Back to Journals » Journal of Inflammation Research » Volume 14
Distinctive Under-Expression Profile of Inflammatory and Redox Genes in the Blood of Elderly Patients with Cardiovascular Disease
Authors Milanesi E, Manda G ![]() , Dobre M
, Dobre M ![]() , Codrici E
, Codrici E ![]() , Neagoe IV, Popescu BO, Bajenaru OA, Spiru L
, Neagoe IV, Popescu BO, Bajenaru OA, Spiru L ![]() , Tudose C
, Tudose C ![]() , Prada GI
, Prada GI ![]() , Davidescu EI
, Davidescu EI ![]() , Piñol-Ripoll G
, Piñol-Ripoll G ![]() , Cuadrado A
, Cuadrado A ![]()
Received 27 October 2020
Accepted for publication 22 December 2020
Published 18 February 2021 Volume 2021:14 Pages 429—442
DOI https://doi.org/10.2147/JIR.S280328
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Ning Quan
Elena Milanesi,1 Gina Manda,1 Maria Dobre,1 Elena Codrici,1 Ionela Victoria Neagoe,1 Bogdan Ovidiu Popescu,1– 3 Ovidiu Alexandru Bajenaru2,4 †, Luiza Spiru,2,5 Catalina Tudose,2,6 Gabriel-Ioan Prada,2,7 Eugenia Irene Davidescu,2,3 Gerard Piñol-Ripoll,8 Antonio Cuadrado1,9– 12
1“Victor Babes” National Institute of Pathology, Bucharest, 050096, Romania; 2Clinical Neurosciences, Geriatrics and Gerontology Departments, “Carol Davila” University of Medicine and Pharmacy, Bucharest, 020021, Romania; 3Neurology Department, Clinical Hospital Colentina, Bucharest, 020125, Romania; 4Neurology Department, University Emergency Hospital, Bucharest, 050098, Romania; 5The Excellence Memory Center and Longevity Medicine, “Ana Aslan” International Foundation,, Bucharest, 050064, Romania; 6Section II, “Prof. Dr. Al. Obregia” Psychiatry Clinical Hospital & the Memory Center of the Romanian Alzheimer Society, Bucharest, 041914, Romania; 7Section IV, “Ana Aslan” National Institute of Gerontology and Geriatrics, Bucharest, 011241, Romania; 8Unitat Trastons Cognitius, Hospital Universitari Santa Maria-IRBLLeida, Lleida, 25198, Spain; 9Department of Endocrine Physiology and Nervous System, Instituto de Investigaciones Biomédicas “Alberto Sols” UAM-CSIC, Madrid, 28029, Spain; 10Department of Biochemistry, Faculty of Medicine, Autonomous University of Madrid, Madrid, 28049, Spain; 11Neuroscience Section, Instituto de Investigación Sanitaria La Paz (IdiPaz), Madrid, 28046, Spain; 12Centro de Investigación Biomédica en Red Sobre Enfermedades Neurodegenerativas (CIBERNED), ISCIII, Madrid, 28031, Spain
†Prof. Ovidiu A. Bajenaru passed away on 03.09.2020
Correspondence: Gina Manda
“Victor Babes” National Institute of Pathology, 99-101 Splaiul Independentei, Bucharest, 050096, Romania
Tel +40 744246887
Fax +40 21 3194528
Email [email protected]
Antonio Cuadrado
Instituto de Investigaciones Biomédicas “Alberto Sols” UAM-CSIC, C/Arturo Duperier 4, Madrid, 28029, Spain
Tel +34 638 29 65 85
Fax +34 915 85 44 01
Email [email protected]
Purpose: Chronic low-grade inflammation and oxidative stress are present in most of the pathologic mechanisms underlying non-communicable diseases. Inflammation and redox biomarkers might therefore have a value in disease prognosis and therapy response. In this context, we performed a case–control study for assessing in whole blood the expression profile of inflammation and redox-related genes in elderly subjects with various comorbidities.
Patients and Methods: In the blood of 130 elderly subjects with various pathologies (cardiovascular disease, hypertension, dyslipidemia including hypercholesterolemia, type 2 diabetes mellitus), kept under control by polyvalent disease-specific medication, we investigated by pathway-focused qRT-PCR a panel comprising 84 inflammation-related and 84 redox-related genes.
Results: The study highlights a distinctive expression profile of genes critically involved in NF-κB-mediated inflammation and redox signaling in the blood of patients with cardiovascular disease, characterized by significant down-regulation of the genes NFKB2, NFKBIA, RELA, RELB, AKT1, IRF1, STAT1, CD40, LTA, TRAF2, PTGS1, ALOX12, DUOX1, DUOX2, MPO, GSR, TXNRD2, HSPA1A, MSRA, and PDLIM1. This gene expression profile defines the transcriptional status of blood leukocytes in stable disease under medication control, without discriminating between disease- and therapy-related changes.
Conclusion: The study brings preliminary proof on a minimally invasive strategy for monitoring disease in patients with cardiovascular pathology, from the point of view of inflammation or redox dysregulation in whole blood.
Keywords: aging-related diseases, cardiovascular disease, inflammation, NF-κB signaling, redox metabolism, oxidative stress
Introduction
The growing prevalence of non-communicable diseases (NCDs), especially in elderly individuals, determines the need for an accelerated search of biomarkers that could improve disease prognosis, monitoring, and therapeutic engagement. Extensive evidence indicates a critical role of low-grade chronic inflammation and oxidative stress as active participants in the pathological mechanisms underlying NCDs such as cardiovascular disease (CVD), hypertension, type 2 diabetes mellitus (T2DM), and dyslipidemia.1–5 Underlying mechanisms are cellular senescence,6 mitochondrial dysfunction,7 defective autophagy and mitophagy of misfolded or oxidized proteins,8 activation of inflammasomes by cell debris and misplaced “self” molecules,9 dysregulation of the ubiquitin–proteasome system,10 activation of the DNA damage response11, and dysbiosis.12
A multitude of stimuli can trigger chronic inflammatory responses in innate immune cells, mainly in macrophages through cognate receptors of Pathogen-Associated Molecular Patterns (PAMPs) and Damage-Associated Molecular Patterns (DAMPs) that are signaling mostly via the NF-κB pathway.1,13 A persistently increased NF-κB activity can drive senescence of immune cells that acquire an altered transcriptional phenotype, resulting in increased production of inflammatory factors such as IL-6, IL-8, IL-7, MCP-2, MIP-3, ICAM, Il-1α, and IL-β.14 Concurrently, a persistent alteration of the redox metabolism occurring in elderly individuals, due to an increased production of reactive oxygen species (ROS) by mitochondrial and cytoplasmatic sources, or to defective antioxidant protection15 is a risk factor for NCDs.16 The ensuing low-grade redox dysregulation alters signaling responses to growth and trophic factors, cytokines, chemokines, etc., leading to cellular senescence and chronic inflammation.17 Various comorbidities, such as obesity, hypertension, and T2DM contribute to exacerbate the deleterious action of this low-grade pro-inflammatory phenotype characterizing elderly individuals.18
The highly reactive circulating leukocytes reflect quite closely both systemic and localized inflammatory and redox-mediated mechanisms. Moreover, the recruitment of dysfunctional blood leukocytes into diseased tissues can amplify these pathological processes, hence worsening disease evolution.19 Being easy to obtain from venous blood, the functional status of circulating leukocytes represents a window for detecting immune abnormalities in NCDs related to inflammation and redox metabolism.
This study aimed to identify gene expression changes that might underlie pathologic processes in elderly patients with various NCDs. We analyzed the expression of 168 inflammation- and redox-related genes in the blood of a cohort of 130 elderly patients with diseases kept under clinical management by polyvalent medication. The detected gene expression profile was consistent with a suppression of inflammatory and redox-mediated processes as adaptive mechanisms aimed at limiting disease progression or relapse.
