")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Discovery and evaluation of asymmetrical monocarbonyl analogs of curcumin as anti-inflammatory agents
Authors Zhang Y, Zhao C , He W, Wang Z, Fang Q, Xiao B, Liu Z, Liang G, Yang S
Received 23 November 2013
Accepted for publication 15 January 2014
Published 4 April 2014 Volume 2014:8 Pages 373—382
DOI https://doi.org/10.2147/DDDT.S58168
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Yali Zhang,1,2,* Chengguang Zhao,1,2,* Wenfei He,2,* Zhe Wang,2 Qilu Fang,2 Bing Xiao,2 Zhiguo Liu,2 Guang Liang,2 Shulin Yang1
1School of Environmental and Biological Engineering, Nanjing University of Science and Technology, Nanjing, Jiangsu, People's Republic of China; 2Chemical Biology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, University Town, Wenzhou, Zhejiang, People's Republic of China
*These authors contributed equally to this work
Abstract: Sepsis is a systemic inflammatory response syndrome and is mainly caused by lipopolysaccharides (LPS) – a component of the cell walls of gram-negative bacteria, via toll-like receptor 4–mitogen-activated protein kinases/nuclear factor-kappa B-dependent proinflammatory signaling pathway. Here, we synthesized 26 asymmetric monocarbonyl analogs of curcumin and evaluated their anti-inflammatory activity by inhibiting the LPS-induced secretion of tumor necrosis factor-α and interleukin-6 in mouse RAW264.7 macrophages. Five active compounds (3a, 3c, 3d, 3j, and 3l) exhibited dose-dependent inhibition against the release of tumor necrosis factor-α and interleukin-6, and they also showed much higher chemical stability than curcumin in vitro. The anti-inflammatory activity of analogs 3a and 3c may be associated with their inhibition of the phosphorylation of extracellular signal-regulated kinase and the activation of nuclear factor-kappa B. In addition, 3c exhibited significant protection against LPS-induced septic death in vivo. These results indicate that asymmetrical monocarbonyl curcumin analogs may be utilized as candidates for the treatment of acute inflammatory diseases.
Keywords: sepsis, inflammatory cytokines, anti-inflammation, quantitative structure–activity relationship
Introduction
Sepsis is a systemic inflammatory response syndrome caused by severe infection.1,2 It is a common condition in patients with gram-negative bacterial infections, with a mortality rate as high as 50%–80%.3 Sepsis and septic shock are characterized by multiorgan failure, caused chiefly by a dysregulated immune response to the infection.4,5 Lipopolysaccharides (LPS), a component of the cell walls of gram-negative bacteria, are the principal mediators of the pathophysiology of septic shock.6 Toll-like receptors, notably toll-like receptor 4, recognize endotoxin LPS and trigger downstream signaling cascades, including the activation of mitogen-activated protein kinases (MAPKs) and nuclear factor-kappa B (NF-κB), and the subsequent upregulation of the gene expression of a variety of inflammatory mediators.7 Inflammatory cytokines, such as tumor necrosis factor (TNF)-α and interleukin (IL)-6, and their related pathways have received much attention as drug targets for treating sepsis.6 Inhibition of the overexpression of inflammatory cytokines by suppressing the activation of MAPKs and NF-κB is a classic strategy for the treatment of sepsis.7,8 For instance, natural products such as kukoamine B, polysaccharides, and a series of chalcone derivatives, have been shown to attenuate systemic inflammatory response syndrome and increase survival rates via the inhibition of MAPKs and NF-κB in LPS-induced sepsis mouse models.8–10
Due to the resistance of such diseases to conventional treatments, as well as the side effects of presently available anti-inflammatory drugs, there is a pressing need for the development of novel anti-inflammatory drugs. Natural products are a valuable source of novel bioactive anti-inflammatory agents. Curcumin, a natural bioactive compound, is derived from the root of Curcuma longa.11 Among natural anti-inflammatory products, curcumin has been paid special attention due to the fact that C. longa has been used as anti-inflammatory traditional medicine for about 2,000 years. Recent evidence has demonstrated that curcumin exhibits potent anti-inflammatory activities, which may help to prevent or even treat sepsis, as well as cancer and diabetes.11–13 Curcumin showed a protective effect in sepsis-induced acute lung injury and organ dysfunction in a rat model.11 The effect of curcumin was studied in patients with rheumatoid arthritis, inflammatory eye diseases, inflammatory bowel disease, chronic pancreatitis, psoriasis, and cancers.13 However, pharmacokinetic defects, such as low bioavailability, fast metabolism, and poor chemical stability significantly limit the clinical application of curcumin.14,15 For the purpose of finding novel derivatives with increased systemic bioavailability and enhanced pharmacological activity, chemical modifications of curcumin have been attempted.16–18 Among the analogs and derivatives of curcumin, much attention has been paid to the monocarbonyl analogs in which the beta-diketone moiety that contributes to the fast degradation and metabolism of the compound is removed.19,20 Since curcumin possesses a symmetrical structure, our group has previously reported several series of symmetrical monocarbonyl analogs of curcumin with improved pharmacokinetic profiles and increased anti-inflammatory activity.10,16–20 As a continuation of our study, 26 asymmetrical monocarbonyl analogs of curcumin were synthesized and their anti-inflammatory activities were evaluated in mouse RAW264.7 macrophages. These asymmetrical analogs exhibited good chemical stabilities in a phosphate buffer. Further, the effects of two representative analogs with anti-inflammatory activity on the MAPKs/NF-κB pathway and in septic animal models were studied.
