work in regions related to genes as defined in the Infinium Human Methylation 450 Bead Chip manifest v. 1.2, including TSS1500, TSS200, 5’UTR, 1stExon, Gene body, 3’UTR, and Unknown regions as well as genomic regions related to CpG islands (CGI), including N-shelf, N-shore, Island, S-shore, S-shelf, and Opensea (both according to UCSC coordinates). We also assessed the distribution of identified methylation changes within regions defined in the Ensembl database,31 including Promoters, Promoter Flanking Region, TF binding site, open chromatin, CTCF binding site, Enhancer and Unknown regions. The background was set as the region in which all the processed CpGs were annotated.
Gene Set Enrichment Analysis
To approximate physiological processes potentially affected by identified methylation changes, we performed the Gene Set Enrichment Analysis (GSEA) using the “GENE2FUNC” function of Functional Mapping and Annotation of Genome-Wide Association Studies platform (FUMA GWAS).32
UCSC Genome Browser Analysis
The functional context of the loci that displayed identical methylation changes in all analyzed datasets was assessed using UCSC Genes,33 Open Regulatory Annotation (ORegAnno),34 GeneHancer,35 and UCSC CpG Islands track36 from the UCSC Genome Browser on Human (GRCh37/hg19) (https://genome.ucsc.edu/).
Results
FM and CFS Specific Methylation Changes
Overall, our analysis identified 1256 DMPs (hypomethylated: 940,74.84%; hypermethylated: 316, 25.16%) in peripheral blood from FM patients, 510 DMPs (108, 21.18% hypomethylated and 402, 78.82% hypermethylated) in T CD3+ cells from CFS patients, and 1751 DMPs (947 CpGs, 54.08% hypomethylated and 804, 45.92% hypermethylated) in PBMCs of CFS patients (the list of the identified DMPs with annotations are shown in Supplementary Tables 1–3). The unsupervised hierarchical clustering based on DMPs that we identified for each dataset showed a clear separation between patients and controls (Figure 1a–c), indicating that, although methylation changes observed in both syndromes were small (between 5% and 10%) (Supplementary Figure 1), all patients in each of the studies displayed these changes. The identified methylation changes overlapped only to a certain extent with previously identified methylation changes in each of the datasets we used. In patients with FM, 16.55% of the methylation changes that we identified with the new analysis framework were those previously identified and for the CFS study based on T CD3+ and CFS based on PBMC cells, common changes accounted to 10.1% and only 1.01%, respectively. The magnitude of the changes was also different between each of these analyses, with the largest absolute methylation changes observed in the analysis of PBMCs from CFS patients (12.16% of identified methylation changes were higher than 10%), significantly smaller in the case of peripheral blood from FM patients (1.75% of methylation changes higher than 10%) and T CD3+ from CFS cases (7.25% of the identified changes over 10%). This may indicate that different cell types harbor distinct methylation changes. Nevertheless, regardless of the analyzed cell type, the genes annotated to the top hits in each of the analyses were those with described functions in neurological processes (Supplementary Table 4).
 |
Figure 1 Heatmaps based on of beta values at identified DMPs, illustrating unsupervised hierarchical clustering of samples from each of the analyzed cohorts. Rows and columns of each heatmap indicate specific DMPs and samples, respectively. The color on heatmap indicates the level of methylation at specific CpG sites and samples (from blue: lower to red: higher). This analysis demonstrates clear separation of case (darker color) and control groups (lighter color) in all cohorts, including: (a) FM peripheral blood (green), (b) CFS T CD3+ (violet), and (c) CFS PBMC cohort (Orange). |
FM and CFS Specific Methylation Changes Affect Regions Indirectly Involved in Transcription Regulation
Next, to approximate physiological function of the identified methylation changes, we analyzed the enrichment of these changes in specific genomic regulatory regions (Figure 2).
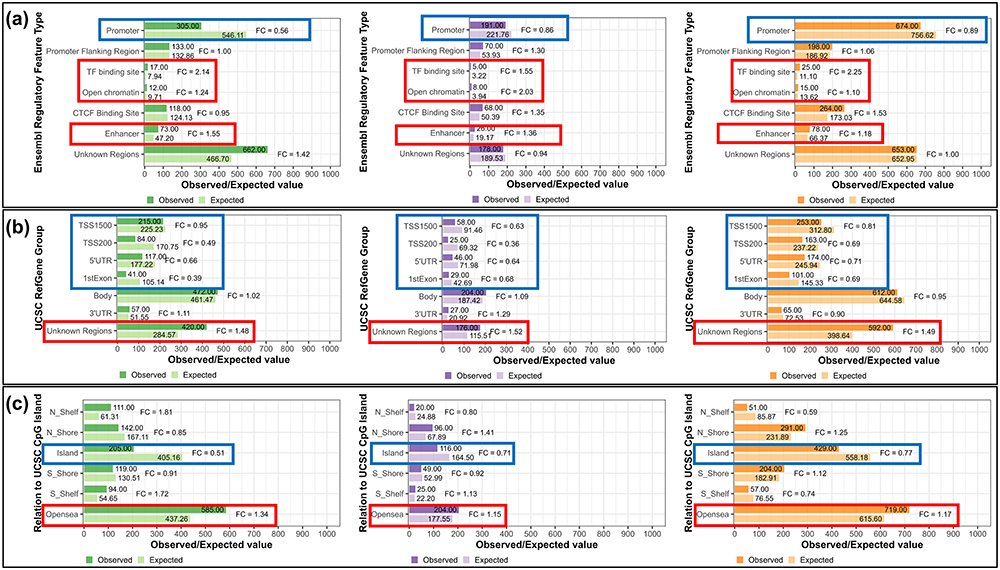 |
Figure 2 The results of the enrichment analysis of DMPs identified in each of the analyzed cohorts within genomic regions, including (a) Ensembl regulatory regions, (b) Gene elements (UCSC), and (c) UCSC CpG Island related elements. Green, violet, and Orange color indicate results for FM peripheral blood, CFS T CD3+ and CFS PBMC cohort, respectively. In each of the figures, the lighter bar illustrates expected number of CpG sites within assessed region (with informative probes obtained after data processing selected as a background in analyzed cohorts: FM peripheral blood: 411,150, CFS T CD3+: 411,537, CFS PBMC: 404,545) and the darker bar shows the observed number of DMPs that were annotated to specific regions. The red box indicates enrichment, whereas the blue box indicates depletion of DMPs within a specific region. This analysis revealed that in each of the analyzed cohorts methylation changes occur predominantly within regions not directly involved in gene expression regulation ((a) – TF binding site, Open chromatin and Enhancer, (b) – Unknown regions, (c) – Opensea) and that they are depleted within promoter-associated regions ((a) – Promoter, (b) – TSS1500, TSS200, 5’UTR and 1stExon, (c) – Island). |
