")
Back to Journals » Infection and Drug Resistance » Volume 10
Diversity and evolution of drug resistance mechanisms in Mycobacterium tuberculosis
Authors Al-Saeedi M, Al-Hajoj S
Received 20 June 2017
Accepted for publication 3 August 2017
Published 13 October 2017 Volume 2017:10 Pages 333—342
DOI https://doi.org/10.2147/IDR.S144446
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sahil Khanna
Mashael Al-Saeedi, Sahal Al-Hajoj
Department of Infection and Immunity, Mycobacteriology Research Section, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
Abstract: Despite the efficacy of antibiotics to protect humankind against many deadly pathogens, such as Mycobacterium tuberculosis, nothing can prevent the emergence of drug-resistant strains. Several mechanisms facilitate drug resistance in M. tuberculosis including compensatory evolution, epistasis, clonal interference, cell wall integrity, efflux pumps, and target mimicry. In this study, we present recent findings relevant to these mechanisms, which can enable the discovery of new drug targets and subsequent development of novel drugs for treatment of drug-resistant M. tuberculosis.
Keywords: Mycobacterium tuberculosis, antibiotic resistance, compensatory evolution, epistasis, efflux pumps, fitness cost
Corrigendum for this paper has been published
Introduction
Tuberculosis (TB) is a significant public health concern with a high disease burden and mortality rate.1 The disease is caused by members of the Mycobacterium tuberculosis complex (MTBC), a group of closely related human-adapted (M. tuberculosis [MTB] and Mycobacterium africanum) and animal-adapted (Mycobacterium bovis, Mycobacterium mungi, Mycobacterium pinnipedii, Mycobacterium microti, and Mycobacterium caprae) strains, as well as smooth tuberculosis bacilli (Mycobacterium canettii).2 Although MTBC species have a remarkable range of mammalian hosts and morphologies, their genomes exhibit ≥99% homology, providing evidence that they evolved from a single ancestor in Africa 70,000 years ago.2–4 However, following advances in agriculture and animal domestication, MTB emerged as a human pathogen that has caused millions of deaths and continues to threaten human health globally.5,6 Successful control and prevention of MTB infection requires tools for a rapid and accurate diagnosis, as well as strategies for effective treatment. Current antibiotics used to treat MTB infection are isoniazid (INH), rifampicin (RIF), pyrazinamide (PZA), and ethambutol (EMB).7,8 Misuse or misadministration of drugs can facilitate the emergence of drug-resistant strains via compensatory evolution, epistasis, and clonal interference phenomena that modulate MTB fitness. These various mechanisms of adaptation have led to the evolution of different drug-resistant levels of MTB strains, including multidrug resistant (MDR; resistance to INH and RIF), extensively drug resistant (XDR; resistance to fluoroquinolones [FQs] and one of the injectable aminoglycosides [AGs]), and totally drug resistant (TDR; resistance to all known drugs).9,10
Therefore, the aim of this review was to highlight recent findings related to mechanisms implicated in the emergence of MTB drug resistance and outline some possible drug targets to contribute to efforts aimed at discovering novel TB treatment.
Development of drug-resistant MTB
MTB diversity and drug resistance
Genomic comparisons have increased our understanding of MTBC diversity. For example, analyses of single nucleotide polymorphisms (SNPs) and the presence or absence of deletion regions (MTB-deleted region 1 or regions of difference) within MTBC genomes have identified several lineages and sub-lineages with distinct characteristics and distributions.2 For instance, lineage 2 (East Asian) and 4 (Euro-American) strains are widely distributed, while lineage 3 (Central Asian and East African Indian) strains are restricted to particular regions in Asia and Africa. These three lineages are characterized by deleted regions and are denoted as “modern lineages.” In contrast, lineage 1 (Indo-Oceanic) strains are common in the Indian Ocean region and the Philippines. This lineage, together with lineages 5 and 6 (West African), and animal lineages comprise the “ancient lineages” and have no deletions characteristic of the modern lineages.2,11 Finally, lineage 7 is considered as an intermediate lineage and has recently reported in Ethiopia.12
Genomic differences among MTBC lineages impact their capability to cause disease and develop drug resistance. For instance, members of the modern lineages are associated with greater disease burden and drug resistance than the ancient lineages, likely due to a high rate of accumulating spontaneous mutations during replication.13,14 Epidemiological studies have illustrated that lineage 2 strains have a higher rate of developing resistance ranging from 1.6 × 10−5 to 5.4 × 10−3 than lineage 1.15
Despite the accumulation of spontaneous mutations among MTBC lineages, their specific (and perhaps unique) drug resistance mechanisms remain unknown. However, increased mutations in DNA repair system and SOS response genes within lineage 2 genomes enhance the possibility of these strains developing resistance and generating mutator phenotype.14,16 Nonetheless, these findings cannot sufficiently explain several enigmas in these strains, such as how the same mutation among MTBC can generate a different level of resistance. Moreover, how do some strains tolerate certain mutations better than others, and why are some strains more strongly associated with infectious transmission and outbreaks? These questions could be resolved by studies of compensatory evolution, epistasis, and clonal interference, as these mechanisms could have large impact on mutation rate acceleration and MTB fitness modulation.17–21
