Back to Journals » OncoTargets and Therapy » Volume 13
Diosgenin and GSK126 Produce Synergistic Effects on Epithelial–Mesenchymal Transition in Gastric Cancer Cells by Mediating EZH2 via the Rho/ROCK Signaling Pathway
Authors Liu S, Rong G, Li X, Geng L, Zeng Z, Jiang D, Yang J, Wei Y
Received 6 November 2019
Accepted for publication 3 April 2020
Published 8 June 2020 Volume 2020:13 Pages 5057—5067
DOI https://doi.org/10.2147/OTT.S237474
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Nicola Silvestris
Shanshan Liu,* Guihong Rong,* Xia Li, Lijun Geng, Zhineng Zeng, Dongxiang Jiang, Jun Yang, Yesheng Wei
Department of Clinical Laboratory, The Affiliated Hospital of Guilin Medical University, Guilin, Guangxi 541001, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yesheng Wei
Department of Clinical Laboratory, The Affiliated Hospital of Guilin Medical University, No. 15, Lequn Road, Xiufeng District, Guangxi 541001, People’s Republic of China
Tel +86 773 2822 730
Email [email protected]
Background: Diosgenin, a natural steroidal saponin isolated from Trigonella foenum-graecum, has been reported to exert anti-cancer effects. Inhibitors of enhancer of zeste homology 2 (EZH2) have been widely used in treatment of cancers. However, the effects of combined treatment with diosgenin and an EZH2 inhibitor on gastric cancer (GC) cells, and the mechanism for those effects are not fully understood.
Methods: AGS and SGC-7901 gastric cancer cells were treated with diosgenin (0 to 8 μM), followed by treatment with either diosgenin or an EZH2 inhibitor, GSK126 alone. Afterwards, an EZH2 overexpression plasmid and Rho inhibitor, GSK429286A was involved in cells. Cell proliferation, cell cycle distribution, and cell apoptosis, migration, and invasion were examined by CCK-8 assays, flow cytometry, and transwell assays. Western blotting was performed to detect the relative levels of protein expression.
Results: Treatment with diosgenin alone caused a dose-dependent decrease in the cell viability, and combined treatment with an EZH2 inhibitor plus GSK126 caused a further significant decrease. A further analysis revealed that treatment with either diosgenin or GSK126 alone induced significant increases in G0/G1 cell cycle arrest and apoptosis, and combined treatment with both agents induced further increases in those parameters. In addition, combined treatment with diosgenin and GSK126 synergistically induced even stronger effects on impaired cell proliferation, G0/G1 phase arrest, and cell apoptosis when compared to treatment with either diosgenin or GSK126 treatment alone. At the molecular level, we demonstrated that inhibition of Rho/ROCK signaling by combined treatment with diosgenin and GSK126 could downregulate the expression of epithelial–mesenchymal transition (EMT)-related molecules. We also found that EZH2 overexpression reversed the anti-tumor effect of diosgenin by inducing cell survival, blocking G1-phase arrest, and promoted EMT. While, these biological properties were further reversed by GSK429286A.
Conclusion: Collectively, combined treatment with diosgenin and GSK126 produced even more significant effects on GC cell inhibition by targeting EZH2 via Rho/ROCK signaling-mediated EMT, which might be a therapeutic strategy for improving the poor therapeutic outcomes obtained with GSK126 monotherapy.
Keywords: gastric cancer, diosgenin, EZH2, Rho/ROCK, EMT
Introduction
Gastric cancer (GC) is one of the leading causes of cancer-related deaths worldwide, and especially in China.1,2 While new surgical techniques and chemotherapy regimens have gradually improved the clinical outcomes of GC patients, the five-year survival rate of advanced GC patients has not improved, mainly due to the high rates of GC metastasis and recurrence.3,4 Therefore, there remains an urgent need to explore the biological mechanisms that drive GC metastasis, and develop new target-specific treatments for GC.
Numerous molecules have been reported to contribute to cancer cell invasion and metastasis, and the epithelial–mesenchymal transition (EMT) process can enhance the ability of cancer cells to migrate and invade.5 The EMT is a complex process characterized by the loss of epithelial markers (e.g. E-cadherin and β-catenin) and an upregulation of mesenchymal markers (e.g., N-cadherin and vimentin).6 Previous studies demonstrated that RhoA and its downstream kinase Rho-kinase (ROCK) are able to mediate cell matrix formation in diabetic nephropathy.7,8 Furthermore, the RhoA/ROCK signaling pathway was previously demonstrated to be associated with EMT.9 For example, ART1 was shown to regulate the EMT process in colon carcinoma via the RhoA/ROCK1/AKT/β-catenin pathway.10 Zhang et al11 showed that the RhoA/ROCK signaling pathway might mediate EMT induced by TGF-β1 in rat peritoneal mesothelial cells.11 However, it remains unclear whether EMT regulated by RhoA/ROCK signaling that involved in GC metastasis.
Diosgenin is a natural steroidal saponin isolated from the plant Trigonella foenum-graecum, which has been reported to exert anti-inflammatory,12 antioxidant,13 and anti-cancer effects.14 Accumulating evidence suggests that diosgenin can induce apoptosis, DNA damage, and activate mitochondrial signaling pathways.15,16 Enhancer of zeste homology 2 (EZH2), a member of the polycomb gene family,17 is overexpressed in many malignancies and could possibly serve as a prognostic factor in certain malignancies.18,19 Some selective EZH2 inhibitors have been reported to exert potent antitumor activity when used in the treatment of cancers.20 Among those inhibitors, GSK126 is a highly selective EZH2 inhibitor and also a newly synthesized S-adenosylmethionine competitor.21 Several studies conducted with solid tumor cell lines have suggested that GSK126 suppresses cell migration and angiogenesis, but has a limited therapeutic effect when used alone.22–24 Therefore, we hypothesized that a combination of GSK126 and diosgenin might be more potent for attenuating GC metastasis when compared to using GSK126 or diosgenin alone.
To test our hypothesis, we first analyzed the effects of diosgenin or GSK126 treatment alone on GC cell proliferation, cell cycle progression, apoptosis, migration, and invasion, and then compared those results with those obtained when using diosgenin combined with GSK126. Furthermore, we investigated whether inhibition of both EZH2 and EGFR would produce a synergistic effect on EMT expression in GC cells by inhibiting RhoA/ROCK signaling.