According to the Ensembl regions, DMPs identified in each of the analyses were enriched in TF binding sites, open chromatin, and enhancers (Figure 2a, red boxes) and were depleted in promoters (Figure 2a, blue box). The direction of enrichment was not consistent between datasets for the Promoter Flanking Region, and CTCF Binding Site (Figure 2a). Similarly, analysis of UCSC gene regions showed that identified DMPs were consistently enriched in unknown regions (Figure 2b, red box) and depleted in TSS1500, TSS200, 5’UTR, and 1st Exon (Figure 2b, blue box). We also analyzed the enrichment of the identified DMPs within regions in relation to CpG Islands, and as expected, CpG islands (Figure 2c, blue box) were depleted of these changes. The N_Shelf, N_Shore, S_Shore, and S_Shelf regions have different directions of the enrichment throughout three datasets (Figure 2c) and we also observed enrichment of identified DMPs in OpenSea (Figure 2c, red box).
Overall, the analysis of genomic annotation of the identified methylation changes suggests uniformly across all analyzed patient groups that those changes do not affect the promoters of genes, but rather affect regions that do not directly regulate the transcription of genes.
Methylation Changes in FM and CFS Appear to Affect Similar Biological Processes
As already mentioned, the analysis of each dataset included in our study resulted in different numbers of DMPs, and only four of the identified methylation changes were common across all analyses. Interestingly, however, when we performed GSEA based on the genes to which the identified methylation changes were annotated, the physiological processes associated with these methylation changes were remarkably coherent between all analyzed datasets (Supplementary Table 5).
Analysis of the association of the identified gene set signatures with tissue-specific gene expression profiles showed that genes annotated to methylation changes identified in both CFS patient groups were expressed in several pain perception-related brain structures, including the amygdala, anterior cingulate cortex, cerebellum, and nucleus accumbens basal ganglia. Studies in which these neurological structures were previously associated with pain have involved both rodents37 and humans.38 The same analysis for FM did not show significant results.
In the GSEA based on the “GO molecular functions” category, the term common for gene sets from all the analyses that we performed was “GO_CALCIUM_ION_BINDING”, which was the only term identified in the analysis of T CD3+ and PBMC cells from CFS patients and peripheral blood cells from FM patients. This process is essential for voltage-gated calcium channels (VGCCs), and their malfunction is widely related to pain physiology. A study of mice with neuronal overexpression of Cacna2d1 (encoding a subunit of VGCCs) revealed that upregulation of this gene correlates with enhanced activity of VGCCs in sensory neurons and with hyperexcitability of dorsal horn neurons, both of which are crucial for pain modulation.39 Moreover, a study of chronic constriction injury to the infraorbital nerve (CCI-ION) rat model showed that CCI-ION correlated with elevated levels of Cavα2δ1 protein in trigeminal ganglion neurons and increased excitatory synaptogenesis compared to non-injured rats.40 VGCCs have also been implicated in pain hypersensitivity, as an injection of T-type VGCC antagonists has been shown to reverse CFA-induced allodynia in rats.41 VGCCs have also been used as therapeutic targets for the treatment of neuropathic pain. For example, gabapentin, a drug that binds to the α2δ-1 VGCC subunit and indirectly blocks injury-evoked synaptogenesis, was shown to decrease α2δ-1 protein levels in spinal cord tissue collected from neuropathic pain model rats after spinal nerve ligation (SNL), but not in spinal cord tissue from sham rats.42
In the GSEA based on the “GO cellular components” category, “GO_INTRINSIC_COMPONENT_OF_PLASMA_MEMBRANE” was the ontology term common to all the datasets. This term was also the only term identified in the analysis of T CD3+ cells from patients with CFS and one of the two terms identified in the analysis of peripheral blood cells from patients with FM. Aberrations in the excitability of the intrinsic membrane in neurons play an important role in chronic pain development; for example, a recent study showed that SNL-induced neuropathic pain model mice exhibited increased intrinsic excitability of thalamic neurons in vitro, when compared to sham mice.43
Furthermore, the top statistically significant ontology terms in the “GO biological processes” category that were common to all three analyses of genes associated with methylation changes in both FM and CFS included terms related to cell adhesion and homophilic cell adhesion via “plasma membrane adhesion molecules”, “cell adhesion via plasma membrane adhesion molecules”, “cell adhesion”, and “biological adhesion”. Altered serum concentrations of 11 angiogenic factors, including soluble platelet endothelial cell adhesion molecule-1 (sPECAM-1), have been observed in patients with acute and subacute pain syndromes, such as postherpetic neuralgia, low back pain, and trigeminal neuralgia, compared with healthy controls.44 sPECAM-1 is a protein closely related to neuroinflammation, as PECAM-1 transcript was recently found to be overexpressed in the brain tissue of patients with multiple sclerosis compared to controls without neuropathological conditions.45
