")
Back to Journals » OncoTargets and Therapy » Volume 14
Dihydroartemisinin as a Sensitizing Agent in Cancer Therapies
Authors Li Q , Ma Q, Cheng J, Zhou X, Pu W, Zhong X , Guo X
Received 19 December 2020
Accepted for publication 18 March 2021
Published 13 April 2021 Volume 2021:14 Pages 2563—2573
DOI https://doi.org/10.2147/OTT.S297785
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Qingrong Li,1– 3,* Qiang Ma,1– 3,* Jibing Cheng,1– 3 Xi Zhou,1– 3 Wenjie Pu,1– 3 Xiaowu Zhong,1– 3 Xiaolan Guo1– 3
1Department of Clinical Laboratory, Affiliated Hospital of North Sichuan Medical College, Nanchong, 637000, People’s Republic of China; 2Translational Medicine Research Center, North Sichuan Medical College, Nanchong, 637000, People’s Republic of China; 3Department of Laboratory Medicine, North Sichuan Medical College, Nanchong, 637000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaolan Guo
Department of Clinical Laboratory, Affiliated Hospital of North Sichuan Medical College Tel +86-817-2262302
Fax +86-817-2282052
Email [email protected]
Abstract: Cancer is one of the major threats to human health. Although humans have struggled with cancer for decades, the efficacy of treatments for most tumors is still very limited. Dihydroartemisinin (DHA) is a derivative of artemisinin, a first-line antimalarial drug originally developed in China. Beyond the anti-malarial effect, DHA has also been reported to show anti-inflammatory, anti-parasitosis, and immune-modulating properties in vitro and in vivo. Furthermore, an increasing number of studies report that DHA possesses anticancer activities on a wide range of cancer types both in vitro and in vivo, as well as enhances the efficacy of chemotherapy, targeted therapy, and even radiotherapy. However, the mechanisms of DHA on different tumors differ in various ways. In this review, we intend to summarize how DHA sensitizes cancer cells to anti-cancer therapies, highlight its molecular mechanisms and pharmacological effects in vitro and in vivo as well as in current clinical trials, and discuss potential issues concerning DHA. Hopefully, more attention will be paid to DHA as a sensitizer for cancer therapy in the future.
Keywords: dihydroartemisinin, anti-tumor drugs, sensitizer, molecular mechanism
Introduction
Cancer is one of the leading causes of death worldwide, with an estimated 18.1 million new cases and 9.6 million deaths globally in 2018.1 Artemisinin is a colorless needle-like crystal extracted from the stems and leaves of the composite inflorescence plant Artemisia annua, which is a sesquiterpene lactone compound containing a peroxy group. The peroxy group in its molecular structure has alternating O-C-O-C segments that can be fractured for chemical synthesis of artemisinin derivatives. Dihydroartemisinin (DHA) is an artificial semi-synthetic derivative obtained after the reduction of artemisinin. DHA retains the antimalarial active group and contains a hydroxyl group, which greatly enhances its antimalarial effects.2 DHA has many advantages compared to artemisinin, such as higher water solubility, higher efficacy, easier absorption, wider distribution, quicker excretion and metabolism, higher efficiency, and lower toxicity. The oral bioavailability of DHA is 10 times more than that of artemisinin, and its antimalarial effect is 4–8 times greater compared with artemisinin.3 In the presence of ferrous ions, the peroxide bridge in DHA breaks and results in the production of cytotoxic reactive oxygen species (ROS), which is considered to be one of the mechanisms behind the anti-tumor and antimalarial effects of DHA.4 Data shows that DHA can kill tumor cells by inducing cell cycle arrest or apoptosis,5,6 preventing tumor angiogenesis,7 inhibiting tumor invasion and metastasis.8 Moreover, no obvious toxicity has been found in normal cells treated with DHA, which reveals that DHA is a potential ideal anti-tumor drug for cancer therapy. Recent studies showed that DHA revealed anti-cancer activities in various tumors and was used as a sensitizer for some cancer therapies as well.
In this review, we summarized studies on the synergistic effect of DHA in tumor therapies to explain that DHA has the potential value to improve the therapeutic effect of clinical anti-cancer drugs, including chemotherapy (Figure 1), targeting therapy, and radiotherapy (Figure 2).