Materials and Methods
Drugs and Reagents
Diosgenin and GSK126 (> 98% purity) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Shanghai Hanxiang Life Technology Ltd. (Shanghai, China), respectively. Both agents were dissolved in dimethyl sulfoxide and stored as 100 mM stock solutions for use in in vitro studies. RPMI-1640 medium and FBS were purchased from Hyclone (GE Healthcare, Chicago, IL, USA). A Cell Counting Kit-8 (CCK-8) Kit was purchased from Sigma-Aldrich and an Annexin V/Propidium Iodide (PI) Apoptosis detection Kit was purchased from BD Biosciences (San Jose, CA, USA). RIPA buffer, a BCA protein assay kit and an enhanced chemiluminescence kit were obtained from Beyotime Institute of Biotechnology (Shanghai, China). All antibodies were purchased from Abcam (Cambridge, UK), ProteinTech (Chicago, IL, USA), or Santa Cruz Biotechnology (Dallas, TX, USA). Transwell chambers (8 µm pore size) were obtained from Corning Inc., (Corning, NY, USA).
Cell Culture and Treatment
Human GC cell lines MGC-803, AGS, and SGC-7901, as well as the gastric epithelial cell line GES-1, were provided by The Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). All the cell lines were routinely cultured in RPMI-1640 medium supplemented with 10% FBS in a humidified chamber containing 5% CO2 at 37°C. For the in vitro studies, AGS and SGC-7901 cells were seeded into 96-well plates at a density of 3 × 103 cells/well, and then treated with diosgenin and/or GSK126 at the indicated concentrations and time points. In this study, GSK126 was used at concentration of 8 μM. While GSK429286A was used at concentration of 10 μM.
RNA Extraction and Quantitative PCR
Total cellular RNA was extracted using TRIzol reagent (Takara, Tokyo, Japan), and cDNA was synthesized using a Prime Script PTMP RT reagent kit (Perfect Real Time, Takara). Each quantitative PCR analysis was performed in triplicate on an ABI 7900HT Fast Real-Time PCR system (Applied Biosystems, Foster City, CA, USA) and using SYBR Premix Ex Taq (Takara) according to the manufacture’s protocol. The primer sequences used in this study were as follows: EZH2 forward: 5′-AATCAGAGTACATGCGACTGAGA-3′ and reverse: 5′-GCTGTATCCTTCGCTGTTTCC-3′; GAPDH forward: 5′-TGTTCGTCATGGGTGTGAAC-3′ and reverse: 5′-ATGGCATGGACTGTGGTCAT-3′. Relative levels of EZH2 expression were calculated using the 2−ΔΔCq method and normalized to those for GAPDH.
CCK-8 Assay
GC cells were seeded into triplicate wells of 96-well plates at a density of 1.0 × 104 cells per well, and then incubated with 100 µL of CCK-8 solution for 2 h at 37°C. The absorbance of each well was measured at 450 nm with a microplate reader (Norcross, GA, USA). IC50 values were calculated using Graphpad Prism 5 software.
Flow Cytometry Analysis
GC cells from different treatment groups were reseeded into 6-well plates, washed twice with ice-cold PBS, and then fixed with 70% ethanol at −20°C overnight. After being washed twice with PBS, the cells were harvested by centrifugation at 1000 rpm for 5 min. For cell cycle analysis, the harvested cells were stained in the dark for 30 min with 500 µL of PI, and subsequently analyzed with a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). For apoptosis analysis, harvested cells were stained with 5 μL of FITC Annexin V and 5 μL of PI for 15 min at room temperature, and then analyzed with a FACScan flow cytometer (BD Biosciences) equipped with FlowJo version 10.0.7 software (FlowJo, Franklin Lakes, NJ, USA).
Transwell Assays
Cell invasion and migration assays were performed using Transwell filter chambers (8-µm pore size, Corning, USA) with and without a Matrigel coating, respectively. In brief, approximately 5 × 104 cells in 200 µL of FBS-free medium were added to the upper chamber and 500 µL of medium containing 10% FBS was added to the lower chamber. After 24 h of incubation at 37°C, cells that had migrated into the lower chamber were fixed with methanol for 10 min and then stained with 0.1% crystal violet for 20 min. The stained cells in five randomly selected microscopic fields were visualized and counted under a light microscope (Olympus, Japan).
Western Blot Analysis
Cells were harvested, lysed in RIPA buffer, and then centrifuged at 12,000 rpm for 30 min. The amount of total protein in each extract was quantified with a BCA protein assay kit. Next, an equal amount of protein from each extract was separated on a 10% SDS-PAGE gel, and the separated protein bands were transferred onto PVDF membranes (Millipore Corp, Burlington, MA, USA). Next, the membranes were blocked with 5% skim milk and then incubated with primary antibodies against EZH2, H3K27m3, RhoA, ROCK, E-cadherin, N-cadherin, vimentin, fibronectin and GAPDH, followed by incubation with a horseradish peroxidase-conjugated secondary antibody. The immunostained protein bands were visualized with an enhanced chemiluminescence kit, and staining intensity was quantified using Image J software (Ver. 1.48, National Institutes of Health, USA).
Statistical Analysis
The experimental data were analyzed using SPSS Statistics for Windows, Version 17.0 (Chicago, IL, USA), and results are expressed as the mean ± standard deviation (SD) of data obtained from at least three experiments. Differences among groups were evaluated using one-way ANOVA, followed by Tukey’s post hoc test. P-values < 0.05 were considered to be statistically significant.
Results
EZH2 Expression Was Significantly Increased in the GC Cell Lines
EZH2 expression in GC cells was determined by using quantitative PCR. As shown in Figure 1A, the levels of EZH2 mRNA expression in the GC cell lines (MGC-803, AGS, and SGC-7901) were significantly higher than those in the GES-1 gastric epithelial cell line (p < 0.05, p < 0.01). Furthermore, Western blot studies confirmed those results (Figure 1B). Notably, AGS and SGC-7901 cells exhibited higher levels of EZH2 expression than MGC-803 cells; therefore, AGS and SGC-7901 cells were used in the following in vitro experiments.