Also interestingly, we found that three of the motifs targeted by human mature RNA, including “ATATGCA_MIR448”, “CGGGACCA_MIR133A_MIR133B”, and “TCCAGAG_MIR518C” were associated with the methylation signatures identified in each of the analyses and were the only terms identified in analysis of T CD3+ cells. Higher expression of microRNA448 in the spinal cord tissue of rats after CCI of the sciatic nerve has been associated with neuropathic pain development by decreasing the microglial expression of SIRT1, a neuroprotective target of microRNA448, and with higher spinal cord levels of inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, when compared to sham rat tissue counterparts.46 Also, lower expression of this microRNA in PC-12 cells induced the expression of Janus kinase 1 (JAK1), which is involved in the JAK/STAT signaling pathway, regulating diverse processes, such as cell migration, cellular responses, inflammation, and angiogenesis.47
The plasma expression levels of microRNA133A in patients with acute chest pain were higher in patients diagnosed with acute myocardial infarction than in those diagnosed with other diagnoses of chest pain and in the plasma of healthy controls.48 This mRNA was also overexpressed in the serum of patients undergoing coronary artery bypass grafting due to unstable angina pectoris, compared to patients undergoing aortic valve replacement due to aortic stenosis.49
MicroRNA133B was shown to be overexpressed in the dorsal root ganglion of rats that did not develop chronic pain after peripheral nerve injury in comparison to its tissue counterpart in sham rats.50 A negative correlation between microRNA-133B thalamic, plasma, and cerebrospinal fluid expression levels and the severity of allodynia has also been observed in rats with central post-stroke pain (CPSP) and selective overexpression of miR-133b-3p. These findings were confirmed in a study that included patients with CPSP who exhibited decreased plasma levels of circulating miR-133b-3p when compared with control participants without pain.51 Moreover, a study investigating the mechanisms underlying the regulation of neuronal differentiation by morphine reported a morphine-induced decrease in miR-133b expression in zebrafish model embryos and immature mammalian neurons, suggesting that morphine acts as an analgesic agent by inducing dopaminergic neuron differentiation in mammals.52
The last mRNA identified across all analyzed datasets, microRNA518C, was not associated with pain. However, another member of the microRNA518 family, MIR518b, has been shown to be abnormally expressed in patients with complex regional pain syndrome in terms of their response to plasma exchange treatment.53
Interestingly also, enrichment analysis of genes annotated to DMPs that we identified in PBMCs of CFS patients returned only processes, aberrations of which may result in the development of chronic pain syndromes (Supplementary Table 5).
Methylation Changes Common for FM and CFS Annotate to Loci Occupied by Genes Potentially Involved in Pain Regulation
Our analysis also identified four CpG sites, cg08155325 (chr5:140857813), cg22838050 (chr19:872690), cg02454025 (chr1:11042201), and cg19643109 (chr6:160697626), displaying uniform methylation level changes in all studded data sets.
cg08155325 is located in the 1st intron of various transcripts of protocadherin gamma (PCDHG) family genes. Protocadherins are neural adhesion proteins that play a crucial role in the establishment and function of specific cell–cell trans-interactions between neurons and between neurons and astrocytes. In addition, these proteins promote dendrite growth and arbor complexity.54,55 For example, gamma protocadherins are involved in the patterning of axon terminals.56 Moreover, PCDHG genes regulate the generation of functional neural circuits in the brain,57 including the modulation of dendrite arborization, neuronal survival, and synaptogenesis.58 Moreover, the expression of the PCDHGA2 gene has been shown to be essential for the establishment of neuronal connections and signal transduction at the synaptic membrane in the neocortex, hippocampus, cerebellum, and olfactory bulb via cadherin-related neuronal receptors and binding to tyrosine kinase Fyn.59 The expression of PCDHGA11 in hippocampal neurons of congenitally helpless animals was significantly higher (with a 2.6-fold change) than that in non-learned helpless rats, and its overexpression could affect transduction signaling and lead to depression.60 Whole-exome sequencing of blood DNA revealed deletion of the PCDHGB1 gene in two patients with dystonia-plus phenotype61 characterized by fatigue and headache symptoms also occurring in CFS.62 Moreover, mice deficient in PCDHG gene cluster lacks inhibitory interneurons in the cortex and cerebellum and displays motor deficits and seizure.63
Interestingly, the U343 cell-line knockout for PCDHGC3 gene displayed downregulation of the several genes from WNT signaling pathway. This is a major pathway involved in embryogenesis, and its disruption is common in cancer.64 In addition, the cg08155325 locus is a binding site for several transcription factors (TF) described in the JASPAR Transcription Factors Database, including RXRA:VDR, SOX12, SOX14, and PHOX2B, and according to the ORegAnno database CTCF binds to two bases close to this locus. Most of these TF do not appear to be involved in the physiology of pain, but PHOX2B is expressed in cranial neurons. Moreover, research on recombinant mouse embryos shows that PHOX2B is essential for neuronal development of the trigeminal, superior, and jugular ganglia and the trigeminal sensory nuclei and damage to these brain parts leads to pain sensation in the oro-facial region,65 a symptom observed in FM and CFS.66