Compensatory evolution and MTB drug resistance
Drug concentration is a primary determinant of resistance-associated mutations during drug therapy.22 Mutations develop when the drug concentration is not optimal, although mutations impose a fitness cost on bacteria that targets genes encoding essential biological functions, often leading to reduced bacterial growth, survival, and virulence.23,24 Contrary to fitness cost concept, several studies have documented that some frequently transmitted MTB strains undergo low- or no-cost mutations but exhibit high level of resistance to drugs.17,19,20 Thus, these strains may harbor resistance mechanisms developed through compensatory evolution, which can modulate MTB fitness (Figure 1).25,26
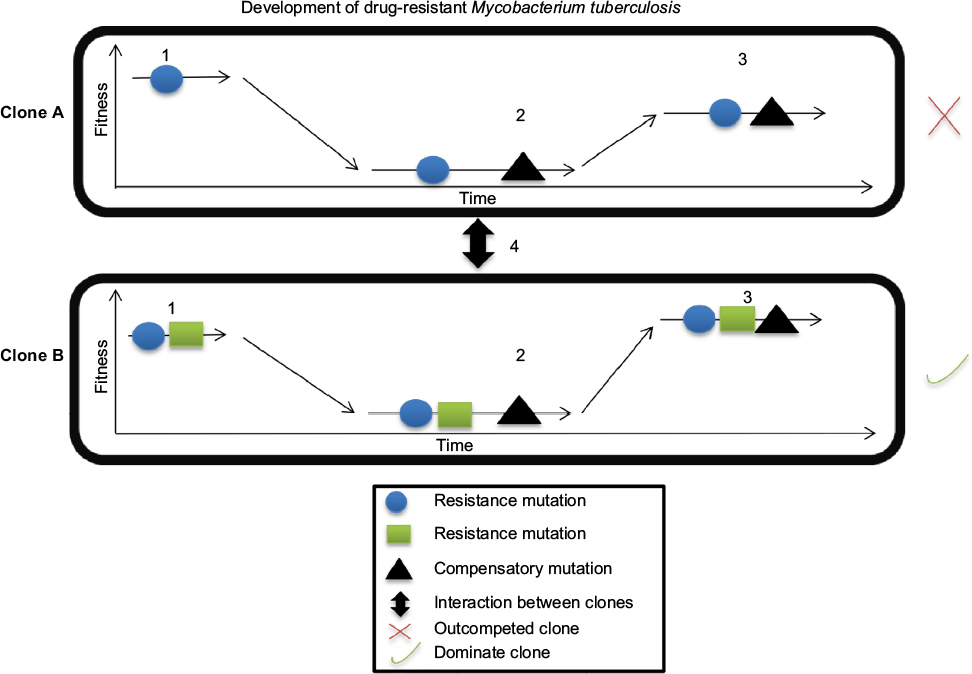 | Figure 1 Development of drug resistance. Notes: 1, when MTB acquires mutations during therapy, they reduce MTB fitness (clones A and B). 2, an acquired secondary mutation restores MTB fitness. 3, the epistatic interaction between mutations improves MTB fitness and maintains drug resistance within the specific MTB genetic background. 4, clonal interference determines clone fate via competition, which leads to emergence of the most dominate clone with drug resistance in the population and elimination of the clone with a lower mutational effect. Abbreviation: MTB, Mycobacterium tuberculosis. |
Compensatory evolution is mediated by the acquiring of a second mutation that minimizes the deleterious effect (resistance cost) of the original mutation. This mechanism allows MTB to increase its fitness without losing the resistance phenotype.27 Compensatory evolution can develop from either additional or alternative mutations that occur in intra- or extragenic loci.28
In MTB, compensatory evolution associated with INH resistance occurs when a mutation in the regulatory region of ahpC leads to overexpression of alkyl hydroperoxide reductase (AhpC), which may compensate for the fitness cost of Ser315Thr mutation in katG, normally encodes a catalase-peroxidase, converting INH into a bioactive form.29
Compensatory evolution of RIF resistance has also been reported.30,31 In one study, mutations in rpoB, which encodes the β subunit of RNA polymerase, were detected in 95% of clinical isolates and conferred a high level of RIF resistance, but have also been associated with noticeable fitness cost.17,30,32 However, S531L mutation was seen in most MDR isolates that exhibited a no- or low-cost fitness effect. This phenomenon is explained by the acquisition of compensatory mutations in neighboring rpoA and rpoC genes, which can mitigate the fitness cost of S531L.33–36 Comas et al found that up to 30% of MDR cases in high MTB burden countries carried mutations in rpoA and rpoC, suggesting they may play a role in spreading MDR strains in these countries.33 Interestingly, a recent study revealed the compensatory role of an intragenic V615M mutation, located in rpoB gene, with respect to RIF resistance-associated rpoB mutations. The study showed that V615M mutation can modulate RNA polymerase bridge helix structure and contribute to an increased rate of transcription elongation, thus compensating for defective RNA polymerase activity associated with S531L mutation.37
In addition to compensatory mutations, alternative mechanisms of fitness compensation may exist. For example, Freihofer et al38 found that altered gene regulation can also reduce the deleterious effects of certain genetic mutations. The study found that emergence of the A1408G mutation in the 16S rRNA gene is accompanied by overexpression of tlyA, which encodes a methyltransferase, resulting in methylation of neighboring 16S rRNA position C1409 and increased MTB fitness. The identification of non-mutational mechanisms could provide a new strategy for optimizing current treatment regimens through inhibition of the compensatory event, leading to disruption of the stabilization of drug resistance transmission.
Role of epistasis and genetic background in MTB drug resistance
Epistasis occurs when several mutations interact with each other to express new advantageous traits for an organism and are often necessary for bacteria to modify their fitness cost.39 During epistatic interactions, the effect of multiple mutations is greater or less than the effect of the individual mutation and can lead to either beneficial or deleterious phenotypes.7 Thus, epistasis is classified as 1) positive (antagonistic), 2) negative (synergistic), or 3) sign.40
In positive epistasis, the net fitness of the interactions is higher than expected (Figure 2).83 A study has reported the role of positive epistasis in drug resistance development in MTB. Borrell et al41 identified a particular combination of mutations in rpoB and gyrA that conferred resistance to RIF and ofloxacin (OFX). The study showed that a gyrA D94G mutation is associated with improving deleterious fitness effects. Thus, gyrA D94G is correlated with positive epistasis in MTB, and it is frequently occurred within XDR strains.