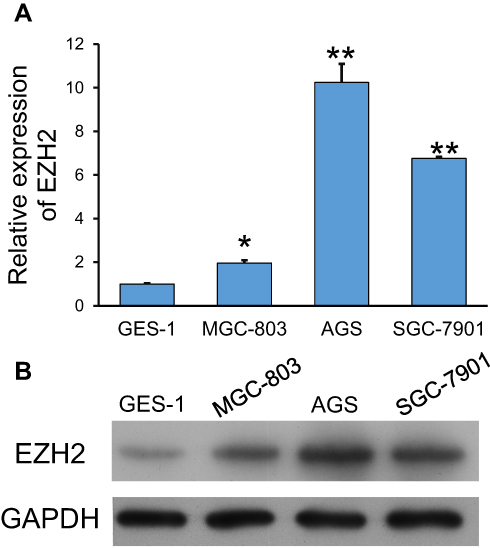 |
Figure 1 Expression of EZH2 in GC cell lines. (A, B) Quantitative PCR (A) and Western blotting (B) were used to examine EZH2 expression in GC cell lines (MGC-803, AGS, and SGC-7901) and the gastric epithelial cell line, GES-1. GAPDH served as an internal control. *p < 0.05, **p < 0.01, as compared with GES-1 cells. |
Diosgenin Treatment Decreased GC Cell Proliferation in a Concentration-Dependent Manner
To investigate the effect of diosgenin on GC cells, the effect of different doses of diosgenin on human AGS and SGC-7901 cell proliferation was determined using the CCK-8 assay. As shown in Figure 2A, the inhibitory effect of diosgenin on AGS and SGC-7901 cell proliferation gradually increased in a dose-dependent manner when compared with the proliferation of untreated control cells. The IC50 of AGS and SGC-7901 to GSK126 was 38.48±1.43 μM and 35.05±1.13 μM (Figure 2B). The IC50 values for the anti-proliferative effect of diosgenin on AGS and SGC-7901 cells were 20.02 μM and 17.40 μM, respectively (Figure 2C). Based on those results, diosgenin concentrations of 10.01 μM and 8.7 μM (50% of the IC50 values) were chosen as the optimal concentrations to be used for subsequent treatment of AGS and SGC-7901 cells, respectively.
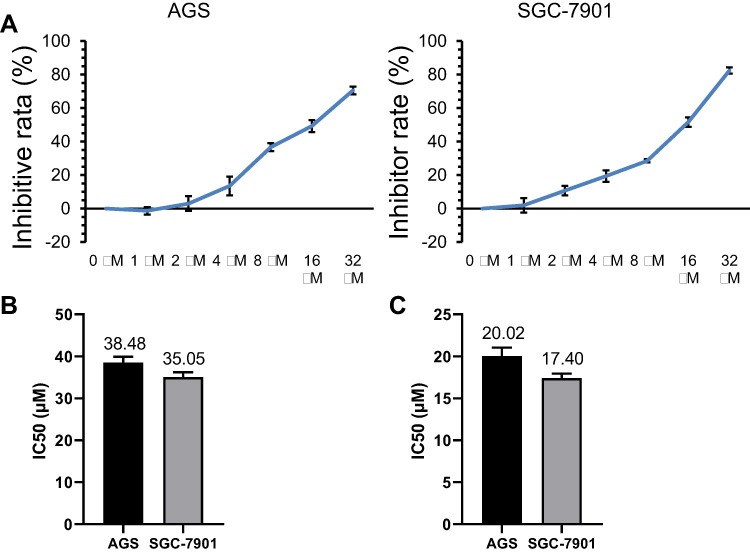 |
Figure 2 Concentration-dependent anti-proliferative effect of diosgenin on AGS and SGC-7901 cells. (A) CCK-8 assays were performed to evaluate the effect of different concentrations of diosgenin on the proliferation of AGS and SGC-7901 cells. (B, C) The IC50 values of AGS and SGC-7901 to GSK126 (B) and diosgenin (C) were calculated measured by using CCK8 kit assay , respectively. |
Combined Treatment with Diosgenin and GSK126 Synergistically Inhibited GC Cell Proliferation
Next, the proliferation of AGS and SGC-7901 cells was determined by CCK-8 assays performed at specified time points after the cells had been treated with diosgenin and an EZH2 inhibitor (8 μM GSK126) either alone or in combination. As shown in Figure 3A, either diosgenin or GSK126 significantly inhibited cell proliferation at 24, 48, and 72 h, respectively (p < 0.05, p < 0.01). Interestingly, the combination of diosgenin and GSK126 caused a further inhibition AGS cell proliferation (p < 0.01). Similar results were also found for SGC-7901 cells (Figure 3B, p < 0.05, p < 0.01). Meanwhile, we selected 48 h as the optimal cell treatment time point.
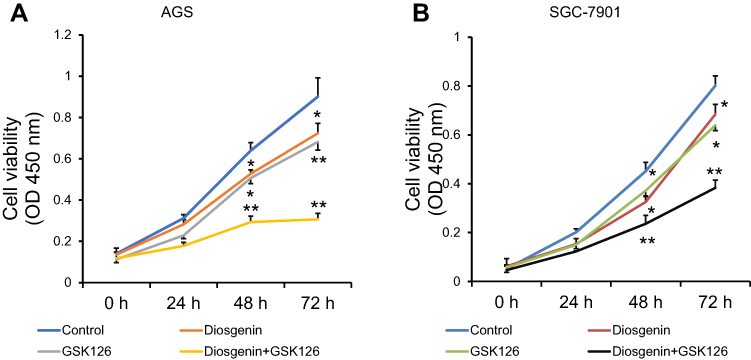 |
Figure 3 A combination of diosgenin and GSK126 produced a synergistic effect on GC cell proliferation. CCK-8 assays were performed to evaluate the proliferation of (A) AGS and (B) SGC-7901 cells after treatment with either diosgenin or GSK126 alone, or in combination. *p < 0.05, **p < 0.01, as compared with control cells. |
Combined Treatment with Diosgenin and GSK126 Synergistically Induced GC Cell Cycle Arrest and Apoptosis
It is now widely accepted that cell cycle distribution and apoptosis play important roles in cell proliferation, and especially in tumor progression. Therefore, we examined whether treatment with GSK126 and/or diosgenin would affect cell cycle progression and apoptosis in GC cells. As shown in Figure 4A, when compared with control cells, the percentage of cells in G0/G1 phase was obviously increased, while the percentage of S phase cells was obviously decreased among cells treated with either GSK126 or diosgenin alone. Furthermore, among cells that received combined treatment with GSK126 plus diosgenin, the percentage of cells in G0/G1 phase was remarkably increased and the percentage of cells in S phase was decreased when compared with control cells or cells treated with either GSK126 or diosgenin alone (AGS cells: p < 0.05, p < 0.01) (SGC-7901 cells: p < 0.05 p < 0.01, p < 0.001). In addition, flow cytometry results (Figure 4B) showed that the percentages of apoptotic AGS cells in the control, diosgenin, and GSK126 groups were 5.4% ± 1.14%, 12.43% ± 0.80%, and 16.63% ± 0.49%, respectively, whereas the percentage of apoptotic AGS cells in the diosgenin plus GSK126 treatment group was 33.03% ± 2.36%. We also found that treatment with GSK126 or diosgenin alone significantly increased the cell apoptosis rate (p < 0.05), and that increase was further enhanced by combined treatment with diosgenin plus GSK126 (p < 0.05, p < 0.01). These results showed that combined treatment with diosgenin and GSK126 synergistically induced G0/G1phase arrest and apoptosis in AGS and SGC-7901 cells.