cg22838050 is located within the binding site of EGR1 transcription factor and enhancers within MED16 and CFD genes. CFD is a crucial regulator of inflammation, as it is involved in the activation of the innate immune response67 and has been shown to be upregulated in dorsal root ganglia (DRG) neurons of rats with paclitaxel-induced peripheral neuropathy.68 Increased EGR1 expression in anterior cingulate cortex neurons was observed in response to chronic inflammatory pain, similar to that in the spinal dorsal horn, and was significantly (p < 0.001) reduced in Egr1-knockout mice.69 Also, increased levels of EGR1 protein have been reported in a study of rat neurons of the insular cortex after nerve injury.70 EGR1 expression is significantly reduced in the blood of patients with Irritable Bowel Syndrome (IBS) after placebo treatment, and reduced levels of EGR1 protein could be a biomarker for IBS, a syndrome with gastrointestinal symptoms similar to those observed in FM and CFS.71
cg02454025 is localized in the TSS200 of C1orf127 and RARA::RXRG TF binding sites. Interestingly, missense mutations in C1orf127 have been detected in patients with primary ciliary dyskinesia (PCD), but the precise function of this gene remains unknown.72 RARA and RXRG were overexpressed in DRG (RARA) and trigeminal ganglion (RXRG) neurons of recombinant Nav1.8Cre/Rosa26fsTRAP mice in a study on ribosome-bound, sensory neuron-specific, and nociceptor-enriched RNAs, suggesting the involvement of these genes in pain development.73
Lastly, the cg19643109 is localized in IRF3 TF binding site and inhibition of IRF3 with dexmedetomidine in rats was associated with reduction of visceral pain,74 and this type of pain is frequently reported by both FM cases4 and CFS patients.75
Discussion
Genetic studies have shown only a weak association of the genetic background with fibromyalgia, and chronic fatigue syndrome. This suggests that epigenetic changes may play key roles in the development of these diseases. To date, only three studies have reported genome-wide methylation changes in fibromyalgia and chronic fatigue syndrome, but the results of these studies are coherent to a very limited extent. We re-analyzed datasets from those studies with a uniform data analysis framework and additional data analysis modules, such as cell fraction and batch effect corrections, which were not always used in previous analyses and are currently considered state-of-the-art in blood-based EWAS studies.
Our analysis identified different numbers of methylation changes in each dataset, the majority of which were not found in previous studies. However, GSEA based on genes annotated to those changes consistently in all datasets associated those changes with physiological processes, disturbances of which may lead to the symptoms reported by patients with fibromyalgia and chronic fatigue syndrome. The discrepancies between methylation signatures that we identified can at least partly be attributed to the type of cells used, study design, and data quality. Despite these limitations, the surprising coherence of physiological processes associated with the signatures identified in each of the analyses clearly suggests that methylation changes may play a key role in the development of syndromes characterized by chronic pain.
We were unable to access data from five other studies on genome-wide methylation changes in FM,76 CFS,77–79 and chronic widespread pain.80 However, we were able to access the list of genes annotated to methylation changes from these studies. Although none of these studies were based on a unified data analysis pipeline that we implemented, there were common genes between these analyses and our study, despite the fact that each of those analyses found different numbers of genes (75 genes in T CD4+ cells in study,77 6835 genes in PBMCs in study,78 and 829 genes in PBMCs in study79). In addition, closer analysis of the data from these studies showed that the poor overlap of our results with data from two of those studies was likely attributed to the low magnitude of the methylation changes identified in those studies, which was below 5%80 and in our analyses, we only considered methylation changes higher than 5% as meaningful in the context of the limitations of bead array technology. Also in one of the previous studies, DMPs were identified using M-values (a derivative of the beta values used in our data analysis), and we were not able to directly compare our results with the results reported in that study.76
Additionally, we identified four studies that analyzed methylation changes in FM and CFS using the NGS platform, of which two used the RRBS protocol81,82 and two others used targeted bisulfite sequencing.83,84 Despite the different technologies used and different hypotheses tested, there were common genes between our study and RRBS-based studies. Our results did not include any genes identified by targeted bisulfite NGS sequencing. However, these studies reported smaller methylation differences than those we aimed to identify in our data analysis.
There are also a number of studies that analyzed methylation changes at single genes in FM and CFS.85–92 However, in most of these studies, pyrosequencing was used for methylation screening and identified gene-specific methylation changes were smaller than 5%; thus, they could not have been identified in our analysis. Nevertheless, we identified differentially methylated probes within the BDNF gene that were reported to undergo hypermethylation in patients with CFS and comorbid FM in one of the gene-centric methylation studies.92
Conclusion
Overall, our results, especially when analyzed in the context of results reported in similar studies that utilized different data analysis and experimental approaches, clearly indicate that symptoms of chronic pain may be caused by the methylation changes, but further and better controlled studies are required to elaborate the role of DNA methylation in the development of chronic pain syndromes.
