")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Discovery and evaluation of novel anti-inflammatory derivatives of natural bioactive curcumin
Authors Zhang Y, Jiang X, Peng K, Chen C, Fu L, Wang Z, Feng J, Liu Z, Zhang H, Liang G, Pan Z
Received 24 June 2014
Accepted for publication 31 July 2014
Published 4 November 2014 Volume 2014:8 Pages 2161—2171
DOI https://doi.org/10.2147/DDDT.S69914
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Yali Zhang,1,* Xin Jiang,2,* Kesong Peng,1 Chengwei Chen,3 Lili Fu,1 Zhe Wang,1 Jianpeng Feng,1 Zhiguo Liu,1,4 Huajie Zhang,1 Guang Liang,1 Zheer Pan3
1Chemical Biology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 2The Children’s Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, People’s Republic of China; 3Department of Orthopedic Surgery, The First Affiliated Hospital, Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 4Wenzhou Undersun Biotchnology Co. Ltd., Wenzhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Abstract: Curcumin is a natural active product that has various pharmacological activities such as anti-inflammatory effects. Here, we report the synthesis and evaluation of 34 monocarbonyl curcumin analogs as novel anti-inflammatory agents. Among the analogs, the symmetrical heterocyclic type displayed the strongest inhibition of lipopolysaccharide (LPS)-stimulated expression of pro-inflammatory cytokines in macrophages. Analogs S1–S5 and AS29 reduced tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) production in a dose-dependent manner and also displayed excellent stability and low cytotoxicity in vitro. In addition, analog S1 dose-dependently inhibited LPS-induced extracellular signal-regulated kinase (ERK) phosphorylation. Furthermore, analogs S1 and S4 displayed a significant protective effect on LPS-induced septic death in mouse models, with 40% and 50% survival rates, respectively. These data demonstrate that the heterocyclic monocarbonyl curcumin analogs have potential therapeutic effects in acute inflammatory diseases.
Keywords: curcumin, anti-inflammation, sepsis, macrophage, cytokine
Introduction
Inflammation is part of a complex biological response that is induced by harmful stimulation, such as burns, pathogens, and chemical irritants. Over-expressed inflammatory cytokines are involved in the pathological processes of a number of diseases.1,2 In particular, sepsis, a systemic inflammatory response syndrome characterized by cytokine storm and multi-organ failure that occurs during severe infection, is believed to be related to an exacerbated release of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), and interleukin-6 (IL-6).3,4 Excessive or sustained release of inflammatory cytokines leads to serious consequences, including septic shock.5 Cytokines also play a vital role in the development of sepsis.6–9 The development of novel therapeutic agents that block pro-inflammatory pathways or inhibit the over-expression of cytokines has been the focus of researchers investigating the treatment of inflammatory disease.10–12
Curcumin is a natural yellow product derived from the turmeric rhizome of the herb Curcuma Longa (Figure 1). Curcumin has shown to be non-toxic and exhibits various biological activities such as anti-oxidant, anti-inflammatory, anti-carcinogenic, anti-diabetic, anti-bacterial, anti-fungal, anti-viral, anti-fibrotic and anti-ulcer effects.13–17 Curcumin treatment has been observed to have both preventive and therapeutic anti-inflammatory effects in various animal models.18 In addition, more than 65 clinical trials of curcumin have finished and curcumin is proven to be safe.19 Despite the effective activity of curcumin on many cellular targets linked to a variety of diseases, low bioavailability and weak stability have significantly limited its clinical application.20 To find new analogs with increased bioavailability and enhanced pharmacological activity, researchers have attempted the chemical modification of curcumin.21–23 Among these analogs, monocarbonyl analogs have received much attention as potential curcumin analogs, and the beta-diketone moiety, which is believed to be responsible for the weak metabolism, has been removed.24,25 Our group has adopted the strategy of synthesis of monocarbonyl curcumin analogs to overcome these limitations (Figure 1). Based on the aforementioned studies, and as a part of our ongoing research, 34 novel monocarbonyl analogs of curcumin were synthesized, and their cytokine-inhibitory and anti-sepsis effects were studied.
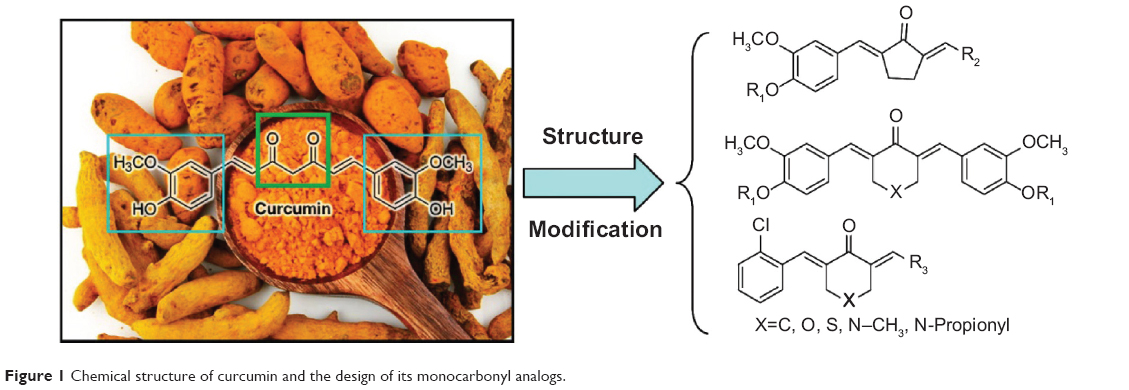 | Figure 1 Chemical structure of curcumin and the design of its monocarbonyl analogs. |
Materials and methods
Chemical experimental procedures
The synthesis of enamide 2 and 7
To a stirred solution of cyclopentanone or piperidine or pyrone (20 mM) and p-TSA (200 mg) in cyclohexane (20 mL) was added morpholine (30 mM), and the mixture reaction was refluxed for four hours at 90°C. Subsequently, the solvent was removed under reduced pressure to give various activated enamine intermediates 2 or 7 as yellow viscous oils, and they were used directly without further purification.
General procedure for synthesis of S1–S5
To a stirred solution of Vanilline (10 mM) in ethanol (EtOH) (12 mL) was added N-propionyl 4-piperidone, pyrone, or thiopyrone (5 mM) at room temperature, and hydrogen chloride (HCl) (gas) was bubbled into the mixture until the reaction was completed. Then the reaction mixture was poured into cold water (25 mL) to yield a colored precipitate. After filtration, the resultant colored solid was washed with water and used immediately for the next step without further purification. To a solution of the crude product in tetrahydrofuran (THF) (10 mL) was added propionyl or isobutyryl chloride (10 mM) in the presence of triethylamine (Et3N), resulting in S1–S5. The resulting crude products were purified by chromatography over silica gel using petroleum/ethyl acetate (EtOAc) as the eluent.