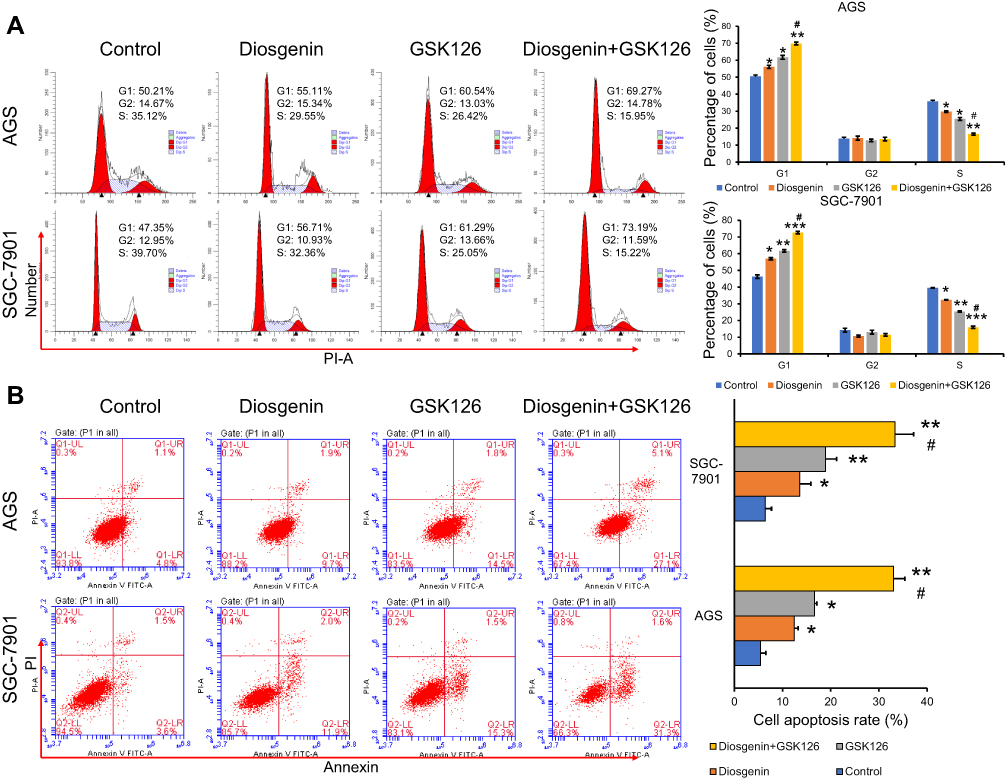 |
Figure 4 The effects of combined treatment with diosgenin and GSK126 on cell cycle progression and apoptosis in GC cells. AGS and SGC-7901 cells were treated with diosgenin or GSK126 alone, or in combination, respectively. (A) Flow cytometry with PI staining was used to detect the cell cycle distribution of AGS and SGC-7901 cells. (B) Flow cytometry with Annexin V/PI double staining was used to detect the cell cycle distribution of AGS and SGC-7901 cells. *p < 0.05, **p < 0.01, ***p < 0.001, as compared with control; #p < 0.05, as compared with diosgenin or GSK126 treatment. |
Combined Treatment with Diosgenin and GSK126 Synergistically Induced GC Cell Migration and Invasion
We next performed Transwell assays to evaluate the effects of GSK126 and/or diosgenin on cell migration and invasion. As shown in Figure 5A, treatment with either diosgenin or GSK126 alone significantly reduced the numbers of migrated and invasive AGS cells (p < 0.05, p < 0.01). Furthermore, combined treatment with diosgenin and GSK126 produced even greater reductions in the numbers of migrated and invasive AGS cells (p < 0.05, p < 0.01). Similarly, treatment with either diosgenin or GSK126 alone induced a notable decrease in the numbers of migrated and invasive SGC-7901 cells, whereas treatment with the combination of diosgenin and GSK126 induced an even further decrease in the numbers of migrated and invasive SGC-7901 cells (Figure 5B, p < 0.05, p < 0.01).
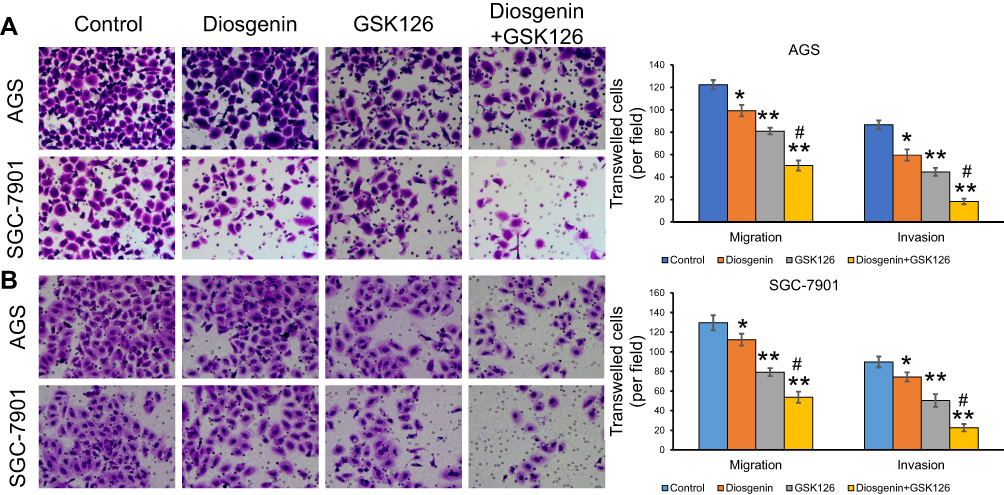 |
Figure 5 The effects of combined treatment with diosgenin and GSK126 on the migration and invasion of GC cells. AGS and SGC-7901 cells were treated with diosgenin or GSK126 alone, or in combination, respectively. Transwell assays were performed to evaluate the migration and invasion capabilities of (A) AGS and (B) SGC-7901 cells. *p < 0.05, **p < 0.01, as compared with control; #p < 0.05, as compared with diosgenin or GSK126 treatment. |
Effects of Combined Treatment with Diosgenin and GSK126 on Rho/ROCK Signaling and Epithelial–Mesenchymal Transition
To elucidate the potential mechanisms governing the suppression of cell proliferation, migration, and invasion by the combination of diosgenin and GSK126, we monitored the expression of signal molecules associated with Rho/ROCK signaling in AGS and SGC-7901 cells. We first confirmed that EZH2 expression was significantly downregulated after treatment with either diosgenin or GSK126 alone, and even further downregulated after combined treatment with diosgenin plus GSK126 (p < 0.05). We found that both RhoA and ROCK expression were slightly decreased after treatment with either diosgenin or GSK126 alone, but notably reduced after treatment with diosgenin plus GSK126 (Figure 6A). Because Rho/ROCK signaling is indispensable for cell migration and invasion (factors that might mediate epithelial–mesenchymal transition), we explored whether inhibition of Rho/ROCK signaling by the combination of diosgenin and GSK126 could affect the expression of EMT-associated molecules. As expected, treatment with either diosgenin or GSK126 alone induced an obvious increase in E-cadherin expression and decreases in N-cadherin, vimentin, and fibronectin expression. These effects were further enhanced by the combination of diosgenin and GSK126 (Figure 6B).