Data Sharing Statement
All analyzed datasets are publicly available in the Gene Expression Omnibus database under accession numbers GSE85506, GSE156792, GSE93266, and GSE102504.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by the Polish Return Grant Program of the Polish National Agency for Academic Exchange (grant ID: PPN/PPO/2018/1/00088).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Moyano S, Kilstein JG, Alegre de Miguel C. New diagnostic criteria for fibromyalgia: here to stay? Reumatol Clin. 2015;11(4):210–214. doi:10.1016/j.reuma.2014.07.008
2. Galvez-Sánchez CM, Reyes del paso GA. Diagnostic criteria for fibromyalgia: critical review and future perspectives. J Clin Med. 2020;9(4):1219. doi:10.3390/jcm9041219
3. Wolfe F, Clauw DJ, Fitzcharles MA, et al. The American College of Rheumatology preliminary diagnostic criteria for fibromyalgia and measurement of symptom severity. Arthritis Care Res. 2010;62(5):600–610. doi:10.1002/acr.20140
4. Sarzi-Puttini P, Giorgi V, Marotto D, Atzeni F. Fibromyalgia: an update on clinical characteristics, aetiopathogenesis and treatment. Nat Rev Rheumatol. 2020;16(11):645–660. doi:10.1038/s41584-020-00506-w
5. Bateman L, Bested AC, Bonilla HF, et al. Myalgic encephalomyelitis/chronic fatigue syndrome: essentials of diagnosis and management. Mayo Clin Proc. 2021;96(11):2861–2878. doi:10.1016/j.mayocp.2021.07.004
6. Bested AC, Marshall LM. Review of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: an evidence-based approach to diagnosis and management by clinicians. Rev Environ Health. 2015;30(4):223–249. doi:10.1515/reveh-2015-0026
7. Borchers AT, Gershwin ME. Fibromyalgia: a critical and comprehensive review. Clin Rev Allergy Immunol. 2015;49(2):100–151. doi:10.1007/s12016-015-8509-4
8. Queiroz LP. Worldwide epidemiology of fibromyalgia. Curr Pain Headache Rep. 2013;17(8):356. doi:10.1007/s11916-013-0356-5
9. Lim EJ, Ahn YC, Jang ES, Lee SW, Lee SH, Son CG. Systematic review and meta-analysis of the prevalence of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME). J Transl Med. 2020;18(1):100. doi:10.1186/s12967-020-02269-0
10. Arnold LM, Hudson JI, Hess EV, et al. Family study of fibromyalgia. Arthritis Rheum. 2004;50(3):944–952. doi:10.1002/art.20042
11. Dutta D, Brummett CM, Moser SE, et al. Heritability of the fibromyalgia phenotype varies by age. Arthritis Rheumatol. 2020;72(5):815–823. doi:10.1002/art.41171
12. Rahman MS, Winsvold BS, Chavez Chavez SO, et al. Genome-wide association study identifies RNF123 locus as associated with chronic widespread musculoskeletal pain. Ann Rheum Dis. 2021;80(9):1227–1235. doi:10.1136/annrheumdis-2020-219624
13. Gloor Y, Matthey A, Sobo K, et al. Uncovering a genetic polymorphism located in huntingtin associated protein 1 in modulation of central pain sensitization signaling pathways. Front Neurosci. 2022;16:807773. doi:10.3389/fnins.2022.807773
14. Buchwald D, Herrell R, Ashton S, et al. A twin study of chronic fatigue. Psychosom Med. 2001;63(6):936–943. doi:10.1097/00006842-200111000-00012
15. Walsh CM, Zainal NZ, Middleton SJ, Paykel ES. A family history study of chronic fatigue syndrome. Psychiatr Genet. 2001;11(3):123–128. doi:10.1097/00041444-200109000-00003
16. Hickie I, Kirk K, Martin N. Unique genetic and environmental determinants of prolonged fatigue: a twin study. Psychol Med. 1999;29(2):259–268. doi:10.1017/s0033291798007934
17. Smith AK, Fang H, Whistler T, Unger ER, Rajeevan MS. Convergent genomic studies identify association of GRIK2 and NPAS2 with chronic fatigue syndrome. Neuropsychobiology. 2011;64(4):183–194. doi:10.1159/000326692
18. Tanigawa Y, Li J, Justesen JM, et al. Components of genetic associations across 2138 phenotypes in the UK Biobank highlight adipocyte biology. Nat Commun. 2019;10(1):4064. doi:10.1038/s41467-019-11953-9
19. Canela-Xandri O, Rawlik K, Tenesa A. An atlas of genetic associations in UK Biobank. Nat Genet. 2018;50(11):1593–1599. doi:10.1038/s41588-018-0248-z
20. Ciampi de Andrade D, Maschietto M, Galhardoni R, et al. Epigenetics insights into chronic pain: DNA hypomethylation in fibromyalgia-a controlled pilot-study. Pain. 2017;158(8):1473–1480. doi:10.1097/j.pain.0000000000000932
21. Herrera S, de Vega WC, Ashbrook D, Vernon SD, McGowan PO. Genome-epigenome interactions associated with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Epigenetics. 2018;13(12):1174–1190. doi:10.1080/15592294.2018.1549769
22. de Vega WC, Herrera S, Vernon SD, McGowan PO. Epigenetic modifications and glucocorticoid sensitivity in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). BMC Med Genomics. 2017;10(1):11. doi:10.1186/s12920-017-0248-3
23. de Vega WC, Erdman L, Vernon SD, Goldenberg A, McGowan PO. Integration of DNA methylation & health scores identifies subtypes in myalgic encephalomyelitis/chronic fatigue syndrome. Epigenomics. 2018;10(5):539–557. doi:10.2217/epi-2017-0150
24. Sarzi-Puttini P, Giorgi V, Atzeni F, et al. Fibromyalgia position paper. Clin Exp Rheumatol. 2021;39(3):186–193. doi:10.55563/clinexprheumatol/i19pig
