Back to Journals » Cancer Management and Research » Volume 11
Disulfiram inhibits epithelial–mesenchymal transition through TGFβ–ERK–Snail pathway independently of Smad4 to decrease oral squamous cell carcinoma metastasis
Authors Bu W ![]() , Wang Z, Meng L, Li X, Liu X, Chen Y, Xin Y, Li B, Sun H
, Wang Z, Meng L, Li X, Liu X, Chen Y, Xin Y, Li B, Sun H
Received 30 December 2018
Accepted for publication 7 April 2019
Published 1 May 2019 Volume 2019:11 Pages 3887—3898
DOI https://doi.org/10.2147/CMAR.S199912
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Harikrishna Nakshatri
Wenhuan Bu, 1* Zilin Wang, 1,* Lin Meng, 1,* Xing Li, 2 Xinchen Liu, 1 Yumeng Chen, 1 Ying Xin, 3 Baoquan Li, 4 Hongchen Sun 1
1Department of Oral Pathology, School and Hospital of Stomatology, Jilin University, 130000 Changchun, People’s Republic of China; 2School and Hospital of Stomatology, China Medical University, 110000 Shenyang, People’s Republic of China; 3Department of Oral Pathology, Hospital of Stomatology, Xi’an Jiaotong University, Xi’an, 710000, People’s Republic of China; 4Department of Temporomandibular Joint, School and Hospital of Stomatology, Jilin University, 130000 Changchun, People’s Republic of China
*These authors contributed equally to this work
Purpose: Smad4 loss is highly related to poor prognosis and decreased patient survival in oral squamous cell carcinoma (OSCC), suggesting that agents that target both Smad4-mutated and Smad4 wild-type cells could treat OSCC more effectively. Disulfiram (Dsf) has anticancer activity through a variety of mechanisms, including inhibition of epithelial–mesenchymal transition (EMT). It remains unclear whether Dsf has the same effect on Smad4-mutated and Smad4 wild-type OSCC or not and what mechanism is involved.
Methods: Effect of Dsf on TGFβ 1-induced EMT in CAL27 (Smad4 mutation) and SCC25 (Smad4 wild-type) cells were evaluated through analyzing changes in morphology, expression of EMT markers, and migration and invasion of cells. The ERK-pathway inhibitor U0126 was used to confirm TGFβ–ERK–Snail pathway–mediated cell behavior. Dsf’s effects on tumor growth and metastasis in vivo were examined through a subcutaneous xenograft mouse model and an intravenous tumor mouse model.
Results: Dsf inhibited TGFβ 1-induced EMT through suppression of morphological change, EMT-marker expression, and cell migration and invasion in both CAL27 and SCC25. Phosphorylation of ERK and expression of Snail were blocked by Dsf treatment. Like Dsf, U0126 had a similar effect on EMT of CAL27 and SCC25. Dsf also reduced tumor growth and metastasis in vivo, accompanied by decreased expression of EMT markers in tumors.
Conclusion: These results indicated that Dsf inhibited EMT of OSCC in vitro and in vivo independently of Smad4 through suppression of the TGFβ–ERK–Snail pathway, suggesting the broad-spectrum anticancer potential of Dsf for clinical use against OSCC.
Keywords: disulfiram, epithelial–mesenchymal transition, Smad4 mutation, oral squamous cell carcinoma
Corrigendum for this paper has been published
Further corrigendum has been published
Introduction
Oral cancer, with a large majority of cases (~90%) being oral squamous cell carcinoma (OSCC),1 is the sixth-commonest malignancy globally.2 Although diversified prognostic tools were contrived to improve the poor survival rates among patients with OSCC — for example, the eighth edition of the cancer staging manual published by the American Joint Committee on Cancer in 2016 incorporated two new parameters: depth of invasion and extranodal extension in the staging system for OSCC — the 5-year survival rate of OSCC patients remains stagnated at approximately 40%–50%.3,4 In 2018, >300,000 (2% of all cancer cases) people were newly diagnosed with cancer of the lip and oral cavity, which makes oral cancer a leading cause of cancer death, especially among men in South Asia.5
Numerous studies have demonstrated that epithelial–mesenchymal transition (EMT) is implicated in promoting carcinoma invasion and metastasis in multiple kinds of cancer,6 including oral cancer.7 EMT is a highly conserved cellular program in which polarized immotile epithelial cells convert to motile mesenchymal cells.8 It is a fundamental process for morphogenesis during embryonic development, tissue remodeling and wound healing, which involves a variety of signaling pathways (eg, TGFβ, Wnt, Notch) and is characterized by downregulated expression of epithelial proteins, such as E-cadherin, and upregulated expression of mesenchymal protein, such as vimentin.9 Furthermore, it has been proved that decreased E-cadherin and increased vimentin are closely associated with recurrence and death in OSCC patients.10 Also, transcription factors, such as Snail, Slug, ZEB1, ZEB2, Twist1, and Twist2, responsible for suppressing E-cadherin expression, are related to EMT.11
As a genetic disease, cancer usually accumulates genetic and epigenetic aberrations,12 which cause heterogeneity of cancer, leading to worse clinical outcomes. Smad4 is a strong tumor suppressor, the loss or inactivation of which is common in tumorigenesis.13 Loss of chromosome 18q, a region encoding Smad4, is commonly found in human head and neck SCC (HNSCC),14 which is associated with decreased survival rates. Research on Smad4 expression in human HNSCC showed that 86% of tumors and 67% of adjacent nonmalignant mucosa had >50% Smad4 reduction and a Smad4-knockout mouse model showed increased HNSCC susceptibility.15 As an important signaling molecule in the TGFβ pathway, Smad4 plays an ambiguous role in TGFβ-induced EMT, dependent on cell type. In NMuMG mouse mammary gland epithelial cells, Smad4 loss abolished TGFβ-induced EMT,16 while in Colo-357 pancreatic tumor cells, Smad4 was dispensable in TGFβ-induced EMT.17 However, it is still unknown whether Smad4 is essential for TGFβ-induced EMT in OSCC.