Materials and methods
Chemical synthesis
All chemical reagents were obtained from Sigma-Aldrich (St Louis, MO, USA), Fluka (Buchs, Switzerland), and Aladdin (Beijing, People’s Republic of China). Silica gel (GF254) for thin-layer chromatography and column chromatography (100–200 mesh and 200–300 mesh) were obtained from Aladdin. Melting points were tested on a Fisher–Johns melting apparatus (Thermo Fisher Scientific, Waltham, MA, USA). Electron-spray ionization mass spectra (ESI-MS) data were determined on a Bruker esquire HCT™ spectrometer (Bruker Corporation, Billerica, MA, USA). The proton nuclear magnetic resonance (1H NMR) spectra data was recorded on a 600 MHz spectrometer (Bruker Corporation). All compounds were furnished by the aldol condensation of substituted aromatic aldehydes and intermediators (E)-4-(o-hydroxy)but-3-en-2-one (2a) or (E)-4-(p-chlorine)but-3-en-2-one (2b) under base conditions, respectively. The detailed synthesis and spectral characterization of new or unreported compounds are described in the Supplementary material.
Quantitative structure–activity relationship analysis
The methods and software used for the quantitative structure–activity relationship (SAR) model establishment and analysis (including descriptor calculation and selection, multiple linear regression analysis, and related software) were described in our previous publication.10
Animals
Male C57BL/6 mice weighing 18–22 g were obtained from the Animal Center of Wenzhou Medical College (Wenzhou, People’s Republic of China). Animals were housed at a constant room temperature with a 12-hour/12-hour light–dark cycle and fed with a standard rodent diet and water. The animals were acclimatized to the laboratory for at least 7 days before being used in the experiments. Protocols involving the use of animals were approved by the Wenzhou Medical College’s Animal Policy and Welfare Committee (approval documents: 2009/APWC/0031).
Reagents
LPSs were purchased from Sigma (St Louis, MO, USA). In addition, eBioscience, Inc. (San Diego, CA, USA) was the source of the mouse IL-6 enzyme-linked immunosorbent assay (ELISA) kit and mouse TNF-α ELISA kit. Anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH), anti-IκBα, (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha), and anti-extracellular signal-regulated kinase (ERK) antibody were obtained from Santa Cruz Biotechnology, Inc., (Santa Cruz, CA, USA); anti-p-P38, anti-P38, anti-p-Jun N-terminal kinase (JNK), and anti-JNK were obtained from Cell Signaling Technology, Inc., (Danvers, MA, USA).
Cells
Mouse RAW264.7 macrophages were obtained from the American Type Culture Collection (Manassas, VA, USA). RAW264.7 macrophages were incubated in Dulbecco’s Modified Eagle’s Medium (Gibco®; Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco®; Life Technologies), 100 U/mL of penicillin, and 100 mg/mL of streptomycin at 37°C with 5% CO2.
Detection of TNF-α and IL-6 in medium by ELISA
RAW264.7 macrophages were treated with LPS (0.5 μg/mL) in the presence or absence of compounds for 24 hours. The culture media were collected and centrifuged (1,000 rpm) at 4°C for 5 minutes, and the supernatant was collected. TNF-α and IL-6 levels in the medium were determined with an ELISA kit (eBioScience, Inc.) according to the manufacturer’s instructions. The total amount of the inflammatory factor in the medium was normalized to the total protein quantity of the viable cell pellets.
Ultraviolet-visible absorption spectra of curcumin and its analogs
Absorbance readings were taken from 250–600 nm using a SpectraMax® M5 (Molecular Devices LLC, Sunnyvale, CA, USA). A stock solution of 1 mM curcumin or active compounds 3a, 3c, 3d, 3j, and 3l were prepared and diluted by phosphate buffer (pH 7.4), containing 5% dimethyl sulfoxide (DMSO), to a final concentration of 20 μM. The ultraviolet absorption spectra were collected for over 25 minutes at 5-minute intervals at 25°C. All spectral measurements were carried out in a 1 cm path-length quartz cuvette.