25. Morris TJ, Butcher LM, Feber A, et al. ChAMP: 450k chip analysis methylation pipeline. Bioinformatics. 2014;30(3):428–430. doi:10.1093/bioinformatics/btt684
26. Tian Y, Morris TJ, Webster AP, et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics. 2017;33(24):3982–3984. doi:10.1093/bioinformatics/btx513
27. Campagna MP, Xavier A, Lechner-Scott J, et al. Epigenome-wide association studies: current knowledge, strategies and recommendations. Clin Epigenetics. 2021;13(1):214. doi:10.1186/s13148-021-01200-8
28. Teschendorff AE, Breeze CE, Zheng SC, Beck S. A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies. BMC Bioinf. 2017;18(1):105. doi:10.1186/s12859-017-1511-5
29. McGregor K, Bernatsky S, Colmegna I, et al. An evaluation of methods correcting for cell-type heterogeneity in DNA methylation studies. Genome Biol. 2016;17(1):84. doi:10.1186/s13059-016-0935-y
30. Bińkowski J, Taryma-Leśniak O, Łuczkowska K, et al. Epigenetic activation of antiviral sensors and effectors of interferon response pathways during SARS-CoV-2 infection. Biomed Pharmacother. 2022;153:113396. doi:10.1016/j.biopha.2022.113396
31. Cunningham F, Allen JE, Allen J, et al. Ensembl 2022. Nucleic Acids Res. 2022;50(D1):D988–D995. doi:10.1093/nar/gkab1049
32. Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. doi:10.1038/s41467-017-01261-5
33. Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank: update. Nucleic Acids Res. 2004;32(Database issue):D23–D26. doi:10.1093/nar/gkh045
34. Lesurf R, Cotto KC, Wang G, et al. ORegAnno 3.0: a community-driven resource for curated regulatory annotation. Nucleic Acids Res. 2016;44(D1):D126–D132. doi:10.1093/nar/gkv1203
35. Fishilevich S, Nudel R, Rappaport N, et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database. 2017;2017. doi:10.1093/database/bax028
36. Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196(2):261–282. doi:10.1016/0022-2836(87)90689-9
37. Bliss TV, Collingridge GL, Kaang BK, Zhuo M. Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat Rev Neurosci. 2016;17(8):485–496. doi:10.1038/nrn.2016.68
38. Shi H, Yuan C, Dai Z, Ma H, Sheng L. Gray matter abnormalities associated with fibromyalgia: a meta-analysis of voxel-based morphometric studies. Semin Arthritis Rheum. 2016;46(3):330–337. doi:10.1016/j.semarthrit.2016.06.002
39. Li CY, Zhang XL, Matthews EA, et al. Calcium channel alpha2delta1 subunit mediates spinal hyperexcitability in pain modulation. Pain. 2006;125(1–2):20–34. doi:10.1016/j.pain.2006.04.022
40. Li KW, Yu YP, Zhou C, et al. Calcium channel α2δ1 proteins mediate trigeminal neuropathic pain states associated with aberrant excitatory synaptogenesis. J Biol Chem. 2014;289(10):7025–7037. doi:10.1074/jbc.M114.548990
41. Harding EK, Dedek A, Bonin RP, Salter MW, Snutch TP, Hildebrand ME. The T-type calcium channel antagonist, Z944, reduces spinal excitability and pain hypersensitivity. Br J Pharmacol. 2021;178(17):3517–3532. doi:10.1111/bph.15498
42. Chen J, Li L, Chen SR, et al. The α2δ-1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep. 2018;22(9):2307–2321. doi:10.1016/j.celrep.2018.02.021
43. Zhang WT, Sha WL, Zhu Q, Wu XB, He C. Plasticity of neuronal excitability and synaptic balance in the anterior nucleus of paraventricular thalamus after nerve injury. Brain Res Bull. 2022;188:1–10. doi:10.1016/j.brainresbull.2022.07.008
44. Yang X, Yuan C, Wang H, et al. Changes in serum angiogenic factors among patients with acute pain and subacute pain. Front Mol Neurosci. 2022;15:960460. doi:10.3389/fnmol.2022.960460
45. Wimmer I, Tietz S, Nishihara H, et al. PECAM-1 stabilizes blood-brain barrier integrity and favors paracellular T-cell diapedesis across the blood-brain barrier during neuroinflammation. Front Immunol. 2019;10:711. doi:10.3389/fimmu.2019.00711
46. Chu Y, Ge W, Wang X. MicroRNA-448 modulates the progression of neuropathic pain by targeting sirtuin 1. Exp Ther Med. 2019;18(6):4665–4672. doi:10.3892/etm.2019.8165
47. Wang L, Zhu K, Yang B, Cai Y. Knockdown of Linc00052 alleviated spinal nerve ligation-triggered neuropathic pain through regulating miR-448 and JAK1. J Cell Physiol. 2020;235(10):6528–6535. doi:10.1002/jcp.29465
48. Ke-Gang J, Zhi-Wei L, Xin Z, et al. Evaluating diagnostic and prognostic value of plasma miRNA133a in acute chest pain patients undergoing coronary angiography. Medicine. 2016;95(17):e3412. doi:10.1097/MD.0000000000003412
49. Miyamoto S, Usami S, Kuwabara Y, et al. Expression patterns of miRNA-423-5p in the serum and pericardial fluid in patients undergoing cardiac surgery. PLoS One. 2015;10(11):e0142904. doi:10.1371/journal.pone.0142904
50. Norcini M, Choi D, Lu H, et al. Intrathecal injection of miR-133b-3p or miR-143-3p prevents the development of persistent cold and mechanical allodynia following a peripheral nerve injury in rats. Neuroscience. 2018;386:223–239. doi:10.1016/j.neuroscience.2018.06.040
51. Guo X, Lu J, Yan M, et al. MicroRNA-133b-3p targets purinergic P2X4 receptor to regulate central poststroke pain in rats. Neuroscience. 2022;481:60–72. doi:10.1016/j.neuroscience.2021.10.015