Disulfiram (Dsf), which has been used for alcohol dependence since the 1940s,18 has shown anticancer activity for four decades and been widely researched.19 Dsf plays its anticancer role through blockage of proteasome activity,20 induction of cell apoptosis,21 inhibition of invasion and angiogenesis,22 suppression of stem cell–like properties,23 and reduction of drug resistance.24 Besides acting on cancer cells, Dsf can affect tumor stromata, such as macrophages, resulting in disturbed cancer progression.25 In the field of metastasis, research has shown that Dsf suppresses invasion of tumor cells through inhibition of MMP2 and MMP9 activity.26 In addition, it has been reported that Dsf inhibited TGFβ-induced EMT in breast cancer cells and tumor growth in a xenograft model.27 Although Dsf’s anticancer activity has been proved in a variety of cancers, previous studies ignored the existence of different mutants in cancer. It remains unclear whether Dsf has the same effect on Smad4-mutated and Smad4 wild-type OSCC cells or not and what mechanism is involved.
In this study, we discovered that Dsf inhibited EMT both in Smad4-mutated and Smad4 wild-type OSCC cells. This inhibition of EMT was attributed to downregulation of the TGFβ–ERK–Snail pathway independently of Smad4, leading to decreased tumor growth and reduced metastasis in vivo.
Methods
Reagents
Dsf and U0126 were purchased from MCE (USA) and dissolved in DMSO. TGFβ1 was purchased from Proteintech (USA) and dissolved in 4 mM HCl containing 0.1% endotoxin-free human serum albumin to a concentration of 0.5–1.0 mg/mL. Primary antibodies against E-cadherin, vimentin, and GAPDH were purchased from Proteintech, antibodies to ERK and p-ERK from Cell Signaling Technology (USA), antibodies to Snail from Abcam (UK), and antibodies to Ki67 from Santa Cruz Biotechnology (USA).
Cell culture
The human OSCC cell lines CAL27 and SCC25 were obtained from Nanjing Keygen Biotech (China). These cells were grown in DMEM ( Thermo Fisher Scientific, USA) containing 10% FBS (Biological Industries, Israel) with 100 U/mL penicillin and 100 μg/mL streptomycin (HyClone, USA) in a 37°C humidified incubator containing 5% CO2.
Cytotoxicity assay
Cells were seeded at 104/well in a 96-well plate overnight, followed by incubation with Dsf at different concentrations for 24 hours. Then, 10 μL CCK8 solution was added for 2 hours at 37°C. OD450 values were obtained with a microplate reader (Bio-Rad, USA). Percentage cell viability was calculated compared to control wells without Dsf.
Cell-apoptosis analysis
Apoptosis analysis was performed with an annexin V APC apoptosis-analysis kit (Tianjin Sungene Biotech, China). Briefly, cells from different groups were collected, washed with cold PBS, resuspended in 100 μL of binding buffer. Annexin V APC (5 μL) and 5 μL propidium iodide solution were added to the cell solution for 15 minutes at room temperature, followed by flow-cytometry analysis.
Cell-cycle assessment
Cells from different groups were collected before fixing them with cold ethanol for 1 hourr at room temperature. Cells were washed with cold PBS, followed by resuspension in 0.5 mL propidium iodide–RNase staining solution (Tianjin Sungene Biotech, China). After incubation for 30 minutes at room temperature, samples were analyzed by flow cytometry.
Immunofluorescence staining
Cells were cultured in six-well plates and treated with different drugs. After being washed with PBS twice, cells were fixed with 4% paraformaldehyde for 20 minutes at room temperature. Then, they were permeabilized with 0.1% Triton X-100 and blocked with 5% BSA, followed by incubation with primary antibody over night at 4°C. After incubation with Cy3-labeled goat antirabbit IgG antibody (Proteintech) for 1 hour at room temperature in the dark, cells were counterstained with DAPI. Images were collected using fluorescence microscopy (Olympus, Japan).
Western blot
Cells were washed with cold PBS twice and lysed in RIPA buffer containing protease inhibitor and phosphatase inhibitor. Then, the lysate was centrifuged for 20 minutes and the supernatants retained. Protein samples were separated by 8%–12% SDS-PAGE and transferred onto polyvinylidene difluoride membranes. After being blocked with 5% BSA in TBST, membranes were incubated with primary antibodies overnight at 4°C and subsequently incubated with secondary antibodies (Proteintech). The signals were detected with an enhanced chemiluminescence–detection reagent. Results were normalized to the internal control — GAPDH.
In vitro migration and invasion assays
The upper chamber of the transwell (Corning Costar, USA) was coated with Matrigel (BD, USA) for invasion assays and without Matrigel for migration assays. Cells were pretreated with Dsf for 24 hours before simulation of TGFβ1 and Dsf for 6 days, then seeded in the upper chamber (5×104/well) with DMEM while supplying DMEM containing 10% FBS in the lower chamber. After incubation for 24 hours, cotton swabs were used to remove cells in the upper chamber and invaded cells that had traversed to the reverse side of the membrane were fixed with 4% paraformaldehyde and stained with gentian violet.
Animal experiments
Male BALB/c nude mice 4–6 weeks old (Beijing Vital River Laboratory Animal Technology, China) were housed in pathogen-free conditions. For the subcutaneous tumor model, CAL27 cells (5×106/100 μL DMEM per mouse) were subcutaneously injected in the right flank of mice. When the average tumor volume had reached 100 mm3, the mice were randomly divided into two groups, which were treated with vehicle (saline only) or Dsf (50 mg/kg) twice a week by intragastric administration for 38 days. Tumor volume was measured at the same time. For the intravenous tumor model, CAL27 cells were pretreated with Dsf and TGFβ1 in vitro, then 5×106 cells/100 μL DMEM per mouse) were injected into the caudal vein, followed by 2 months' housing. Then, these mice were killed, and tumors and organs collected were fixed, paraffin-embedded, sectioned, and stained with H&E.