Western blot
After treatment with compounds and LPS, RAW264.7 macrophages were washed with phosphate buffered saline and harvested with cell lysis. After being centrifuged at 12,000 rpm at 4°C for 10 minutes, the protein concentration was determined. After the sample loading buffer was added, protein samples were electrophoresed and then transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA). Each membrane was blocked for 1.5 hours at room temperature and incubated with specific primary antibodies against p-ERK (1:300), ERK (1:300), p-P38 (1:1,000), P38 (1:1,000), p-JNK (anti-p-Jun N-terminal kinase) (1:1,000), JNK (1:1,000), IkBα (1:300), and GAPDH (1:1,000) at 4°C overnight. Following three washes with Tris-buffered saline, the nitrocellulose membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hour, and visualized using enhanced chemiluminescence reagents (Bio-Rad Laboratories). The amounts of the proteins were analyzed using ImageJ analysis software version 1.38e (National Institutes of Health, Bethesda, MD, USA) and normalized to their respective controls.
Assay of cellular NF-κB p-65 translocation
RAW264.7 macrophages were pretreated with DMSO, 3a or 3c (at 10 μM) for 30 minutes, and then treated with or without 0.5 μg/mL of LPS for an additional hour, respectively. The cells were immunofluorescence-labeled according to the manufacturer’s instructions using a Cellular NF-κB p65 Translocation Kit (Beyotime Institute of Biotechnology, Nantong, People’s Republic of China). P65 protein and nuclei fluoresce were stained red and blue, respectively, and they can be simultaneously viewed by fluorescence microscope (Nikon Corporation, Tokyo, Japan) at an excitation wavelength of 350 nm for 4′,6-diamidino-2-phenylindole, and 540 nm for Cy3. To create a two-color image, the red and blue images were overlaid, producing purple fluorescence in areas of colocalization.
LPS-induced inflammatory mortality in mice
The compounds were first dissolved with macrogol-15-hydroxystearate (a nonionic solubilizer for injection from BASF [Ludwigshafen, Germany]) with or without medium-chain triglycerides (MCT) from BASF in a water bath at 37°C. The concentration of the compounds was 2 mg/mL. The concentration of the solubilizer ranged from 5%–10% and that of MCT ranged from 0.5%–2% in the final solution. For the vehicle, the mixture of solubilizer and MCT was prepared at 10% and 2%, respectively. Male C57BL/6 mice weighing 18–22 g were pretreated with compound 3c (10 mg/kg) in a water solution by intravenous injection 15 minutes before the intraperitoneal injection of LPS (25 mg/kg). The control animals received a similar volume (200 μL) of the vehicle. The mortalities were recorded for 7 days.
Results and discussion
Chemistry
The synthesis and structures of compounds 3a–3m and 4a–4m are shown in Figure 1. Briefly, 20% NaOH was added dropwise to a solution of either o-hydroxybenzaldehyde (1a) or p-chlorobenzaldehyde (1b) in acetone to obtain the intermediate compound, (E)-4-(o-hydroxy)but-3-en-2-one (2a) or (E)-4-(p-chlorine)but-3-en-2-one (2b). The α,β-unsaturated ketone 2a or 2b reacted with different substituted aromatic aldehydes in basic conditions through aldol condensation to obtain the asymmetrical monocarbonyl curcumin analogs, 3a–4m or 4a–4m. Different substituents with opposing electronic properties in the benzene rings were designed to investigate anti-inflammatory activity and were used to discuss the SAR. The yields of pure products were in the range of 12%–96%. The structures of all compounds were characterized using 1H NMR and ESI-MS.
 | Figure 1 Design and chemical structures of asymmetric MACs. |
Inhibition of cytokine production induced by LPS
The secretion of cytokines, such as TNF-α and IL-6, induced by bacterial LPS plays an important role in inflammatory conditions.6,21 Inhibition of LPS-induced TNF-α and IL-6 release was used for the evaluation of the anti-inflammatory activities of curcumin analogs. Hence, the anti-inflammatory effects of 26 synthetic compounds were evaluated in LPS-stimulated RAW264.7 macrophages with curcumin as a positive control. Briefly, macrophages were pretreated with compounds (10 μM) for 30 minutes, and then activated with LPS (0.5 μg/mL) or DMSO alone (negative control) for 24 hours. Culture medium was collected by centrifugation and the cytokines were measured by ELISA. As shown in Figure 2, the preliminary screening showed that the majority of curcumin analogs exhibited stronger inhibitory ability than that of curcumin. According to the results, the inhibitory effect against IL-6 may be enhanced when an electron-donating hydroxyl group is positioned in the left phenyl ring, while an electron-withdrawing chlorine in the left phenyl ring has little effect. However, this conclusion is not true of TNF-α inhibition, based on our data. In the right ring, the existence of a methoxy group led to increased anti-inflammatory activity regardless of IL-6 or TNF-α inhibition. According to the performance of 3l and 3m, furan and thiophene as the right moiety are also favorable in increasing the inhibition of IL-6 and TNF-α. Compounds 3a, 3c, 3d, 3j, and 3l showed the highest anti-inflammatory activity among all of the tested compounds. Hence, these five compounds were selected as the candidate compounds for further studies.