General procedure for the synthesis of AS1–AS15, AS28, and AS29
The stirred solution of 3 or 9 (2 mM) in EtOH (25 mL) was added to the various aldehydes (2 mM) at room temperature. Subsequently, HCl (gas) was bubbled into the mixture for 30 minutes, and the mixture was stirred for 3 to 5 hours at 50°C–70°C. The mixture was then poured into ice water and extracted with EtOAc (3×20 mL). The combined organic layers were washed with 10% sodium bicarbonate (NaHCO3), dried over anhydrous magnesium sulfate (MgSO4), filtered, and concentrated in vacuo. Flash chromatography of the residue over silica gel using EtOAc/petroleum as the eluent gave AS1–AS15, AS28 and AS29.
General procedure for the synthesis of AS16–AS27
Similar to the procedure described for the preparation of AS1–AS15 with the exception of using a different aldehyde, all the phenolic hydroxyls of the resulting products (1 mM) were estered by dropping the propionyl chloride (10 mM) in the presence of Et3N (0.25 mL) at 0°C, using THF (10 mL) as the solvent. The reaction mixture was stirred at room temperature overnight. Thin layer chromatography demonstrated that the reaction had been completed. After removing the solvent, the resultant residue was chromatographed over silica gel using EtOAc/petroleum, as the eluent generated the desired products AS16–AS27.
Cell line and reagents
Mouse macrophages and human normal hepatic HL-7702 cells were obtained from the American Type Culture Collection, Manassas, VA, USA. Cell culture reagents were obtained from Gibco Life Technologies, Grand Island, NY, USA. Fetal bovine serum (obtained from HyClone, Logan, UT, USA) was heat-inactivated for thirty minutes at 65°C. Lipopolysaccharide (LPS) and chemical reagents were purchased from Sigma–Aldrich, St Louis, MO, USA. Anti-p-ERK and anti-ERK antibody was obtained from Santa Cruz Biotechnology, Santa Cruz, CA, USA; anti-p-p38, anti-p38, anti-p-JNK and anti-JNK were from Cell Signaling Technology, Danvers, MA, USA. Curcumin and compounds S1–S5, and AS1–AS29 were dissolved in dimethyl sulfoxide (DMSO) for in vitro experiments.
Animals
Male C57BL/6 mice weighing 18–22 g were obtained from the Animal Center of Wenzhou Medical University (Wenzhou, People’s Republic of China). Animals were housed at constant room temperature with a 12:12 hour light:dark cycle, fed with a standard rodent diet, and provided with sufficient water. The animals were allowed to adapt to the laboratory environment for at least 7 days before their use in experiments. Protocols involving the use of animals were approved by the Wenzhou Medical University Animal Policy and Welfare Committee.
Enzyme-linked immunosorbent assay
Mouse macrophages were incubated in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, Eggenstein, Germany) and replenished with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin at 37°C with 5% CO2. Cells were pretreated with compounds for 30 minutes, then treated with LPS (0.5 μg/mL) for 24 hours. After treatment, the culture media and cells were collected separately. The levels of tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) in the media were determined by enzyme-linked immunosorbent assay (ELISA) using mouse TNF-α and mouse IL-6 ELISA kits (eBioScience, San Diego, CA). The total quantity of the inflammatory factor in the media was standardized to the total protein amount of the viable cell pellets.
Western blot
Macrophages were treated with 0.5 μg/mL LPS for 20 minutes before incubation with or without compounds for 30 minutes. For the preparation of total cell proteins, macrophages were washed with PBS and lysed. The proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), and were electrophoretically transferred onto nitrocellulose membrane. The membranes were first incubated with the primary antibodies, then incubated with the horseradish peroxidase (HRP)-coupled secondary antibodies. Detection was performed with enhanced chemiluminescence reagents, and the protein bands were quantified by densitometry using ImageJ image processing program (US National Institutes of Health, Washington, DC, USA).
Methyl thiazolyl tetrazolium assay
HL-7702 cells were seeded into 96-well plates at a density of 5,000 cells per well in Roswell Park Memorial Institute 1640 medium, and replenished with 5% heat-inactivated serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were incubated at 37°C in 5% CO2 for 24 hours. Cells were cultured with DMSO or 10 μM tested compounds for 24 hours before the methyl thiazolyl tetrazolium (MTT) assay. A fresh solution of MTT (5 mg/mL) prepared in phosphate buffer solution was added to each single well. The plate was then incubated in a CO2 incubator for 4 hours, cells dissolved with 150 μL DMSO, and read for optical density at 490 nm. Viability was defined as the ratio (expressed as a percentage) of absorbance of treated cells to DMSO treated cells.
In vivo study method
Compounds were firstly dissolved with macrogol 15 hydroxystearate (a nonionic solubilizer [BASF, Ludwigshafen, Germany]) for injection with or without medium chain triglycerides (MCT), from BASF, in a water bath at 37°C. The concentration of compounds was 2 mg/mL. The concentration of solubilizer ranged from 5%–10%, and MCT 0.5%–2% in the final solution. For the vehicle, the mixture of solubilizer and MCT was prepared at 10% and 2%, respectively. Male C57BL/6 mice weighing 18–22 g were pretreated with compound S1 or S4 (10 mg/kg) in a water solution by intravenous injection 15 minutes before the intraperitoneal injection of LPS (15 mg/kg). Control animals received a similar volume (200 μL) of vehicle. Body weight change and mortality were recorded for 7 days.
The stability experiment of active analogs
Compounds were dissolved in DMSO, and the concentration was 10 mM. The stock solution was diluted 10 times, and then added 1 μL diluent into an Eppendorf tube which was assembled with 99 μL phosphate buffered saline (PBS). After that we could obtain the miscible liquid with the concentration of 10 μM and add it into the cuvette which was read for optical density at 250–600 nm. The absorbance was read at set intervals (0, 5, 10, 15, 20, and 25 minutes).
Statistical analysis
The results are presented as means ± standard error of the means (SEMs). Student’s t-test was employed to analyze the differences between sets of data. Statistics were performed using GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA, USA). P-values less than 0.05 (P<0.05) were considered indicative of significance. All experiments were repeated at least three times.