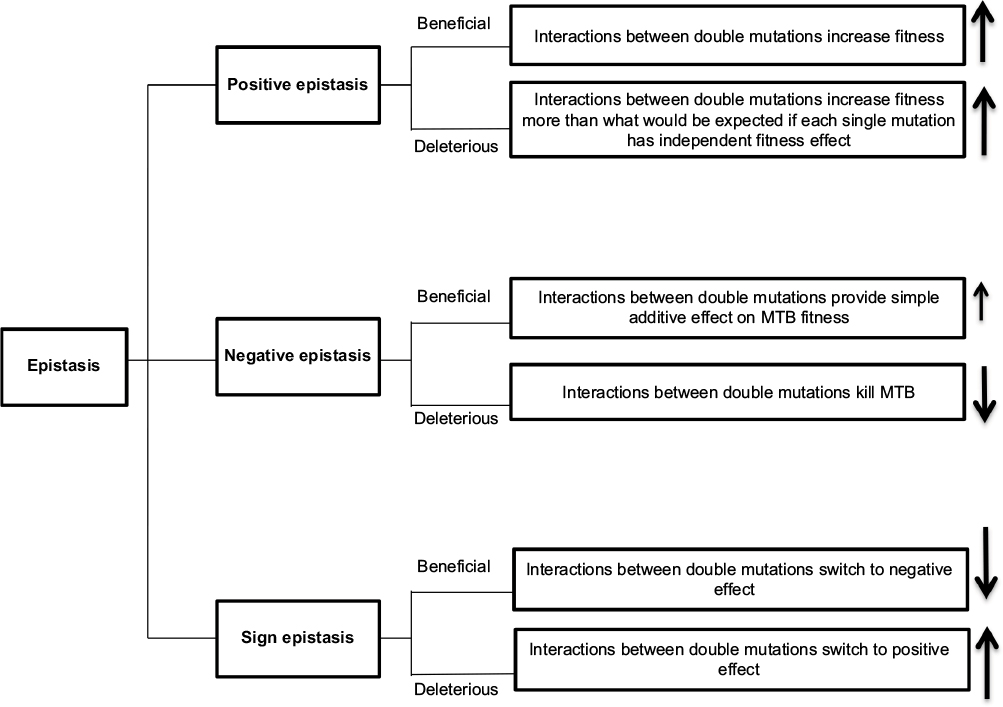 | Figure 2 Forms of epistatic interaction between mutations. Note: (↑) indicated high MTB fitness and (↓) indicated low MTB fitness. Abbreviation: MTB, Mycobacterium tuberculosis. |
Contrary to positive epistasis, negative epistasis is characterized by fitness lower than expected after mutation interactions. In bacteria, interactions between beneficial mutations provide a simple additive effect on fitness, while interactions between deleterious mutations are lethal (Figure 2).83 However, in sign epistasis, the fitness of mutations depends on the genetic background of the bacteria. These mutations can be deleterious, beneficial, or neutral. Reciprocal sign epistasis is an extreme form of the interactions in which beneficial mutations together exert a negative effect or when deleterious mutations become positive (Figure 2).40,42 The interaction between compensatory and drug resistance mutations is an example of sign epistasis.43 When compensatory mutations occur in a susceptible genetic background, they become deleterious. Thus, the acquisition of resistance mutations promotes the epistatic interaction between compensatory and acquired resistance mutations, which results in the emergence and maintenance of the resistance phenotype in that particular bacterial genetic background.44
In addition, increasing evidence supports the role of sign epistasis in MTBC diversity.43 This is mainly due to the epistatic interactions between the mutations within the MTBC genetic background, the acquired resistance mutations, and the compensatory mutations.45 Fenner et al46 found that a mutation in either katG or inhA confers different levels of resistance. Lineage 2 strains carry mutations in both katG (high INH resistance) and inhA (low INH resistance) genes and show different levels of drug resistance compared to lineage 1 bacteria, in which only the inhA mutation has been identified. Similarly, when bacteria from different lineages were exposed to the same dose of RIF, they exhibited different fitness costs and resistance levels.17 These data support the role of epistasis interactions between MTBC genetic background and acquired mutations that confer various levels of resistance across MTBC lineages.
Dynamics of clonal interference in MTB
Depending on the bacterial population size, mutation rate (U), and distribution of fitness effects, various mutations can simultaneously develop in a single population.47 In this situation, clonal interference may occur and significantly impact resistance development in the population. When two distinct resistance mutations develop independently within distinctive bacterial individuals, they compete with each other. Thus, a clone with a greater mutation effect outcompetes a clone with smaller mutation effects, which is then eliminated from the population (Figure 1).48
The intra-host evolution of MTB provides a straightforward model for understanding clonal interference in vivo. Some studies have reported competition between MTB clones in a single patient sample. Sun et al47 examined seven isolates from three patients; the first patient was free from MTB drug resistance, but after 19 months of treatment, four independent mutations were detected: three mutations in katG and one mutation in the regulatory region of inhA. After 5 months, most of the mutations reverted, and only one mutation in katG was detected. The second patient harbored MTB with a mutation in rpoB (L533P) but was still sensitive to RIF. After 18 months, the L533P mutation was replaced with a second mutation in rpoB (H526Y), leading to RIF-resistant MTB. The third patient was a relapsed case of MTB with two unfixed mutations of ethA (L35R and A341E) after 11 months of treatment that showed no change in EMB resistance status. These observations illustrate how the competition and interchange between resistance-related mutations can lead to MDR. Similarly, Eldholm et al49 followed an XDR-TB patient for 3.5 years and performed genome sequencing of nine isolates from the same patient. They observed a high level of heterogeneity in the isolate population: 35 mutations were identified, including 20 transient and 15 fixed mutations. Eventually, 12 mutations were determined to be drug resistance related, although only seven of these mutations reached fixation stage. This observation indicates that the competition between high and low effective resistance mutations lead to high resistance.