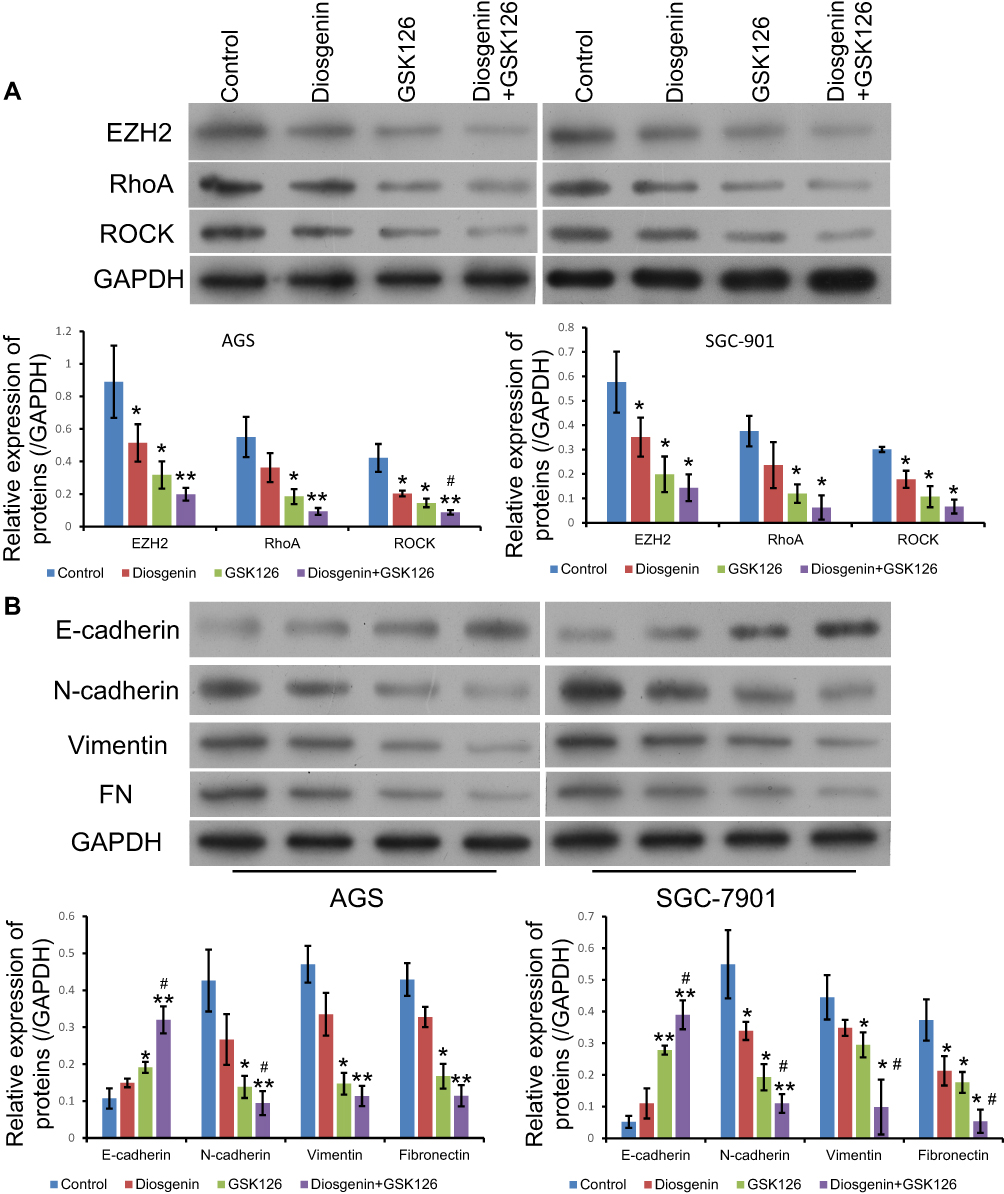 |
Figure 6 The effects of combined treatment with diosgenin and GSK126 on Rho/ROCK signaling and the epithelial–mesenchymal transition. AGS and SGC-7901 cells were treated with diosgenin or GSK126 alone, or in combination, respectively. (A) Western blot assays were performed to detect the expression of EZH2, RhoA, and ROCK in AGS cells. (B) The levels of E-cadherin, N-cadherin, vimentin, and fibronectin expression in AGS cells were measured by Western blotting *p < 0.05, **p < 0.01, as compared with control; #p < 0.05, as compared with diosgenin or GSK126 treatment. |
Diosgenin Inhibited Malignant Biological Properties Through EZH2/RhoA Pathway
To confirm the mechanism of anti-tumor effect of diosgenin in gastric cancer, an EZH2 overexpression vector and RhoA inhibitor, GSK429286A were included in the experiment. As shown in Figure 7A, Apoptosis induced by diosgenin treatment was reversed by introduction of EZH2 overexpression in AGS and SGC-7901. While a RhoA inhibitor, GSK429286A, contributed to the reversal of the inhibitive effect of EZH2 on cell apoptosis (Figure 7A). Stimulation of diosgenin lead to G1 phase arrest, together with decrease of S-phase distribution in AGS and SGC-7901 cells, and then the effect of diosgenin was reversed in response to EZH2 overexpression. However, GSK429286A blocked the effect of EZH2 overexpression by inducing G1 phase arrest and decreasing S phase distribution (Figure 7B). Western blot assay showed that EZH2 expression was elevated in cells treated with diosgenin and EZH2 overexpression plasmid. However, its expression was not affected due to GSK429286A (Figure 7E and F).