Results and discussion
Chemistry
The synthetic routes of the symmetrical and asymmetrical monocarbonyl analogs of curcumin (MACs) are shown in Figure 2. The synthesis of the symmetrical curcumin analogs S1–S5 began with the key Claisene–Schmidt condensation reaction between vanillin and corresponding heterocyclic ketone, including piperidone, pyrone and thiopyrone. All of the phenolic hydroxyls of the products were then estered with propionyl or isobutyryl chloride in the presence of Et3N. The asymmetrical monocarbonyl curcumin derivatives (AS1–AS29) were synthesized also through the aldol condensation strategy. Commercially available cyclopentanone 3 or heterocyclic ketone 4 were activated with morpholine, and the resulting enamines 2 and 7 were condensed with vanillin and 2-chlorobenzaldehyde, respectively, to afford the desired unilateral unsaturated ketones. Subsequently, AS1–AS15, AS28, and AS29 were obtained by constructing the second unsaturated ketone in an acidic environment. Finally, propionylation of the phenolic hydroxyls was carried out in the presence of Et3N to complete the synthesis of AS16–AS27. All compounds were purified by recrystallization or column chromatography. Their structures, determined by spectral data from electrospray mass spectrometry (ESI-MS) and proton nuclear magnetic resonance (1H-NMR) spectroscopy, are shown in Figure 2. Spectral data can be found in the Supplementary material. Before biological experiments were carried out, the purity of all compounds was detected by high-performance liquid chromatography (HPLC), as shown in the Supplementary material.
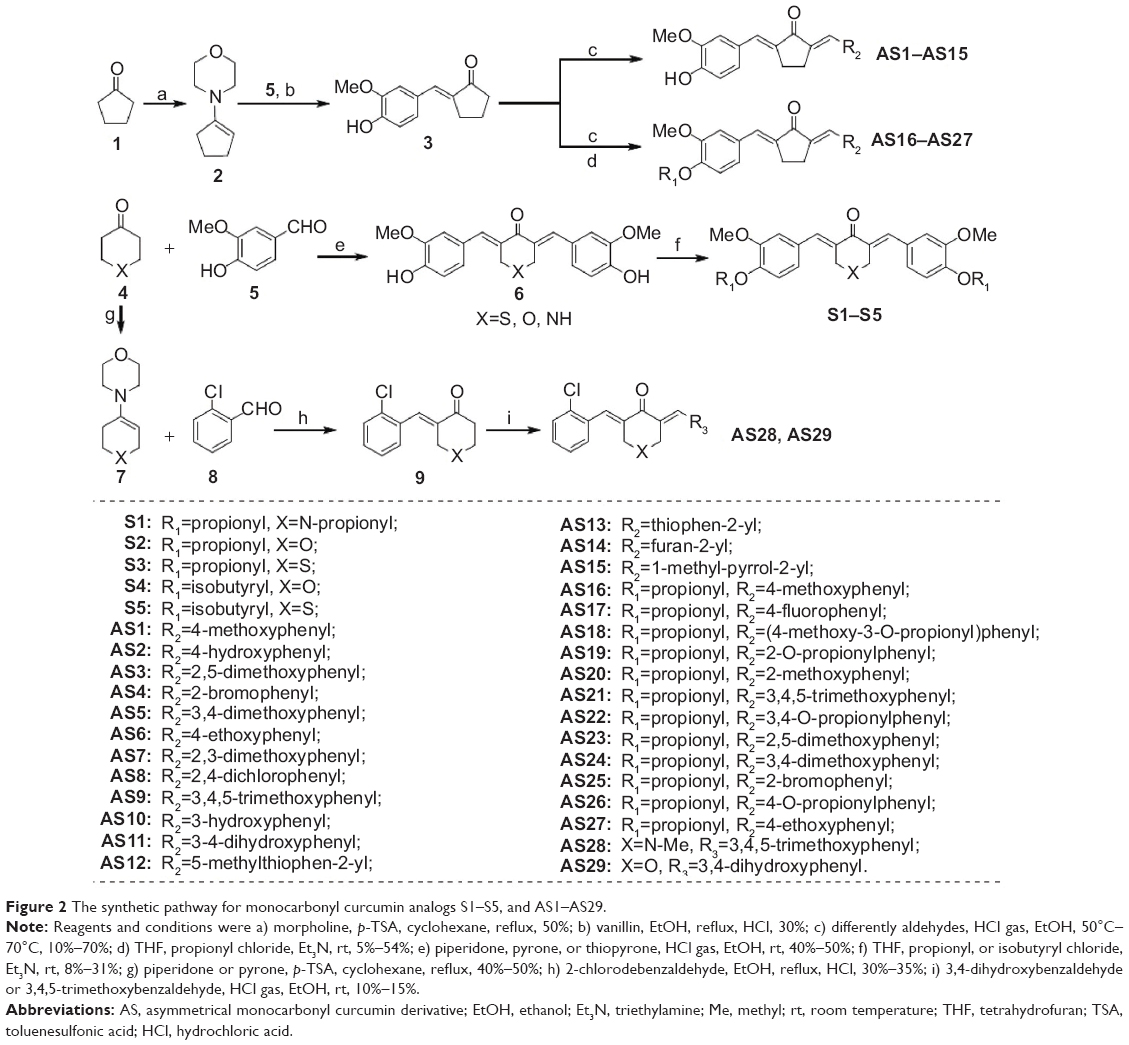 | Figure 2 The synthetic pathway for monocarbonyl curcumin analogs S1–S5, and AS1–AS29. |
Compounds inhibited LPS-induced TNF-α and IL-6 production
TNF-α and IL-6 are two well-known pro-inflammatory cytokines. It has been well demonstrated that they play important roles in the pathological development of many inflammatory diseases.26,27 To investigate whether the 34 synthetic analogs have anti-inflammatory activity, their ability to reduce LPS-stimulated TNF-α and IL-6 production was determined in macrophages. After pretreatment with compounds for 30 minutes, macrophages were treated with LPS for 24 hours. The levels of cytokines in the culture media were determined by ELISA. The results of the anti-inflammatory assay of two series of monocarbonyl curcumin analogs are shown in Figure 3. Initial screening showed that most of the analogs attenuated the LPS-induced TNF-α and IL-6 secretion at a dosage of 10 μM. Several of the synthetic analogs exhibited a more potent inhibitory ability than curcumin. In particular, the heterocyclic analogs (S1–S5, and AS29) showed stronger inhibition than the others, with a range of 59.5%–83.4%. Compound S5, which has a thiopyrone skeleton, displayed the greatest activity, with an inhibition ratio of 98.7%. As for the cyclopenta-containing monocarbonyl analogs, compound AS12, which has a heterocyclic-methylthiophen R2 substitution-group, also showed an inhibition ratio over 50% on IL-6 production. Of the asymmetrical analogs, only the 4-piperidone derivative AS29 exhibited a strong inhibitory effect. Therefore, the design of novel monocarbonyl curcumin analogs with a symmetric and heterocyclic scaffold may improve the anti-inflammatory activity of this group of compounds.