Animal experiments were conducted according to the animal care guidelines of Jilin University. Animal use in this study was approved by the Institutional Animal Care and Use Committee of Jilin University, Changchun, China (permit SYXK 2018-0006).
Immunohistochemistry
Immunohistochemistry was performed with formalin-fixed, paraffin-embedded tumor-sample sections. After being deparaffinized and rehydrated, sections were stained with primary antibodies for 2 hours at room temperature. Then, sections were incubated with biotinylated secondary antibody, followed by a horseradish peroxidase–conjugated streptavidin–biotin complex. After developqment in diaminobenzidine, these sections were observed under light microscopy.
Statistical analysis
All in vitro experiments were repeated at least three times. All data are expressed as means ± SEM. Comparisons among multiple group were made by one-way ANOVA followed by Tukey post hoc analysis. P<0.05 was used for statistical significance.
Results
Dsf suppressed TGFβ1-induced EMT in OSCC cells
TGFβ1 was used to induce EMT in OSCC CAL27 (Smad4-mutated) and SCC-25 (Smad4 wild-type) cells in vitro.28 To eliminate the interference of cell death and inhibition of cell proliferation in TGFβ1-induced EMT, we initially determined noncytotoxic Dsf concentration by CCK8 assay (
Morphological change, which is indicative of EMT, was initially observed (Figure 1, A and E). TGFβ1 induction endowed CAL27 and SCC25 with fibroblast-like mesenchymal morphology characterized by increased aspect ratio in cells (Figure 1, B and F). Dsf treatment inhibited TGFβ1-induced morphological change in a dose-dependent manner. With increased Dsf concentration, cells kept an epithelial-like appearance, even when stimulated by TGFβ1. To illuminate further whether Dsf could ameliorate abnormal expression of EMT-related proteins, we performed Western blot assays and immunofluorescence staining (Figure 1, C, D, G, and H). As expected, Dsf blocked TGFβ1-induced upregulation of the mesenchymal marker vimentin, as well as downregulation of the epithelial marker E-cadherin. These results indicated that Dsf can inhibit TGFβ1-induced EMT in both CAL27 and SCC25.
 | Figure 1 Dsf inhibited TGFβ1-induced EMT in OSCC cells. Notes: EMT in OSCC cells was induced by TGFβ1 stimulation for 6 days. Dsf was added to cells 24 hours before TGFβ1 induction to inhibit EMT. (A, E) Morphological changes in OSCC Smad4-mutated CAL27 and Smad4 wild-type SCC25 cells after stimulation with TGFβ1 (10 ng/mL) alone or combined with Dsf (5, 10, 20 μM) were observed under light microscopy. Bar 100 μm. (B, F) Aspect ratios of cells were quantified. Values represent mean ± SEM. (C, G) Western blot was performed to detect the expression of E-cadherin, vimentin and Snail in cells treated with TGFβ1 (10 ng/mL) alone or combined with Dsf (5, 10, 20 μM); quantitative results of Western blot from three independent experiments shown as mean ± SEM. (D, H) Expression of E-cadherin and vimentin examined through immunofluorescence assay by fluorescence microscopy in cells treated with TGFβ1 (10 ng/mL) alone or combined with Dsf (20 μM). *P<0.05; **P<0.01; ***P<0.001; bar 20 μm.Abbreviations: Dsf, disulfiram; OSCC, oral squamous cell carcinoma; EMT, epithelial–mesenchymal transition. |
Dsf inhibited TGFβ1-induced migration and invasion in OSCC cells
Increased cell migration and invasion are two characteristics of cells in EMT. Transwell assays were used to evaluate the effect of Dsf on migration and invasion in CAL27 and SCC25 cells. As shown in Figure 2, A and E, stimulation of TGFβ1 enhanced migration of CAL27 and SCC25 cells, while this enhancement was suppressed by Dsf in a dose-dependent manner. With increased Dsf concentration, the number of migrated cells decreased (Figure 2, B and F). Matrigel was coated on the upper chamber to simulate basement membrane in invasion assays. The results revealed that Dsf decreased the number of cells invading through Matrigel-coated filters, although TGFβ1 increased cell invasion (Figure 2, C, D, G, and H). These findings demonstrated that Dsf can inhibit migration and invasion induced by TGFβ1 in both CAL27 and SCC25 cells.