Patients and Methods
Subjects
Elderly individuals (n=130), 67- to 81-year-old, were randomly recruited from 3 neurology, 1 psychiatry, and 2 geriatric clinics. The inclusion criterion was age above 60 years, and exclusion criteria comprised: (i) acute inflammatory reactions and infection in the last 30 days prior to the study inclusion; (ii) history of any type of cancer and autoimmune diseases; (iii) acute episodes of morbidities during the last year before being recruited in the present study. All the recruited subjects, complying with the above-mentioned inclusion/exclusion criteria, were clinically evaluated for the presence of NCDs. Patients presented one or several morbidities, comprising hypertension (HT), cardiovascular diseases (CVD), dyslipidemia (DL), including hypercholesterolemia (HC) and Type 2 diabetes mellitus (T2DM) as described in Table 1A. Disease was kept under control with polyvalent medication. For analyzing gene expression data, the subjects presenting a type of disease constituted the case group, while all the other subjects in the cohort, not presenting that specific disease, constituted the control group (Figure 1, Table 1B). The study was conducted in accordance with the Declaration of Helsinki, and was approved by the local ethics committees of participating clinics, as follows: Clinical Hospital Colentina, Bucharest, Romania – 12/11.05.2017; University Emergency Hospital, Bucharest, Romania – 56457/07.11.2017; “Prof. Dr. Al. Obregia” Psychiatry Clinical Hospital, Bucharest, Romania – 3/17.05.2017; “Ana Aslan” National Institute of Gerontology and Geriatrics, Bucharest, Romania – 299.1101.2018; Hospital Arnau de Vilanova de Lleida, Lleida, Spain – CE 1218. Written informed consent was obtained from all study participants at the moment of recruitment and blood collection.
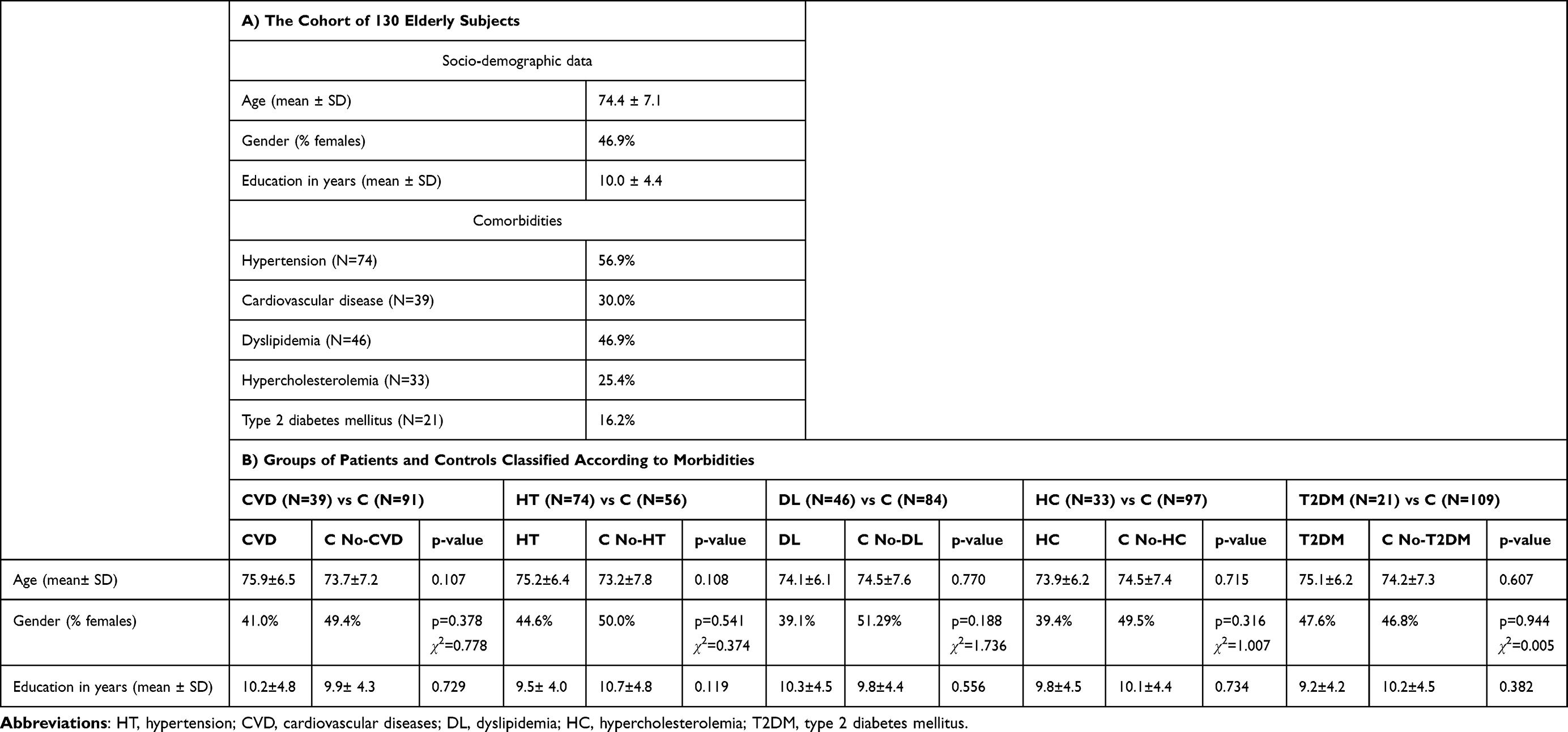 |
Table 1 Socio-Demographic Features and Comorbidities in the Investigated Cohort of 130 Elderly Subjects (A) and in the Groups of Patients and Controls Classified According to Morbidities (B) |
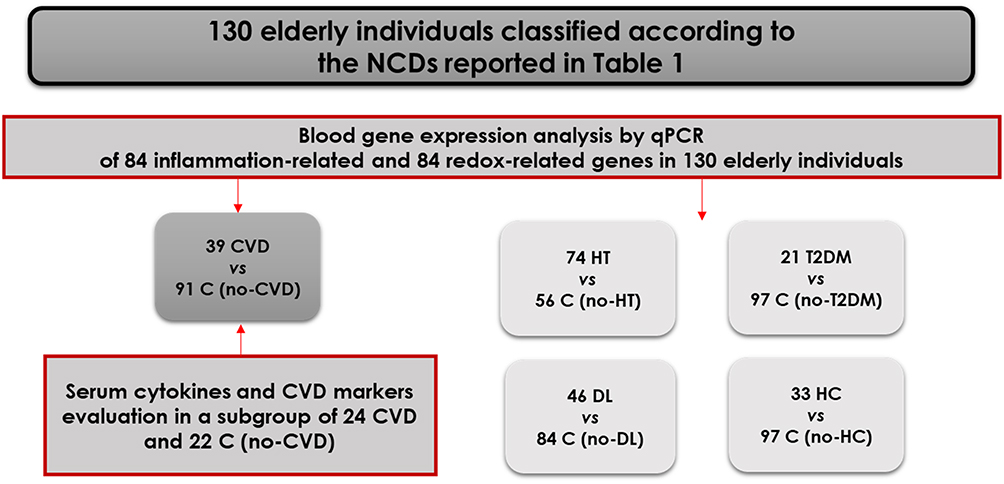 |
Figure 1 Classification of the 130 recruited elderly patients in case and control groups, according to comorbidities (CVD-cardiovascular diseases, HT-hypertension, DL-dyslipidemia, HC-hypercholesterolemia, T2DM- Type 2 diabetes mellitus). The subjects presenting a type of comorbidity constituted the case group, while all the other subjects in the cohort, not presenting that specific comorbidity, constituted the control group (C). |
Blood Samples
For all the recruited subjects, venous blood (2.5 mL) was collected in PAXgene Blood RNA Tubes (Qiagen) for RNA isolation and gene expression studies. Tubes with blood were kept at room temperature for 24 h, transferred to −20°C for another 24 h, and, finally, stored at −80°C until use.