52. Sanchez-Simon FM, Zhang XX, Loh HH, Law PY, Rodriguez RE. Morphine regulates dopaminergic neuron differentiation via miR-133b. Mol Pharmacol. 2010;78(5):935–942. doi:10.1124/mol.110.066837
53. Ramanathan S, Douglas SR, Alexander GM, et al. Exosome microRNA signatures in patients with complex regional pain syndrome undergoing plasma exchange. J Transl Med. 2019;17(1):81. doi:10.1186/s12967-019-1833-3
54. Peek SL, Mah KM, Weiner JA. Regulation of neural circuit formation by protocadherins. Cell Mol Life Sci. 2017;74(22):4133–4157. doi:10.1007/s00018-017-2572-3
55. Pancho A, Aerts T, Mitsogiannis MD, Seuntjens E. Protocadherins at the crossroad of signaling pathways. Front Mol Neurosci. 2020;13:117. doi:10.3389/fnmol.2020.00117
56. Molumby MJ, Keeler AB, Weiner JA. Homophilic protocadherin cell-cell interactions promote dendrite complexity. Cell Rep. 2016;15(5):1037–1050. doi:10.1016/j.celrep.2016.03.093
57. Hirayama T, Yagi T. Regulation of clustered protocadherin genes in individual neurons. Semin Cell Dev Biol. 2017;69:122–130. doi:10.1016/j.semcdb.2017.05.026
58. Mah KM, Houston DW, Weiner JA. The γ-Protocadherin-C3 isoform inhibits canonical Wnt signalling by binding to and stabilizing Axin1 at the membrane. Sci Rep. 2016;6(1):31665. doi:10.1038/srep31665
59. Yagi T, Takeichi M. Cadherin superfamily genes: functions, genomic organization, and neurologic diversity. Genes Dev. 2000;14(10):1169–1180. doi:10.1101/gad.14.10.1169
60. Garafola CS, Henn FA. A change in hippocampal protocadherin gamma expression in a learned helpless rat. Brain Res. 2014;1593:55–64. doi:10.1016/j.brainres.2014.08.071
61. Li LX, Huang JH, Pan LZ, Zhang XL, Pan YG, Jin LJ. Whole-exome sequencing identified rare variants in PCDHGB1 in patients with adult-onset dystonia. Mov Disord. 2022;37(5):1099–1101. doi:10.1002/mds.28965
62. Matsui T, Ii K, Hojo S, Sano K. Cervical neuro-muscular syndrome: discovery of a new disease group caused by abnormalities in the cervical muscles. Neurol Med Chir. 2012;52(2):75–80. doi:10.2176/nmc.52.75
63. Carriere CH, Wang WX, Sing AD, et al. The γ-protocadherins regulate the survival of GABAergic interneurons during developmental cell death. J Neurosci. 2020;40(45):8652–8668. doi:10.1523/JNEUROSCI.1636-20.2020
64. Feldheim J, Wend D, Lauer MJ, et al. Protocadherin Gamma C3 (PCDHGC3) is strongly expressed in glioblastoma and its high expression is associated with longer progression-free survival of patients. Int J Mol Sci. 2022;23(15):8101. doi:10.3390/ijms23158101
65. D’Autréaux F, Coppola E, Hirsch MR, Birchmeier C, Brunet JF. Homeoprotein Phox2b commands a somatic-to-visceral switch in cranial sensory pathways. Proc Natl Acad Sci U S A. 2011;108(50):20018–20023. doi:10.1073/pnas.1110416108
66. Robinson LJ, Durham J, Newton JL. A systematic review of the comorbidity between temporomandibular disorders and chronic fatigue syndrome. J Oral Rehabil. 2016;43(4):306–316. doi:10.1111/joor.12367
67. Gavriilaki E, Papakonstantinou A, Agrios KA. Novel Insights into Factor D Inhibition. Int J Mol Sci. 2022;23(13):7216. doi:10.3390/ijms23137216
68. Sun W, Yang S, Wu S, et al. Transcriptome analysis reveals dysregulation of inflammatory and neuronal function in dorsal root ganglion of paclitaxel-induced peripheral neuropathy rats. Mol Pain. 2022:17448069221106167. doi:10.1177/17448069221106167
69. Ko SW, Vadakkan KI, Ao H, et al. Selective contribution of Egr1 (zif/268) to persistent inflammatory pain. J Pain. 2005;6(1):12–20. doi:10.1016/j.jpain.2004.10.001
70. Han J, Kwon M, Cha M, et al. Plasticity-related PKMζ signaling in the insular cortex is involved in the modulation of neuropathic pain after nerve injury. Neural Plast. 2015;2015:601767. doi:10.1155/2015/601767
71. Wang RS, Lembo AJ, Kaptchuk TJ, et al. Genomic effects associated with response to placebo treatment in a randomized trial of irritable bowel syndrome. Front Pain Res. 2021;2:775386. doi:10.3389/fpain.2021.775386
72. Alsamri MT, Alabdouli A, Iram D, et al. A study on the genetics of primary ciliary dyskinesia. J Clin Med. 2021;10(21):5102. doi:10.3390/jcm10215102
73. Megat S, Ray PR, Tavares-Ferreira D, et al. Differences between dorsal root and trigeminal ganglion nociceptors in mice revealed by translational profiling. J Neurosci. 2019;39(35):6829–6847. doi:10.1523/JNEUROSCI.2663-18.2019
74. Li J, Tang H, Tu W. Mechanism of dexmedetomidine preconditioning on spinal cord analgesia in rats with functional chronic visceral pain. Acta Cir Bras. 2022;37(2):e370203. doi:10.1590/acb370203
75. Lakhan SE, Kirchgessner A. Gut inflammation in chronic fatigue syndrome. Nutr Metab. 2010;7(1):79. doi:10.1186/1743-7075-7-79
76. Menzies V, Lyon DE, Archer KJ, et al. Epigenetic alterations and an increased frequency of micronuclei in women with fibromyalgia. Nurs Res Pract. 2013;2013:795784. doi:10.1155/2013/795784
77. Brenu EW, Staines DR, Marshall-Gradisnik SM. Methylation profile of CD4+ T cells in chronic fatigue syndrome/myalgic encephalomyelitis. J Clin Cell Immunol. 2014;5(228):10–4172.