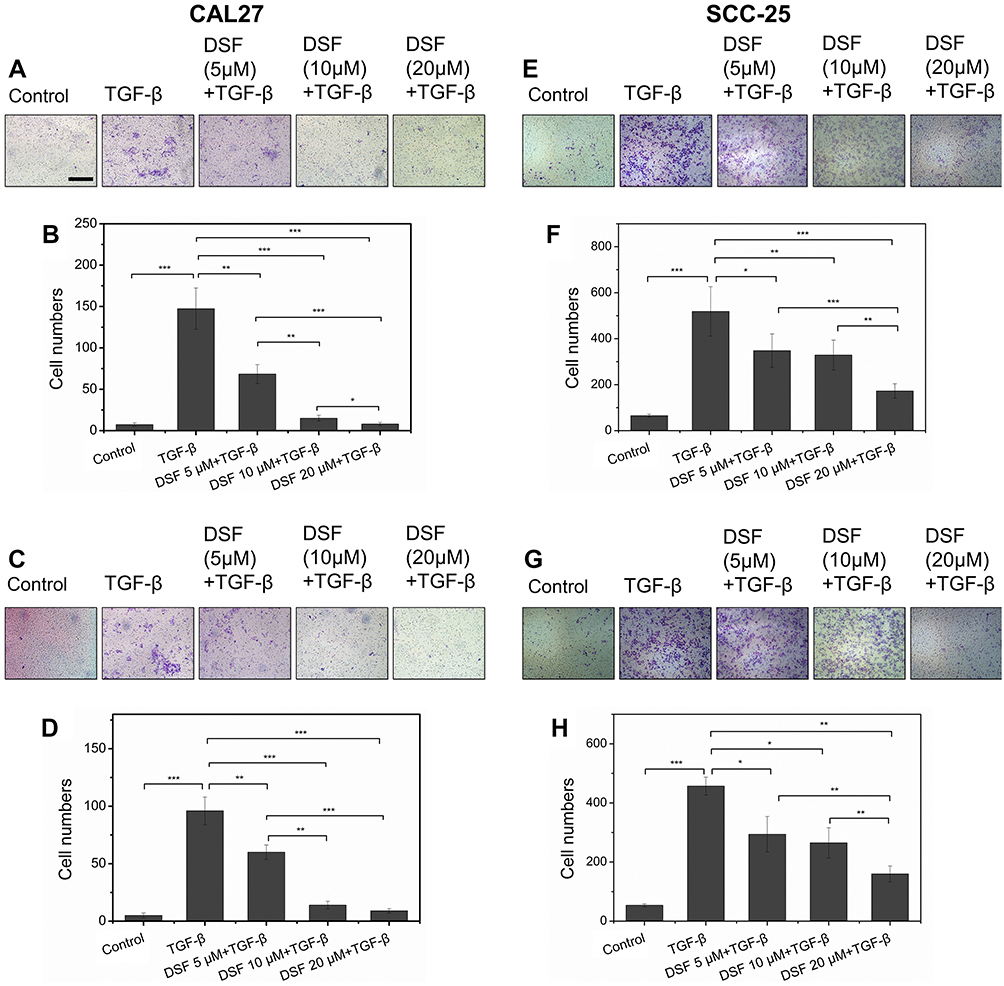 | Figure 2 Dsf suppressed TGFβ1-induced migration and invasion in OSCC cells. Notes: (A, E) Transwell-based migration assays were performed to check the effect of Dsf on cell migration of Smad4-mutated CAL27 and Smad4 wild-type SCC25 cells exposed to TGFβ1 (10 ng/mL). (B, F) Quantification of cell migration. (C, G) Transwell-based invasion assays were performed to check the effect of Dsf on cell invasion of Smad4-mutated CAL27 and Smad4 wild-type SCC25 cells exposed to TGFβ1 (10 ng/mL). (D, H) Quantification of cell invasion. Values represent mean ± SEM. *P<0.05; **P<0.01; ***P<0.001; bar 300 μm.Abbreviation: Dsf, disulfiram; OSCC, oral squamous cell carcinoma. |
Dsf inhibited TGFβ–ERK–Snail pathway in OSCC cells independently of Smad4
TGFβ can induce EMT through both Smad-dependent and Smad-independent pathwas. Smad4 loss disturbs the Smad-dependent pathway. Based on the results in Figures 1 and 2, showing Dsf blocked TGFβ1 induced EMT in both Smad4-mutated CAL27 and Smad4 wild-type SCC25 cells, we believe a Smad-independent pathway was involved in the inhibitory effect of Dsf on EMT. Phosphorylation of ERK was necessary in TGFβ1-induced EMT in murine mammary-gland epithelial cells. Therefore, we checked the change in ERK activation by Western blot. Results showed that TGFβ1 significantly promoted phosphorylation of ERK in 15 minutes, then phosphorylation decreased, while this effect was blocked by Dsf (Figure 3, A and D). We also analyzed downstream signaling of ERK, which is responsible for TGFβ1-induced EMT. As an important transcriptional factor in EMT, Snail has been reported to be upregulated by ERK and TGFβ. As shown in Figure 1, C and G , TGFβ1 stimulated the expression of Snail, but Dsf suppressed this promoting effect. All these results were similar in Smad4-mutated and Smad4 wild-type OSCC cells.
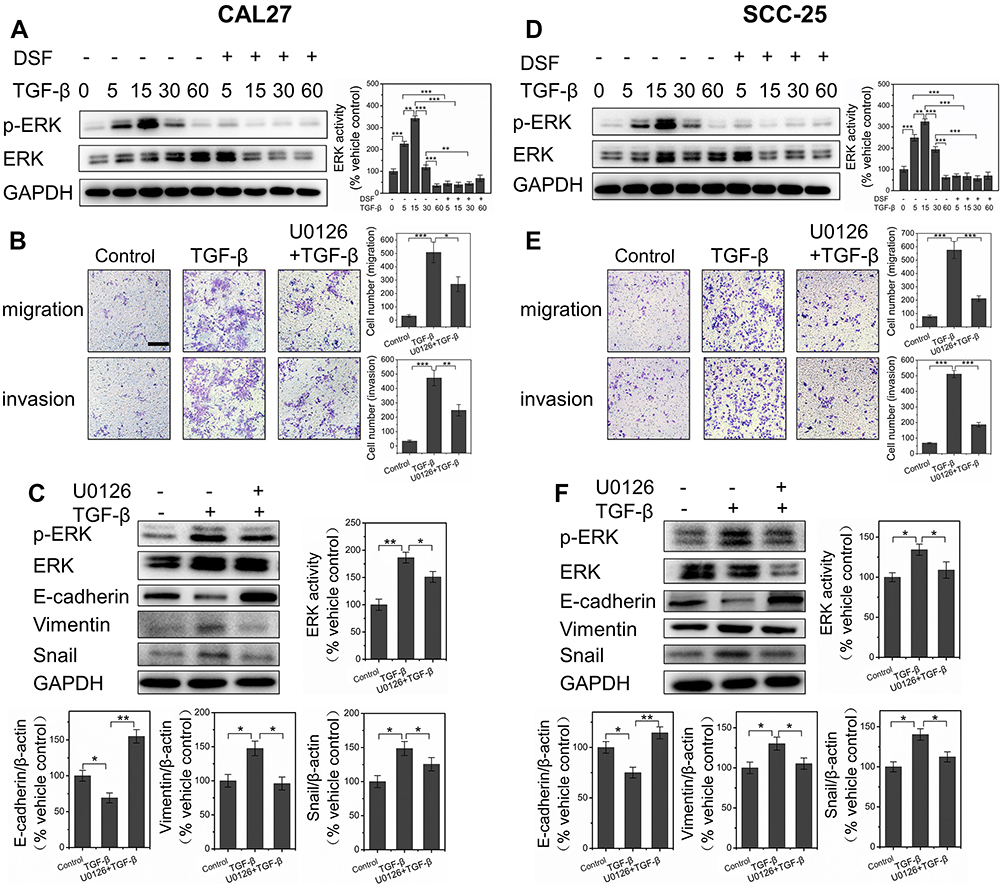 | Figure 3 Dsf regulated EMT of OSCC cells through TGFβ–ERK–Snail pathway independently of Smad4. Notes: (A, D) Western blot results of p-ERK and ERK derived from Smad4-mutated CAL27 and Smad4 wild-type SCC25 stimulated by TGFβ1 for different periods (5, 15, 30, 60 minutes) or combined with Dsf (20 μM) pretreatment for 24 hours; quantitative results of Western blot from three independent experiments shown as mean ± SEM. Direct role of TGFβ–ERK–Snail pathway investigated using U0126, a chemical inhibitor of MEK1/2 and upstream activator of ERK. (B, E) Transwell-based migration and invasion assays were performed after cells had been exposed to TGFβ1 (10 ng/mL) alone or