Back to Journals » ImmunoTargets and Therapy » Volume 14
Directed-Complement Activation as a Novel Immunotherapeutic Approach for HER2-Breast Cancer
Authors Seguin-Devaux C ![]() , Brandus B, Plesseria JM, Iserentant G, Servais JY, Pitiot A, Kanli G, Behrmann I, Schober R, Zimmer J, Cohen JH, Dervillez X
, Brandus B, Plesseria JM, Iserentant G, Servais JY, Pitiot A, Kanli G, Behrmann I, Schober R, Zimmer J, Cohen JH, Dervillez X
Received 4 March 2025
Accepted for publication 28 July 2025
Published 5 September 2025 Volume 2025:14 Pages 979—995
DOI https://doi.org/10.2147/ITT.S517584
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Sarah Wheeler
Carole Seguin-Devaux,1,* Bianca Brandus,1,* Jean-Marc Plesseria,1 Gilles Iserentant,1 Jean-Yves Servais,1 Aubin Pitiot,1 Georgia Kanli,2 Iris Behrmann,3 Rafaëla Schober,1 Jacques Zimmer,1 Jacques HM Cohen,4 Xavier Dervillez1,3
1Department of Infection and Immunity, Luxembourg Institute of Health, Esch sur Alzette, Luxembourg; 2Department of Oncology, Luxembourg Institute of Health, Luxembourg, Luxembourg; 3Department of Life Sciences and Medicine, University of Luxembourg, Belval, Luxembourg; 4LRN EA4682, University of Reims Champagne Ardennes, Reims, France
*These authors contributed equally to this work
Correspondence: Carole Seguin-Devaux, Luxembourg Institute of Health, Department of Infection and Immunity, 29 rue Henri Koch, Esch-sur-Alzette, L-4354, Luxembourg, Tel +352 26970-224, Email [email protected]
Purpose: Directing selective complement activation towards tumor cells is an attractive strategy to promote their elimination. We have generated complement-activating multimeric immunotherapeutic complexes (CoMiX), stimulating either the alternative pathway (via Factor H Related protein 4 (FHR4)) or the classical pathway (via triple Fc dimers) on HER2-expressing tumor cells.
Methods: We used the C‐terminal α‐chain multimerizing scaffold of the C4 binding protein (C4bp) to generate CoMiX-FHR4 as well as CoMiX-Fc with 2 different anti-HER2 VHH, VHH(T) and VHH(P), recognizing trastuzumab- or pertuzumab-competing epitopes, respectively. The different CoMiX were compared in vitro for C3b and C5b9 depositions, complement-dependent cytotoxicity (CDC), and their ability to activate NK cells and phagocytosis by macrophages. We further explored their therapeutic efficacy on human BT474 tumor xenografts established in nude mice.
Results: CoMiX-FHR4/VHH(T) and -FHR4/VHH(P) lead to the highest C3b and C5b9 depositions and CDC on BT474 tumor cells (p< 0.0001), both individually and in combinations with their CoMiX-Fc counterparts, surpassing the low complement activating capacity of trastuzumab and pertuzumab. All CoMiX induced BT474 cell death and phagocytosis of tumor cells by macrophages while CoMiX-Fc also stimulated NK cell activation. In human BT474 xenografts sensitive to trastuzumab, CoMiX induced a massive C3b deposition 6 hours after injection. CoMiX-FHR4 reduced the tumor volume compared to controls (p< 0.05) but to a lesser extent than trastuzumab (p< 0.001) while CoMiX-VHH(P)/Fc led to a tumor volume reduction similar to pertuzumab. Combinations of two CoMiX-FHR4 or two CoMiX-Fc were more potent, similarly to the combination of trastuzumab and pertuzumab, leading to increased NK cell infiltration in xenografts. Importantly, CoMiX-FHR4 was still active against trastuzumab-resistant xenografts, delaying tumor growth and inducing a large NK cell infiltration.
Conclusion: We showed here that directed complement activation on tumor cells is an alternative to therapeutic antibodies for future combination therapies upon resistance to standard-of-care treatment.
Keywords: immunotherapy, complement-dependent cytotoxicity, NK cells, phagocytosis, xenograft model
Introduction
Recombinant monoclonal immunoglobulin gamma (IgG) antibodies (Abs) have become a major weapon in cancer immunotherapy.1 The classical complement pathway is activated when antibody–antigen complexes are formed on cell surfaces, followed by binding of the complement protein C1q to the C-terminal half of the CH2 domain of Fc.2 A few therapeutic antibodies (Abs) targeting CD20, such as ofatumumab and daratumumab, efficiently lead to complement-dependent cytotoxicity (CDC) in vivo and therapeutic success.3,4 Trastuzumab, the first humanized mAb approved for breast cancer treatment, inhibits the human epidermal growth factor receptor 2 (EGFR2) and EGFR signaling pathways;5 Pertuzumab binds to a distinct epitope on HER2, blocking HER2 heterodimerization but includes the same IgG1-Fc domain as trastuzumab triggering antibody-dependent cell-mediated cytotoxicity (ADCC).6,7 Nevertheless, many patients do not respond to anti-HER2-Abs due to inherited or acquired resistance.8 Fc bioengineering or antibody drug-conjugation combining anticancer drug, such as trastuzumab-emtansine (T-DM1) or trastuzumab–deruxtecan (T-DXd), are the main approaches used to improve their efficacy.9,10 Complement activation represents, however, a substantial part to improve the overall biological activity of Abs used in cancer immunotherapy.
All three-complement pathways, activated by antibodies or by structural elements on target cells or pathogens, converge at the level of C3, which is cleaved by C3 convertase to produce the anaphylatoxin C3a and the component C3b, followed by the sequential recruitment and activation of C5.11 The assembly of the terminal complement proteins C5b–C9, the membrane attack complex (MAC), results in pore formation and subsequent lysis of the target cell. C3b degradation products on tumor targets (iC3b, C3dg and C3d) are recognized by effector cells through CD11b and CD11c, leading to complement-dependent cell cytotoxicity (CDCC) or phagocytosis (CDCP), which allows the clearance of target cells. In addition, complement activation releases pro-inflammatory mediators such as C3a and C5a, the latter upregulating the Fcγ receptors (FcγR) on phagocytes to enhance phagocytosis but also tumor cytolysis. The alternative complement pathway can be harnessed against tumor progression.12,13 Factor H-related protein 4 activates complement by serving as a platform for the assembly of alternative pathway C3 convertase by competing with factor H (FH) for C3b binding.14 We have developed a novel anti-tumor complement-mediated strategy by directing the activator of the alternative pathway FHR4 towards HER2-overexpressing cancer cells.15 We used the oligomerization scaffold of the C-terminal domain of the α-chain of the C4b-binding protein (C4bp) to enable the formation of hexa- and heptamers16 for the generation of complement-activating multimeric immunotherapeutic complexes (CoMiX). CoMiX displayed multivalent FHR4 complement effector moieties and elicit C3b deposition, MAC formation, and CDC by overcoming the complement inhibitory threshold on cancer cells.15
In the present work, we have extended the use of the C4bpα multimerizing scaffold to express CoMiX-Fc that assemble as functional triple Fc-dimers. We used 2 different anti-HER2 VHH, VHH(T) and VHH(P), recognizing trastuzumab- or pertuzumab-competing HER2 epitopes, respectively, to generate 2 types of CoMiX-FHR4 [FHR4/VHH(T) and FHR4/VHH(P)] and 2 types of CoMiX-Fc [VHH(T)/Fc and VHH(P)/Fc]. CoMiX-FHR4 and CoMiX-Fc were evaluated in vitro, individually or in combination, and their therapeutic efficacy was further assessed on nude mouse xenografts of human HER2-expressing BT474 breast cancer cell lines, sensitive or not to trastuzumab. Our study made proof of concept that recruitment of directed complement on cancer cells in vivo is an effective strategy to inhibit tumor growth. Once the mode of action and pharmacokinetics of CoMiX will have been deeply characterized, this approach might be used in clinical conditions of immunotherapeutic failure with standard-of-care treatment.
Materials and Methods
Ethics Approval
The study was approved by the LIH Institutional Review Board (PoC CoMIX-2017) and conducted in accordance with the Declaration of Helsinki. Peripheral blood mononuclear cells (PBMCs) were provided by healthy donors of the Luxembourg Red Cross giving their PBMCs for research in an anonymized way without the requirement of written inform consent. The study was approved by the Luxembourg Red Cross under the project’s number approval LIH-2018-0005. Animal experiments were approved by the animal welfare committee of the Luxembourg Institute of Health (DII-2018-15) and the Ministry of Agriculture of Luxembourg (LUPA2019/82) following the guidelines of the Directive 2010/63/EU of the European Parliament and of the Council on the protection of animals used for scientific purposes.