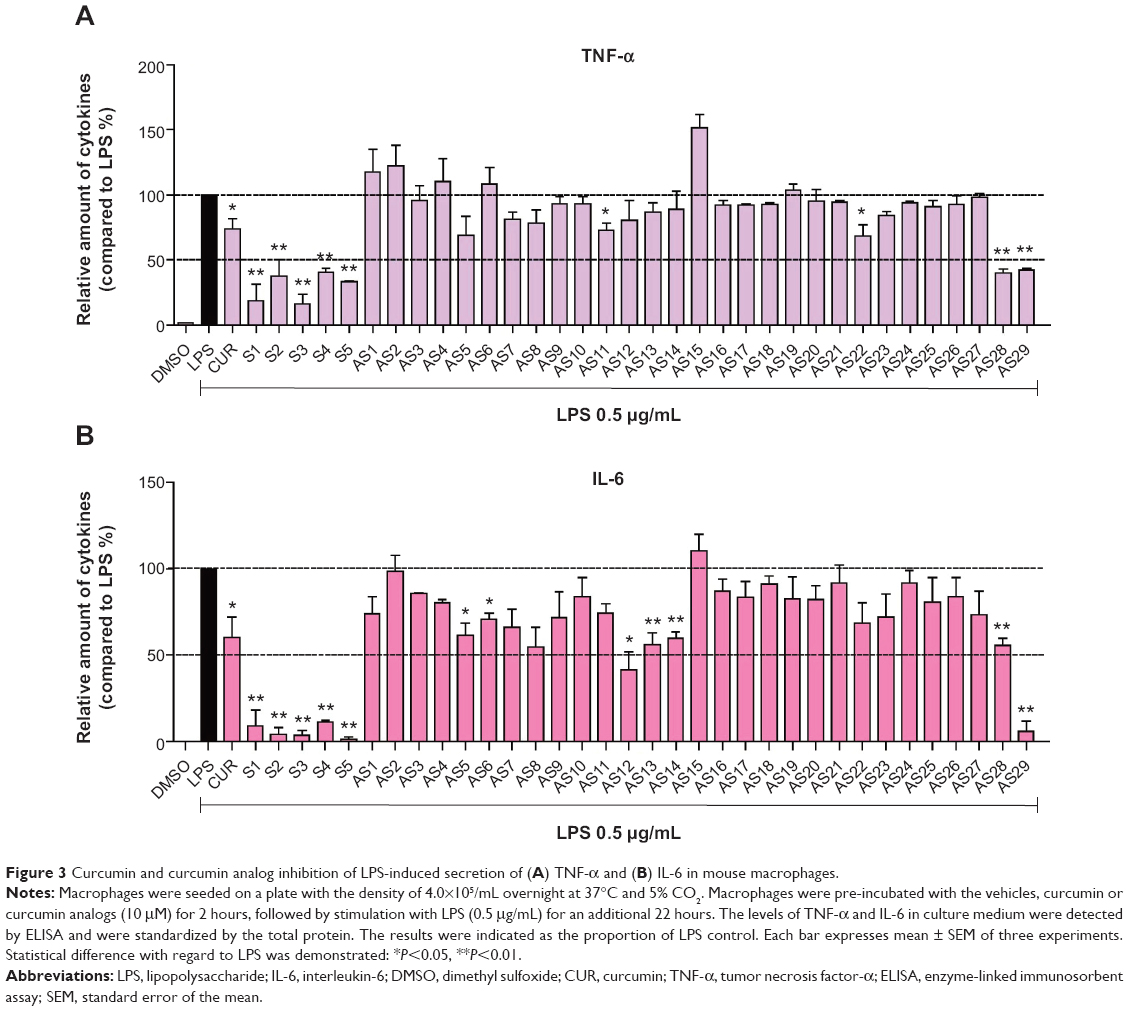 | Figure 3 Curcumin and curcumin analog inhibition of LPS-induced secretion of (A) TNF-α and (B) IL-6 in mouse macrophages. |
Quantitative structure–activity relationship study
A quantitative structure–activity relationship (QSAR) study was carried out to demonstrate the effects of various substituents on the anti-inflammatory activity of the analogs. To obtain a QSAR model, the molecular structures of all the curcumin analogs were built with Maestro Version 9.1 (Schrödinger, New York, NY, USA) and represented by a series of molecular descriptors. All calculations were based on the semi-empirical parameterized model 6 (PM6) method. A detailed description of the QSAR study is presented in the Supplementary material. Scatter plots showing predicted activities versus experimental values are displayed in Figure 4. Statistically significant models, Equation 1 (Eq1) and Equation 2 (Eq2), containing three different variables, were obtained for the anti-TNF-α and anti-IL-6 activities of the compounds, respectively. Both models possessed relatively high regression coefficients: R2=0.86 for TNF-α inhibition and R2=0.89 for IL-6 inhibition. To further estimate the QSAR model, leave-one-out (LOO) cross-validation was performed. The Q-squares from LOO validation were 0.85 and 0.88 for TNF-α inhibition model and IL-6 inhibition model, respectively. LOO cross-validation manifested the final models as robust and satisfactory. The high-quality QSAR results indicated that the anti-inflammatory activities of curcumin analogs are highly correlated with their chemical structures. Comprehensive consideration of the structure–activity relationship (SAR) and QSAR results showed that molecular symmetry and electronegativity may play a crucial role in the anti-inflammatory activity of these monocarbonyl curcumin analogs.