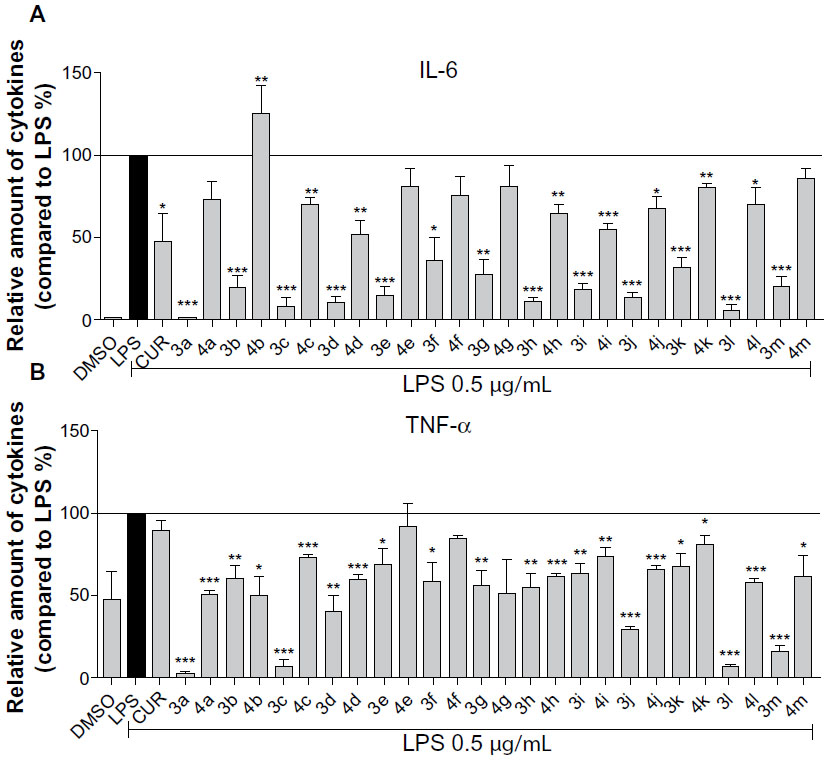 | Figure 2 Synthetic asymmetric monocarbonyl CUR analogs inhibited LPS-induced TNF-α and IL-6 secretion in RAW264.7 macrophages. |
Quantitative SAR of these analogs
To further explore the SAR of these compounds and to assess the effects of different substituents on the biological activity, a quantitative SAR (QSAR) model was calculated. During a QSAR model study, the structure properties of compounds are often represented by their molecular descriptors.22 Statistically significant models with three variables were derived for anti-TNF-α and IL-6 activity, respectively. The scatter plot of predicted values versus experimental values is illustrated in Figure 3. The statistically significant models, equation 1 and equation 2, were obtained for anti-TNF-α and anti-IL-6 activities of compounds with relatively high regression coefficients (R2) of 0.78 and 0.93, respectively. The variables in the equation 1 model contained the topological property (Moran), geometrical molecular descriptor (Weighted Holistic Invariant Molecular Descriptor [WHIM]), and quantum chemistry descriptor (electronic Eigenvalue descriptors [EEVA]), while in the equation 2 model, the variables contained the geometrical molecular descriptors (H-GEometry Topology and Atom-Weights AssemblY [GETAWAY] and R-GETAWAY descriptors) and quantum chemistry descriptor (EEVA). H-GETAWAY1 (weighted by atomic van der Waals radii) and R-GETAWAY2 (weighted by atomic sanderson electronegativity radii) were proposed by Consonni et al.23 The QSAR results indicate that molecular electronegativity may play a crucial role in the anti-inflammatory activity of the asymmetrical monocarbonyl curcumin analogs.
 | Figure 3 Plots of predicted activity against the corresponding experimental activity on TNF-α and IL-6 inhibition. |
Active compounds inhibit the LPS- induced cytokine release in a dose- dependent manner
To further investigate the anti-inflammatory activities of these analogs of curcumin, RAW264.7 macrophages were pretreated with these compounds at concentrations of 2.5 μM, 5 μM, and 10 μM for 30 minutes, and they were subsequently incubated with or without LPS (0.5 μg/mL) for 24 hours. Figure 4 shows that the compounds alone cannot induce the production of TNF-α and IL-6, while the presence of LPS produced a significant increase in the secretion of proinflammatory cytokines compared to DMSO. The active compounds exhibited dose-dependent inhibitory activities against LPS-induced TNF-α and IL-6 production; in particular, the inhibition rate of IL-6 is greater than 50%, even at a concentration of 5 μM. This further illustrates that these compounds have the potential to be used as anti-inflammatory agents.