Transfection and Clone Selection
The plasmid design was described in the Supplemental Material and Methods and in Supplementary Figure 1. All CoMiX were generated from HEK293T cells transfected with 4 μg of recombinant plasmid DNA using Lipofectamine-3000 (Invitrogen, Thermo Fisher Scientific BVBA, Merelbeke, Belgium) following the manufacturer’s instructions. CoMiX-FHR4 were produced as previously described15 using double co-transfection whereas CoMiX-Fc, hinge deficient CoMiX-Fc (Δhinge), and multimeric VHH(T) control, lacking any effector functions, were produced using single transfection. Transfected clones were cultured and selected by puromycin (Westburg, Leusden, The Netherlands). Protein expression in the supernatants was analyzed by sandwich ELISA. Briefly, 100 ng/well goat anti-human Fc (Abcam, Cambridge, UK, #ab97221) or rabbit anti-His (Bethyl, Antwerpen, Belgium, #A190214A) polyclonal antibodies (pAbs) were immobilized onto a NUNC MaxiSorp™ 96-well flat-bottom polystyrene plate overnight at 4°C. After blocking with 5% (w/v) bovine serum albumin in PBS, each clone supernatant was incubated for 1 hour at 4°C. CoMiX-Fc were detected with a HRP-conjugated goat anti-human Fc pAb (Abcam, #ab97225), CoMiX-FHR4 and VHH controls were detected with a HRP-conjugated mouse anti-His mAb (Sigma-Aldrich). The plate was revealed with OPD (o-phenylenediamine dihydrochloride)/H2O2 substrate and absorbance was measured at 492 nm on a spectrophotometer. Clones with the highest production were expanded in cellSTACKs® (Corning) in complete DMEM medium (10% FBS, Penicillin/Streptomycin and L-Glutamine). After selection of the clones, clones were produced during 3 cycles of 48h in serum-free optiMEM followed by 24h in complete DMEM medium. Opti-MEM supernatants were precleaned by centrifugation (4000 rpm/10 min) and filtered using 0.22 µm PVDF vacuum filter units (GE-Healthcare, Leuven, Belgium). The protocols of protein production and purification are described in the Supplemental Material and Methods as well as cell culture conditions and the list of all antibodies. The molecular pattern of CoMiX molecules was confirmed by SDS/PAGE followed by either Western blotting or SYPRO Ruby gel staining, as detailed in the Supplemental Material and Methods.
Flow Cytometry Analysis
HER2-positive BT474 cancer cells were incubated for 1 hour at 4°C with 3-fold serial dilutions of 15 µg/well CoMiX, VHH controls and Abs, or 7.5 µg/well of two molecules for combinations, and incubated with 25% normal human serum (NHS) diluted in two different gelatin veronal buffers (GVB++: 141 mM NaCl, 0.3 mM CaCl2, 1 mM MgCl2, 0.1% gelatin, 1.8 mM Na-barbital and 3.1 mM barbituric acid, pH 7.3–7.4) (GVB+: same composition but with 3 mM MgCl2 and 5 mM EGTA without CaCl2) for 30 minutes at 37°C. Complement activation was detected with mouse anti-human C3/C3b/iC3b monoclonal antibody (mAb) (Cedarlane, Sanbio B.V., Uden, The Netherlands) or mouse anti-human C5b9 mAb (Novus, USA, Centennial) and Live/Dead (L/D) staining (Invitrogen), followed by goat anti-mouse IgG AF647 Ab (Invitrogen) staining. The samples were fixed with 1% PFA and analyzed with a BD LSR Fortessa™ cell analyzer. The percentage of dead cells was calculated by dividing the number of L/D-positive cells by the total number of cells analyzed. To analyze the NK cell activating properties of CoMiX, we incubated BT474 cells with 5-fold diluted CoMiX starting at 20 µg/condition for 1 hour at 4°C. Cells were then co-incubated with human CD16 expressing NK cells (NK92humCD16 ATCC, PTA-8836) for 4 hours at 37°C, and then stained with BV421™ anti-human CD107a (Biolegend) antibody as a degranulation marker. GolgiStop™ (BD Biosciences) and GolgiPlug™ (BD Biosciences) were added after 1 hour of incubation.
CoMiX-Mediated Cellular Phagocytosis of BT474 Cells
M2 macrophages were derived from monocytes of PBMCs from healthy donors of the Luxembourg Red Cross as previously described.15 Macrophages were plated into Lab-Tek 8-chamber slides (Nunc, Thermo Fisher Scientific) and stained with PKH26 (Sigma-Aldrich). BT474 cells were stained with CFSE (Invitrogen, Thermo Fisher) and then incubated for 30 min with 20 µg/mL of controls, CoMiX, trastuzumab, pertuzumab or their combinations in GVB++ supplemented with 25% C5-deficient NHS to prevent lysis. CFSE-stained complement-coated tumor cells were washed, added to the KHP26-stained M2 macrophages (2:1) and incubated for 18 hours at 37°C/5% CO2 in complete RPMI medium. Cells were fixed with 1% PFA, stained with DAPI and analyzed by confocal microscopy on the CSU-W1 Spinning Disk/High Speed Widefield confocal microscope lens X4. Three series of 10 pictures were taken for each experimental condition. Phagocytosis was evaluated on 400 macrophages for each condition.
In vivo Model: Human BT474 Breast Cancer Xenografts
Mice were housed in specific pathogen-free facilities with food and water supplied ad libitum. Five weeks old female BALB/c nude mice (Janvier Labs, Le Genest-Saint-Isle, France) (± 25 g) were anesthetized with isoflurane and injected subcutaneously into the mammary fat pads with 5×106 (1:1) Matrigel-mixed trastuzumab-sensitive BT474 cells. Tumor growth was measured with calipers and their volumes calculated (Volume = (Length x Width x Width)/2). After tumors reached 60 mm3 (in 30–40 days), mice were randomly divided into 11 groups (5 mice/group). Each group was treated 9 times with 100 µg of each molecule, or their combinations (50 µg of each of the two molecules). Tumors were measured with calipers every second or third day for 25 days or until the tumor size reached 1000 mm3. Magnetic Resonance Imaging (MRI) was performed on one representative mouse of each group. Mice were sacrificed at day 11 post-treatment and placed in tubes containing cold solution of 4% PFA. MRI was performed on a 3T preclinical horizontal bore scanner (MR Solutions, Guilford, UK), equipped with a quadrature volume coil designed for rodent head imaging, with a 17 cm horizontal bore. The heads were placed in a tube with fluorinert (3M, MN, USA). Anatomical FSE T2w (Spatial Resolution: 0.15 × 0.15 × 1mm; Number of Slices: 30; TE\TR: 68\7000 ms; Number of Averages: 1) were used to calculate the tumor volumes per animal which was calculated by the ImageJ software.17 Finally, FHR4/VHH(P) and its combination with FHR4/VHH(T) were further explored in xenografts of trastuzumab-resistant BT474 cells (ATCC-CRL-3247) using the same animal protocol (4 groups, 5 mice/group) as described above.
Analysis of Immune Responses in Xenografts
To analyze complement activation in xenografts, a single dose of 100 µg of the combination of CoMiX-FHR4 [50 µg FHR4/VHH(T) + 50 µg FHR4/VHH(P)], CoMiX-Fc [50 µg VHH(T)/Fc + 50 µg VHH(P)/Fc], the VHH(T) control or the combination of each commercial antibody was injected intravenously into the lateral tail vein of 8 weeks old female BALB/c nude mice (4 mice/molecule). One or six hours after injection, mice were sacrificed by cervical dislocation. The tumors were surgically removed, placed in OCT cryomold, and instantly frozen in liquid nitrogen for immunofluorescence analysis (Supplemental Material and Methods). For the protocol of BT474 xenografts sensitive to trastuzumab, mice were sacrificed after 25 days or when the tumor size reached 1000 mm3. Xenografts were surgically removed, placed in 4% PFA overnight, stored in 60% ethanol before shipping for immunohistochemistry staining at HistoWiz, Inc. (detailed in Supplemental Material and Methods). Frequencies of CD45 and NK cells were measured by ImageJ and the IHC support tool using statistical color models.17 For the protocol of BT474 xenografts resistant to trastuzumab, xenografts were collected at the end of the treatments (Day 11) with three mice in each group. Cryosections were performed as described above, stained with a rat anti-mouse NKp46/NCR1 mAb (R&D Systems, clone # 29A1.4) and revealed using a donkey AF568-conjugated anti-rat pAb (Abcam).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism version 10.0.0 for Windows (GraphPad Software, Massachusetts USA). For all in vitro experiments and for immunohistochemistry, multiple groups receiving the different single CoMiX or combination treatments were compared using one-way ANOVA analysis of variance and post-hoc Tukey’s tests. For studies in mice, an appropriate sample size (n = 5) was calculated during the study design to obtain groups with a difference of tumor growth of 20% considering a common standard deviation of 5% using a bilateral Student’s T-test based on a 95% confidence level. Mice with different tumor sizes were randomized between the groups. The groups were compared using one way ANOVA analysis of variance followed by unpaired Student’s T-test. A p value < 0.05 was considered as statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Results
Molecular Design of CoMiX
We used the oligomerization scaffold of the C-terminal domain of the C4bpα chain to enable the formation of hexa- and heptamers. As described in Figure 1, Fc-based complement activating multimeric immunotherapeutic complexes (CoMiX-Fc) display a multivalent targeting function derived from trastuzumab (VHH(T)) or pertuzumab (VHH(P)), a monovalent tracking function (eGFP), and three dimeric C4bpα--Fc regions (effector function) engineered with a dual hinge region between the C4bpα-scaffold and the IgG1 CH2-CH3: VHH(T)/Fc and VHH(P)/Fc. To evaluate the importance of the Fc dimers, Fc-based multimers without hinge (VHH(T)/Fc Δhinge) were produced. The two FHR4-based CoMiX (CoMiX-FHR4) include the FHR4 effector function instead of the Fc regions without hinge and the targeting function VHH(T) or VHH(P). As a control, VHH(T) without effector function was also generated.