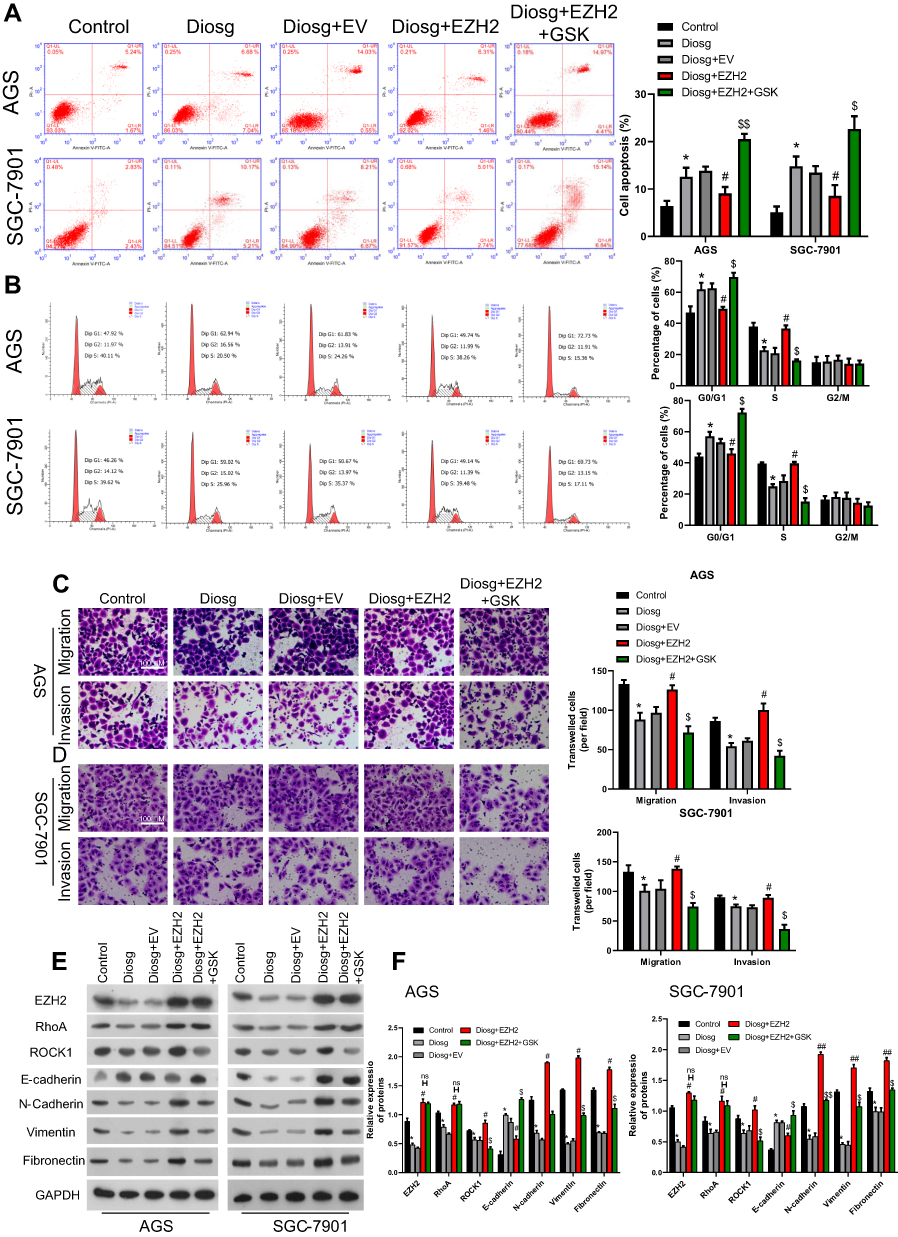 |
Figure 7 Diosgenin inhibited malignant biological properties through EZH2/RhoA pathway. (A, B) Flow cytometry with PI staining was used to detect the cell cycle distribution of AGS (A) and SGC-7901 (B). (C, D) Transwell assays were performed to evaluate the migration and invasion capabilities of (C) AGS and (D) SGC-7901 cells. (E, F) Western blot assay was used to measure EZH2, RhoA/ROCK1 and EMT related proteins. *p<0.05 vs control; #p<0.05, ##p<0.01 vs diosgenin; $p<0.05, $$p<0.01 vs diosgenin+EZH2. |
In order to confirm the effect of involving of EZH2/RhoA pathway in diosgenin treatment in AGS and SGC-7901, Transwell assay was performed to evaluate the EMT character in response to EZH2 overexpression and GSK429286A. As shown in Figure 7C and D, cell mobility in AGS and SGC-7901 was significantly inhibited by diosgenin induction, and then reversed by EZH2 overexpression. While exposure to GSK429286A blocked the promotive effect EZH2 on cell mobility (Figure 7C and D). Results of Western blot showed that N-cadherin, Vimentin, and Fibronectin expression was inhibited by diosgenin in AGS and SGC-7901, and reversed by EZH2 overexpression. Finally, enhancive effect of EZH2 was blocked by GSK429286A (Figure 7E and F).