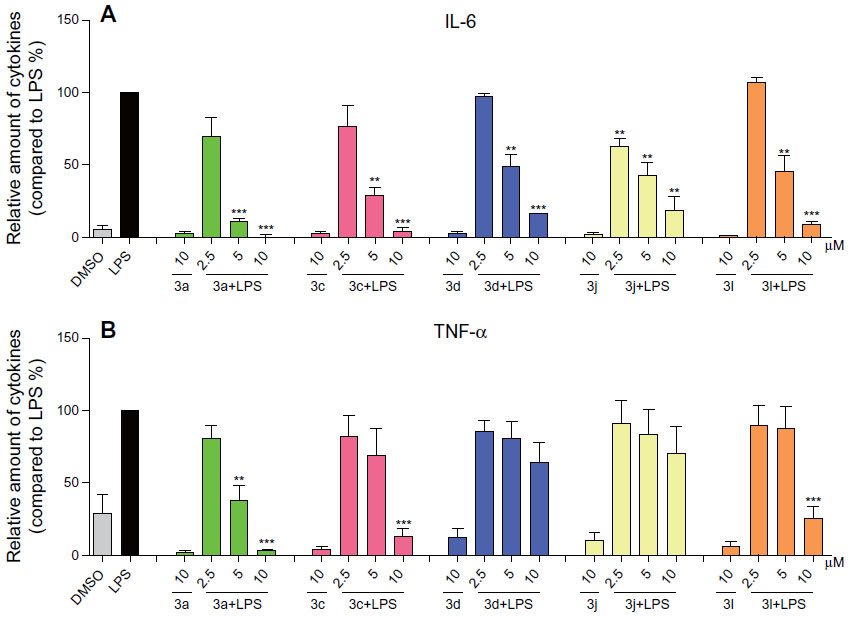 | Figure 4 Active compounds dose-dependently inhibited LPS-induced TNF-α and IL-6 secretion in RAW264.7 macrophages. |
The chemical stabilities of active compounds and curcumin
Clinical trials of curcumin have been limited by curcumin’s poor stability. Wang et al24 found that 90% of curcumin degraded within 30 minutes in phosphate buffer (pH 7.4). Here, we tested the active compounds 3a, 3c, 3d, 3j, and 3l in phosphate buffer (pH 7.4), containing 5% DMSO, using an absorption spectrum assay. As shown in Figure 5, the optical density values of the maximal absorption peak of the leading compound, curcumin, decreased over time. However, within 25 minutes, the absorption peak of the active compounds exhibited no significant change. These results indicate that the active compounds are chemically more stable than curcumin in vitro.
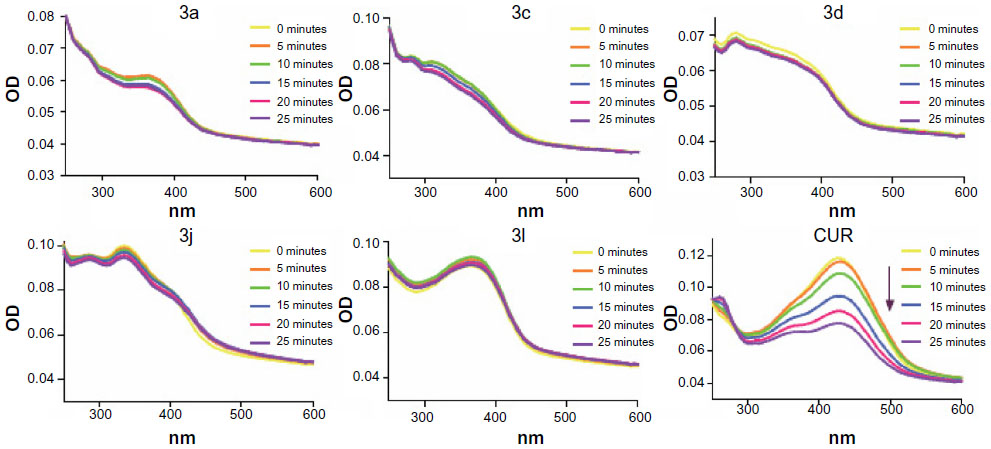 | Figure 5 Ultraviolet-visible absorption spectra of CUR, 3a, 3c, 3d, 3j, 3l, and CUR in phosphate buffer (pH 7.4) containing 5% dimethyl sulfoxide. |
Active compounds inhibited the LPS- induced activation of NF-κB
To identify the possible signaling pathways responsible for the production of proinflammatory cytokines that are reduced by the studied compounds, we first examined the effect of the active compounds on NF-κB known to be activated by LPS. NF-κB, a nuclear transcriptional factor, plays a key role in regulating immune response and inflammation.25 Under normal conditions, NF-κB is sequestered in the cytoplasm as an inactive complex, bound to the inhibitory κB proteins such as IκBα. In response to LPS, IκB kinase β phosphorylates cytoplasmic IκBα. The phosphorylated IκBα is subjected to ubiquitylation followed by proteasome-mediated degradation, after which NF-κB moves into the nucleus to promote the transcription of inflammatory genes.26,27 Since the degradation of IκB plays an important role in mediating