 |
Figure 1 Design of CoMiX-FHR4, CoMiX-Fc, and the controls VHH(T) and VHH(T)/Fc Δhinge. All constructs are co-transfected with the eGFP.C4bpβ construct, leading to the covalent association of a single eGFP tracking function with the multimeric fusion C4bpα core. We used VHH(T) and VHH(P), recognizing trastuzumab- or pertuzumab-competing HER2 epitopes, respectively, to generate 2 types of CoMiX-FHR4, FHR4/VHH(T) and FHR4/VHH(P), or 2 types of CoMiX-Fc VHH(T)/Fc or VHH(P)/Fc. C4bpα.His8x VHH(T) is the control multimeric molecule with no effector function, and VHH(T)/Fc Δhinge is the control molecule of VHH(T)/Fc without a hinge region that allows the formation of triple Fc dimers. |
The molecular pattern of the produced multimeric immunoconjugates was analyzed by Western blot (Supplementary Figure 2). Under non-reducing conditions, FHR4/VHH(T) and FHR4/VHH(P) display seven bands representing the number of FHR4 units in the complexes. All CoMiX-Fc showed a tracking function (eGFP.C4bpβ) with a size of 50 kDa and an anti-HER2-VHH targeting function derived from trastuzumab (VHH(T)) or pertuzumab (VHH(P)), with a size of 40 or 30 kDa, respectively.
CoMiX Promote C3b Deposition, MAC Formation, and Direct Killing of BT474 Tumor Cells
The effect of CoMiX-Fc and CoMiX-FHR4 on complement activation was first analyzed, individually or in combination, by measuring C3b and C5b9 depositions as well as CDC on the HER2 expressing BT474 tumor cells using flow cytometry (Figure 2, Supplementary Figure 3) in the presence of 25% NHS. Dose-dependent responses of C3b and C5b9 depositions were observed (Supplementary Figure 4). The combination of both CoMiX constructs significantly enhanced C3b and C5b9 depositions as well as CDC as compared to the single administrations. At their highest concentration (Figure 2), CoMiX-FHR4 molecules led to the highest C3b deposition (Figure 2A) and CDC (Figure 2C), both when tested individually or in combination (p< 0.0001). CoMiX-Fc were less efficient to facilitate C3b deposition and CDC than CoMiX-FHR4 but surpassed the low complement activating capacity of trastuzumab and pertuzumab (Figure 2A and C). C5b9 deposition (Figure 2B) mediated by CoMiX-FHR4 and CoMiX-Fc harboring VHH(T) was more potent than the combination with CoMiX-Fc harboring VHH(P) (Figure 2B, p< 0.0001). Importantly, all molecules exceeded the C5b9 deposition induced by trastuzumab and pertuzumab (p< 0.05). Combinations of CoMiX-Fc with CoMiX-FHR4 significantly enhanced C3b and C5b9 depositions as well as CDC as compared to CoMiX-Fc alone (p< 0.0001). Overall, all CoMiX with the VHH(T) targeting system activate the complement system more efficiently than their VHH(P) counterparts. The correlation between C3b deposition and the percentage of dead cells (at 15 µg/well) is depicted in Figure 2D. Trastuzumab and pertuzumab, when used individually, have little effect on C3b deposition and cell death. However, when combined, they elicit an increased C3b deposition and a slight increase in cell death (Figure 2D). Both CoMiX-FHR4 constructs demonstrated the highest complement activating properties. CoMiX-Fc had a similar effect on complement activation as the combination of trastuzumab and pertuzumab. The killing activities of the different combinations of CoMiX-FHR4 with CoMiX-Fc exceeded the individual capacity of all molecules. Regarding CDC, FHR4/VHH(T) alone was superior to the combination of the two CoMiX-Fc (p<0.05). Taken together, these data indicate that FHR4/VHH(T) are the most efficient CoMiX in facilitating C3b deposition and the direct killing of tumor cells.
 |
Figure 2 CoMiX induce complement activation on BT474 tumor cells. (A) Flow cytometry analysis of C3b/iC3b deposition detected with mouse anti-human C3b mAb and a secondary goat anti-mouse IgG Ab conjugated with AF647, (B) membrane attack complex (MAC) formation was analyzed using anti-C5b9 mAb followed by PE-conjugated anti-mouse IgG pAb. MAC-formation was highest when CoMiX-Fc and CoMiX-FHR4 were combined, and (C) CDC on BT474 tumor cells incubated with 15 µg/well of multimeric immunotherapeutic complexes. The percentage of dead cells was calculated by dividing the number of live/dead-positive (dead) cells with the total number of analyzed cells. Data are presented as mean values ±SD of n = 3 independent experiments. Statistical analysis was performed using a one-way ANOVA and post-hoc Tukey’s test. (D) A linear correlation between C3b deposition (MFI) and the percentage of dead cells was observed (15 µg/well applied). Consistent with C3b deposition and MAC-formation, CoMiX-Fc and CoMiX-FHR4 significantly increased the percentage of dead cells compared to control multimers and Abs. (E and F) CoMiX-FHR4 activate the alternative complement pathway, whereas CoMiX-Fc facilitate classical pathway activation. C3b deposition (E) and CDC (F) of BT474 tumor cells incubated with saturating concentrations (15 µg/well) of CoMiX and control mAbs, individually or in combinations. 25% NHS diluted in either GVB++ (Orange bars) or GVB+ buffer (green bars) was added for 30 minutes at 37°C. Data are presented as mean values ±SD of n = 3 independent experiments. Statistical analysis was performed using a two-way ANOVA test, assessing the GVB++ and GVB+ conditions for each of the molecules. All comparisons between GVB++ and GVB+ reached statistical significance (p < 0.0001). * p < 0.05 **p < 0.01, ***p < 0.001, ****p < 0.0001. |
Mechanisms-of-Action of CoMiX-FHR4 and CoMiX-Fc
To assess which complement pathway can be activated by CoMiX, we used 25% NHS diluted in either GVB++ or GVB+ buffer. GVB++ buffer contains Ca2+ and Mg2+ ions, allowing the initiation of all three pathways of the complement cascade. The absence of Ca2+ ions in the GVB+ buffer selectively inhibits the classical and lectin pathways, hence only the alternative pathway can become active. As shown in Figure 2E and F and Supplementary Figure 5A and B, under all conditions the C3b deposition (Figure 2E) and the percentage of dead cells (Figure 2F) were significantly reduced in GVB+ compared to GVB++ buffer (p< 0.0001). Interestingly, the complement activating properties of CoMiX-FHR4 were reduced only by half in GVB+ buffer whereas CoMiX-Fc and antibodies completely lost their activating effects (Figure 2E and F). Thus, FHR4-CoMiX facilitate the activation of the alternative pathway, while CoMiX-Fc act as classical pathway activators.