Alternative mechanisms implicated in drug resistance
MTB shows intrinsic resistance to different drugs through various mechanisms, including cell wall or membrane impermeability and efflux pump action. Mutations can enhance intrinsic resistance, generate new proteins that inactivate the drug or block interactions with its target, or alter the target to prevent its recognition by the drugs (Figure 3).50
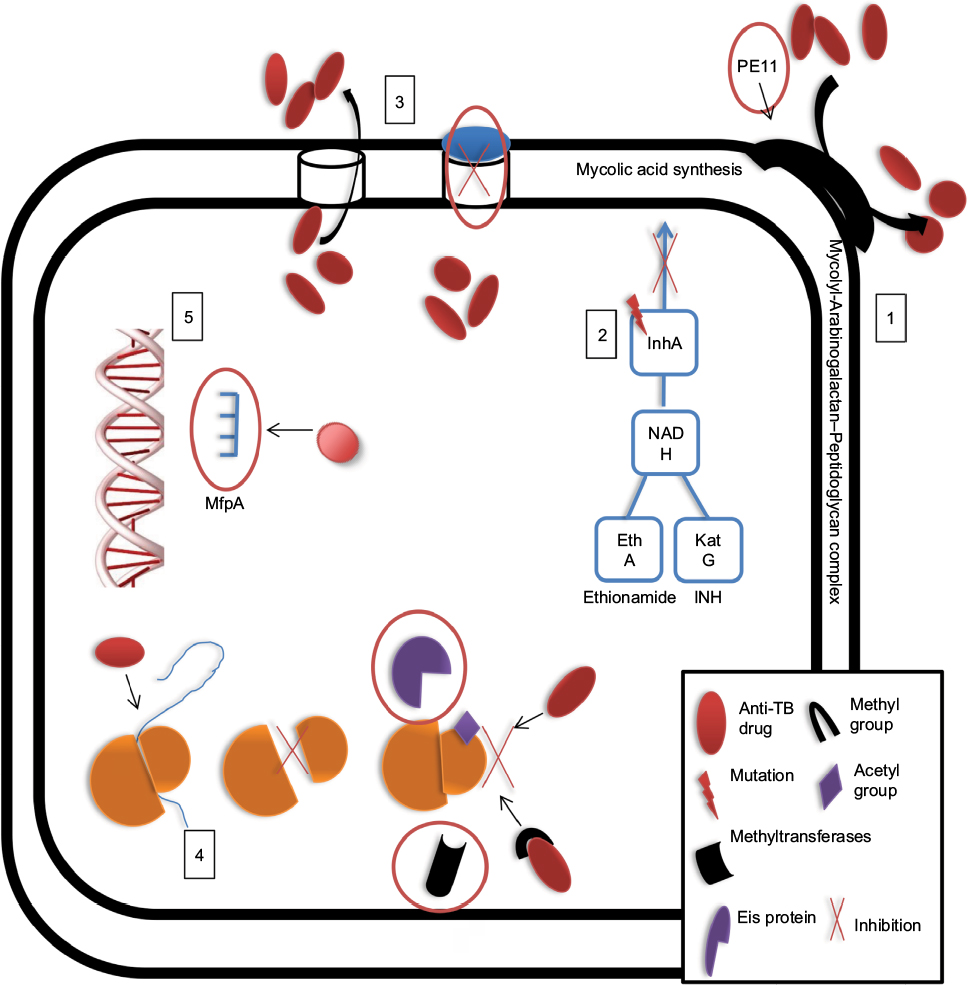 | Figure 3 MTB can exhibit resistance to drugs via: 1, intrinsically decreased permeability of the cell wall; 2, acquisition of mutations that block drug entry; 3, extrusion drugs via efflux pumps; 4, modification of the drug or its target, or 5, target mimicry. Note: Possible drug targets are indicated by red circles. Abbreviations: INH, isoniazid; MTB, Mycobacterium tuberculosis; TB, tuberculosis. |
Impermeability of the MTB cell wall and drug resistance
The MTB cell wall structure is unique due to mycolyl-arabinogalactan–peptidoglycan complexes and free glycolipids (e.g., trehalose dimycolate, PPE family proteins, and phthiocerol dimycocerosates), forming a hydrophobic barrier that limits the entrance of various drugs.51 Several studies have identified the essential genes, enzymatic activities, and cellular components that increase resistance levels by decreasing cell wall permeability. For example, the expression of monooxygenase (mymA) operon is regulated by virS, which maintains mycolic acid composition and enhances cell wall integrity.52 Mutations in virS-mymA lead to increased cell wall permeability and diffusion of INH, RIF, PZA, and ciprofloxacin inside the cells.53–56
The PE11 protein of MTB plays a role in cell wall maintenance and is a putative lipase/esterase involved in cell wall remodeling.57 Expression of PE11 in M. smegmatis mc2155 modulates cell wall morphology, composition, aggregation, and pellicle formation and mediates resistance to INH, RIF, EMB, vancomycin, and ampicillin. These findings suggest that upregulation of PE11 in MTB reduces penetration of antibiotics during active TB.58
Moreover, MTB can preserve cell wall integrity through acquired mutations that lead to overexpression of genes encoding enzymes involved in cell wall synthesis. For example, pro-drugs, INH and ethionamide, which share a similar mechanism of action, are converted into their bioactive forms (isonicotinic-acyl radicals and 2-ethyl-4-amidopyridine, respectively) by katG and ethA gene products. These bioactive forms react with nicotinamide adenine dinucleotide to form nicotinamide adenine dinucleotide adducts that bind to enoyl-acyl carrier protein reductase (encoded by inhA), a key enzyme involved in fatty acid synthase II system, and inhibit mycolic acid synthesis.50,59 Therefore, mutations in inhA prevent binding of INH and ethionamide to their targets and confer resistance to these drugs.60 Furthermore, arabinosyl transferases (which link peptidoglycan with an outer mycolic acid layer to form the mycolyl-arabinogalactan–peptidoglycan complex) may be a target for EMB, a drug that inhibits arabinosyl transferase activity and causes increased cell wall permeability.61 When emb genes acquire mutations, overexpression of emb genes and increased EMB proteins can overcome certain levels of the drug. A specific mutation in embB at codon 306 may correlate with EMB resistance, and mutations in cell wall synthesis-associated genes aftA and ubiA lead to overexpression of embC and EmbCAB substrates and subsequent resistance to EMB.62–64 In addition, novel mutations at embA G43C and G554N and at embB S412P have found to confer a high level of resistance to EMB among MTB isolates.65,66
Overall, these findings highlight the role of MTB cell wall maintenance in intrinsic and acquired drug resistance. Thus, identification of novel proteins (e.g., PE11) that increase the cell wall integrity could facilitate their use as promising drug targets for MTB treatment.