the activation of NF-κB, we evaluated the effects of active compounds on IκBα degradation in LPS-stimulated RAW264.7 cells. Thus, we examined the effect of the active compounds on NF-κB signaling. As shown in Figure 6A, all of the five compounds at 10 μM markedly reversed the LPS-induced IκBα degradation to the control level. Meanwhile, the most active compounds, 3a (Figure 6B) and 3c (Figure 6C), dose-dependently reduced the LPS-induced IκBα degradation, though 3c at 5 μM has no significant effect. Figure 6D showed that LPS obviously induced the translocation of NF-κB p65 from the cytosol to the nucleus (red for p65 and blue for the nuclei). Pretreatment with 3a and 3c attenuated NF-κB p65 translocation, and compound 3c exhibited stronger activity than 3a. These results demonstrate that the active compounds inhibit the LPS-induced activation of NF-κB, and that NF-κB inactivation may be involved in the anti-inflammatory actions of these compounds.
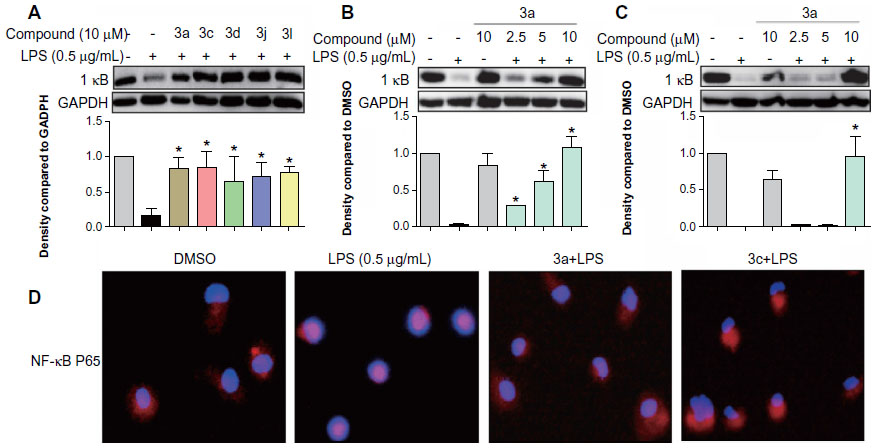 | Figure 6 Active compounds inhibited LPS-induced NF-κB activation in RAW264.7 macrophages. |
Active compounds inhibited the LPS-induced phosphorylation of MAPKs
MAPKs are a family of serine/threonine kinases involved in a variety of cellular processes such as inflammation, cell growth/differentiation, and cell survival/death.28–30 Three types of MAPKs, have been identified: ERK; c-JNK; and p38 MAPK. As shown in Figure 7A and B, exposure to LPS for 20 minutes significantly increased the phosphorylation of ERK, p38, and JNK. The active compounds 3a, 3c, 3d, and 3l notably reduced LPS-induced ERK phosphorylation, while 3j has no obvious effect. However, none of these compounds had any effect on the LPS-induced phosphorylation of p38 and JNK. These results indicate that the active compounds inhibit LPS-induced ERK phosphorylation, and ERK may also be a signaling target involved in the anti-inflammatory actions of these compounds.