NK cells bind IgG antibodies through their FcγRIIIa receptors (CD16), leading to the destruction of opsonized target cells via ADCC. HER2-positive BT474 tumor cells were incubated with CoMiX and the human CD16 expressing NK cell line (NK92humCD16) (Figure 3A and B and Supplementary Figure 5). The expression of the degranulation marker CD107a was highly upregulated on NK cells by VHH(P)/Fc and trastuzumab while VHH(T)/Fc alone elicited only half of the effect (p <0.0001, Figure 3A). Without hinge (VHHT/Fc Δhinge), the percentage of CD107a-positive NK cells was strongly reduced when compared to CoMiX-Fc, indicating that the hinge region is mandatory for the functional assembly of multiple Fc-dimers. Pertuzumab had a lower capacity to induce degranulation as compared to CoMiX VHH(P)/Fc (p < 0.0001). CoMiX-FHR4 did not significantly affect CD107a expression. In concordance, IFN-γ expression was significantly enhanced by VHH(T)/Fc and VHH(P)/Fc as well as by trastuzumab and pertuzumab as compared to CoMiX-FHR4 (p < 0.0001, Figure 3B).
 |
Figure 3 CoMiX-Fc enables NK cell activation and BT474 phagocytosis mediated by M2 macrophages. (A) HER2-positive BT474 tumor cells incubated with 15 μg/well of CoMiX were co-incubated with NK92humCD16 cells. The expression of the CD107a degranulation marker was analyzed by flow cytometry. (B) The intracellular accumulation of the cytokine IFN-γ was likewise analyzed by flow cytometry. Data are presented as mean values ±SD of the triplicates of one representative experiment of three independent experiments. Statistical analysis was performed using a one-way ANOVA and post-hoc Tukey’s test. (C) Complement-dependent macrophage-mediated phagocytosis of human BT474 cells. CFSE-stained BT474 tumor cells were incubated with controls, CoMiX, trastuzumab + pertuzumab or their combination with 25% C5-deficient human serum to prevent lysis. The percentage of phagocytic macrophages was measured. Data are presented as mean values ± SD out of n = 3 series of 10 confocal images for each condition. Statistical analysis was performed using a one-way ANOVA and post-hoc Tukey’s test. (D) Left: Confocal microscopy images of BT474 phagocytosis by M2 macrophages when incubated with CoMiX VHH(T)/Fc. The white arrows show the phagocytic macrophages (red) having engulfed BT474 cell(s) that harbor a green or yellowish color. Scale bar represents 50 µm. Right: Details of phagocytic macrophage (red) at higher magnification (60× oil immersion). Scale bar represents 10 µm. (E) Immunofluorescent staining of tumor sections collected 1 or 6 hours after injection of CoMiX-FHR4 (upper panel), CoMiX-Fc (intermediate panel), PBS (lower panel) and the controls VHH(T) and trastuzumab + pertuzumab (C3 staining 6 hours after injection). CoMiX were visualized with either a rabbit anti-His mAb followed by the goat anti-rabbit IgG Fc AF568- or a goat anti-human IgG AF647-conjugated antibody. Complement activation was visualized using the polyclonal rabbit anti-C3d antibody followed by AF568-conjugated anti-rabbit IgG. *p < 0.05, **p < 0.01, ****p < 0.0001. |
We previously showed that FHR4 induces complement-dependent macrophage-mediated phagocytosis of BT474 tumor cells.15 We next compared the effects of CoMix-Fc and CoMiX-FHR4 on macrophage-mediated phagocytosis using C5-deficient serum to inhibit CDC (Figure 3C and D). As previously described, CoMiX-FHR4 VHH(T) enhanced phagocytosis of BT474 target cells to the same extent as trastuzumab (around 20%, p <0.05). CoMiX VHH(P)/Fc, but not CoMiX VHH(T)/Fc, increased the number of BT474 cells phagocytosed by macrophages up to 30% (p < 0.005) (Figure 3C). All combinations between CoMiX and antibodies strongly enhanced phagocytosis by around 40% (p < 0.01), except for the trastuzumab+pertuzumab and trastuzumab+FHR4/VHH(P) combinations. We also confirm here that the hinge region is mandatory for functional Fc/FcγR interactions to stimulate phagocytosis by macrophages.
CoMiX-FHR4 and CoMiX-Fc Reduce Tumor Progression in vivo
In a xenograft mouse model of BT474 cells (sensitive to both trastuzumab and pertuzumab), we first checked the ability of CoMiX-FHR4 and CoMiX-Fc to infiltrate the tumor tissue and activate the mouse complement; immunohistochemistry of tumor sections is shown in Figure 3E. One hour after injection, both CoMiX were detected mainly at the tumor periphery and co-localized with C3b-deposition (left panels). 6 hours after injection, the whole tumor was infiltrated with CoMiX-FHR4 (middle panels), and complement activation occurred within the whole tumor tissue (right panels). Although the Fc staining of CoMiX-Fc was much weaker than the His staining of CoMiX-FHR4, CoMiX-Fc activate the classical complement pathway (middle right panel) within the xenograft to the same degree as the combination of trastuzumab and pertuzumab (lower right panel).
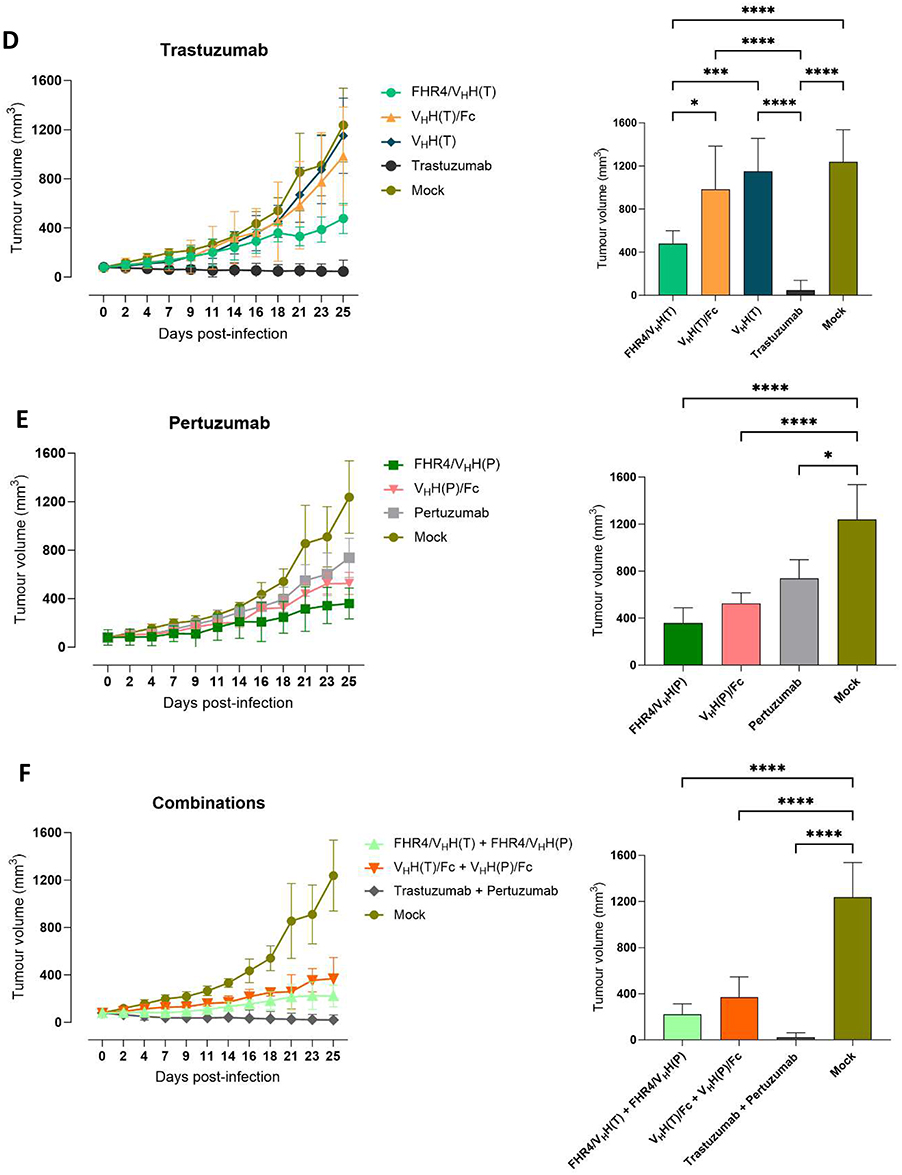
The effect of CoMiX was further analyzed in vivo in the xenograft mouse model as described in Figure 4A. No toxicity associated with the administration of CoMiX was revealed by monitoring body weight loss and clinical symptoms. Tumor volumes were tracked for 15 days after the last drug injection, and MRI was performed for representative mice after sacrifice (Figure 4B). As shown in Figure 4C, both CoMiX-FHR4 significantly reduced the tumor volume compared to PBS (mock, p < 0.05) as well as their combination (p< 0.01). VHH(T)/Fc had no significant effect on tumor growth whereas VHH(P)/Fc and its combination with VHH(T)/Fc decreased tumor growth significantly (p<0.05). When analyzing tumor growth (Figure 4D-F, left panels) or tumor volume at sacrifice (Figure 4D-F, right panels), both CoMiX-FHR4, alone or in combination, had less effect on tumor growth than trastuzumab although this was not significant at sacrifice (p> 0.05) (Figure 4D). VHH(P)/Fc had a greater effect than pertuzumab (Figure 4E). Although the combination of trastuzumab and pertuzumab was the most effective one, tumor volumes at sacrifice were not significantly different from the ones observed upon treatment with CoMiX-FHR4 or CoMiX-Fc combinations (p> 0.05) (Figure 4F).