78. Trivedi MS, Oltra E, Sarria L, et al. Identification of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome-associated DNA methylation patterns. PLoS One. 2018;13(7):e0201066. doi:10.1371/journal.pone.0201066
79. de Vega WC, Vernon SD, McGowan PO, Ballestar E. DNA methylation modifications associated with chronic fatigue syndrome. PLoS One. 2014;9(8):e104757. doi:10.1371/journal.pone.0104757
80. Burri A, Marinova Z, Robinson MD, et al. Are epigenetic factors implicated in chronic widespread pain? PLoS One. 2016;11(11):e0165548. doi:10.1371/journal.pone.0165548
81. Helliwell AM, Sweetman EC, Stockwell PA, Edgar CD, Chatterjee A, Tate WP. Changes in DNA methylation profiles of myalgic encephalomyelitis/chronic fatigue syndrome patients reflect systemic dysfunctions. Clin Epigenetics. 2020;12(1):167. doi:10.1186/s13148-020-00960-z
82. Helliwell AM, Stockwell PA, Edgar CD, Chatterjee A, Tate WP. Dynamic epigenetic changes during a relapse and recovery cycle in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Int J Mol Sci. 2022;23(19):11852. doi:10.3390/ijms231911852
83. Gerra MC, Carnevali D, Ossola P, et al. DNA methylation changes in fibromyalgia suggest the role of the immune-inflammatory response and central sensitization. J Clin Med. 2021;10(21):4992. doi:10.3390/jcm10214992
84. Gerra MC, Carnevali D, Pedersen IS, et al. DNA methylation changes in genes involved in inflammation and depression in fibromyalgia: a pilot study. Scand J Pain. 2021;21(2):372–383. doi:10.1515/sjpain-2020-0124
85. Vangeel E, Van Den Eede F, Hompes T, et al. Chronic fatigue syndrome and DNA hypomethylation of the glucocorticoid receptor gene promoter 1f region: associations with hpa axis hypofunction and childhood trauma. Psychosom Med. 2015;77(8):853–862. doi:10.1097/PSY.0000000000000224
86. Falkenberg VR, Whistler T, Murray JR, Unger ER, Rajeevan MS. Acute psychosocial stress-mediated changes in the expression and methylation of perforin in chronic fatigue syndrome. Genet Epigenet. 2013;5:1–9. doi:10.4137/GEG.S10944
87. Polli A, Hendrix J, Ickmans K, et al. Genetic and epigenetic regulation of Catechol-O-methyltransferase in relation to inflammation in chronic fatigue syndrome and Fibromyalgia. J Transl Med. 2022;20(1):487. doi:10.1186/s12967-022-03662-7
88. Vangeel EB, Kempke S, Bakusic J, et al. Glucocorticoid receptor DNA methylation and childhood trauma in chronic fatigue syndrome patients. J Psychosom Res. 2018;104:55–60. doi:10.1016/j.jpsychores.2017.11.011
89. Achenbach J, Rhein M, Glahn A, Frieling H, Karst M. Leptin promoter methylation in female patients with painful multisomatoform disorder and chronic widespread pain. Clin Epigenetics. 2022;14(1):13. doi:10.1186/s13148-022-01235-5
90. Achenbach J, Rhein M, Gombert S, et al. Childhood traumatization is associated with differences in TRPA1 promoter methylation in female patients with multisomatoform disorder with pain as the leading bodily symptom. Clin Epigenetics. 2019;11(1):126. doi:10.1186/s13148-019-0731-0
91. Falkenberg VR, Gurbaxani BM, Unger ER, Rajeevan MS. Functional genomics of serotonin receptor 2A (HTR2A): interaction of polymorphism, methylation, expression and disease association. Neuromolecular Med. 2011;13(1):66–76. doi:10.1007/s12017-010-8138-2
92. Polli A, Ghosh M, Bakusic J, et al. DNA methylation and brain-derived neurotrophic factor expression account for symptoms and widespread hyperalgesia in patients with chronic fatigue syndrome and comorbid fibromyalgia. Arthritis Rheumatol. 2020;72(11):1936–1944. doi:10.1002/art.41405
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.