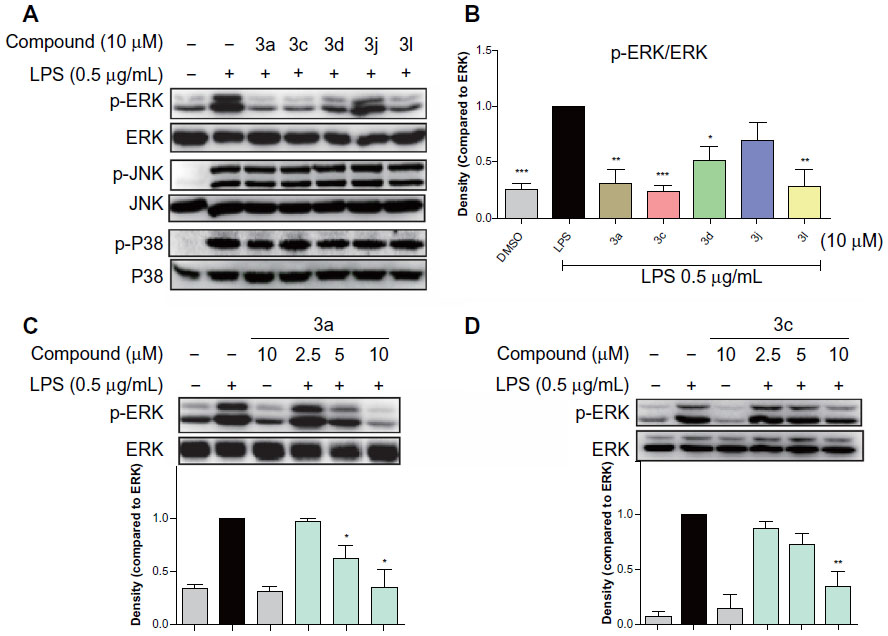 | Figure 7 Active compounds inhibited LPS-induced phosphorylation of MAPKs in RAW264.7 macrophages. |
To determine the dose-dependent effects of the most active compounds, 3a and 3c, RAW264.7 cells were pretreated with the compounds at doses of 2.5 μM, 5 μM, or 10 μM for 30 minutes. LPS was then added for a further 20 minutes. In the negative control, the drugs were incubated for a further 20 minutes without LPS. As shown in Figure 7C and D, 3a and 3c alone do not have an effect on the phosphorylation of ERK; however, both dose-dependently attenuated LPS-induced ERK phosphorylation. These data confirm that 3a and 3c can inhibit LPS-induced ERK phosphorylation and may, therefore, be potential ERK inhibitors.
Active compound 3c inhibited LPS-induced mortality
We further investigated whether the active compound 3c was able to attenuate endotoxin shock in vivo. As a major endotoxin, LPS from gram-negative bacteria has been implicated as a major cause of sepsis.8 Before the animal study, we tested the acute toxicity of compounds 3a and 3c, which indicated that the single oral administration of 3c at 1 g/kg was nontoxic in mice (data not shown). As such, 3c was selected for this experiment and prepared as an aqueous solution for intravenous injection. Mice were injected with LPS at the dosage of 25 mg/kg intraperitoneally 15 minutes after the intravenous injection of 10 mg/kg of 3c, and the survival rate was monitored for 7 days. As shown in Figure 8, 100% of the animals treated with LPS alone died within 3 days as a result of septic shock. At the end of the experiment, the mice treated with 3c showed a 60% survival rate. These data suggest that 3c exhibits anti-inflammatory activity in vivo.
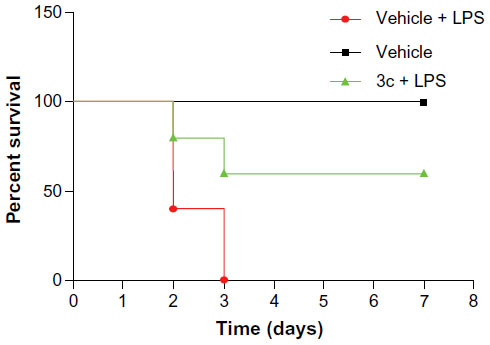 | Figure 8 3c attenuated LPS-induced septic shock in vivo. |
Conclusion
In summary, we synthesized 26 asymmetric curcumin analogs and confirmed their structures by 1H NMR and ESI-MS. Evaluation of the in vitro bioactivity of these analogs showed their inhibition of LPS-induced production of the proinflammatory cytokines, TNF-α and IL-6, in mouse RAW264.7 macrophages, and most analogs exhibited stronger anti-inflammatory activity compared with the leading compound curcumin. The active compounds, 3a, 3c, 3d, 3j, and 3l, presented dose-dependent inhibition on TNF-α and IL-6. Meanwhile, these active compounds showed excellent chemical stabilities in phosphate buffer (pH 7.4). Furthermore, the ERK and NF-κB pathways were found to be involved in the anti-inflammatory activity of the most active compounds, 3a and 3c. Lastly, intravenous injection of 10 mg/kg of 3c attenuated LPS-induced mortality in vivo. All of these results indicate that asymmetrical monocarbonyl curcumin analogs may be utilized as candidates for the treatment of acute inflammatory diseases.
Acknowledgments
Financial support was provided by the National Natural Science Funding of China (Grants 21272179 and 81202462), High-Level Innovative Talent Funding of Zhejiang Department of Health (GL), Zhejiang Natural Science Funding (Grant LQ12H30002), Grant from the Zhejiang Department of Health (2012KYA129), the Zhejiang Zhejiang Key Group Project in Scientific Innovation (2010R50042), and the Research Development Fund of Wenzhou Medical University (QTJ13003). The project was sponsored by the China Postdoctoral Science Foundation (grants 20090461121 and 201003591).
Disclosure
The authors report no conflicts of interest in this work.