For a subgroup of 46 elderly subjects, venous blood was collected in SSTII Advance vacutainers for serum separation and investigation of cardiovascular markers and several cytokines, chemokines, and growth factors in serum by xMAP protein multiplexing.
Gene Expression
Total RNA was isolated from the blood collected in PAXgene tubes according to the manufacturer’s protocol. RNA purity and quantity were spectrophotometrically assessed using a NanoDrop 2000 equipment (Thermo Scientific). Reverse transcription was performed with the RT2 First Strand Kit (Qiagen) using 400 ng of RNA. The expression of 84 key genes involved in redox responses and of 84 genes related to inflammatory processes (Supplementary Table 1) was evaluated with RT2 Profiler™ PCR Array Human Oxidative Stress Plus (PAHS-065Y, Qiagen) and RT2 Profiler™ PCR Array Human NF-κB Signalling Pathway (PAHS-025Z, Qiagen), respectively. The SYBR Green chemistry on an ABI-7500 fast instrument (Applied Biosystems) was applied. The geometric mean of two housekeeping genes (HPRT1 and RPLP0) was used to normalize the expression level of each transcript. These two reference genes were selected among five candidates that are available in each array (ACTB, B2M, GAPDH, HPRT1, and RPLP0), using the RefFinder algorithm (http://leonxie.esy.es/RefFinder/).20 Gene expression levels were calculated as 2−ΔCT values. Fold Change (FC) in gene expression was calculated as the 2−ΔCT mean values in patients divided by 2−ΔCT mean values in controls. Results are presented as Fold Regulation (FR) as follows: when the FC value was above 1, FR was equal to FC, and results were reported as fold up-regulation; when the FC value was less than 1, FR was expressed as the negative inverse of FC, and results were reported as fold down-regulation.
Protein Multiplexing in Serum
Through protein multiplexing (xMAP array analysis) various cytokines/chemokines/growth factors and CVD markers were assessed in the serum of 24 CVD patients and 22 no-CVD controls, for which serum was available. The xMAP array analysis was performed according to the manufacturer’s protocols, and the multiplex data acquisition was done using a Luminex® 200™ system. Serum cytokines, chemokines, and growth factor levels were determined using the following kits from R&D Systems: 1) the Human XL Cytokine Discovery Premixed Kit (R&D Systems), with an analyte-specific bead set addressing CCL11/Eotaxin, CCL2/JE/MCP-1, CCL3/MIP-1alpha, CCL4/MIP-1beta, CD40 ligand/TNFSF5, CX3CL1/Fractalkine, CXCL10/IP10/CRG-2, EGF, G-CSF, IL-13, IL-15, IL-1ra/IL-1F3, IL-3, and IL-7; 2) the Human High Sensitivity Cytokine Base A, addressing TNFα, VEGF, IL-2, IL-1β, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12p70, GM-CSF, and IFNγ. The concentration of the serum CVD markers (CK_MB, TROPONIN1, CXCL6, CXCL16, Endocan1, FABP3, FABP4, PGF, OSM (Supplementary Table 2) was evaluated using the MILLIPLEX MAP Human Cardiovascular Disease (CVD) Magnetic Bead Panel 1 (Merck Millipore). Briefly, the beads were incubated for several hours (depending on the kit) with standards, controls, and samples in a 96-well plate. All further incubations with the Biotin-Antibody Cocktail and with Streptavidin phycoerythrin were performed at room temperature, in the dark, under shaking at 800 rpm. Multiplex data analysis was performed using xPONENT 4.2 software (Millipore) and the calibration curves were generated with a 5-parameter logistic fit. Duplicate samples were used for all serum specimens, and the mean concentrations per duplicate samples were used for statistical analysis.
Statistical Analysis
For statistical analysis, subjects presenting a particular type of NCD were grouped and were compared with the rest of the subjects from the study cohort which did not have that particular NCD (control group) (Figure 1 and Table 1B). Differences in gene or protein expression levels between groups were evaluated with the Mann–Whitney U-test, since the data were not normally distributed (Kolmogorov–Smirnov, p<0.05). Only changes in gene expression with absolute FR values >1.8 and p values <0.005 were considered significant in this study. Correlations between gene expression levels were calculated using the Pearson coefficient and were considered significant with a coefficient r > 0.7 (positive correlation) and p≤0.001.
Results
The expression levels of 168 genes involved in inflammation and redox metabolism were analyzed in a cohort of 130 elderly individuals whose demographic and clinical parameters are described in Table 1. The investigated patients presented one or more chronic morbidities that were controlled with polyvalent medication. Analysis was performed according to each of the NCDs registered in the investigated cohort of 130 elderly patients (Table 1A), by comparing the data from subjects presenting a particular morbidity with the data from subjects not having that particular disease (control group) (Table 1B).
We identified a distinctive gene expression profile in CVD patients (N=39) as compared to the corresponding controls, that was not found in the groups of patients presenting primarily hypertension (N=74), dyslipidemia (N=46), including hypercholesterolemia (N=33), or T2DM (N=21) (Supplementary Table 3).
The investigated CVD patients had atherosclerosis (N=21) (carotid atherosclerosis-14, mesenteric artery disease-1, coronary artery disease-6), venous chronic insufficiency (N=2), arrhythmia (N=8) (atrial fibrillation-6, paroxysmal tachycardia-1, sinus node dysfunction-1), angina pectoris (N=5), congestive heart failure (N=2), and cardiomyopathy (N=1). The disease was kept under control by medication as shown by the registered serum concentrations of various CVD biomarkers that were in the normal range (Supplementary Table 2).
The gene expression profile in the blood of CVD patients was characterized by significant down-regulation of several inflammatory genes (Figure 2). These genes are part of the NF-κB system (Figure 2A: NFKB2, NFKBIA, RELA, and RELB), or participate in signaling pathways interacting with NF-κB (Figure 2B: AKT1, IRF1, and STAT1), are involved in receptor-mediated signaling through NF-κB (Figure 2C: CD40, LTA, and TRAF2) and in arachidonic acid metabolism (Figure 2D: ALOX12 and PTGS1). We have further investigated the impact of inflammatory genes down-regulation on the level of 15 pro-inflammatory cytokines/chemokines/growth factors in serum from a sub-group of elderly subjects (24 CVD patients and 22 no-CVD controls). Down-regulation of the inflammation genes identified in 39 CVD patients and 91 controls (Figure 2) was reproduced also in the smaller sub-group for which cytokines were measured (data not shown). All investigated pro-inflammatory factors, excepting IL-1β, were in the normal range. The concentration of IL-1β was decreased to 67% in CVD patients as compared to no-CVD controls (CVD patients: 0.44 pg/mL, no-CVD subjects: 0.75 pg/mL, p=0.002). We found no significant correlations (r>0.5, p<0.05) between the IL-1β decrease and the down-regulated genes in CVD patients.
 |
Figure 2 Inflammation-related genes differentially expressed in the blood of 39 CVD patients versus 91 controls. The reported genes are involved in: (A) NF-kB signaling, (B) Other signaling pathways, (C) Receptor-mediated signaling, (D) Metabolism of arachidonic acid. Gene expression levels are expressed as mean relative fold of change versus controls and standard error of the mean (SEM). The p-value was calculated using the Mann–Whitney U-test (***p<0.001). |
In parallel to inflammatory genes down-regulation, we observed under-expression of several redox genes in the blood of CVD patients (Figure 3), that are involved in reactive oxygen species (ROS) production (Figure 3A: DUOX1/2 and MPO), glutathione (GSR) and thioredoxin (TXNRD2) metabolism (Figure 3B), oxidative damage repair (Figure 3C; HSPA1A and MSRA), and adaptors for cytoskeletal proteins (Figure 3D: PDLIM1).