Figure 4 Continued. Figure 4 CoMiX FHR4/VHH(T), FHR4/VHH(P), and VHH(P)/Fc efficiently reduce tumor growth in a similar way as pertuzumab, but not as trastuzumab. (A) Experimental design of the measurement of mammary fat pad volume of subcutaneous xenografts in the presence of different CoMiX or control antibodies. BT474 cells were injected into the mammary fat pads of mice. When the tumor volume reached ~60 mm3, the mice were injected intravenously into the lateral tail vein with 100 µg of CoMiX or control antibodies, or with 50 µg of each molecule in case of combination treatments. The injection was repeated 8 times, on days 1, 2, 3, 4, 7, 8, 9, and 10 after the first injection. The tumors were measured every second or third day with calipers. (B) MRI images of a representative tumor for each group of mice sacrificed at the end of the protocol or when the tumor’s size reached 1000 mm3. (C) The effects of CoMiX [FHR4/VHH(T), FHR4/VHH(P), VHH(T)/Fc, VHH(P)/Fc, VHH(T)] and two therapeutic antibodies (trastuzumab and pertuzumab) were evaluated individually and in combination. The treatment duration is indicated for all groups in green, as mentioned for the first FHR4/VHH(T) group on the left side of the graph. (D–F) The growth of the tumors (curve, left panels) and its size at time of sacrifice (bars, right panels) was further analyzed between all groups that received the same anti-HER-2 epitope (CoMiX or therapeutic antibody): (D) Trastuzumab, (E) Pertuzumab, and (F) combination of the two epitopes. The groups were compared using the unpaired Student’s T-test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.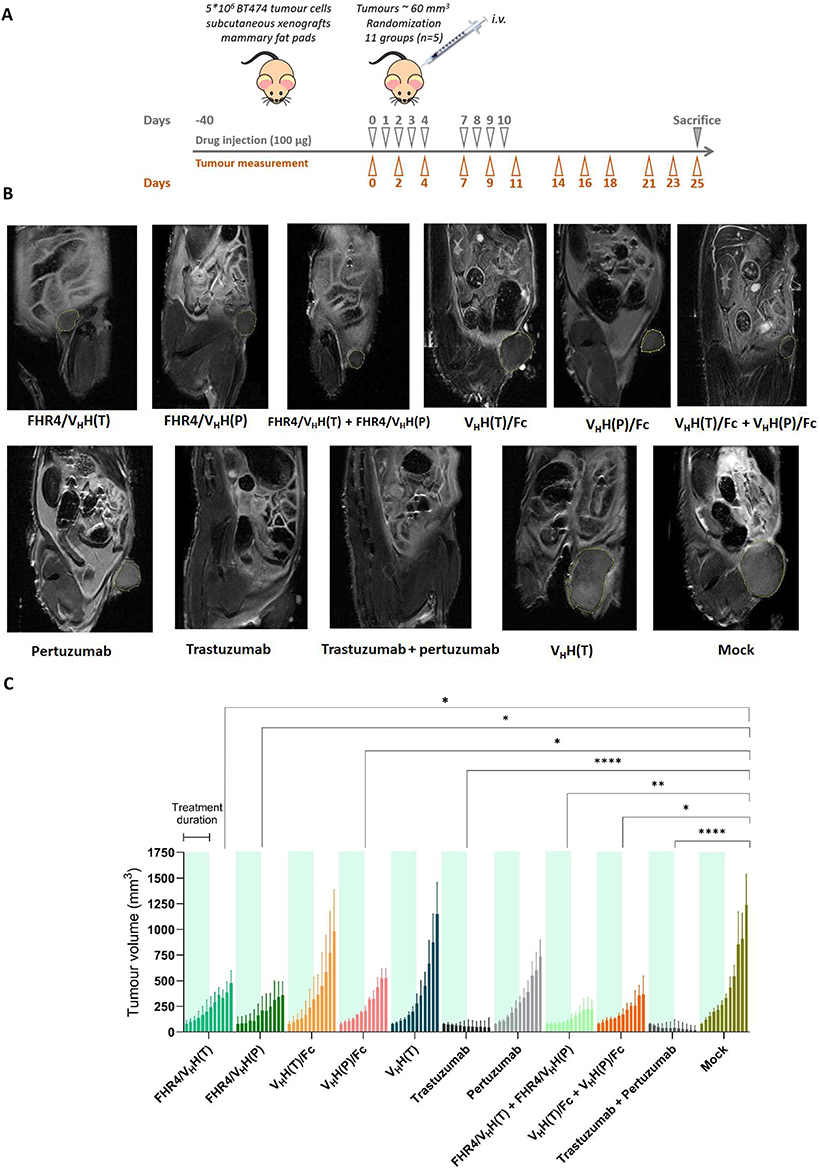
A large leukocyte infiltration was revealed in xenografts of mice treated with trastuzumab or with all combinations; to a lesser extent in xenografts from the FHR4-VHH(T)-, FHR4-VHH(P)- or VHH(P)/Fc-treated mice (left panels, Figure 5A). The NK cell activating receptor NKp46 involved in the elimination of target cells was consistently detected in those infiltrations (right panels, Figure 5A), up to a level of 16.7% of cells upon treatment with trastuzumab and up to 14.5% upon treatment with the combination of FHR4-VHH(T) and FHR4-VHH(P) (Figure 5B). Moreover, a similar frequency of NK cells appeared in trastuzumab-, or in FHR4-VHH(T)- or in FHR4-VHH(P)-treated mice. Although variable anti-HER2 antibodies concentrations were measured in the sera of the mice (Supplementary Figure 6), a trend towards higher anti-HER2 antibodies in trastuzumab, VHH(P)/Fc-, FHR4/VHH(T)-, and (trastuzumab+pertuzumab)-treated mice was depicted as compared to control mice.
 |
Figure 5 CoMiX stimulate leukocyte and NK cells infiltration. Mice with HER-2 positive xenografts were treated with the different CoMiX, trastuzumab, pertuzumab or their combination as presented in Figure 4A. Tumors were excised and stained. (A) From single and combination treatments, representative pictures of immunohistochemistry staining (40X) of leukocytes (left panels), using an anti-mouse CD45 antibody, or of NK cells (middle panels), using a mouse NKp46/Ncr1 antibody, revealed by an HRP secondary antibody. (B) CD45- and NKp46-positive cells, expressed as percentage of total cells, are shown for each group of mice. Data are represented as the mean values ± SEM of 10 slides from 3 xenografts of each group. Statistical analysis was performed using a one-way ANOVA and post-hoc Tukey’s test (*p < 0.05). |
While trastuzumab is extremely effective at the early stages of treatment, cells quickly develop various resistance mechanisms.8 We thus explored the effects of CoMiX FHR4/VHH(P) and its combination with CoMiX FHR4/VHH(T) (Figure 6A) on xenografts of a trastuzumab-resistant BT474 cell line (ATCC-CRL-3247). Injecting trastuzumab for 10 days increased the tumor growth of trastuzumab-resistant cancer cells as compared to the controls, as shown previously.18–20 In contrast, CoMiX-FHR4 significantly reduced their growth during treatment and up to 25 days after treatment initiation when the tumor relapsed. No difference regarding the tumor growth was observed for the treatment with CoMiX FHR4/VHH(P) alone or in combination with CoMiX FHR4/VHH(T). At Day 11, a massive NK cell infiltration was observed in the tumors treated with FHR4/VHH(P), alone or in combination with FHR4/VHH(T) (Figure 6B), as shown previously in trastuzumab-sensitive xenografts, whereas the infiltration was much weaker in tumors treated with trastuzumab. Taken together, our in vivo data showed that CoMiX-FHR4 and CoMiX-Fc lead to a fast complement deposition resulting in the recruitment of NK cells. CoMiX may therefore offer a novel treatment alternative for tumors that have become resistant to trastuzumab.