Action of efflux pumps and drug resistance
Efflux pumps are natural drug barriers widely distributed in both prokaryotic and eukaryotic cell walls. These pumps maintain cellular hemostasis and regulate exchange of nutrients across the cell membrane.67 They are classified into six major families based on their energy source, size, and substrates. These include the ATP-binding cassette (ABC), small multidrug resistance (SMR), resistance nodulation division (RND), major facilitator superfamily (MFS), multidrug toxic compound extrusion (MATE), and drug metabolite transporter (DMT) superfamily.68 All but except MATE and DMT efflux pumps are specific to MTB strains.69 Drug resistance mediated by efflux pumps depends on their basal expression and by drug-induced gene expression or overexpression that result from acquired mutations.70
Recently, several studies have used whole-genome sequencing to identify relevant mutations in efflux pump-associated genes that confer resistance. Li et al70 examined the expression level of efflux pump genes within MDR isolates and found that at least one efflux pump was overexpressed in eight out of nine isolates, suggesting that this system contributed to the development of resistance to multiple drugs. Interestingly, one MDR isolate carried a mutation in rpoB that conferred RIF resistance but intact katG, inhA, and oxyR-ahpC, indicating that efflux pumps, rather than mutations alone, were responsible for the INH resistance. An additional study showed that mutations within the Rv0678-encoded transcription repressor of MmpL5 (RND family) led to overexpression of the MmpL5 pump and resistance to clofazimine.71 Similarly, Kanji et al72,73 examined SNPs associated with Rv2688c, Rv0194, Rv2936 (drrA), Rv2937 (drrB), and Rv1634, which encode pumps from the ABC and MFS transporter families, within XDR strains. The study showed significant levels of gene expression compared to susceptible and H37Rv strains, thereby demonstrating the importance of efflux pumps in drug resistance development in XDR isolates. Another study explored the expression of Rv2686c, Rv2687c, Rv2688c, Rv0933c, and Rv1258c within MDR and XDR isolates and found high expression of the Rv0933c-encoded PstB pump (ABC transporter family) in response to FQ treatment, suggesting that the expression of pstB is associated with resistance to FQ. In addition, MFS Tap efflux pumps showed a high level of expression, suggesting a correlation with kanamycin resistance.74 Expression of Rv1258c-encoded Tap pumps is regulated by transcription activator WhiB7, which is more highly expressed following a point mutation in the 5′ untranslated region of whiB7.75–78
These findings confirm that efflux pumps strongly contribute to developing drug resistance in MTB, thus highlighting efflux pumps as potential candidate targets for novel MTB drugs. Diverse synthetic and plant-derived molecules that act as efflux pump inhibitors (EPIs) have recently been identified, all of which exert different levels of efflux pump inhibition in MTB.79 When these inhibitors bind to efflux pumps, they increase drug retention inside the cytoplasm, restore the drug’s activity, and prevent the selection of resistant mutants.79 These results occur when EPIs act as a single drug such as an SQ109 inhibitor, or enhances the efficacy of certain drug combinations such as timcodar.80,81 More recently, Kumar et al82 found that synthesis and design of hybrid EPIs by fusion of Verapamil™ with phenothiazines enhances inhibition of efflux pumps and may enable identification of novel molecules with a high efficacy for killing MTB.
Modification of drugs and their respective targets
An additional mechanism implicated in MTB drug resistance is the modification of either the drug or its target through specific enzymes. These enzymes are often enable acetylation or methylation of the drug or its target to prevent recognition and interaction between the two. One study found that MTB and M. bovis carry erm37, which encodes an rRNA methyltransferase that blocks interactions between macrolides and the ribosome.83 MTB also expresses enhanced intracellular survival (EIS) proteins, which are homologs of AG acetyltransferases.84 These proteins can acetylate multiple sites of AGs, resulting in their inactivation.85 Houghton et al87 found that EIS proteins can also protect MTB against capreomycin.86 A more recent study conducted by Warrier et al determined that MTB can methylate drugs via N-methylation, such as inactivation of the “14” drug at the N-5 position via a methyltransferase encoded by Rv0560c.93
Target mimicry and drug resistance
Target mimicry is a novel mechanism developed by MDR strains to detoxify FQ drugs, which target DNA gyrase. When FQs bind to DNA gyrase, they inhibit DNA replication, repair, and transcription.88 A previous study showed that FQ resistance develops through the Mycobacteria FQ resistance protein A (MfpA).89 MfpA can resemble the shape, size, and surface of the DNA double helix, suggesting that MfpA mimics the structure of MTB DNA.90,91 Once MfpA binds to DNA gyrase, the protein prevents FQ binding and protects MTB from the drug’s action.92
Conclusion