Discussion
To the best of our knowledge, this study is the first to provide evidence that the suppressive effects of diosgenin or GSK126 on GC cell proliferation, migration, and invasion can be significantly enhanced by co-treatment with both agents. Consistent with this finding, Katona et al25 showed that combined treatment with EZH2 and an epidermal growth factor receptor (EGFR) inhibitor caused a significant decrease in the proliferation of colon cancer cells and increased their rate of apoptosis.25 Furthermore, another study showed that the apoptosis rate and amount of DNA damage in glioblastoma cells were further enhanced by co-treatment with an EZH2 inhibitor plus a histone deacetylase inhibitor.26 In addition, Yang et al27 demonstrated that inhibition of both EZH2 and EGFR produced a synergistic effect on GC cell apoptosis by increasing autophagy.27
Diosgenin, the major steroidal saponin in the fenugreek seed, has been reported to exert in vitro anti-cancer effects in a variety of cancer cells.28 For example, diosgenin inhibited proliferation and induced caspase-dependent apoptosis in skin squamous cell carcinoma cells.29 In addition, diosgenin has also demonstrated potent anti-cancer activity in colon cancer,30 breast cancer,31 and myeloid leukemia cells.32 Similarly, a study by Mao et al32 showed that diosgenin could significantly suppress BGC-823 cell invasion and survival in a hypoxic mimic microenvironment.33 GSK126, a novel EZH2 inhibitor, has been shown to affect tumor cell function in various types of cancers. GSK126 effectively reduced the levels of methylated histone 3 (H3K27me3) in MM.1S and LP1 cells,23 and inhibition of EZH2 by GSK126 inhibited cell migration and angiogenesis in solid tumors.34 Treatment of neuroblastoma cells with EZH2-specific inhibitors (GSK126 and EPZ6438) resulted in G0/G1 arrest and decreased cell survival.35 Therefore, we might speculate that combined treatment with diosgenin and GSK126 should produce a stronger suppressive effect on tumor cellular function. As expected, combined treatment with diosgenin plus GSK126 significantly enhanced the suppressive effects of diosgenin or GSK126 alone on cell proliferation, migration, and invasion. Consistent with those findings, diosgenin was previously shown to enhance the efficiency of Adriamycin in Adriamycin-resistant breast cancer cells by regulating cell survival and apoptosis.36
We also explored the molecular mechanisms by which the combination of diosgenin and GSK126 affects GC cell proliferation and invasion. ROCK, including its two subtypes (ROCK1 and ROCK2) belongs to the serine/threonine family of protein kinases, and is the downstream effector protein of RhoA.37 The RhoA/ROCK signaling pathway is known to be activated in ovarian cancer,38 hepatocarcinoma,39,40 and breast cancer.41 We found that the RhoA/ROCK signaling pathway was significantly suppressed by the combination of diosgenin and GSK126. The RhoA/ROCK signaling pathway was previously demonstrated to be associated with the EMT process.9 Therefore, we examined whether downregulation of the RhoA/ROCK signaling pathway would affect the expression of EMT-related molecules associated with cell migration and invasion. Our results revealed that a combination of diosgenin and GSK126 produced synergistic suppressive effects on GC cell migration and invasion, and also altered the expression of EMT markers, including E-cadherin, N-cadherin, vimentin, and fibronectin. In line with our findings, previous studies have shown that activation of the RhoA/ROCK pathway contributes to the EMT process and metastasis of liver40 and colon cancers.10 These results suggest that the synergic effect of the combined use of diosgenin and GSK126 on GC cell survival is partially regulated by RhoA/ROCK-mediated EMT.
In summary, our study is the first to show that a combination of diosgenin and GSK126 produces stronger suppressive effects on GC cell proliferation, migration, and invasion, when compared to the effects produced by treatment with diosgenin or GSK126 alone. Moreover, we further found these effects might result from targeting EZH2 via RhoA/ROCK-mediated EMT expression. These findings might contribute to the development of new strategies for improving the less than satisfactory therapeutic outcomes achieved with GSK126 monotherapy.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
2. Chen D, You X, Pan Y, Liu Q, Cao G. TRIM37 promotes cell invasion and metastasis by regulating SIP1-mediated epithelial-mesenchymal transition in gastric cancer. Onco Targets Ther. 2018;11:8803–8813. doi:10.2147/OTT.S178446
3. Xu W, Yang Z, Lu N. Molecular targeted therapy for the treatment of gastric cancer. J Exp Clin Cancer Res. 2016;35:1. doi:10.1186/s13046-015-0276-9
4. Jiang M, Shi L, Yang C, et al. miR-1254 inhibits cell proliferation, migration, and invasion by down-regulating Smurf1 in gastric cancer. Cell Death Dis. 2019;10(1):32. doi:10.1038/s41419-018-1262-x
5. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. doi:10.1038/nrm3758
6. Tanabe S, Aoyagi K, Yokozaki H, Sasaki H. Gene expression signatures for identifying diffuse-type gastric cancer associated with epithelial-mesenchymal transition. Int J Oncol. 2014;44(6):1955–1970. doi:10.3892/ijo.2014.2387
7. Peng F, Wu D, Gao B, et al. RhoA/Rho-kinase contribute to the pathogenesis of diabetic renal disease. Diabetes. 2008;57(6):1683–1692. doi:10.2337/db07-1149
8. Kolavennu V, Zeng L, Peng H, Wang Y, Danesh FR. Targeting of RhoA/ROCK signaling ameliorates progression of diabetic nephropathy independent of glucose control. Diabetes. 2008;57(3):714–723. doi:10.2337/db07-1241
9. Bhowmick NA, Ghiassi M, Bakin A, et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12(1):27–36. doi:10.1091/mbc.12.1.27
10. Song GL, Jin CC, Zhao W, et al. Regulation of the RhoA/ROCK/AKT/β-catenin pathway by arginine-specific ADP-ribosytransferases 1 promotes migration and epithelial-mesenchymal transition in colon carcinoma. Int J Oncol. 2016;49(2):646. doi:10.3892/ijo.2016.3539
11. Zhang H, Liu X, Liu Y, Yi B, Yu X. Epithelial–mesenchymal transition of rat peritoneal mesothelial cells via Rhoa/Rock pathway. In Vitro Cell Dev Biol Anim. 2011;47(2):165–172. doi:10.1007/s11626-010-9369-0
12. Yamada T, Hoshino M, Hayakawa T, et al. Dietary diosgenin attenuates subacute intestinal inflammation associated with indomethacin in rats. Am J Physiol. 1997;273(2 Pt 1):G355–G364. doi:10.1152/ajpgi.1997.273.2.G355
13. Son IS, Kim JH, Sohn HY, Son KH, Kim JS, Kwon CS. Antioxidative and hypolipidemic effects of diosgenin, a steroidal saponin of yam (Dioscorea spp.), on high-cholesterol fed rats. Biosci Biotechnol Biochem. 2007;71(12):3063–3071. doi:10.1271/bbb.70472
14. Hou R, Zhou QL, Wang BX, Tashiro S, Onodera S, Ikejima T. Diosgenin induces apoptosis in HeLa cells via activation of caspase pathway. Acta Pharmacol Sin. 2004;25(8):1077–1082.