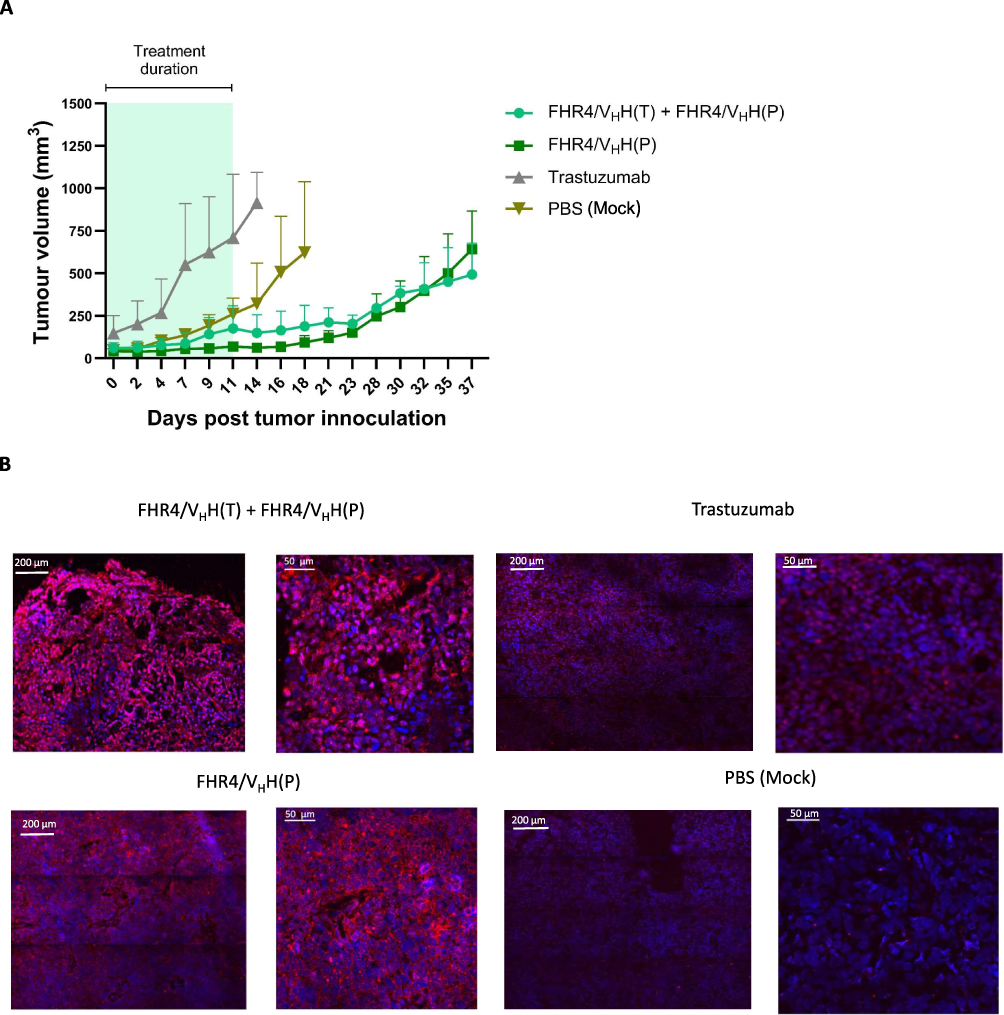 |
Figure 6 CoMiX FHR4/VHH(T) and FHR4/VHH(P) exert their anti-tumor effect on trastuzumab-resistant BT474 cells. (A) Trastuzumab-resistant BT474 cells were injected into the mammary fat pads of female BALB/c nude mice. When the tumor volume reached ~60 mm3, the mice were injected with 100 µg of CoMiX-FHR4, trastuzumab, or PBS, as described in Figure 5A. The tumors were measured every second or third day until day 37 of the study or until meeting the endpoint of 10,000 mm3. (B) Immunofluorescent stainings of NK cells in trastuzumab-resistant BT474 tumor xenografts performed just after the end of the treatment (at D+11). Cryosections were stained with a mouse NKp46/NCR1 antibody, revealed using a donkey anti-rat AF568-conjugated pAb, and analyzed by confocal microscope (lens X40) monitored by the Nikon NIS-Elements software. One representative image and its magnified view are shown for tumors treated with combined CoMiX-FHR4 [FHR4/VHH(T) + FHR4/VHH(P)] (upper left panels), tumors treated with CoMiX FHR4/VHH(P) (lower left panels), tumors treated with trastuzumab (upper right panels), and tumors treated with PBS (mock, lower right panels). |
Discussion
HER2-targeted therapies have changed the paradigm for patients and significantly improved their prognosis over the past few decades.21,22 For most patients with advanced or metastatic HER2-positive breast cancer, the combination of trastuzumab with pertuzumab has become standard of care.23 Breast cancer remains nevertheless a clinical challenge due to emerging resistance to anti-HER2 therapies, leading to disease progression. New HER2-targeted drugs, such as tucatinib and trastuzumab deruxtecan, have raised survival rates in cancers with de novo stage IV disease or cancers that inevitably recur.24 The therapeutic landscape for HER2 breast cancers has significantly grown with 20% of new drugs having received approval over the past two decades.25 Phase 3 trials were conducted for second line options with the evaluation of antibody-drug conjugates but also of other targets including PI3KCA mutations, PD-L1 and CDK4/6 offering novel combination strategies with improved outcomes. However, novel effective therapies are still needed in both early and metastatic settings.
Immunotherapy with directed complement activation is a promising approach for solid cancer.26 The activation of the classical pathway contributes to the therapeutic efficacy of Rituximab27–29 and was further enhanced with ofatumumab and daratumumab.4 Factor H is a key complement regulator that can be hijacked by bacteria30–32 or cancer cells33 to evade complement-mediated cytotoxicity.29,34 Factor H-related proteins 1 to 5 have different activating or inhibiting effects.35–37 We have previously shown that BT474 tumor cells establish a complement inhibitory threshold, that can be overcome by high valences of FHR4 when directed to the surface of tumor cells.15 By lacking the regulatory domains of FH, but not the surface binding region of the protein, FHR4 competes with FH for binding of C3b, iC3b, and C3d.15 Fc regions, however, mediate several effector functions, including classical complement pathway activation38 as well as the activation of immune cells (phagocytes, NK cells) via various Fc receptors.39,40
Here, we showed that both CoMiX-FHR4 and CoMiX-Fc have a complement activating capacity on HER2-positive BT474 cancer cells, inducing direct lysis of tumor cells. CoMiX-FHR4 had a superior effect in vitro on CDC when compared to CoMiX-Fc and Abs (Figure 2). CoMiX-FHR4 serve as a platform for the assembly of the C3-convertase of the alternative pathway whereas CoMiX-Fc activate the classical pathway, largely more than trastuzumab or pertuzumab. This greater effect might be explained by the dual hinge region, providing Ab-like moieties that are more distant from the Fc region, hampering the natural whipping out of complement activating molecules by the flexibility of the Fab fragment on the Fc fragment in natural IgG. Therefore, the combination of CoMiX-Fc with CoMiX-FHR4 resulted in enhanced complement activation. Since VHH(T) and VHH(P) moieties recognize different epitopes, their effect was also additive when two CoMiX-FHR4 or CoMiX-Fc constructs were combined. Alternatively, CoMiX-Fc activate NK cells and induce complement-mediated phagocytosis of BT474 cells by M2 macrophages. VHH(P)/Fc was able to activate NK cells and complement-mediated phagocytosis with greater efficacy than pertuzumab, while VHH(T)/Fc had a similar effect than trastuzumab.
In BT474 xenografts sensitive to trastuzumab, the diffusion of CoMiX into the tumor was fast, and the tumor demonstrated massive complement activation 6h after systemic injection. FHR4/VHH(T), FHR4/VHH(P), and VHH(P)/Fc reduced tumor growth but not VHH(T)/Fc that showed less efficiency than VHH(P)/Fc regarding the phagocytosis by macrophages and the degranulation of NK cells in vitro (Figure 4) and little leukocyte infiltration in xenografts. Trastuzumab was the most potent treatment, indicating that complement activation alone is less efficient in limiting tumor growth than the additive effects of cell signaling inhibition coupled with ADCC/ADCP. Besides its cytostastic effect, the immunological engagement of trastuzumab mediates its clinical efficacy via ADCC elicited by NK cells or macrophages, resulting in cancer cell killing, but also through activation of macrophages, neutrophils, and dendritic cells, modulating the adaptive immunity. This was supported by the large leukocyte infiltration observed in xenografts of mice treated with trastuzumab but not with pertuzumab. Activated NK cells expressing NKp46 were highly represented among the leukocytes. FHR4/VHH(T) or FHR4/VHH(P) induced less leukocyte infiltration than trastuzumab, but similar NK cells recruitment suggests that the directed activation of complement on cancer cells might stimulate enough NK cell responses. Of note, VHH(P)/Fc was as potent as pertuzumab in reducing tumor growth in vivo, probably due to its greater efficacy to activate NK cells and complement-mediated phagocytosis, as shown in vitro, but also by the increased presence of NK cells in xenografts as compared to pertuzumab.
Taken together, it is tempting to speculate that NK cell activation or macrophage-mediated tumor cell phagocytosis plays a more significant role in reducing tumor growth in vivo than complement-induced lysis. This might explain that both CoMiX-FHR4 are less potent in vivo than trastuzumab. Moreover, we cannot exclude that the binding affinity of CoMiX for mouse complement proteins and for mouse responder cells may be different than for human cells in the host in vivo, which may explain the differences among the mechanisms responsible for the efficacy of the compounds in vivo and in vitro. This will be further investigated in another work, once pharmacokinetics data will be known, using humanized mice having functional NK cells and adaptive immunity such as Tg NSG tg-huIL-15 mice to recapitulate the complexity of human tumor-immune interactions and investigate the key role of C3b deposition. Nude mice were indeed chosen here to establish human HER2+ xenograft and show the direct effects of CoMiX and tumor killing by complement without the confounding influence of adaptive immune responses.