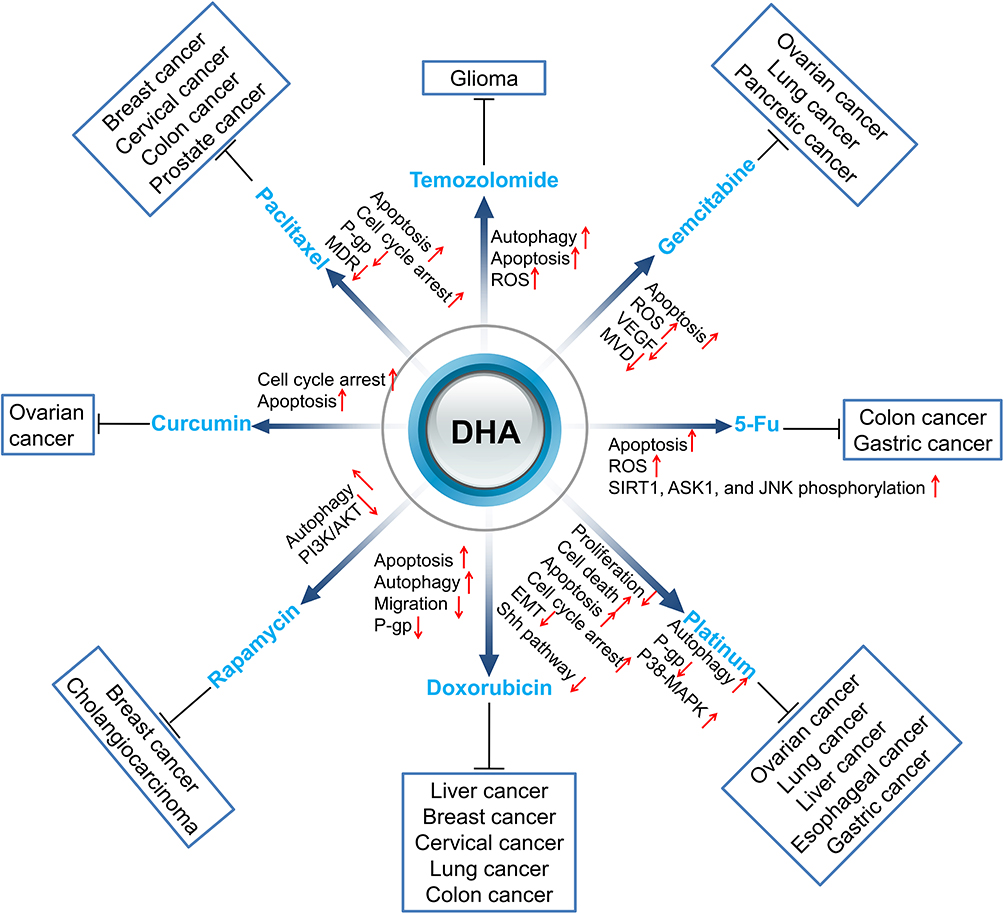 |
Figure 1 Sensitization of DHA to chemotherapy drugs. Red arrows ↑ and ↓ indicate proteins or pathways up-regulated and down-regulated by DHA to elevate the toxicity of other chemotherapy drugs to cancer cells. |
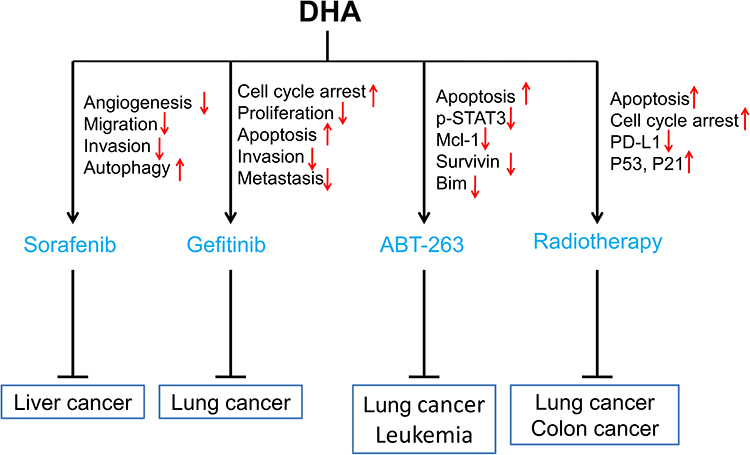 |
Figure 2 Sensitization of DHA to targeted drugs and radiotherapy. Red arrows ↑ and ↓ indicate proteins or pathways up-regulated and down-regulated by DHA to elevate the toxicity of targeting drugs or radiotherapy to cancer cells. |
In vitro Evidence of DHA Positive Effects on Chemosensitization
DHA Enhances the Anti-Cancer Activity of Alkylating Agents
Alkylating agents, also known as biological alkylating agents, are a class of cell cycle non-specific drugs. The most used alkylating agents in cancer patients are temozolomide, nitrogen mustard, etc. The alkyl group in the molecular structure is the common feature of this kind of drug. Alkyl groups can be alkylated with nucleophilic groups in DNA, RNA, or proteins in cancer cells, which can form cross-links or cause a depurination effect and finally induce DNA damage. In the next cell cycle, the miscoding of the base pair results in DNA structural and functional damage and eventually induces cell death in severe cases.