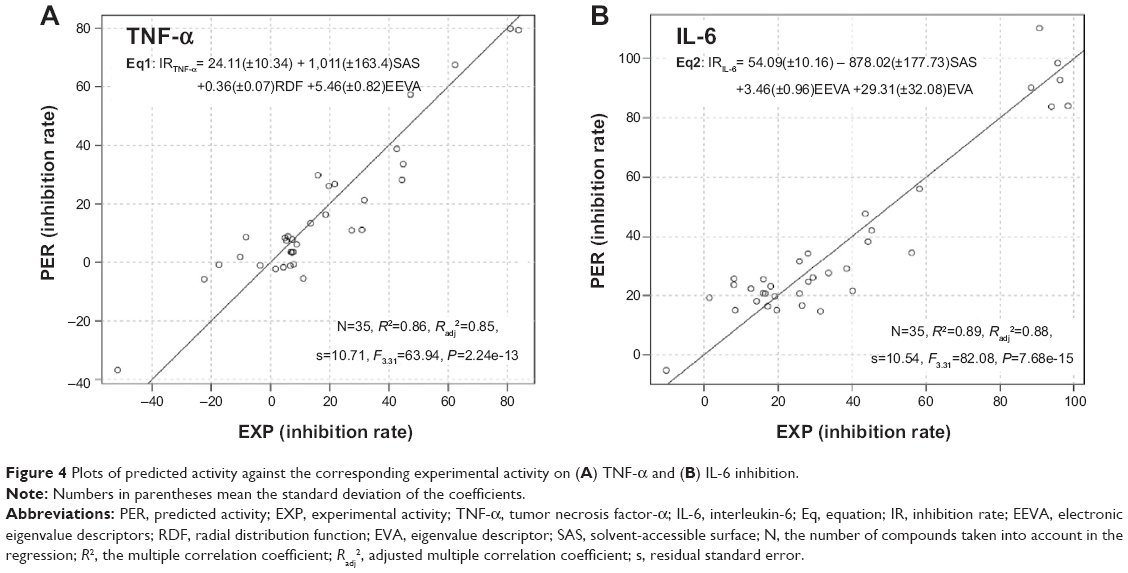 | Figure 4 Plots of predicted activity against the corresponding experimental activity on (A) TNF-α and (B) IL-6 inhibition. |
Active compounds dose-dependently inhibited LPS-induced cytokines production
The dose-dependent inhibitory effects of six of the above-mentioned compounds, S1–S5 and AS29, on LPS-induced TNF-α and IL-6 production were studied. After pretreatment with analogs at a series of concentrations (1.0, 2.5 and 5 μM) for 30 minutes, cells were treated with LPS (0.5 μg/mL) for 24 hours. An ELISA assay was used to determine the amount of TNF-α and IL-6 released. As shown in Figure 5, all six compounds exhibited good, dose-dependent reduction of LPS-induced TNF-α and IL-6 production. This result further demonstrates the potential of these analogs as anti-inflammatory agents.
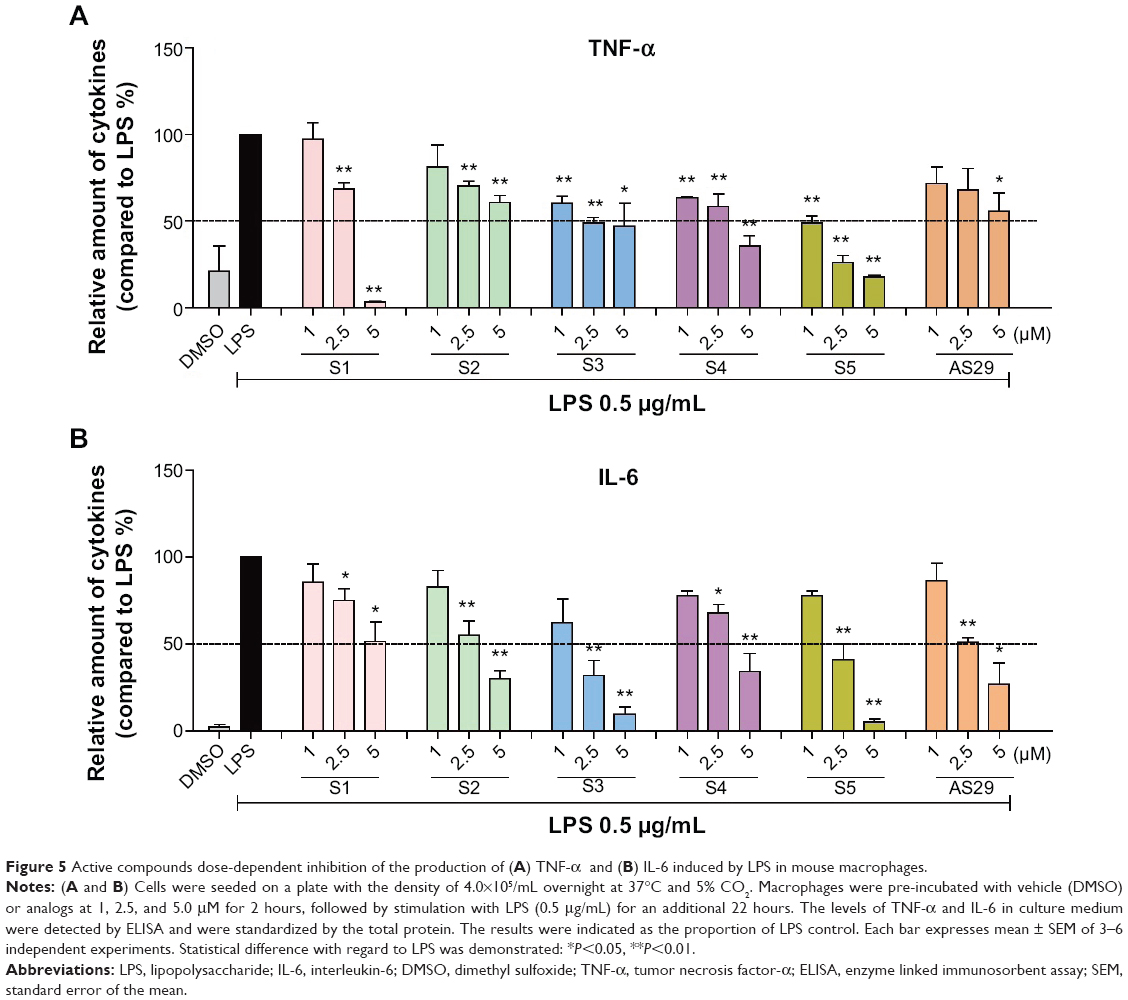 | Figure 5 Active compounds dose-dependent inhibition of the production of (A) TNF-α and (B) IL-6 induced by LPS in mouse macrophages. |
S1 attenuated LPS-induced ERK phosphorylation
In the lipopolysaccharide–toll-like receptor 4 (LPS–TLR4) inflammatory pathway, NF-κB, a nuclear transcriptional factor, and mitogen-activated protein kinases (MAPKs) play key roles in regulating inflammatory cytokine gene expression.28 Curcumin has been demonstrated to reduce a variety of gene transcriptions via suppressing the activation of NF-κB and MAPKs.29 To identify the possible signaling pathways involved in the anti-inflammatory effects of compounds S1–S5 and AS29, we first examined the effect of these six compounds on LPS-activated NF-κB and MAPKs. Three types of MAPKs have been identified: extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK. Under resting conditions, NF-κB is bound to inhibitory κB proteins such as IκB-α, which sequester NF-κB in cytoplasm as an inactive complex. In response to LPS, IκBα is subjected to ubiquitylation, followed by proteasome-mediated degradation, after which NF-κB moves into the nucleus to promote the transcription of inflammatory genes.30 Thus, we used a Western blot to detect IκB degradation and the phosphorylation of ERK, JNK, and p38 in LPS-stimulated macrophages with or without the pretreatment of these compounds. As illustrated in Figure 6A, the addition of LPS for 20 minutes resulted in obvious phosphorylation of MAPKs (ERK, P38, and JNK) and degradation of IκB-α. Pretreatment with the anti-inflammatory active analogs S1–S5 and AS29 at 10 μM did not reduce the MAPK phosphorylation and IκB-α degradation, with the exception of S1, which significantly inhibited the LPS-induced phosphorylation of ERK. To determine the dose-dependent effect of S1, macrophages were pretreated with doses of 2.5, 5, or 10 μM for 30 minutes. LPS was then added, and the macrophages were incubated for an additional 20 minutes. As shown in Figure 6B, S1 dose-dependently attenuated LPS-induced ERK phosphorylation. These data confirm that S1 can inhibit LPS-induced ERK phosphorylation and may therefore be a potential ERK inhibitor. These results indicate that there may be a mechanistic difference among these curcumin analogs, although they are derived from the same structural lead.