The ongoing evolution of resistance mechanisms among MTB population is a serious concern. In this review, we highlighted the major mechanisms that lead to drug resistance. These mechanisms include compensatory evolution, epistasis, clonal interference, decreased cell wall permeability, overexpression of efflux pumps, drug/target modification, and target mimicry. These mechanisms allow MTB to modulate their fitness, enhance their transmissibility, and stabilize the resistance phenotype within their population. Understanding of these mechanisms enable researchers to identify novel drug targets in order to develop effective drugs.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. Global tuberculosis report 2016. Geneva: World Health Organization; 2016. Available from: http://www.who.int/tb/publications/global_report/en/. Accessed October 4, 2017. | ||
Brites D, Gagneux S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol Rev. 2015;264(1):6–24. | ||
Gutierrez MC, Brisse S, Brosch R, et al. Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis. PLoS Pathog. 2005;1(1):19. | ||
Hershberg R, Lipatov M, Small PM, et al. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 2008;6(12):0060311. | ||
Comas I, Coscolla M, Luo T, et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45(10):1176–1182. | ||
Delogu G, Sali M, Fadda G. The biology of Mycobacterium tuberculosis infection. Mediterr J Hematol Infect Dis. 2013;5(1):070. | ||
Fonseca JD, Knight GM, McHugh TD. The complex evolution of antibiotic resistance in Mycobacterium tuberculosis. Int J Infect Dis. 2015;32:94–100. | ||
Combs DL, O’Brien RJ, Geiter LJ. USPHS tuberculosis short-course chemotherapy trial 21: effectiveness, toxicity, and acceptability. The report of final results. Ann Intern Med. 1990;112(6):397–406. | ||
Espinal MA, Kim SJ, Suarez PG, et al. Standard short-course chemotherapy for drug-resistant tuberculosis: treatment outcomes in 6 countries. JAMA. 2000;283(19):2537–2545. | ||
Gunther G. Multidrug-resistant and extensively drug-resistant tuberculosis: a review of current concepts and future challenges. Clin Med. 2014;14(3):279–285. | ||
Coscolla M, Gagneux S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin Immunol. 2014;26(6):431–444. | ||
Coll F, McNerney R, Guerra-Assuncao JA, et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat Commun. 2014;5:4812. | ||
Borrell S, Gagneux S. Infectiousness, reproductive fitness and evolution of drug-resistant Mycobacterium tuberculosis. Int J Tuberc Lung Dis. 2009;13(12):1456–1466. | ||
McGrath M, Gey van Pittius NC, van Helden PD, Warren RM, Warner DF. Mutation rate and the emergence of drug resistance in Mycobacterium tuberculosis. J Antimicrob Chemother. 2014;69(2):292–302. | ||
Merker M, Kohl TA, Roetzer A, et al. Whole genome sequencing reveals complex evolution patterns of multidrug-resistant Mycobacterium tuberculosis Beijing strains in patients. PLoS One. 2013;8(12):e82551. | ||
Dos Vultos T, Mestre O, Rauzier J, et al. Evolution and diversity of clonal bacteria: the paradigm of Mycobacterium tuberculosis. PLoS One. 2008;3(2):0001538. | ||
Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJ. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science. 2006;312(5782):1944–1946. | ||
Billington OJ, McHugh TD, Gillespie SH. Physiological cost of rifampin resistance induced in vitro in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1999;43(8):1866–1869. | ||
Davies AP, Billington OJ, Bannister BA, Weir WR, McHugh TD, Gillespie SH. Comparison of fitness of two isolates of Mycobacterium tuberculosis, one of which had developed multi-drug resistance during the course of treatment. J Infect. 2000;41(2):184–187. | ||
Sander P, Springer B, Prammananan T, et al. Fitness cost of chromosomal drug resistance-conferring mutations. Antimicrob Agents Chemother. 2002;46(5):1204–1211. | ||
Bottger EC, Springer B, Pletschette M, Sander P. Fitness of antibiotic-resistant microorganisms and compensatory mutations. Nat Med. 1998;4(12):1343–1344. | ||
Andersson DI, Hughes D. Persistence of antibiotic resistance in bacterial populations. FEMS Microbiol Rev. 2011;35(5):901–911. | ||
Spratt BG. Antibiotic resistance: counting the cost. Curr Biol. 1996;6(10):1219–1221. | ||
Olofsson SK, Cars O. Optimizing drug exposure to minimize selection of antibiotic resistance. Clin Infect Dis. 2007;1(45):S129–S136. | ||
Gagneux S, Burgos MV, DeRiemer K, et al. Impact of bacterial genetics on the transmission of isoniazid-resistant Mycobacterium tuberculosis. PLoS Pathog. 2006;2(6):16. | ||
van Doorn HR, de Haas PE, Kremer K, Vandenbroucke-Grauls CM, Borgdorff MW, van Soolingen D. Public health impact of isoniazid-resistant Mycobacterium tuberculosis strains with a mutation at amino-acid position 315 of katG: a decade of experience in The Netherlands. Clin Microbiol Infect. 2006;12(8):769–775. | ||
Andersson DI, Hughes D. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol. 2010;8(4):260–271. | ||
Andersson DI, Levin BR. The biological cost of antibiotic resistance. Curr Opin Microbiol. 1999;2(5):489–493. | ||
Sherman DR, Mdluli K, Hickey MJ, et al. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science. 1996;272(5268):1641–1643. | ||
Ramaswamy S, Musser JM. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber Lung Dis. 1998;79(1):3–29. | ||
Somoskovi A, Parsons LM, Salfinger M. The molecular basis of resistance to isoniazid, rifampin, and pyrazinamide in Mycobacterium tuberculosis. Respir Res. 2001;2(3):164–168. | ||
Mariam DH, Mengistu Y, Hoffner SE, Andersson DI. Effect of rpoB mutations conferring rifampin resistance on fitness of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2004;48(4):1289–1294. | ||