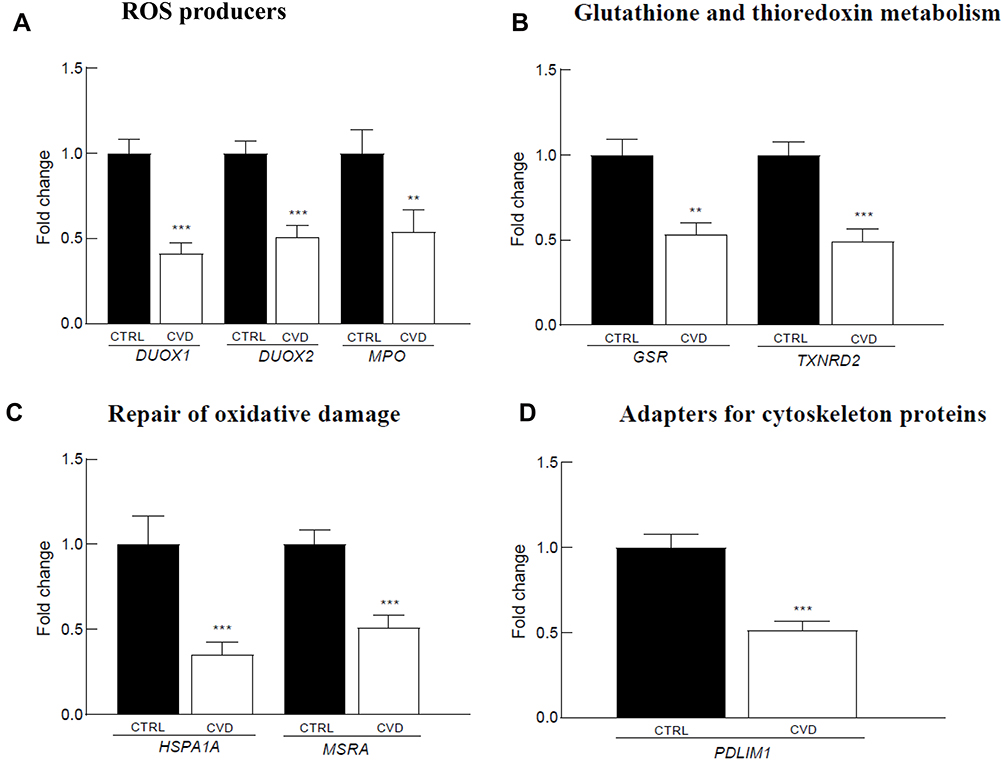 |
Figure 3 Redox-related genes differentially expressed in the blood of 39 CVD patients versus 91 controls. The reported genes are involved in: (A) ROS producers, (B) Glutathione and thioredoxin metabolism, (C) Repair of oxidative demage, D. Adapters for cytoskeleton proteins. Gene expression levels are expressed as mean relative fold of change versus control and standard error of the mean (SEM). The p-value was calculated using the Mann–Whitney U-test (**p<0.01; ***p<0.001). |
The Pearson correlation analysis performed on the inflammatory and redox genes that were down-regulated in CVD patients indicated significant intra-pathway connections for most of these inflammatory (Figure 4) and redox (Figure 5) genes. However, no significant association was evidenced for the LTA gene in the inflammatory pathway (Figure 4B), and for the GSR gene in the redox pathway (Figure 5A), suggesting that mechanisms other than those analyzed in our arrays underlie their down-regulation. Intra-pathway correlations were more abundant in the group of CVD patients than in the control group, therefore providing a distinctive CVD signature. For instance, NFKBIA was correlated with several genes in the NF-κB-related signaling pathway in the case of CVD patients (Figure 4A), while no significant connections were found in the control group (Figure 4B). Likewise, more associations centered on the RELA gene were evidenced in CVD patients as compared with the control group (Figure 4A and B). In the case of redox genes, two intra-pathway correlations were registered for the MPO gene in CVD patients (Figure 5A), but no significant connections were observed in controls (Figure 5B).
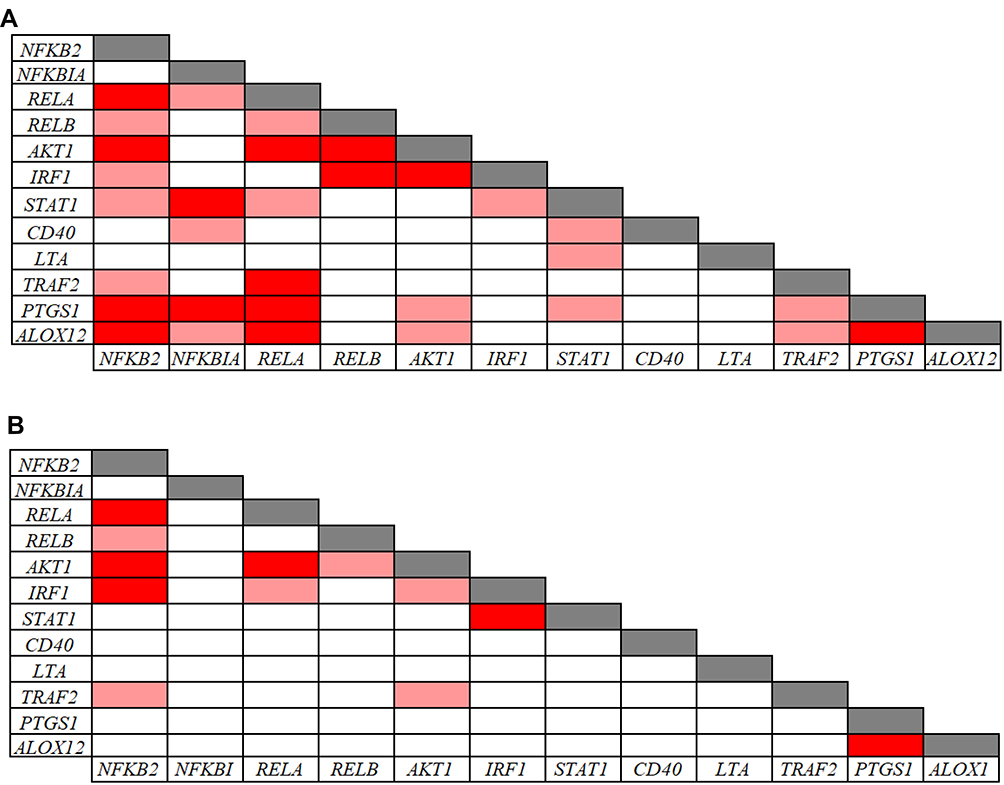 |
Figure 4 Pearson correlations among inflammation-related gene expression levels in 39 CVD patients (A) and in 91 controls (B). Correlations with p-values < 0.001 and r values > 0.7 (light red: 0.7 < r < 0.8; dark red: r ≥ 0.8) are presented. |
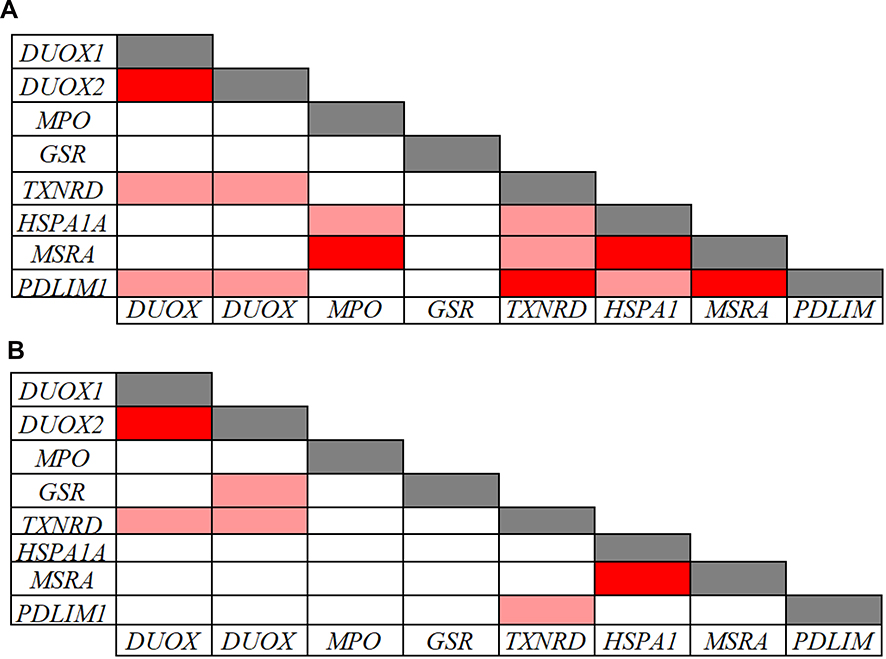 |
Figure 5 Pearson correlations among redox related-gene expression levels in 39 CVD patients (A) and in 91 controls (B). Correlations with p-values < 0.001 and r values > 0.7 (light red: 0.7 < r < 0.8; dark red: r ≥ 0.8) are presented. |
When analyzing the inter-pathway correlations, multiple associations between inflammatory and redox genes were detected in the group of CVD patients (Figure 6A), with the inflammatory NFKB2, RELA, TRAF2, PTGS1, and ALOX12 genes showing the highest number of inter-pathway associations. Regarding redox genes, TXNRD2, HSPA1A, MSRA, and PDLIM1 showed multiple connections with inflammatory genes in CVD patients (Figure 6A). Comparing CVD patients with controls, we found far more inter-pathway correlations in the CVD group (Figure 6A and B). For instance, no correlations of the NFKB2, RELA, IRF1, AKT1, STAT1, CD40, LTA, and TRAF2 genes with redox genes were detected in controls, while multiple connections were evidenced in CVD patients. These results provide a particular transcriptional signature of inflammatory and redox genes identified in the whole blood of CVD patients.