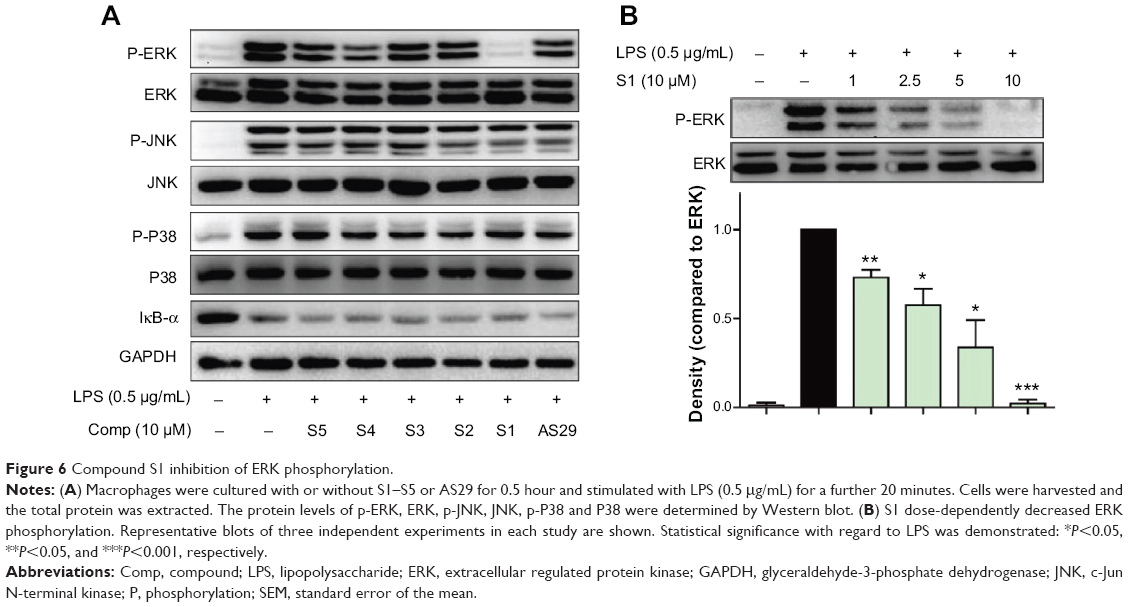 | Figure 6 Compound S1 inhibition of ERK phosphorylation. |
The toxicity and stability of active compounds
As a natural active product, curcumin exhibits no toxicity and is very safe in clinical trials. Before the in vivo experiment, an MTT assay was used to test the cytotoxicity of six active analogs, S1–S5 and AS29, in the human normal hepatic cell line HL-7702. As shown in Figure 7A, all six analogs showed low toxicity at a concentration of 10 μM, indicating that they are relatively safe. However, the clinical applications of curcumin have been significantly limited by its poor chemical stability.20 The monocarbonyl analogs of curcumin are designed to improve chemical stability by removing the central β-diketone moiety. Thus, the stability of the new synthetic analogs in phosphate buffer (pH 7.4) containing 5% DMSO were tested here using the ultraviolet-visible (UV-visible) absorption spectra at different times (0, 5, 10, 15, 20 and 25 minutes). The optical density (OD) values of the maximal absorption peak of curcumin decreased quickly, and analogs S1, S3, S4, S5, and AS29 showed much higher stability than curcumin, while S2 slightly decomposed during the incubation (Figure 7B–H). Meanwhile, the half-life of the most active compounds S1 and S4 were detected by HPLC in vitro. As shown in Figure S1, the half-lives of S1 (>120 minutes) and S4 (>240 minutes) are longer than the compound curcumin (<90 minutes). These results indicate that the active compounds are chemically more stable than curcumin in vitro.