combined with U0126 (20 μM); quantification of cell migration and invasion shown as means ± SEM. (C, F) Western blot results of p-ERK, ERK, E-cadherin, vimentin and Snail from cells exposed to TGFβ1 (10 ng/mL) alone or combined with U0126 (20 μM); quantitative results of Western blot from three independent experiments shown as mean ± SEM. *P<0.05; **P<0.01; ***P<0.001; bar 200 μm.Abbreviations: Dsf, disulfiram; OSCC, oral squamous cell carcinoma; EMT, epithelial–mesenchymal transition. |
To further make sure that Dsf inhibited EMT through inhibition of the TGFβ–ERK–Snail pathway, U0126, a chemical inhibitor of MEK1/2-upstream activator of ERK, was used. Our data showed that U0126 suppressed the TGFβ–ERK–Snail pathway, demonstrated by reduced expression of p-ERK and Snail (Figure 3, C and F). Corresponding with the effect of Dsf on EMT, U0126 treatment resulted in increased expression of E-cadherin and decreased levels of vimentin (Figure 3, C and F). Also, U0126 obviously blocked TGFβ1-induced migration and invasion (Figure 3, B and E). These results provide solid evidence of Dsf’s anti-EMT activity through the TGFβ–ERK–Snail pathway independently of Smad4.
Dsf suppressed metastasis in vivo
EMT is highly associated with tumor metastasis, so we investigated antimetastasis activity of Dsf in vivo. CAL27 cells were pretreated with TGFβ1 for 6 days in vitro to trigger EMT or pretreated with Dsf (20 μM) for 24 hours before TGFβ1 stimulation to inhibit EMT in vitro, then intravenously injected into nude mice through the caudal vein. Surprisingly, we found macroscopic subcutaneous tumors on buttocks in the control and TGFβ1 groups, with tumors in the TGFβ1 group larger than the control group, while only one mouse (n=3) was found with macroscopic tumors in the Dsf group, which was much smaller (Figure 4A). Results from immunohistochemistry of these tumors showed that pretreatment with TGFβ1 caused stronger expression of vimentin and Snail compared with the control group, whereas this upregulation was suppressed by Dsf pretreatment (Figure 4C). A large number of macroscopic metastatic nodules on the lung were observed in the TGFβ1 group, despite few megascopic metastases in the control and Dsf-treatment groups (Figure 4B). H&E staining was performed to check intrapulmonary metastasis further. Metastatic nodules were distributed sporadically in the control group, while widely distributed in the TGFβ group and scarcely distributed in the Dsf group (Figure 4B). Taken together, these results indicate that Dsf inhibited OSCC metastasis on account of TGFβ1 induction.