 |
Figure 6 Pearson correlations among inflammation- and redox-related gene expression levels in 39 CVD patients (A) and in 91 controls (B). Correlations with p-values < 0.001 and r values > 0.7 (light red: 0.7 < r < 0.8; dark red: r ≥ 0.8) are presented. |
Discussion
The pathway-focused qRT-PCR analysis in whole blood indicates that the investigated CVD patients, with disease kept under control by medication, are characterized by abnormally low expression levels of several inflammatory and redox genes which generally show good intra- and inter-pathway correlations. Further functional proteomics studies will be required to determine if the observed gene down-regulation correlates with altered inflammatory or redox responses, but we will briefly discuss below their potential implications.
With regard to inflammation, several of the down-regulated genes in CVD patients are involved in the canonical or non-canonical NF-κB pathways (Figure 7). In the canonical NF-κB pathway, we found under-expression of the RELA and NFKBIA genes, encoding the transcription factor p65-NF-κB and its cognate repressor IκBα, respectively. This apparent contradiction is consistent with the fact that NFKBIA contains several κB-responsive elements that participate in a feedback control of the canonical pathway, and therefore the depletion of p65-NF-κB will necessarily lead to the diminution of its repressor.21–23 The canonical NF-κB pathway is partly responsible for the persistent low-grade inflammation that characterizes most chronic diseases, including CVD,13,24 and is also pivotal for mounting efficient immune responses against pathogens.25–27
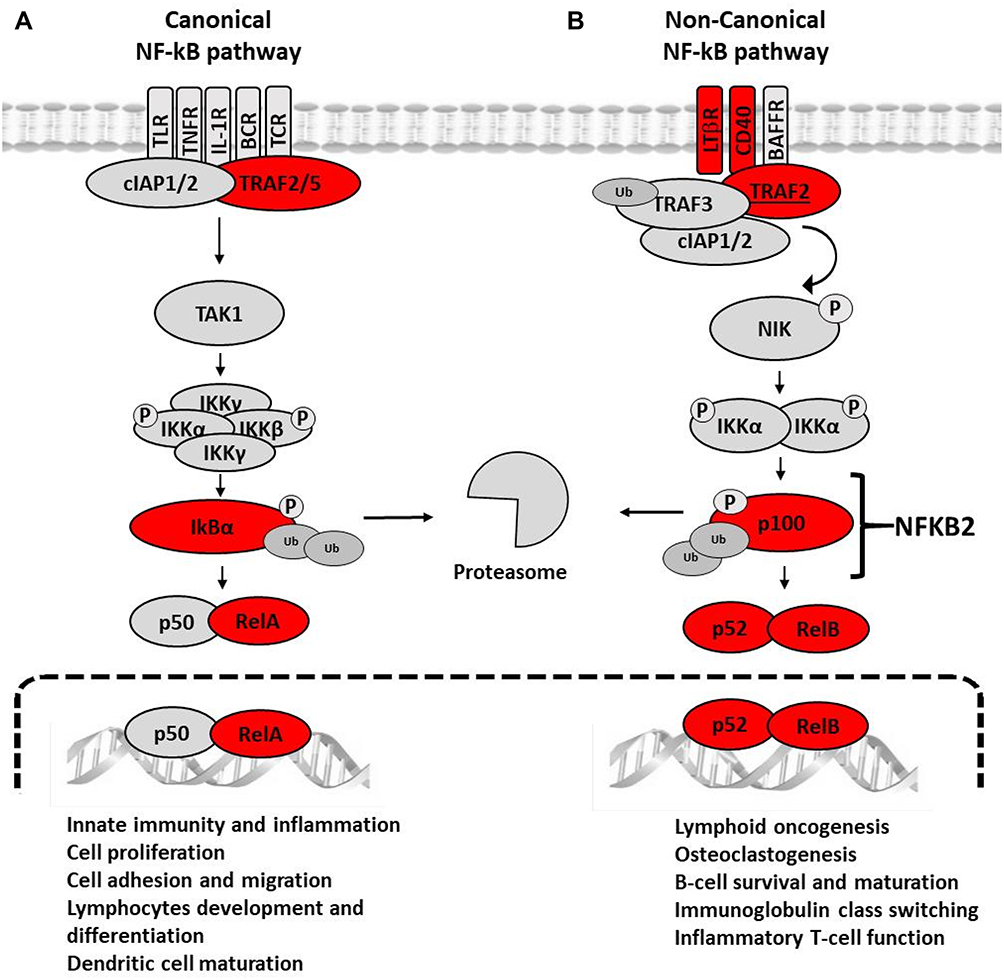 |
Figure 7 Signaling constituents of the canonical (A) and the non-canonical (B) NF-κB pathways that are under-expressed in CVD patients. In (A), the binding of ligands to particular receptors leads to the activation of an IKK complex (IKKα and/or IKKβ catalytic subunits and two IKKγ molecules). This complex phosphorylates IκBα leading to degradation by the proteasome and consequent formation of the transcriptionally-active p50-RelA complex. In (B) the binding of ligands to particular receptors triggers the phosphorylation and processing of p100 (encoded by NFKB2) into the mature p52 protein and subsequent nuclear translocation of the transcriptionally-active RelB-p52 heterodimer. The products of the genes found differentially expressed the present study in the group of patients with CVD are marked with a red circle. |
The under-expression of genes involved in the non-canonical NF-κB pathway addresses the core genes NFKB2 and RELB, whose products lead to the formation of the transcriptionally active p52/RelB heterodimer. Moreover, the scaffold-encoding gene TRAF2, as well as the CD40 and LTA genes encoding members of the tumor necrosis receptor family was also under-expressed. Accordingly, the immune functions governed by this pathway are expected to be impaired in CVD patients, ie, survival and maturation of B lymphocytes, differentiation of dendritic cells into competent antigen presenting cells, and cellular responses against some RNA viruses.28,29 Altogether, the observed gene under-expression within the NF-κB activation pathways might result to some extent in immune suppression.