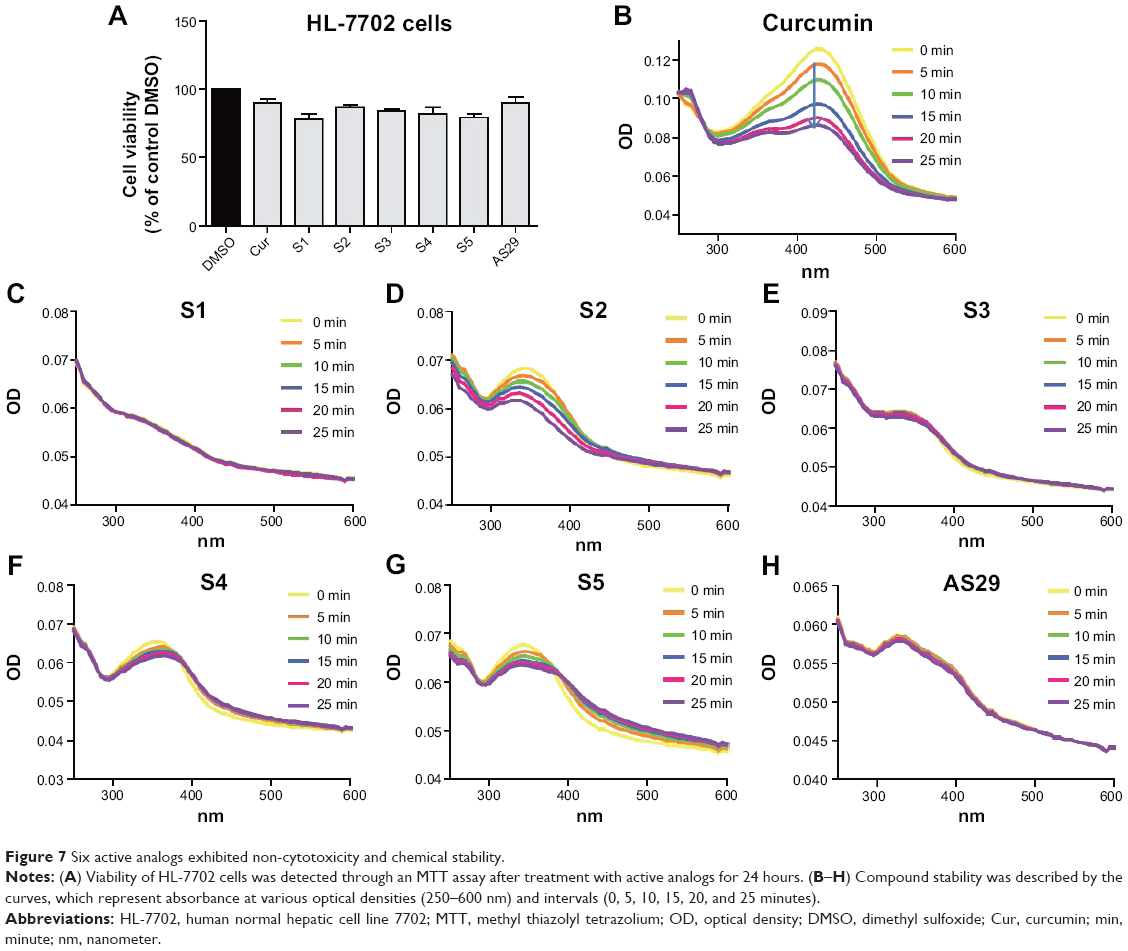 | Figure 7 Six active analogs exhibited non-cytotoxicity and chemical stability. |
Anti-inflammatory effect of S1 and S4 in vivo
LPS has been implicated as an important pathogenic factor for the induction of sepsis, which is characterized by an inflammatory cytokine storm. Curcumin and its analogs have been reported to have therapeutic effects on sepsis and septic shock.31 We have illustrated the inhibitory effects of these analogs on LPS-induced pro-inflammatory cytokine production. We further determined whether two representative compounds, S1 and S4, were able to relieve septic shock through inhibition of LPS-induced inflammatory response in mice. The water-soluble formulations of S1 and S4 were prepared for intravenous injection. Mice were injected intraperitoneally (ip) with LPS at the dosage of 15 mg/kg 15 minutes after the intravenous (iv) injection of 10 mg/kg S1or S4, and their survival rates were monitored for 7 days. The results in Figure 8 show that 100% of the animals injected with LPS alone died within 3–4 days as a result of septic shock. In septic animals administrated with S1 and S4, the survival rates were increased significantly compared with the LPS-alone group (40% in the S1-treated group and 50% in the S4-treated group). Thus, these data suggest that S1 and S4 exhibit anti-inflammatory activity in vivo.
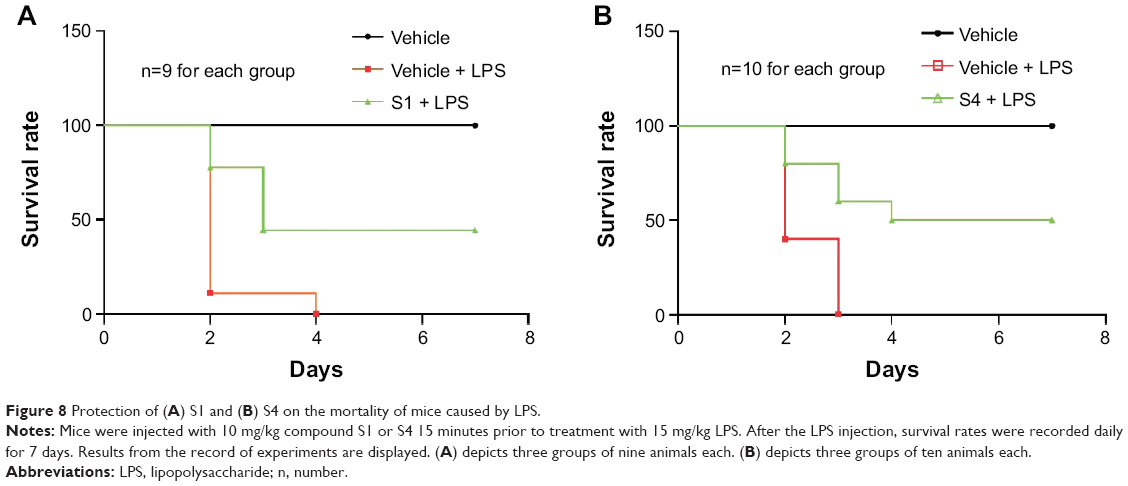 | Figure 8 Protection of (A) S1 and (B) S4 on the mortality of mice caused by LPS. |
Conclusion
In summary, we synthesized a series of symmetrical and asymmetrical analogs of curcumin and estimated their anti-inflammatory activities against LPS-induced TNF-α and IL-6 release in mouse macrophages. The majority of analogs effectively inhibited the LPS-induced production of TNF-α and IL-6. Together with the SAR and QSAR analysis, we found that the monocarbonyl curcumin analogs with symmetric and heterocyclic structures have stronger anti-inflammatory activity. Six compounds with potent anti-inflammatory activity also exhibited excellent chemical stability and low toxicity in vitro. Mechanistically, compound S1 significantly inhibited LPS-induced phosphorylation of ERK. Furthermore, S1 and S4 were selected for in vivo anti-sepsis testing and were found to markedly decrease LPS-induced lethality in septic mice. All of these results indicate that these novel monocarbonyl curcumin analogs may serve as potential agents for the treatment of various inflammatory diseases.
Acknowledgments
Financial support was provided by the National Natural Science Fund of China (Grants 21272179, 81272462, and 81202462); Project of Zhejiang Provincial Key Constructive Subject (Traditional Chinese Medicine, Grant 2012-XK-A28); Qianjiang Talent Project of Zhejiang Province (Grant 2013R10020), and the Zhejiang Key Group Project in Scientific Innovation (Grant 2010R50042).