The combined use of trastuzumab and pertuzumab enhanced their therapeutic efficacy in vivo as well as the combination of CoMiX-FHR4 and of CoMiX-Fc as compared to their single administration. This is probably due to higher CDC and complement-mediated phagocytosis, as observed in vitro, and as reported as a synergistic mode of action through complement activation for the dual therapy trastuzumab and pertuzumab.6 Ultimately, when tested on a trastuzumab-resistant BT474 cell line, FHR4/VHH(P) and the combination of FHR4/VHH(T) and FHR4/VHH(P) delayed significantly tumor growth as compared to trastuzumab which induced an even higher growth rate than the control group, as previously observed18–20 due to the upregulation of angiogenesis factors.41,42 Up to 15 days after the last injection, CoMiX-FHR4 suppressed the growth of trastuzumab-resistant cells before the tumor relapsed, suggesting that no remission was achieved at the end of the treatment or that the cells had become resistant to the treatment such as by HER2 receptor internalization as previously shown for CoMiX-FHR4 internalization in vitro.15 Pharmacokinetics data will further explain whether CoMiX were sufficiently administrated in terms of dosage and timing since all molecules were provided every other day, like the therapeutic antibodies displaying a long half-life (up to 28 days for trastuzumab).
Our in vivo data corroborate the in vitro data by showing a high local activation of the alternative complement pathway in the xenograft after 6 hours of systemic injection. This mechanism-of-action is strictly independent from the HER2 signaling pathway, as we did not observe any CDC in vitro without providing serum or with decomplemented serum up to 24 hours after incubation with CoMiX.15 Complement is currently considered as an essential effector of the cytotoxic anti-tumor responses of Abs but also in antitumor immunity.43 The crosstalk of complement effectors and cellular signaling pathways is known to regulate the T and B cell responses in terms of survival, differentiation, and activation, but also to shape the responses of myeloid cells toward pro-tumoral or immunosuppressive effects.44 A trend towards increased concentrations of HER2 antibodies was detected in sera of mice treated with trastuzumab, VHH(P)/Fc, FHR4/VHH(T), and trastuzumab plus pertuzumab. Induction of antitumor antibodies is more frequently observed in clinically responding patients45 through endocytosis of tumor antigen–containing immune complexes or phagocytosis of antibody-opsonized tumor target cells.
Advances in HER2-positive breast cancer therapy currently rely on antibody–drug conjugates or bi-specific antibodies using two different HER2 epitopes for higher efficacy.46 A future format of CoMiX including the targeting of both HER2 epitopes could improve its effectiveness and avoid the occurrence of resistance. Importantly, the fast recruitment of the complement system is highly unlikely to lead to treatment resistance, in contrast to currently used Abs.8,10 The complement system has a strong potential to stimulate inflammation but is nevertheless tightly regulated. Membrane-anchored complement regulatory proteins (CD46, CD55 and CD59) are overexpressed on host cells to prevent high complement activation.47,48 Several other soluble complement regulatory proteins are also present in body fluids, such as FH, factor H-like protein 1 (FHL1), classical pathway inhibitors like C4b-binding protein (C4bp), C1 inhibitor as well as terminal pathway inhibitors like clusterin or vitronectin. Therefore, the directed C3b activation on targeted tumor cells could represent an alternative and safe anticancer therapy49 since we previously showed that CoMiX-FHR4 do not mediate significant soluble phase complement activation at concentrations that are commonly used in the clinic for therapeutic mAbs.15 Complement components are known to regulate the function of the tumor microenvironment by recruiting cells favoring both tumor-promoting and tumor-suppressing responses;12 a clear understanding of the cellular mechanisms underlying CoMiX effects will, therefore, be crucial for their optimization for clinical development.
Conclusion
Directed- complement activation with CoMiX reduces tumor growth in vivo. CoMiX VHH(P)/Fc were as effective as pertuzumab, and CoMiX-FHR4 might help to circumvent resistance to trastuzumab since their mechanism-of-action differs from conventional therapeutic antibodies. Their combination showed enhanced effects on tumor growth that open the way to explore combination therapies to reach a synergistic mechanism-of-action with Abs.
Abbreviations
Abs, Therapeutic antibodies; ADCC, Antibody-dependent cell-mediated cytotoxicity; C4bp, C4b-binding protein; CDC, Complement-dependent cytotoxicity; CDCC, Complement-dependent cell cytotoxicity; CDCP, Complement-dependent cell phagocytosis; CoMiX, Complement activating Multimetric Immunitherapeutic compleXes; FH, Factor H; FHL1, Factor H-like protein 1; FHR4, Factor-H Related protein 4; GVB, Gelatin veronal buffer; HER-2, Human epidermal growth factor receptor 2; IgG, Immunoglobulin gamma antibodies; L/D, Live/Dead; MAC, Membrane attack complex; MRI, Magnetic Resonance Imaging; NHS, Normal human serum; PBMCs, Peripheral blood mononuclear cells; T-DM1, Trastuzumab-emtansine; T-DXd, Trastuzumab–deruxtecan.
Acknowledgments
We thank the participants who has given their PBMCs for research to the Luxembourg Red Cross and the Luxembourg Red Cross for the collection of the samples. We would like to acknowledge Brigitte Reveil for her technical assistance and Pr Béatrice Clemenceau, University of Nantes, for providing the NK92humCD16 cell line.
Funding
The study was supported by the “Fonds National de la Recherche” (POC17/12252709/COMIX and PRIDE17/11823097/MICROH-DTU for the funding of Bianca Brandus) and the Ministry of Higher Education and Research of Luxembourg(LIH GBB 98000005).
Disclosure
A patent application has been granted in the USA for CoMiX (WO2017202776) by the inventors (C.S.D, J.H.M.C, X.D). The authors have declared that no other conflict of interest exists. This paper has been uploaded to Biorxiv as a preprint: https://www.biorxiv.org/content/10.1101/2024.02.02.578619v1
References
1. Delgado M, Garcia-Sanz JA. Therapeutic monoclonal antibodies against cancer: present and future. Cells. 2023;12(24):2837. doi:10.3390/cells12242837
2. Ain D, Shaikh T, Manimala S, Ghebrehiwet B. The role of complement in the tumor microenvironment. Fac Rev. 2021;10:80. doi:10.12703/r/10-80
3. Golay J, Taylor RP. The role of complement in the mechanism of action of therapeutic anti-cancer mAbs. Antibodies. 2020;9(4):58. doi:10.3390/antib9040058
4. Ovcinnikovs V, Dijkman K, Zom GG, Beurskens FJ, Trouw LA. Enhancing complement activation by therapeutic anti-tumor antibodies: mechanisms, strategies, and engineering approaches. Semin Immunol. 2024;77:101922. doi:10.1016/j.smim.2024.101922
5. Maadi H, Wang Z. A novel mechanism underlying the inhibitory effects of trastuzumab on the growth of HER2-positive breast cancer cells. Cells. 2022;11(24):4093. doi:10.3390/cells11244093
6. Tsao LC, Crosby EJ, Trotter TN, et al. Trastuzumab/pertuzumab combination therapy stimulates antitumor responses through complement-dependent cytotoxicity and phagocytosis. JCI Insight. 2022;7(6). doi:10.1172/jci.insight.155636
7. Jagosky M, Tan AR. Combination of pertuzumab and trastuzumab in the treatment of HER2-positive early breast cancer: a review of the emerging clinical data. Breast Cancer. 2021;13:393–407. doi:10.2147/bctt.S176514
8. Wang ZH, Zheng ZQ, Jia SC, et al. Trastuzumab resistance in HER2-positive breast cancer: mechanisms, emerging biomarkers and targeting agents. Front Oncol. 2022;12:1006429. doi:10.3389/fonc.2022.1006429
9. Singh R, Chandley P, Rohatgi S. Recent advances in the development of monoclonal antibodies and next-generation antibodies. Immunohorizons. 2023;7(12):886–897. doi:10.4049/immunohorizons.2300102
10. Hunter FW, Barker HR, Lipert B, et al. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br J Cancer. 2020;122(5):603–612. doi:10.1038/s41416-019-0635-y
11. Zarantonello A, Revel M, Grunenwald A, Roumenina LT. C3-dependent effector functions of complement. Immunol Rev. 2023;313(1):120–138. doi:10.1111/imr.13147