The down-regulation of the NF-κB pathways in CVD patients was accompanied by the under-expression of several other genes that are under the NF-κB control and/or whose products impinge on the NF-κB system. This was the case of the AKT1 gene.30,31 Moreover, the IRF1 and STAT1 genes, that shape interferon (IFN)-mediated immune responses against viruses and bacteria, were also under-expressed in CVD patients. IRF1, whose transcription is under NF-κB control, encodes a transcriptional regulator of IFN-inducible genes, and STAT1 participates in an amplification loop of IFN.32,33
The suppression of inflammatory pathways in CVD patients is also sustained by the under-expression of genes that are involved in the oxidative metabolism of arachidonic acid. This was the case of the ALOX12 gene, encoding human platelet-type arachidonate 12-lipoxygenase, and the PTGS1 gene encoding the constitutive form of cyclooxygenase (COX-1) that catalyzes the formation of cyclic prostanoids with various effects on platelet activation and aggregation.34–39 In CVD patients, down-regulation of these genes is probably due to the anti-thrombotic medication.40,41 In addition, PTGS1 under-expression may affect the priming of the innate immune system by increasing phagocytosis and ROS-mediated killing of bacteria, while ALOX12 under-expression can impair physiologic inflammatory responses.42,43
Concerning the redox metabolism, several related genes were found under-expressed in CVD patients. These include genes involved in ROS production (DUOX1/2 and MPO), antioxidant response (GSR and TNXRD1), repair of oxidative damage (HSPA1A and MSRA), and regulatory adapter proteins (PDLIM1). The down-regulation of these genes may have consequences on redox signaling as well as on the oxidative antimicrobial defense. For instance, the under-expression of the MPO gene may decrease the ability of blood cells to kill non-specifically invading pathogens through the generated hypochlorous acid.44 Increased MPO levels are also associated with CVD and with a higher mortality risk, while MPO-deficiency was shown to decrease the susceptibility to neutrophil-mediated vascular dysfunction.45,46
Down-regulation of the genes encoding NADPH-oxidases, such as DUOX1 and DUOX2, was reported in the blood of CVD patients.47,48 Although DUOX1/2 expression is high in the thyroid gland, these genes are also induced by pathogens and inflammatory cytokines in immune cells through various receptors in connection with NF-κB signaling.49–52 Therefore, the observed gene under-expression in CVD patients is consistent with the reduction in the levels of the core NF-κB genes and might be related to disease mechanisms and response to therapy, as well as with an impaired antimicrobial defense.53
CVD patients exhibited under-expression of the antioxidant genes GSR and TXNRD2 encoding reductases that regenerate the reduced forms of glutathione and thioredoxin, respectively, hence providing antioxidant protection in CVD.54,55 Other genes identified in the present study are involved in repairing oxidatively damaged, misfolded, and aggregated proteins, namely HSPA1A and MSRA.56–59 Especially MSRA is essential during innate immune responses against intracellular microbes for repairing the oxidative damages inflicted to the host’s proteins by the innate immune response developed against the invading pathogens.60,61
Significant intra-pathway correlations were highlighted in the group of CVD patients for some of the under-expressed inflammatory genes, indicating that the observed changes are most likely integrated in the same signaling networks. In turn, the registered correlations between redox genes seem to indicate only co-occurrence within a disease- or therapy-characteristic pattern and do not appear to reflect functional inter-connection. Some degree of inter-pathway correlations between inflammatory and redox genes was expected, considering that NF-κB activation is under redox control and that NF-κB controls the expression of several antioxidant genes.62,63 Nevertheless, part of the observed correlations between inflammatory and redox genes might derive from independent processes that are either disease-specific or triggered by the medication.
The present study suggests that under-expression of critical inflammation and redox genes in the blood of CVD patients underlies their good clinical evolution under disease-specific medication. Additionally, the normal levels of CVD markers and pro-inflammatory factors in serum, observed in the smaller sub-group of 24 CVD patients versus 22 no-CVD controls, indicated that inflammation seems to be kept under control in these patients, and may account for the favorable disease outcome. Moreover, the down-regulation of IL-1β levels in serum is further sustaining this conclusion. In turn, due to the correlated down-regulation of critical inflammatory and redox genes in blood, the immune competence of CVD patients might be compromised, potentially leading to defective antimicrobial responses. Nevertheless, it is likely that the registered relatively low under-expression of several genes involved in the NF-κB and in redox networks might be insufficient to determine a clinically relevant immune suppression leading to recurrent infections.
Considering that CVD is characterized by low-grade inflammation and oxidative stress, the down-regulation of critical inflammatory and redox genes found in the present study most likely reflects the effect of disease-specific polyvalent medication, comprising angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor antagonists, beta-adrenergic blocking agents, anticoagulant and antiplatelet drugs, diuretics, and statins (Supplementary Figure 1). Some of these drugs exert antioxidant effects that greatly contribute to limit disease progression, therefore giving a new valence to their intended therapeutic use.64,65 For instance, beta-blockers can limit pathologic oxidative stress and redox signaling both in the heart and in the circulating immune cells of patients with heart failure.66 ACE inhibitors prevent vascular superoxide production, increase the bioactivity of nitric oxide, and decrease the activity of various transcription factors such as the ROS-dependent activation of NF-κB in vascular inflammation.67,68 Drug-induced decrease of ROS levels is further impacting on various redox-sensitive signaling pathways and also on gene expression, therefore having an array of consequences broader than those directly related to oxidative stress.69,70
Interestingly, even though both CVD and hypertension were kept under control by medication in the investigated cohorts, and several drugs were used in both disease groups, no significant changes in gene expression were registered in the all-cause hypertension group. This might be due to a distinctive profile of pathological mechanisms in patients with atherosclerosis which dominate in the CVD group. This is possibly resulting also in distinctive or more pronounced effects of medication.
Limitations of the Study
The small number of subjects in each disease group did not allow to stratify patients according to disease sub-categories/stage or therapeutic regimens comprising various combinations of drugs. Based on the results obtained in the present study, broader and more disease- and therapy-focused investigations should be performed for clarifying whether the reported gene expression changes are responsible, at least partly, for keeping disease under control in the context of disease variants and polyvalent medication.
The present study provides data on gene expression changes in whole blood, that might have systemic consequences that have to be comprehensively investigated through a follow-up of the CVD patients. The functional impact of the registered gene expression changes was not thoroughly assessed in this study, except for an analysis of some cardiovascular markers and pro-inflammatory factors in serum. Therefore, further investigations will be required to determine if the observed gene down-regulations result in decreased levels of the proteins encoded by the identified genes and if these cellular disturbances lead to clinically relevant alterations of inflammatory or redox responses.
Conclusions
The present study highlights the down-regulation of several genes involved in NF-kB-mediated inflammation and redox metabolism in the blood of elderly CVD patients with disease kept under control by polyvalent medication. The study strongly suggests that this specific panel of under-expressed inflammation- and redox-related genes can be used for non-invasively monitoring disease progression and patient engagement to medication in CVD.