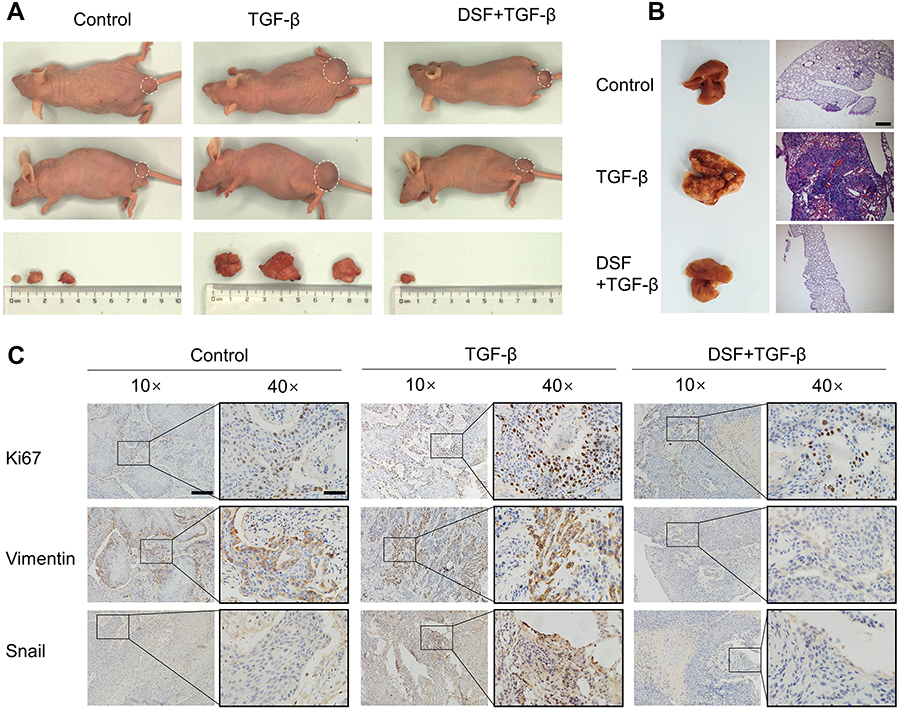 | Figure 4 In vivo anti-metastasis effect of Dsf. Notes: Mice were intravenously injected with CAL27 cells pretreated by TGFβ1 (10 ng/mL) alone or combined with Dsf (20 μM). (A) Typical mouse with macroscopic tumors at the buttocks after injection of CAL27 cells from vertical view (upper panel), lateral view (middle panel), and the tumors. (B) Metastatic tumor nodule on the lung were observed by naked eye (left panel) and H&E staining (right panel, 40× magnification, bar 400 μm). (C) Expression of Ki67, vimentin and Snail in tumor tissue assessed with immunohistochemical staining (100× magnification, bar 200 μm and 400× magnification, bar 50 μm). Abbreviation: Dsf, disulfiram. |
Dsf inhibited OSCC growth
Finally, to investigate the effect of Dsf on OSCC growth, we established a subcutaneous tumor model in BALB/c nude mice with CAL27 cells. As shown in Figure 5, A and B, compared with the control group, tumor volume in the Dsf-treated group was suppressed markedly. To evaluate the proliferation status of tumors, we performed immunohistochemical staining of Ki67 (Figure 5C). Ki67 expression was inhibited significantly in the Dsf-treatment group, which indicated Dsf anticancer activity in OSCC. The EMT status of these tumors was also investigated. We found obvious expression of vimentin and Snail at the infiltrative border in the control group, while expression of these EMT markers was blocked when mice were administered Dsf (Figure 5C), suggesting that Dsf can inhibit tumor growth and EMT in vivo.
 | Figure 5 Dsf inhibited OSCC-tumor growth in vivo. Notes: Mice with subcutaneous xenografts of CAL27 cells were randomly divided into two group (five mice/group): vehicle control (PBS intragastrically) and Dsf (50 mg/kg intragastrically). PBS or Dsf were administered twice a week for 38 days and tumors removed for analysis. (A) Growth curves of tumor size. Values represent mean ± SEM. (B) Typical mouse bearing tumors from vertical view (upper panel), lateral view (middle panel), and the tumors (lower panel). (C) Expression of Ki67, vimentin and Snail in tumor tissue was assessed with immunohistochemical staining (100× magnification, bar 200 μm and 400× magnification, bar 50 μm).Abbreviations: Dsf, disulfiram; OSCC, oral squamous cell carcinoma. |
Discussion
As a key mediator in canonical TGFβ signaling, Smad4 was firstly identified as tumor suppressor in the pancreas.29 Reduced expression of Smad4 was found in five OSCC cell lines compared with normal oral keratinocytes.30 In an immunostaining study of 108 OSCC tissue samples, Smad4 loss was as high as 61.12%, and this loss was significantly associated with more advanced tumor features, such as poorly differentiated tumors, lymph-node metastasis, and decreased survival rates.31 These observations suggest that Smad4 loss or reduction is a common event in OSCC and adverse to OSCC therapy. Therefore, finding a drug that can target Smad4-mutated and Smad4 wild-type OSCC is meaningful for better treatment of patients.
Although the anticancer effect of Dsf has been known in numerous cancers for many years,19 very little is known about this drug’s activity on Smad4-mutated or Smad4 wild-type OSCC. Therefore, we used the Smad4-mutant OSCC cell line CAL27 to establish a subcutaneous xenograft nude mouse model and examined the anticancer effect of Dsf on OSCC with this model. Consistently with previous studies, we found that Dsf obviously inhibited tumor growth. Previous research has also demonstrated that Dsf blocks lung metastasis of human hepatocellular carcinoma cells and liver metastasis of human breast cancer cells in nude mice. As such, we examined the antimetastasis effect of Dsf on OSCC in in an intravenous tumor mouse model. As expected, we found that Dsf suppressed OSCC lung metastasis in vivo. Uncommonly, we observed subcutaneous metastasis at the buttock in the intravenous tumor mouse model. This might be attributable to the specific subcutaneous microenvironment where CAL27 cells prefer to colonize. It has been reported that patients with SCC can suffer subcutaneous metastasis, although this metastasis is relatively rare.32–34 Also, subcutaneous metastasis is usually associated with rapid progression of cancer. Indeed, in our experiment, the TGFβ group had much bigger subcutaneous metastatic tumors compared with the control and Dsf groups, while the Dsf group had only one small subcutaneous metastatic tumor. In conclusion, our data indicate that Dsf is effective in inhibiting highly malignant OSCC growth and metastasis.