Comas I, Borrell S, Roetzer A, et al. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet. 2011;44(1):106–110. | ||
Yang C, Luo T, Shen X, et al. Transmission of multidrug-resistant Mycobacterium tuberculosis in Shanghai, China: a retrospective observational study using whole-genome sequencing and epidemiological investigation. Lancet Infect Dis. 2017;17(3):275–284. | ||
de Vos M, Muller B, Borrell S, et al. Putative compensatory mutations in the rpoC gene of rifampin-resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob Agents Chemother. 2013;57(2):827–832. | ||
Brandis G, Pietsch F, Alemayehu R, Hughes D. Comprehensive phenotypic characterization of rifampicin resistance mutations in Salmonella provides insight into the evolution of resistance in Mycobacterium tuberculosis. J Antimicrob Chemother. 2015;70(3):680–685. | ||
Meftahi N, Namouchi A, Mhenni B, Brandis G, Hughes D, Mardassi H. Evidence for the critical role of a secondary site rpoB mutation in the compensatory evolution and successful transmission of an MDR tuberculosis outbreak strain. J Antimicrob Chemother. 2016;71(2):324–332. | ||
Freihofer P, Akbergenov R, Teo Y, Juskeviciene R, Andersson DI, Bottger EC. Nonmutational compensation of the fitness cost of antibiotic resistance in mycobacteria by overexpression of tlyA rRNA methylase. RNA. 2016;22(12):1836–1843. | ||
Phillips PC. Epistasis--the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet. 2008;9(11):855–867. | ||
Wong A. Epistasis and the evolution of antimicrobial resistance. Front Microbiol. 2017;8:246. | ||
Borrell S, Teo Y, Giardina F, et al. Epistasis between antibiotic resistance mutations drives the evolution of extensively drug-resistant tuberculosis. Evol Med Public Health. 2013;1:65–74. | ||
Trindade S, Sousa A, Xavier KB, Dionisio F, Ferreira MG, Gordo I. Positive epistasis drives the acquisition of multidrug resistance. PLoS Genet. 2009;5(7):24. | ||
Borrell S, Gagneux S. Strain diversity, epistasis and the evolution of drug resistance in Mycobacterium tuberculosis. Clin Microbiol Infect. 2011;17(6):815–820. | ||
Schrag SJ, Perrot V, Levin BR. Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc Biol Sci. 1997;264(1386):1287–1291. | ||
Muller B, Borrell S, Rose G, Gagneux S. The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis. Trends Genet. 2013;29(3):160–169. | ||
Fenner L, Egger M, Bodmer T, et al. Effect of mutation and genetic background on drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2012;56(6):3047–3053. | ||
Sun G, Luo T, Yang C, et al. Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. J Infect Dis. 2012;206(11):1724–1733. | ||
Muller HJ. Some genetic aspects of sex. Am Nat. 1932;66(703):118–138. | ||
Eldholm V, Norheim G, von der Lippe B, et al. Evolution of extensively drug-resistant Mycobacterium tuberculosis from a susceptible ancestor in a single patient. Genome Biol. 2014;15(11):490. | ||
Green KD, Garneau-Tsodikova S. Resistance in tuberculosis: what do we know and where can we go? Front Microbiol. 2013;4:208. | ||
Favrot L, Ronning DR. Targeting the mycobacterial envelope for tuberculosis drug development. Expert Rev Anti Infect Ther. 2012;10(9):1023–1036. | ||
Forrellad MA, Klepp LI, Gioffre A, et al. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. 2013;4(1):3–66. | ||
Singh A, Gupta R, Vishwakarma RA, et al. Requirement of the mymA operon for appropriate cell wall ultrastructure and persistence of Mycobacterium tuberculosis in the spleens of guinea pigs. J Bacteriol. 2005;187(12):4173–4186. | ||
Singh A, Jain S, Gupta S, Das T, Tyagi AK. mymA operon of Mycobacterium tuberculosis: its regulation and importance in the cell envelope. FEMS Microbiol Lett. 2003;227(1):53–63. | ||
Gao LY, Laval F, Lawson EH, et al. Requirement for kasB in Mycobacterium mycolic acid biosynthesis, cell wall impermeability and intracellular survival: implications for therapy. Mol Microbiol. 2003;49(6):1547–1563. | ||
Philalay JS, Palermo CO, Hauge KA, Rustad TR, Cangelosi GA. Genes required for intrinsic multidrug resistance in Mycobacterium avium. Antimicrob Agents Chemother. 2004;48(9):3412–3418. | ||
Singh G, Singh G, Jadeja D, Kaur J. Lipid hydrolyzing enzymes in virulence: Mycobacterium tuberculosis as a model system. Crit Rev Microbiol. 2010;36(3):259–269. | ||
Singh P, Rao RN, Reddy JR, et al. PE11, a PE/PPE family protein of Mycobacterium tuberculosis is involved in cell wall remodeling and virulence. Sci Rep. 2016;6:21624. | ||
Marrakchi H, Laneelle MA, Daffe M. Mycolic acids: structures, biosynthesis, and beyond. Chem Biol. 2014;21(1):67–85. | ||
Banerjee A, Dubnau E, Quemard A, et al. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263(5144):227–230. | ||
Telenti A, Philipp WJ, Sreevatsan S, et al. The emb operon, a gene cluster of Mycobacterium tuberculosis involved in resistance to ethambutol. Nat Med. 1997;3(5):567–570. | ||
Alcaide F, Pfyffer GE, Telenti A. Role of embB in natural and acquired resistance to ethambutol in mycobacteria. Antimicrob Agents Chemother. 1997;41(10):2270–2273. | ||
Safi H, Sayers B, Hazbon MH, Alland D. Transfer of embB codon 306 mutations into clinical Mycobacterium tuberculosis strains alters susceptibility to ethambutol, isoniazid, and rifampin. Antimicrob Agents Chemother. 2008;52(6):2027–2034. | ||
Safi H, Lingaraju S, Amin A, et al. Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-beta-D-arabinose biosynthetic and utilization pathway genes. Nat Genet. 2013;45(10):1190–1197. | ||