Abbreviations
ACTB, Actin Beta; AKT1, AKT Serine/Threonine Kinase 1; ALOX12, Arachidonate 12-Lipoxygenase, 12S Type; B2M, Beta-2-Microglobulin; CD40, CD40 Molecule; DUOX1, Dual Oxidase 1; DUOX2, Dual Oxidase 2; GAPDH, Glyceraldehyde-3-Phosphate Dehydrogenase; GSR, Glutathione-Disulfide Reductase; HPRT1, Hypoxanthine Phosphoribosyltransferase 1; HSPA1A, Heat Shock Protein Family A (Hsp70) Member 1A; IRF1, Interferon Regulatory Factor 1, LTA, Lymphotoxin Alpha; MPO, Myeloperoxidase; MSRA, Methionine Sulfoxide Reductase A; NFKB2, Nuclear Factor Kappa B Subunit 2; NFKBIA, NFKB Inhibitor Alpha; PDLIM1, PDZ and LIM Domain 1, PTGS1, Prostaglandin-Endoperoxide Synthase 1; RELA, RELA Proto-Oncogene, NF-KB Subunit; RELB, RELB Proto-Oncogene, NF-KB Subunit; RPLP0, Ribosomal Protein Lateral Stalk Subunit P0; STAT1, Signal Transducer And Activator Of Transcription 1, TRAF2, TNF Receptor Associated Factor 2; TXNRD2, Thioredoxin Reductase 2.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
Research and publication of the present study was funded by the Romanian Ministry of Education and Research through the European Regional Development Fund, Competitiveness Operational Program 2014–2020 [the REDBRAIN project, ID: P_37_732] and through Programme 1 – Development of the national research-development system [grant 7PFE/2018].
Disclosure
The authors declare that there is no financial interest or other conflict of interest regarding the publication of this paper.
References
1. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14(10):576–590. doi:10.1038/s41574-018-0059-4
2. Siti HN, Kamisah Y, Kamsiah J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vascul Pharmacol. 2015;71:40–56. doi:10.1016/j.vph.2015.03.005
3. Guzik TJ, Touyz RM. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension. 2017;70(4):660–667. doi:10.1161/HYPERTENSIONAHA.117.07802
4. Klisic A, Isakovic A, Kocic G, et al. Relationship between oxidative stress, inflammation and dyslipidemia with fatty liver index in patients with Type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes. 2018;126(06):371–378. doi:10.1055/s-0043-118667
5. Miljkovic M, Stefanovic A, Simic-Ogrizovic S, et al. Association of dyslipidemia, oxidative stress, and inflammation with redox status in VLDL, LDL, and HDL lipoproteins in patients with renal disease. Angiology. 2018;69(10):861–870. doi:10.1177/0003319718780041
6. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75(1):685–705. doi:10.1146/annurev-physiol-030212-183653
7. Payne BAI, Chinnery PF. Mitochondrial dysfunction in aging: much progress but many unresolved questions. Biochim Biophys Acta Bioenerg. 2015;1847(11):1347–1353. doi:10.1016/j.bbabio.2015.05.022
8. Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24(12):604–612. doi:10.1016/j.tig.2008.10.002
9. Marín-Aguilar F, Ruiz-Cabello J, Cordero MD. Aging and the Inflammasomes. Exp Suppl. 2018;108:303–320. doi:10.1007/978-3-319-89390-7_13
10. Saez I, Vilchez D. The mechanistic links between proteasome activity, aging and age-related diseases. Curr Genomics. 2014;15(1):38–51. doi:10.2174/138920291501140306113344
11. Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, Von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8(3):311–323. doi:10.1111/j.1474-9726.2009.00481.x
12. Kamo T, Akazawa H, Suda W, et al. Dysbiosis and compositional alterations with aging in the gut microbiota of patients with heart failure. Bando Y, ed. PLoS One. 2017;12(3):e0174099. doi:10.1371/journal.pone.0174099
13. Tilstra JS, Clauson CL, Niedernhofer LJ, Robbins PD. NF-κB in aging and disease. Aging Dis. 2011;2(6):449–465.
14. Coppé J-P, Patil CK, Rodier F, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. Blagosklonny M V., ed. PLoS One. 2010;5(2):e9188. doi:10.1371/journal.pone.0009188
15. Hishikawa A, Hayashi K, Itoh H. Transcription Factors as Therapeutic Targets in Chronic Kidney Disease. Molecules. 2018;23(5):1123. doi:10.3390/molecules23051123
16. Papaconstantinou J. The role of signaling pathways of inflammation and oxidative stress in development of senescence and aging phenotypes in cardiovascular disease. Cells. 2019;8(11):1383. doi:10.3390/cells8111383
17. Liguori I, Russo G, Curcio F, et al. Oxidative stress, aging, and diseases. Clin Interv Aging. 2018;13:757–772. doi:10.2147/CIA.S158513
18. Furman D, Campisi J, Verdin E, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25(12):1822–1832. doi:10.1038/s41591-019-0675-0
19. IJsselmuiden AJ, Musters RJ, de Ruiter G, et al. Circulating white blood cells and platelets amplify oxidative stress in heart failure. Nat Clin Pract Cardiovasc Med. 2008;5(12):811–820. doi:10.1038/ncpcardio1364
20. Xie F, Xiao P, Chen D, Xu L, Zhang B. miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol Biol. 2012;80(1):75–84. doi:10.1007/s11103-012-9885-2
21. Cheng Q, Cant CA, Moll T, et al. NK-kappa B subunit-specific regulation of the I kappa B alpha promoter. J Biol Chem. 1994;269(18):13551–13557. doi:10.1016/S0021-9258(17)36866-7
22. Huang TT, Miyamoto S. Postrepression activation of NF-κB requires the amino-terminal nuclear export signal specific to IκBα. Mol Cell Biol. 2001;21(14):4737–4747. doi:10.1128/MCB.21.14.4737-4747.2001
23. Mathes E, O’Dea EL, Hoffmann A, Ghosh G. NF-κB dictates the degradation pathway of IκBα. EMBO J. 2008;27(9):1357–1367. doi:10.1038/emboj.2008.73
24. Gordon JW, Shaw JA, Kirshenbaum LA. Multiple Facets of NF-κB in the heart. Circ Res. 2011;108(9):1122–1132. doi:10.1161/CIRCRESAHA.110.226928
25. Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2(1):17023. doi:10.1038/sigtrans.2017.23
26. Dorrington MG, Fraser IDC. NF-κB signaling in macrophages: dynamics, crosstalk, and signal integration. Front Immunol. 2019;10. doi:10.3389/fimmu.2019.00705.
27. Nelson RH, Nelson DE. Signal distortion: how intracellular pathogens alter host cell fate by modulating NF-κB dynamics. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.02962.