EMT is an important progress implicated in cancer metastasis.6 Indeed, we found downregulated expression of the EMT markers vimentin and Snail when Dsf inhibited OSCC metastasis in vivo, suggesting that EMT is involved in the antimetastasis activity of Dsf. As such, we investigated the effect of Dsf on EMT in OSCC cells in vitro. In order to figure out whether the mutation of Smad4 influenced the activity of Dsf in OSCC, we chose Smad4-mutated (CAL27) and Smad4 wild-type (SCC25) OSCC cells.28 EMT is characterized by loss of epithelial cell polarity and acquisition of mesenchymal morphology, accompanied by cell migration, invasion, metastasis, and chemotherapeutic resistance.8,9 The anti-EMT effect of Dsf has been proved in hepatocellular carcinoma and breast cancer.27,35 In line with previous reports, our results showed that Dsf suppressed EMT in OSCC, demonstrated by recovery of epithelial morphology, decreased migration, and invasion potential, and reduced expression of vimentin, independently of Smad4. These findings indicated that Dsf has a broad-spectrum anti-EMT effect in multiple cancers and this effect is unaffected by Smad4 mutation, suggesting most human OSCC tumors will be sensitive to Dsf treatment, even though 61.12% of them are Smad4-mutated, portending powerful clinical translational potential for our findings.
Multiple signaling pathways participate in EMT, among which the TGFβ-signaling pathway has been best studied.9 TGFβ promotes EMT through Smad-dependent and Smad-independent pathways.9 Smad4, the loss of which abrogates the Smad-dependent TGFβ pathway, combines with phosphorylated Smad2/3 after binding of TGFβ to its receptor, then translocates into nuclei to regulate the expression of transcriptional factors implicated in the initiation of EMT.36 Our results showed that Dsf can inhibit EMT without the influence of Smad4 mutation, suggesting a Smad-independent pathway is involved in the anti-EMT activity of Dsf. As one of the Smad-independent TGFβ pathways, the ERK pathway plays a vital role in cancer progression. Activation of this pathway contributes to growth promotion in pancreatic cancers and progression in undifferentiated carcinomas when Smad4 is abolished37,38 and was required in TGFβ1-induced EMT in murine mammary-gland epithelial cells.39 Consistently with previous reports, we found that TGFβ1 induced phosphorylation of ERK in both Smad4-mutated and Smad4 wild-type OSCC cells. The kinetics of ERK phosphorylation by TGFβ induction vary by cell type and culture condition. Activation with delayed response in some cell lines may result from Smad-dependent transcription responses, but rapid activation (5–15 minutes) in other cases suggests independence of transcription.40,41 Like the latter, in our research TGFβ1 induced ERK phosphorylation rapidly at 5 minutes, followed by the most prominent at 15 minutes, further suggesting that this activation of ERK is independent of Smad4. Snail is an important transcriptional factor in EMT, which can bind to adjacent E-boxes in genes encoding epithelial markers, such as E-cadherin, CAR, and occludin, leading to gene repression.42 The ERK pathway participates in the activation of the Snail minimal promoter,43 by which activation of ERK promotes breast cancer cell migration and invasion.44,45 Our results showed that the suppression effect of Dsf on the TGFβ–ERK–Snail pathway in EMT without Smad4 involvement and that inhibition of the TGFβ–ERK–Snail pathway by U0126, a chemical inhibitor of MEK1/2, the upstream activator of ERK, resulted in blocked EMT, consistent with the effect of Dsf. These findings testified that Dsf inhibited EMT in OSCC by targeting TGFβ–ERK–Snail pathway, rising above Smad4. Numerous evidence has proved that the ERK-signaling pathway promotes cell proliferation and cancer metastasis.46 In our experiment, we observed that Dsf suppressed ERK phosphorylation, which may have contributed to Dsf’s inhibition effect on tumor growth and metastasis in vivo.
Conclusion
Dsf triggered an inhibitory effect on EMT in OSCC, in accordance with reduced migration and invasion in vitro, as well as decreased tumor growth and metastasis in vivo. The Smad4-independent pathway TGFβ–ERK–Snail pathway was found to play an essential role in the Dsf-induced restraining of OSCC EMT. Based on these findings, more research is needed to evaluate the anti-EMT effect of Dsf in other gene-mutated cancers and relevant preclinical models, promoting its clinical use against human OSCC.
Acknowledgments
We thank Dr Yuji Mishina from the University of Michigan for his technical support and Dr Jian Xu from the University of Southern California for her critical reading. This study was supported by grants from the National Key R&D Program of China (2016YFC1102800), National Natural Science Foundation of China (881320108011), Provincial Industrial Innovation Project of Jilin Province (2016C044-3), and Jilin Scientific and Technological Development Program (20170101093JC).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96.
2. Warnakulasuriya S. Causes of oral cancer–an appraisal of controversies. Br Dent J. 2009;207(10):471–475.
3. Mascitti M, Rubini C, de Michele F, et al. American Joint Committee on Cancer staging system 7th edition versus 8th edition: any improvement for patients with squamous cell carcinoma of the tongue? Oral Surg Oral Med Oral Pathol Oral Radiol. 2018;126(5):415–423.
4. Rot S, Kaune T, Taubert H, et al. Prognostic impact of mRNA levels of LGR5 transcript variants in OSCC patients. BMC Cancer. 2019;19(1):155.
5. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
6. de Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110.
7. Chaw SY, Abdul Majeed A, Dalley AJ, Chan A, Stein S, Farah CS. Epithelial to mesenchymal transition (EMT) biomarkers–E-cadherin, beta-catenin, APC and Vimentin–in oral squamous cell carcinogenesis and transformation. Oral Oncol. 2012;48(10):997–1006.
8. Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14(6):818–829.
9. Kahata K, Dadras MS, Moustakas A. TGF-β family signaling in epithelial differentiation and epithelial-mesenchymal transition. Cold Spring Harb Perspect Biol. 2018;10(1):a022194.
10. Liu L-K, Jiang X-Y, Zhou -X-X, Wang D-M, Song X-L, Jiang H-B. Upregulation of vimentin and aberrant expression of E-cadherin/beta-catenin complex in oral squamous cell carcinomas: correlation with the clinicopathological features and patient outcome. Mod Pathol. 2010;23(2):213–224.
11. van Roy F. Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat Rev Cancer. 2014;14(2):121–134.