Disclosure
The authors report no conflicts of interest in this work.
References
Kapoor M, Martel–Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2010;7(1):33–42. | ||
McLaren JE, Michael DR, Ashlin TG, Ramji DP. Cytokines, macrophage lipid metabolism and foam cells: implications for cardiovascular disease therapy. Prog Lipid Res. 2011;50(4):331–347. | ||
Abraham E, Singer M. Mechanisms of sepsis-induced organ dysfunction. Critical Care Medicine. 2007;35(10):2408–2416. | ||
Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8(10):776–787. | ||
Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420(6917): 885–891. | ||
Reyes CS, García–Muñoz F, Reyes D, González G, Dominguez C, Domenech E. Role of cytokines (interleukin-1β, 6, 8, tumour necrosis factor-α, and soluble receptor of interleukin-2) and C-reactive protein in the diagnosis of neonatal sepsis. Acta Paediatr. 2003;92(2)221–227. | ||
Abdelhamid AE, Chuang SL, Hayes P, Fell JM. In vitro cow’s milk protein-specific inflammatory and regulatory cytokine responses in preterm infants with necrotizing enterocolitis and sepsis. Pediatr Res. 2011;69(2):165–169. | ||
Mokart D, Merlin M, Sannini A, et al. Procalcitonin, interleukin 6 and systemic inflammatory response syndrome (SIRS): early markers of postoperative sepsis after major surgery. Br J Anaesth. 2005;94(6):767–773. | ||
Weinberg GA, D’Angio CT. The search for new diagnostic tests for neonatal sepsis. J Pediat. 2009;155(5):763–764. | ||
Hu Y, Tong G, Xu W, et al. Anti-inflammatory effects of simvastatin on adipokines in type 2 diabetic patients with carotid atherosclerosis. Diab Vasc Dis Res. 2009;6(4):262–268. | ||
Qiu Y, Yanase T, Hu H, et al. Dihydrotestosterone suppresses foam cell formation and attenuates atherosclerosis development. Endocrinology. 2010;151(7):3307–3316. | ||
Mazor R, Itzhaki O, Sela S, et al. Tumor necrosis factor- α: a possible priming agent for the polymorphonuclear leukocyte-reduced nicotinamide–adenine dinucleotide phosphate oxidase in hypertension. Hypertension. 2010;55(2):353–362. | ||
Chattopadhyay I, Biswas K, Bandyopadhyay U, Banerjee R. Turmeric and curcumin: biological actions and medicinal applications. Current Science. 2004;87(1):44–53. | ||
Suzuki M, Nakamura T, Iyoki S, et al. Elucidation of anti-allergic activities of curcumin-related compounds with a special reference to their anti-oxidative activities. Biol Pharm Bull. 2005;28(8):1438–1443. | ||
Kurup VP, Barrios CS, Raju R, Johnson BD, Levy MB, Fink JN. Immune response modulation by curcumin in a latex allergy model. Clin Mol Allergy. 2007;5:1. | ||
Nazam Ansari M, Bhandari U, Pillai, KK. Protective role of curcumin in myocardial oxidative damage induced by isoproterenol in rats. Hum Exp Toxicol. 2007;26(12):933–938. | ||
Kurup VP, Barrios CS. Immunomodulatory effects of curcumin in allergy. Mol Nutr Food Res. 2008;52(9):1031–1039. | ||
Sugimoto K, Hanai H, Tozawa K, et al. Curcumin prevents and ameliorates trinitrobenzene sulfonic acid-induced colitis in mice. Gastroenterology. 2002;123(6):1912–1922. | ||
Gupta SC, Patchva S, Aggarwal BB. Therapeutic roles of curcumin: lessons learned from clinical trials. AAPS J. 2013;15(1):195–218. | ||
Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4(6):807–818. | ||
Bukhari SN, Lauro G, Jantan I, Bifulco G, Amjad MW. Pharmacological evaluation and docking studies of α, β-unsaturated carbonyl based synthetic compounds as inhibitors of secretory phospholipase A2, cyclooxygenases, lipoxygenase and proinflammatory cytokines. Bioorganic and Medicinal Chemistry. 2014;22(15):4151–4161. | ||
Bukhari SN, Jantan I, Unsal Tan O, Sher M, Naeem–UI–Hassan M, Qin HL. Biological activity and molecular docking studies of curcumin-related α, β-unsaturated carbonyl-based synthetic compounds as anticancer agents and mushroom tyrosinase inhibitors. J Agric Food Chem. Epub 2014, Jun 10. | ||
Wu J, Zhang Y, Cai Y, et al. Discovery and evaluation of piperid-4-one-containing mono-carbonyl analogs of curcumin as anti-inflammatory agents. Bioorg Med Chem. 2013;21(11):3058–3065. | ||
Liang G, Li X, Chen L, et al. Synthesis and anti-inflammatory activities of mono-carbonyl analogues of curcumin. Bioorg Med Chem Lett. 2008;18(4):1525–1529. | ||
Liang G, Shao L, Wang Y, et al. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorg Med Chem. 2009;17(6):2623–2631. | ||
Haase M, Bellomo R, Haase–Fielitz A. Novel biomarkers, oxidative stress, and the role of labile iron toxicity in cardiopulmonary bypass-associated acute kidney injury. J Am Coll Cardiol. 2010;55(19):2024–2033. | ||
Navarro–Gonzalez J, Mora–Fernandez C, Gomez–Chinchon M, Muros M, Herrera H, Garcia J. Serum and gene expression profile of tumor necrosis factor-alpha and interleukin-6 in hypertensive diabetic patients: effect of amlodipine administration. Int J Immunopathol Pharmacol. 2009;23(1):51–59. | ||
Joh EH, Gu W, Kim DH. Echinocystic acid ameliorates lung inflammation in mice and alveolar macrophages by inhibiting the binding of LPS to TLR4 in NF-κB and MAPK pathways. Biochem Pharmacol. 2012;84(3):331–340. | ||
Zhang L, Wu C, Zhao S, et al. Demethoxycurcumin, a natural derivative of curcumin attenuates LPS-induced pro-inflammatory responses through down-regulation of intracellular ROS-related MAPK/NF-κB signaling pathways in N9 microglia induced by lipopolysaccharide. Int immunopharmacol. 2010;10(3):331–338. | ||
Baldwin AS Jr. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. | ||
Xiao X, Yang M, Sun D, Sun S. Curcumin protects against sepsis-induced acute lung injury in rats. J Surg Res. 2012;176(1):e31–e39. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.