28. Hernandez A, Burger M, Blomberg BB, et al. Inhibition of NF-κB during human dendritic cell differentiation generates anergy and regulatory T-cell activity for one but not two human leukocyte antigen DR mismatches. Hum Immunol. 2007;68(9):715–729. doi:10.1016/j.humimm.2007.05.010
29. Sun S-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17(9):545–558. doi:10.1038/nri.2017.52
30. Du K, Tsichlis PN. Regulation of the Akt kinase by interacting proteins. Oncogene. 2005;24(50):7401–7409. doi:10.1038/sj.onc.1209099
31. Bai D, Ueno L, Vogt PK. Akt-mediated regulation of NFκB and the essentialness of NFκB for the oncogenicity of PI3K and Akt. Int J Cancer. 2009;125(12):2863–2870. doi:10.1002/ijc.24748
32. Iwanaszko M, Kimmel M. NF-κB and IRF pathways: cross-regulation on target genes promoter level. BMC Genomics. 2015;16(1):307. doi:10.1186/s12864-015-1511-7
33. Balachandran S, Beg AA. Defining emerging roles for NF-κB in antivirus responses: revisiting the interferon-β enhanceosome paradigm. Rall GF, ed. PLoS Pathog. 2011;7(10):e1002165. doi:10.1371/journal.ppat.1002165
34. Roumeliotis AK, Roumeliotis SK, Panagoutsos SA, et al. Association of ALOX12 gene polymorphism with all-cause and cardiovascular mortality in diabetic nephropathy. Int Urol Nephrol. 2018;50(2):321–329. doi:10.1007/s11255-017-1755-z
35. Dobrian AD, Lieb DC, Cole BK, Taylor-Fishwick DA, Chakrabarti SK, Nadler JL. Functional and pathological roles of the 12- and 15-lipoxygenases. Prog Lipid Res. 2011;50(1):115–131. doi:10.1016/j.plipres.2010.10.005
36. Porro B, Songia P, Squellerio I, Tremoli E, Cavalca V. Analysis, physiological and clinical significance of 12-HETE: A neglected platelet-derived 12-lipoxygenase product. J Chromatogr B. 2014;964:26–40. doi:10.1016/j.jchromb.2014.03.015
37. Humphries ESA, Dart C. Neuronal and cardiovascular potassium channels as therapeutic drug targets. J Biomol Screen. 2015;20(9):1055–1073. doi:10.1177/1087057115601677
38. Awtry EH, Loscalzo J. Aspirin. Circulation. 2000;101(10):1206–1218. doi:10.1161/01.CIR.101.10.1206
39. Matthay MA. Severe sepsis — a new treatment with both anticoagulant and antiinflammatory properties. N Engl J Med. 2001;344(10):759–762. doi:10.1056/NEJM200103083441009
40. Ikei KN, Yeung J, Apopa PL, et al. Investigations of human platelet-type 12-lipoxygenase: role of lipoxygenase products in platelet activation. J Lipid Res. 2012;53(12):2546–2559. doi:10.1194/jlr.M026385
41. Iñiguez MA, Cacheiro-Llaguno C, Cuesta N, Díaz-Muñoz MD, Fresno M. Prostanoid function and cardiovascular disease. Arch Physiol Biochem. 2008;114(3):201–209. doi:10.1080/13813450802180882
42. Stables MJ, Newson J, Ayoub SS, Brown J, Hyams CJ, Gilroy DW. Priming innate immune responses to infection by cyclooxygenase inhibition kills antibiotic-susceptible and -resistant bacteria. Blood. 2010;116(16):2950–2959. doi:10.1182/blood-2010-05-284844
43. Mashima R, Okuyama T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 2015;6:297–310. doi:10.1016/j.redox.2015.08.006
44. Nauseef WM. Myeloperoxidase in human neutrophil host defence. Cell Microbiol. 2014;16(8):1146–1155. doi:10.1111/cmi.12312
45. Ndrepepa G. Myeloperoxidase – a bridge linking inflammation and oxidative stress with cardiovascular disease. Clin Chim Acta. 2019;493:36–51. doi:10.1016/j.cca.2019.02.022
46. Rudolph TK, Wipper S, Reiter B, et al. Myeloperoxidase deficiency preserves vasomotor function in humans. Eur Heart J. 2012;33(13):1625–1634. doi:10.1093/eurheartj/ehr193
47. Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. 2015;12(1):5–23. doi:10.1038/cmi.2014.89
48. Donkó Á, Péterfi Z, Sum A, Leto T, Geiszt M. Dual oxidases. Philos Trans R Soc B Biol Sci. 2005;360(1464):2301–2308. doi:10.1098/rstb.2005.1767
49. Joo J-H, Ryu J-H, Kim C-H, et al. Dual oxidase 2 is essential for the toll-like receptor 5-mediated inflammatory response in Airway Mucosa. Antioxid Redox Signal. 2012;16(1):57–70. doi:10.1089/ars.2011.3898
50. van der Vliet A, Danyal K, Heppner DE. Dual oxidase: a novel therapeutic target in allergic disease. Br J Pharmacol. 2018;175(9):1401–1418. doi:10.1111/bph.14158
51. Ha E-M, Lee K-A, Seo YY, et al. Coordination of multiple dual oxidase–regulatory pathways in responses to commensal and infectious microbes in drosophila gut. Nat Immunol. 2009;10(9):949–957. doi:10.1038/ni.1765
52. Yang HT, Huang YH, Yang GW. Mini review: immunologic functions of dual oxidases in mucosal systems of vertebrates. Braz J Biol. 2019. doi:10.1590/1519-6984.208749
53. Sarr D, Tóth E, Gingerich A, Rada B. Antimicrobial actions of dual oxidases and lactoperoxidase. J Microbiol. 2018;56(6):373–386. doi:10.1007/s12275-018-7545-1
54. Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Circ Physiol. 2007;292(3):H1227–H1236. doi:10.1152/ajpheart.01162.2006
55. Yoshihara E, Masaki S, Matsuo Y, Chen Z, Tian H, Yodoi J. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front Immunol. 2014;4. doi:10.3389/fimmu.2013.00514.
56. Calabrese V, Cornelius C, Cuzzocrea S, Iavicoli I, Rizzarelli E, Calabrese EJ. Hormesis, cellular stress response and vitagenes as critical determinants in aging and longevity. Mol Aspects Med. 2011;32(4–6):279–304. doi:10.1016/j.mam.2011.10.007
57. Korbelik M, Sun J, Cecic I. Photodynamic therapy-induced cell surface expression and release of heat shock proteins: relevance for tumor response. Cancer Res. 2005;65(3):1018–1026.
58. Aridon P, Geraci F, Turturici G, D’Amelio M, Savettieri G, Sconzo G. Protective role of heat shock proteins in Parkinson’s disease. Neurodegener Dis. 2011;8(4):155–168. doi:10.1159/000321548
59. Jiang B, Moskovitz J. The functions of the mammalian methionine sulfoxide reductase system and related diseases. Antioxidants. 2018;7(9):122. doi:10.3390/antiox7090122
60. Drazic A, Miura H, Peschek J, et al. Methionine oxidation activates a transcription factor in response to oxidative stress. Proc Natl Acad Sci. 2013;110(23):9493–9498. doi:10.1073/pnas.1300578110
61. Krishna S, Miller LS. Innate and adaptive immune responses against Staphylococcus aureus skin infections. Semin Immunopathol. 2012;34(2):261–280. doi:10.1007/s00281-011-0292-6
62. Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25(51):6717–6730. doi:10.1038/sj.onc.1209937
63. Morgan MJ, Liu Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21(1):103–115. doi:10.1038/cr.2010.178
64. García N, Zazueta C, Aguilera-Aguirre L. Oxidative stress and inflammation in cardiovascular disease. Oxid Med Cell Longev. 2017;2017:1–2. doi:10.1155/2017/5853238
65. Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. Oxidative stress in atherosclerosis. Curr Atheroscler Rep. 2017;19(11):42. doi:10.1007/s11883-017-0678-6
66. Nakamura K, Murakami M, Miura D, et al. Beta-blockers and oxidative stress in patients with heart failure. Pharmaceuticals. 2011;4(8):1088–1100. doi:10.3390/ph4081088
67. Munzel T, Keaney JF. Are ACE inhibitors a “Magic Bullet” against oxidative stress? Circulation. 2001;104(13):1571–1574. doi:10.1161/hc3801.095585
68. Kortekaas KE, Meijer CA, Hinnen JW, et al. ACE inhibitors potently reduce vascular inflammation, results of an open proof-of-concept study in the abdominal aortic aneurysm. Norata GD, ed. PLoS One. 2014;9(12):e111952. doi:10.1371/journal.pone.0111952
69. Kunsch C, Medford RM. Oxidative stress as a regulator of gene expression in the vasculature. Circ Res. 1999;85(8):753–766. doi:10.1161/01.RES.85.8.753
70. Priya Dharshini LC, Vishnupriya S, Sakthivel KM, Rasmi RR. Oxidative stress responsive transcription factors in cellular signalling transduction mechanisms. Cell Signal. 2020;72:109670. doi:10.1016/j.cellsig.2020.109670
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.