Zhao LL, Sun Q, Liu HC, et al. Analysis of embCAB mutations associated with ethambutol resistance in multidrug-resistant Mycobacterium tuberculosis isolates from China. Antimicrob Agents Chemother. 2015;59(4):2045–2050. | ||
Xu Y, Jia H, Huang H, Sun Z, Zhang Z. Mutations Found in embCAB, embR, and ubiA genes of ethambutol-sensitive and -resistant Mycobacterium tuberculosis clinical isolates from China. Biomed Res Int. 2015;951706(10):31. | ||
Nguyen L. Antibiotic resistance mechanisms in M. tuberculosis: an update. Arch Toxicol. 2016;90(7):1585–1604. | ||
Sun J, Deng Z, Yan A. Bacterial multidrug efflux pumps: mechanisms, physiology and pharmacological exploitations. Biochem Biophys Res Commun. 2014;453(2):254–267. | ||
Liu H, Xie JP. Comparative genomics of Mycobacterium tuberculosis drug efflux pumps and their transcriptional regulators. Crit Rev Eukaryot Gene Expr. 2014;24(2):163–180. | ||
Li G, Zhang J, Guo Q, et al. Efflux pump gene expression in multidrug-resistant Mycobacterium tuberculosis clinical isolates. PLoS One. 2015;10(2):e0119013. | ||
Zhang S, Chen J, Cui P, Shi W, Zhang W, Zhang Y. Identification of novel mutations associated with clofazimine resistance in Mycobacterium tuberculosis. J Antimicrob Chemother. 2015;70(9):2507–2510. | ||
Kanji A, Hasan R, Zaver A, et al. Alternate efflux pump mechanism may contribute to drug resistance in extensively drug-resistant isolates of Mycobacterium tuberculosis. Int J Mycobacteriol. 2016;5(suppl 1): S97–S98. | ||
Kanji A, Hasan R, Zhang Y, et al. Increased expression of efflux pump genes in extensively drug-resistant isolates of Mycobacterium tuberculosis. Int J Mycobacteriol. 2016;5(suppl 1):S150. | ||
Oh TS, Kim YJ, Kang HY, Kim CK, Cho SY, Lee HJ. RNA expression analysis of efflux pump genes in clinical isolates of multidrug-resistant and extensively drug-resistant Mycobacterium tuberculosis in South Korea. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases. 2017;49:111–115. | ||
Reeves AZ, Campbell PJ, Sultana R, et al. Aminoglycoside cross-resistance in Mycobacterium tuberculosis due to mutations in the 5’ untranslated region of whiB7. Antimicrob Agents Chemother. 2013;57(4):1857–1865. | ||
Ainsa JA, Blokpoel MC, Otal I, Young DB, De Smet KA, Martin C. Molecular cloning and characterization of Tap, a putative multidrug efflux pump present in Mycobacterium fortuitum and Mycobacterium tuberculosis. J Bacteriol. 1998;180(22):5836–5843. | ||
Burian J, Ramon-Garcia S, Sweet G, Gomez-Velasco A, Av-Gay Y, Thompson CJ. The mycobacterial transcriptional regulator whiB7 gene links redox homeostasis and intrinsic antibiotic resistance. J Biol Chem. 2012;287(1):299–310. | ||
Morris RP, Nguyen L, Gatfield J, et al. Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2005;102(34):12200–12205. | ||
Song L, Wu X. Development of efflux pump inhibitors in antituberculosis therapy. Int J Antimicrob Agents. 2016;47(6):421–429. | ||
de Knegt GJ, van der Meijden A, de Vogel CP, Aarnoutse RE, de Steenwinkel JE. Activity of moxifloxacin and linezolid against Mycobacterium tuberculosis in combination with potentiator drugs verapamil, timcodar, colistin and SQ109. Int J Antimicrob Agents. 2017;49(3):302–307. | ||
Grossman TH, Shoen CM, Jones SM, Jones PL, Cynamon MH, Locher CP. The efflux pump inhibitor timcodar improves the potency of antimycobacterial agents. Antimicrob Agents Chemother. 2015;59(3):1534–1541. | ||
Kumar M, Singh K, Naran K, et al. Design, synthesis, and evaluation of novel hybrid efflux pump inhibitors for use against Mycobacterium tuberculosis. ACS infectious diseases. 2016;2(10):714–725. | ||
Buriankova K, Doucet-Populaire F, Dorson O, et al. Molecular basis of intrinsic macrolide resistance in the Mycobacterium tuberculosis complex. Antimicrob Agents Chemother. 2004;48(1):143–150. | ||
Zaunbrecher MA, Sikes RD Jr, Metchock B, Shinnick TM, Posey JE. Overexpression of the chromosomally encoded aminoglycoside acetyltransferase Eis confers kanamycin resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2009;106(47):20004–20009. | ||
Chen W, Biswas T, Porter VR, Tsodikov OV, Garneau-Tsodikova S. Unusual regioversatility of acetyltransferase Eis, a cause of drug resistance in XDR-TB. Proc Natl Acad Sci U S A. 2011;108(24):9804–9808. | ||
Houghton JL, Green KD, Pricer RE, Mayhoub AS, Garneau-Tsodikova S. Unexpected N-acetylation of capreomycin by mycobacterial Eis enzymes. J Antimicrob Chemother. 2013;68(4):800–805. | ||
Malone KM, Gordon SV. Antibiotic methylation: a new mechanism of antimicrobial resistance. Trends Microbiol. 2016;24(10):771–772. | ||
Andriole VT. The quinolones: past, present, and future. Clin Infect Dis. 2005;15(41):S113–S119. | ||
Montero C, Mateu G, Rodriguez R, Takiff H. Intrinsic resistance of Mycobacterium smegmatis to fluoroquinolones may be influenced by new pentapeptide protein MfpA. Antimicrob Agents Chemother. 2001;45(12):3387–3392. | ||
Ferber D. Biochemistry. Protein that mimics DNA helps tuberculosis bacteria resist antibiotics. Science. 2005;308(5727):1393. | ||
Hegde SS, Vetting MW, Roderick SL, et al. A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science. 2005;308(5727):1480–1483. | ||
Vetting MW, Hegde SS, Fajardo JE, et al. Pentapeptide repeat proteins. Biochemistry. 2006;45(1):1–10. | ||
Warrier T, Kapilashrami K, Argyrou A, et al. N-methylation of a bactericidal compound as a resistance mechanism in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2016;113(31):E4523–E4530. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.