References
Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101(6):1644–1655. | |
Levy MM, Fink MP, Marshall JC, et al; SCCM/ESICM/ACCP/ATS/SIS. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31(4):1250–1256. | |
Verhoef J, Hustinx WM, Frasa H, et al. Issues in the adjunct therapy of severe sepsis. J Antimicrob Chemother. 1996;38(2):167–182. | |
Uji Y, Yamamoto H, Tsuchihashi H, et al. Adiponectin deficiency is associated with severe polymicrobial sepsis, high inflammatory cytokine levels, and high mortality. Surgery. 2009;145(5):550–557. | |
London NR, Zhu W, Bozza FA, et al. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med. 2010;2(23):23ra19. | |
Riedemann NC, Guo RF, Hollmann TJ, et al. Regulatory role of C5a in LPS-induced IL-6 production by neutrophils during sepsis. FASEB J. 2004;18(2):370–372. | |
Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42(2):145–151. | |
Liu X, Zheng X, Wang N, et al. Kukoamine B, a novel dual inhibitor of LPS and CpG DNA, is a potential candidate for sepsis treatment. Br J Pharmacol. 2011;162(6):1274–1290. | |
Chen H, Qu Z, Fu L, et al. Physicochemical properties and antioxidant capacity of 3 polysaccharides from green tea, oolong tea, and black tea. J Food Sci. 2009;74(6):C469–C474. | |
Wu J, Li J, Cai Y, et al. Evaluation and discovery of novel synthetic chalcone derivatives as anti-inflammatory agents. J Med Chem. 2011;54(23):8110–8123. | |
Leitman IM. Curcumin for the prevention of acute lung injury in sepsis: is it more than the flavor of the month? J Surg Res. 2012;176(1):e5–e7. | |
Weisberg SP, Leibel R, Tortoriello DV. Dietary curcumin significantly improves obesity-associated inflammation and diabetes in mouse models of diabesity. Endocrinology. 2008;149(7):3549–3558. | |
Bisht S, Feldmann G, Soni S, et al. Polymeric nanoparticle-encapsulated curcumin (“nanocurcumin”): a novel strategy for human cancer therapy. J Nanobiotechnology. 2007;5:3. | |
Pan MH, Huang TM, Lin JK. Biotransformation of curcumin through reduction and glucuronidation in mice. Drug Metab Dispos. 1999;27(4):486–494. | |
Rosemond MJ, St John-Williams L, Yamaguchi T, et al. Enzymology of a carbonyl reduction clearance pathway for the HIV integrase inhibitor, S-1360: role of human liver cytosolic aldo-keto reductases. Chem Biol Interact. 2004;147(2):129–139. | |
Zhao C, Cai Y, He X, et al. Synthesis and anti-inflammatory evaluation of novel mono-carbonyl analogues of curcumin in LPS-stimulated RAW 264.7 macrophages. Eur J Med Chem. 2010;45(12):5773–5780. | |
Liang G, Zhou H, Wang Y, et al. Inhibition of LPS-induced production of inflammatory factors in the macrophages by mono-carbonyl analogues of curcumin. J Cell Mol Med. 2009;13(9B):3370–3379. | |
Wu J, Zhang Y, Cai Y, et al. Discovery and evaluation of piperid-4-one-containing mono-carbonyl analogs of curcumin as anti-inflammatory agents. Bioorg Med Chem. 2013;21(11):3058–3065. | |
Liang G, Li X, Chen L, et al. Synthesis and anti-inflammatory activities of mono-carbonyl analogues of curcumin. Bioorg Med Chem Lett. 2008;18(4):1525–1529. | |
Liang G, Shao L, Wang Y, et al. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorg Med Chem. 2009;17(6):2623–2631. | |
Roger T, Froidevaux C, Le Roy D, et al. Protection from lethal gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc Natl Acad Sci U S A. 2009;106(7):2348–2352. | |
Xue Y, Li H, Ung CY, et al. Classification of a diverse set of Tetrahymena pyriformis toxicity chemical compounds from molecular descriptors by statistical learning methods. Chem Res Toxicol. 2006;19(8):1030–1039. | |
Consonni V, Todeschini R, Pavan M. Structure/response correlations and similarity/diversity analysis by GETAWAY descriptors. 1. Theory of the novel 3D molecular descriptors. J Chem Inf Comput Sci. 2002;42(3):682–692. | |
Wang YJ, Pan MH, Cheng AL, et al. Stability of curcumin in buffer solutions and characterization of its degradation products. J Pharm Biomed Anal. 1997;15(12):1867–1876. | |
Xiao C, Ghosh S. NF-kappa B, an evolutionarily conserved mediator of immune and inflammatory responses. Adv Exp Med Biol. 2005;560:41–45. | |
Nelson DE, Ihekwaba AE, Elliott M, et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004; 306(5696):704–708. | |
Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8(11):837–848. | |
Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. | |
Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6(5):322–327. | |
Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92(2):689–737. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.