12. Revel M, Daugan MV, Sautés-Fridman C, Fridman WH, Roumenina LT. Complement system: promoter or suppressor of cancer progression?. Antibodies. 2020;9(4). doi:10.3390/antib9040057
13. Cserhalmi M, Papp A, Brandus B, Uzonyi B, Józsi M. Regulation of regulators: role of the complement factor H-related proteins. Semin Immunol. 2019;45:101341. doi:10.1016/j.smim.2019.101341
14. Jozsi M, Meri S. Factor H-related proteins. Methods Mol Biol. 2014;1100:225–236. doi:10.1007/978-1-62703-724-2_18
15. Seguin-Devaux C, Plesseria JM, Verschueren C, et al. FHR4-based immunoconjugates direct complement-dependent cytotoxicity and phagocytosis towards HER2-positive cancer cells. Mol Oncol. 2019;13(12):2531–2553. doi:10.1002/1878-0261.12554
16. Hofmeyer T, Schmelz S, Degiacomi MT, et al. Arranged sevenfold: structural insights into the C-terminal oligomerization domain of human C4b-binding protein. J Mol Biol. 2013;425(8):1302–1317. doi:10.1016/j.jmb.2012.12.017
17. Schneider CA, Rasband WS, Eliceiri KW. NIH image to imageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi:10.1038/nmeth.2089
18. Wielgos ME, Zhang Z, Rajbhandari R, et al. Trastuzumab-resistant HER2(+) breast cancer cells retain sensitivity to poly (ADP-ribose) polymerase (PARP) inhibition. Mol Cancer Ther. 2018;17(5):921–930. doi:10.1158/1535-7163.Mct-17-0302
19. Sanz-álvarez M, Luque M, Morales-Gallego M, et al. Generation and characterization of trastuzumab/pertuzumab-resistant HER2-positive breast cancer cell lines. Int J Mol Sci. 2023;25(1):207. doi:10.3390/ijms25010207
20. Lu Y, Zi X, Pollak M. Molecular mechanisms underlying IGF-I-induced attenuation of the growth-inhibitory activity of trastuzumab (Herceptin) on SKBR3 breast cancer cells. Int, J, Cancer. 2004;108(3):334–341. doi:10.1002/ijc.11445
21. Patel A, Unni N, Peng Y. The changing paradigm for the treatment of HER2-positive breast cancer. Cancers. 2020;12(8):2081. doi:10.3390/cancers12082081
22. Zeng L, Li W, Chen CS. Breast cancer animal models and applications. Zool Res. 2020;41(5):477–494. doi:10.24272/j.issn.2095-8137.2020.095
23. Martínez-Sáez O, Prat A. Current and future management of HER2-positive metastatic breast cancer. JCO Oncol Pract. 2021;17(10):594–604. doi:10.1200/op.21.00172
24. Swain SM, Shastry M, Hamilton E. Targeting HER2-positive breast cancer: advances and future directions. Nat Rev Drug Discov. 2023;22(2):101–126. doi:10.1038/s41573-022-00579-0
25. Michaeli JCM, Michaeli T, Dario Trapani D, et al. Breast cancer drugs: FDA approval, development time, efficacy, clinical benefits, innovation, trials, endpoints, quality of life, value, and price. Breast Cancer. 2024;31(6):1144–1155. doi:10.1007/s12282-024-01634-x
26. Yuan M, Liu L, Wang C, Zhang Y, Zhang J. The complement system: a potential therapeutic target in liver cancer. Life. 2022;12(10):1532. doi:10.3390/life12101532
27. Harjunpää A, Junnikkala S, Meri S. Rituximab (anti-CD20) therapy of B-cell lymphomas: direct complement killing is superior to cellular effector mechanisms. Scand J Immunol. 2000;51(6):634–641. doi:10.1046/j.1365-3083.2000.00745.x
28. Di Gaetano N, Cittera E, Nota R, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171(3):1581–1587. doi:10.4049/jimmunol.171.3.1581
29. Hörl S, Bánki Z, Huber G, et al. Reduction of complement factor H binding to CLL cells improves the induction of rituximab-mediated complement-dependent cytotoxicity. Leukemia. 2013;27(11):2200–2208. doi:10.1038/leu.2013.169
30. Parente R, Clark SJ, Inforzato A, Day AJ. Complement factor H in host defense and immune evasion. Cell Mol Life Sci. 2017;74(9):1605–1624. doi:10.1007/s00018-016-2418-4
31. Moore SR, Menon SS, Cortes C, Ferreira VP. Hijacking factor H for complement immune evasion. Front Immunol. 2021;12:602277. doi:10.3389/fimmu.2021.602277
32. Józsi M. Factor H family proteins in complement evasion of microorganisms. Front Immunol. 2017;8:571. doi:10.3389/fimmu.2017.00571
33. Saxena R, Gottlin EB, Campa MJ, et al. Complement factor H: a novel innate immune checkpoint in cancer immunotherapy. Front Cell Dev Biol. 2024;12:1302490. doi:10.3389/fcell.2024.1302490
34. Hörl S, Banki Z, Huber G, et al. Complement factor H-derived short consensus repeat 18–20 enhanced complement-dependent cytotoxicity of ofatumumab on chronic lymphocytic leukemia cells. Haematologica. 2013;98(12):1939–1947. doi:10.3324/haematol.2013.089615
35. Csincsi, Á I, Szabó Z, Bánlaki Z, et al. FHR-1 binds to C-reactive protein and enhances rather than inhibits complement activation. J Immunol. 2017;199(1):292–303. doi:10.4049/jimmunol.1600483
36. Reiss T, Rosa TFA, Blaesius K, et al. Cutting edge: FHR-1 binding impairs factor H-mediated complement evasion by the malaria parasite plasmodium falciparum. J Immunol. 2018;201(12):3497–3502. doi:10.4049/jimmunol.1800662
37. González-Alsina A, Martín-Merinero H, Mateu-Borrás M, et al. Factor H-related protein 1 promotes complement-mediated opsonization of Pseudomonas aeruginosa. Front Cell Infect Microbiol. 2024;14:1328185. doi:10.3389/fcimb.2024.1328185
38. Lee CH, Romain G, Yan W, et al. IgG Fc domains that bind C1q but not effector Fcγ receptors delineate the importance of complement-mediated effector functions. Nat Immunol. 2017;18(8):889–898. doi:10.1038/ni.3770
39. Gogesch P, Dudek S, van Zandbergen G, Waibler Z, Anzaghe M. The role of fc receptors on the effectiveness of therapeutic monoclonal antibodies. Int J Mol Sci. 2021;22(16):8947. doi:10.3390/ijms22168947
40. Lu LL, Suscovich TJ, Fortune SM, Alter G. Beyond binding: antibody effector functions in infectious diseases. Nat Rev Immunol. 2018;18(1):46–61. doi:10.1038/nri.2017.106
41. Hori A, Shimoda M, Naoi Y, et al. Vasculogenic mimicry is associated with trastuzumab resistance of HER2-positive breast cancer. Breast Cancer Res. 2019;21(1):88. doi:10.1186/s13058-019-1167-3
42. Morales-Guadarrama G, García-Becerra R, Méndez-Pérez EA, García-Quiroz J, Avila E, Díaz L. Vasculogenic mimicry in breast cancer: clinical relevance and drivers. Cells. 2021;10(7):1758. doi:10.3390/cells10071758
43. Pio R, Ajona D, Ortiz-Espinosa S, Mantovani A, Lambris JD. Complementing the cancer-immunity cycle. Front Immunol. 2019;10:774. doi:10.3389/fimmu.2019.00774
44. Magrini E, Minute L, Dambra M, Garlanda C. Complement activation in cancer: effects on tumor-associated myeloid cells and immunosuppression. Semin Immunol. 2022;60:101642. doi:10.1016/j.smim.2022.101642
45. Taylor C, Hershman D, Shah N, et al. Augmented HER-2 specific immunity during treatment with trastuzumab and chemotherapy. Clin Cancer Res. 2007;13(17):5133–5143. doi:10.1158/1078-0432.Ccr-07-0507
46. Zong HF, Li X, Han L, et al. A novel bispecific antibody drug conjugate targeting HER2 and HER3 with potent therapeutic efficacy against breast cancer. Acta Pharmacol Sin. 2024;45(8):1727–1739. doi:10.1038/s41401-024-01279-8
47. Geller A, Yan J. The role of membrane bound complement regulatory proteins in tumor development and cancer immunotherapy. Front Immunol. 2019;10:1074. doi:10.3389/fimmu.2019.01074
48. Mamidi S, Cinci M, Hasmann M, Fehring V, Kirschfink M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol Oncol. 2013;7(3):580–594. doi:10.1016/j.molonc.2013.02.011
49. Elvington M, Huang Y, Morgan BP, et al. A targeted complement-dependent strategy to improve the outcome of mAb therapy, and characterization in a murine model of metastatic cancer. Blood. 2012;119(25):6043–6051. doi:10.1182/blood-2011-10-383232
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Research Progress of NK Cells in Glioblastoma Treatment
Wu H, Liu Q, Wang F, Gao W, Zhou F, Zhao H
OncoTargets and Therapy 2025, 18:87-106
Published Date: 18 January 2025
Adaptation of Natural Killer Cells to Hypoxia: A Review of the Transcriptional, Translational, and Metabolic Processes
Chang TD, Chen YJ, Luo JL, Zhang C, Chen SY, Lin ZQ, Zhang PD, Shen YX, Tang TX, Li H, Dong LM, Tang ZH, Chen D, Wang YM
ImmunoTargets and Therapy 2025, 14:99-121
Published Date: 18 February 2025
TGF-β Decreases NK Cell Mobility and Cytotoxic Efficacy in Complex in vitro Models of the Leukemia Microenvironment
Švubová V, Janstová L, Jedlička M, Mašínová E, Szabová J, Feglarová T, Kuglerová K, Bosáková V, Brodská B, Boráková K, Kundrát D, Trsová I, Böhmová M, Kuželová K, Hrdý J, Gašová Z, Vydra J, Dostálová Merkerová M, Hortová-Kohoutková M, Frič J
ImmunoTargets and Therapy 2025, 14:589-604
Published Date: 18 June 2025