15. Ying W, Chi-Ming C, Jen-Fu C, Qing-Yu H. Dioscin (saponin)-induced generation of reactive oxygen species through mitochondria dysfunction: a proteomic-based study. J Proteome Res. 2007;6(12):4703–4710. doi:10.1021/pr070399r
16. Lv L, Zheng L, Dong D, et al. Dioscin, a natural steroid saponin, induces apoptosis and DNA damage through reactive oxygen species: a potential new drug for treatment of glioblastoma multiforme. Food Chem Toxicol. 2013;59:657–669. doi:10.1016/j.fct.2013.07.012
17. Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128(4):735–745. doi:10.1016/j.cell.2007.02.009
18. Asangani I, Ateeq B, Qi C, et al. Characterization of the EZH2-MMSET histone methyltransferase regulatory axis in cancer. Mol Cell. 2013;49(1):80–93. doi:10.1016/j.molcel.2012.10.008
19. Yutaka K. Targeting histone methyltransferase EZH2 as cancer treatment. J Biochem. 2014;156(5):249–257. doi:10.1093/jb/mvu054
20. Knutson SK, Warholic NM, Wigle TJ, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110(19):7922–7927. doi:10.1073/pnas.1303800110
21. Mccabe MT, Ott HM, Gopinath G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. doi:10.1038/nature11606
22. Khan M, Walters LL, Li Q, et al. Characterization and pharmacologic targeting of EZH2, a fetal retinal protein and epigenetic regulator, in human retinoblastoma. Lab Invest. 2015;95(11):1278–1290. doi:10.1038/labinvest.2015.104
23. Zeng D, Liu M, Pan J. Blocking EZH2 methylation transferase activity by GSK126 decreases stem cell-like myeloma cells. Oncotarget. 2017;8(2):3396–3411. doi:10.18632/oncotarget.13773
24. Koppens MA, Bounova G, Cornelissen-Steijger P, et al. Large variety in a panel of human colon cancer organoids in response to EZH2 inhibition. Oncotarget. 2016;7(43):69816–69828. doi:10.18632/oncotarget.12002
25. Katona BW, Yuanyuan L, Anqi M, Jian J, Xianxin H. EZH2 inhibition enhances the efficacy of an EGFR inhibitor in suppressing colon cancer cells. Cancer Biol Ther. 2014;15(12):1677–1687. doi:10.4161/15384047.2014.972776
26. Grinshtein N, Rioseco CC, Marcellus R, et al. Small molecule epigenetic screen identifies novel EZH2 and HDAC inhibitors that target glioblastoma brain tumor-initiating cells. Oncotarget. 2016;7(37):59360–59376. doi:10.18632/oncotarget.10661
27. Yang Y, Zhu F, Wang Q, Ding Y, Ying R, Zeng L. Inhibition of EZH2 and EGFR produces a synergistic effect on cell apoptosis by increasing autophagy in gastric cancer cells. Onco Targets Ther. 2018;11:8455–8463. doi:10.2147/OTT.S186498
28. Shishodia S, Aggarwal BB. Diosgenin inhibits osteoclastogenesis, invasion, and proliferation through the downregulation of Akt, I kappa B kinase activation and NF-kappa B-regulated gene expression. Oncogene. 2006;25(10):1463. doi:10.1038/sj.onc.1209194
29. Das S, Dey KK, Dey G, et al. Antineoplastic and apoptotic potential of traditional medicines thymoquinone and diosgenin in squamous cell carcinoma. PLoS One. 2012;7(10):e46641. doi:10.1371/journal.pone.0046641
30. Lepage C, Léger DY, Bertrand J, Martin F, Beneytout JL, Liagre B. Diosgenin induces death receptor-5 through activation of p38 pathway and promotes TRAIL-induced apoptosis in colon cancer cells. Cancer Lett. 2011;301(2):193–202. doi:10.1016/j.canlet.2010.12.003
31. He Z, Chen H, Li G, et al. Diosgenin inhibits the migration of human breast cancer MDA-MB-231 cells by suppressing Vav2 activity. Phytomedicine. 2014;21(6):871–876. doi:10.1016/j.phymed.2014.02.002
32. Jiang S, Fan J, Wang Q, et al. Diosgenin induces ROS-dependent autophagy and cytotoxicity via mTOR signaling pathway in chronic myeloid leukemia cells. Phytomedicine. 2016;23(3):243–252. doi:10.1016/j.phymed.2016.01.010
33. Mao ZJ, Tang QJ, Zhang CA, et al. Anti-proliferation and anti-invasion effects of diosgenin on gastric cancer BGC-823 cells with HIF-1alpha shRNAs. Int J Mol Sci. 2012;13(5):6521–6533. doi:10.3390/ijms13056521
34. Chen YT, Zhu F, Lin WR, Ying RB, Yang YP, Zeng LH. The novel EZH2 inhibitor, GSK126, suppresses cell migration and angiogenesis via down-regulating VEGF-A. Cancer Chemother Pharmacol. 2016;77(4):757–765. doi:10.1007/s00280-016-2990-1
35. Bate-Eya LT, Gierman HJ, Ebus ME, et al. Enhancer of zeste homologue 2 plays an important role in neuroblastoma cell survival independent of its histone methyltransferase activity. Eur J Cancer. 2017;75:63–72. doi:10.1016/j.ejca.2016.12.019
36. Wang C, Huo X, Wang L, et al. Dioscin strengthens the efficiency of adriamycin in MCF-7 and MCF-7/ADR cells through autophagy induction: more than just down-regulation of MDR1. Sci Rep. 2016;6:28403. doi:10.1038/srep28403
37. Yang L, Tang L, Dai F, et al. Raf-1/CK2 and RhoA/ROCK signaling promote TNF-alpha-mediated endothelial apoptosis via regulating vimentin cytoskeleton. Toxicology. 2017;389:74–84. doi:10.1016/j.tox.2017.07.010
38. Wang W, Du H, Liu H, Hu F, Liu G. SMAD specific E3 ubiquitin protein ligase 1 promotes ovarian cancer cell migration and invasion via the activation of the RhoA/ROCK signaling pathway. Oncol Rep. 2019;41(1):668–676. doi:10.3892/or.2018.6836
39. An H, Lin J, Sun H, et al. Biejiajian pills inhibits hepatoma carcinoma cell vasculogenic mimicry by suppressing RhoA/ROCK signaling pathway. Nan Fang Yi Ke Da Xue Xue Bao. 2018;38(8):997–1001. doi:10.3969/j.issn.1673-4254.2018.08.16
40. Chen X, Zhang S, Wang Z, et al. Supervillin promotes epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma in hypoxia via activation of the RhoA/ROCK-ERK/p38 pathway. J Exp Clin Cancer Res. 2018;37(1):128. doi:10.1186/s13046-018-0787-2
41. Li D, Wang H, Ding Y, et al. Targeting the NRF-2/RHOA/ROCK signaling pathway with a novel aziridonin, YD0514, to suppress breast cancer progression and lung metastasis. Cancer Lett. 2018;424:97–108. doi:10.1016/j.canlet.2018.03.029
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.