12. You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22(1):9–20.
13. White RA, Malkoski SP, Wang X-J. TGFβ signaling in head and neck squamous cell carcinoma. Oncogene. 2010;29(40):5437–5446.
14. Kim SK, Fan Y, Papadimitrakopoulou V, et al. DPC4, a candidate tumor suppressor gene, is altered infrequently in head and neck squamous cell carcinoma. Cancer Res. 1996;56(11):2519–2521.
15. Bornstein S, White R, Malkoski S, et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J Clin Invest. 2009;119(11):3408–3419.
16. Deckers M, van Dinther M, Buijs J, et al. The tumor suppressor Smad4 is required for transforming growth factor beta-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 2006;66(4):2202–2209.
17. Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor {beta} (TGF-{beta}) target genes and distinguishes TGF-{beta}-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol. 2005;25(18):8108–8125.
18. Kwentus J, Major LF. Disulfiram in the treatment of alcoholism; a review. J Stud Alcohol. 1979;40(5):428–446.
19. Sauna ZE, Shukla S, Ambudkar SV. Disulfiram, an old drug with new potential therapeutic uses for human cancers and fungal infections. Mol Biosyst. 2005;1(2):127–134.
20. Skrott Z, Mistrik M, Andersen KK, et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature. 2017;552(7684):194–199.
21. Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006;66(21):10425–10433.
22. Shian S-G, Kao Y-R, Wu FY-H, Wu C-W. Inhibition of invasion and angiogenesis by zinc-chelating agent disulfiram. Mol Pharmacol. 2003;64(5):1076–1084.
23. Yip NC, Fombon IS, Liu P, et al. Disulfiram modulated ROS-MAPK and NFκB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br J Cancer. 2011;104(10):1564–1574.
24. Loo TW, Clarke DM. Blockage of drug resistance in vitro by disulfiram, a drug used to treat alcoholism. J Natl Cancer Inst. 2000;92(11):898–902.
25. Terashima Y, Toda E, Itakura M, et al. Targeting FROUNT with disulfiram regulates macrophage responses in cancer. SSRN J. 2018.
26. Cho H-J, Lee T-S, Park J-B, et al. Disulfiram suppresses invasive ability of osteosarcoma cells via the inhibition of MMP-2 and MMP-9 expression. J Biochem Mol Biol. 2007;40(6):1069–1076.
27. Han D, Wu G, Chang C, et al. Disulfiram inhibits TGF-β-induced epithelial-mesenchymal transition and stem-like features in breast cancer via ERK/NF-κB/Snail pathway. Oncotarget. 2015;6(38):40907–40919.
28. Qiu W, Schönleben F, Li X, Su GH. Disruption of transforming growth factor beta-Smad signaling pathway in head and neck squamous cell carcinoma as evidenced by mutations of SMAD2 and SMAD4. Cancer Lett. 2007;245(1–2):163–170.
29. Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271(5247):350–353.
30. Iamaroon A, Pattamapun K, Piboonniyom S-O. Aberrant expression of Smad4, a TGF-beta signaling molecule, in oral squamous cell carcinoma. J Oral Sci. 2006;48(3):105–109.
31. Wang X, Sun W, Bai J, et al. Growth inhibition induced by transforming growth factor-beta1 in human oral squamous cell carcinoma. Mol Biol Rep. 2009;36(5):861–869.
32. Amit A, Edwards CL, Athey P, Kaplan AL. Extensive subcutaneous metastases from squamous cell carcinoma of the cervix in patient with HIV. Int J Gynecol Cancer. 2001;11(1):78–80.
33. Kouvaris JR, Plataniotis GA, Floros DG, Sykiotis CA, Trakadas SJ, Vlahos LJ. A benign-looking subcutaneous metastasis from squamous cell cervical carcinoma: A case report and review of the literature. Int J Gynecol Cancer. 2000;10(6):503–506.
34. Hoffman GR, Hayter JP. Widespread subcutaneous distant metastases from a head and neck squamous cell carcinoma. J Oral Maxillofac Surg. 2002;60(8):954–958.
35. Li Y, Wang L-H, Zhang H-T, et al. Disulfiram combined with copper inhibits metastasis and epithelial-mesenchymal transition in hepatocellular carcinoma through the NF-κB and TGF-β pathways. J Cell Mol Med. 2018;22(1):439–451.
36. Feng X-H DR. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693.
37. Chow JYC, Quach KT, Cabrera BL, Cabral JA, Beck SE, Carethers JM. RAS/ERK modulates TGFbeta-regulated PTEN expression in human pancreatic adenocarcinoma cells. Carcinogenesis. 2007;28(11):2321–2327.
38. Iglesias M, Frontelo P, Gamallo C, Quintanilla M. Blockade of Smad4 in transformed keratinocytes containing a Ras oncogene leads to hyperactivation of the Ras-dependent Erk signalling pathway associated with progression to undifferentiated carcinomas. Oncogene. 2000;19(36):4134–4145.
39. Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia. 2004;6(5):603–610.
40. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584.
41. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19(1):128–139.
42. Vincent T, Neve EPA, Johnson JR, et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. 2009;11(8):943–950.
43. Wang Y, Shi J, Chai K, Ying X, Zhou BP. The role of snail in EMT and tumorigenesis. Curr Cancer Drug Targets. 2013;13(9):963–972.
44. Chen H, Zhu G, Li Y, et al. Extracellular signal-regulated kinase signaling pathway regulates breast cancer cell migration by maintaining slug expression. Cancer Res. 2009;69(24):9228–9235.
45. Hsu Y-L, Hou M-F, Kuo P-L, Huang Y-F, Tsai E-M. Breast tumor-associated osteoblast-derived CXCL5 increases cancer progression by ERK/MSK1/Elk-1/snail signaling pathway. Oncogene. 2013;32(37):4436–4447.
46. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–3310
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.