Temozolomide (TMZ) is an imidazolotetrazine alkylating agent with antitumor activity. In the physiological condition (PH=7.4), TMZ spontaneously converts to 5- (3-Methyltriazen-1yl) imidazole-4-carboxamide (MTIC),9 which exerts cytotoxic effects mainly through alkylation/methylation at the O6 and N2 sites of DNA guanine. Studies show that TMZ induces apoptosis of gliomas by up-regulating caspase-3 and down-regulating AKT and HSP90 on protein levels.10,11 TMZ also induces AMPK activation, which is involved in TMZ-induced glioblastoma cell apoptosis by promoting p53 activation, inhibiting mTORC1 signaling, altering the expression levels of apoptosis-related proteins such as p21, Noxa, Bax, and Bcl-2.12 However, TMZ is prone to drug resistance, and the current study demonstrated that TMZ resistance was the result of a combination of factors, such as DNA damage repair systems like O6-methylguanine DNA methyltransferase, mismatch repair, and base excision repair as well as autophagy or cancer stem cells.13 DHA can induce autophagy and apoptosis in cancer cells, and a synergistic effect was seen when DHA was combined with TMZ for cancer therapies. DHA combined with TMZ inhibited U87MG cell proliferation and induced apoptosis.14 The mechanism was mainly down-regulating the phosphorylated mitogen extracellular kinase (MEK) and extracellular signal-regulated protein kinase (ERK) proteins, and up-regulating the expression of P53 and down-regulating the expression of Bcl-2 and Mcl-1, which finally initiated mitochondrial apoptosis in cancer cells. DHA enhanced the toxic response of cells to TMZ and inhibited the proliferation of glioma cells through autophagy.15 In addition, DHA has also been found to enhance the cytotoxic effect of temozolomide (TMZ) on rat C6 glioma cells by ROS generation. When the concentration of DHA was 5 µmol/L, the cytotoxic effect of TMZ was increased by 321% and significantly enhanced TMZ-induced apoptosis and necrosis.16 Additionally, TMZ alone reduced tumor size by about 55%, while the combination of DHA with TMZ resulted in a tumor size of less than 20% compared to untreated animals.17 In summary, DHA enhances the cytotoxic effect of TMZ by inducing autophagy and ROS, subsequently enhancing the anti-proliferative effect of TMZ on cancer cells in vitro and in vivo. Moreover, DHA could also enhance the anti-tumor effect of TMZ by inducing cancer cell apoptosis.
DHA Improves the Activities of Antimetabolite Anti-Cancer Drugs
Currently, the widely used antimetabolite chemotherapeutic drugs include gemcitabine, 5-fluorouracil (5-FU), cytarabine, and capecitabine. These drugs work by interfering with the essential biosynthetic pathways, perturbing the synthesis of DNA/RNA, or forming deoxyribonucleic acid reductase and deoxyribonucleic acid polymerase, as well as incorporating pseudo structural analogues of pyrimidine/purine into DNA.18
Gemcitabine (GEM) is a novel cytosine nucleoside derivative, activated by deoxycytidine kinase after entering the body and further metabolized by cytosine nucleoside deaminase, which is finally incorporated into DNA and arrests the cell cycle at the G1/S phase. Gemcitabine resistance may be related to epithelial-mesenchymal transition (EMT), DNA damage and repair, abnormal signaling pathways, and the related coding genes, etc.19 Deoxycytidine kinase (dCK) is a key protein that transforms GEM into an effective active form and exerts its efficacy. The mutation in dCK is an important mechanism for cancer cells to acquire drug resistance towards GEM. The mutation E197K in dCK is a key event of GEM resistance, resulting in DNA damage in cancer cells.20 The combination of DHA and GEM has a synergistic anti-ovarian cancer effect.21 ROS generated from DHA inhibits the expression of cellular cytidine deaminase (CDA), reduces GEM inactivation, and finally enhances the anticancer activity of GEM. Zhao and colleagues proved that DHA enhanced the anti-cancer activity of GEM to A549 cells by producing ROS.22 Notably, they demonstrated that, although both GEM and DHA increased ROS production, respectively, the combination of the two drugs did not enhance the ROS level, while the synergistic induction of apoptosis was obvious through the Bak-mediated intrinsic apoptotic pathway and the FAS-caspase-8-mediated extrinsic apoptotic pathway. In addition, they discovered that DHA had a more potent pro-apoptotic effect in gemcitabine resistant A549 cells (A549GR), in which Bax and caspases were involved.23 Wang and colleagues demonstrated that DHA increased the anti-cancer activity of GEM both in vitro and in vivo.24 In pancreatic cancer cells, BxPC-3 and PANC-1, DHA enhanced GEM-induced growth inhibition and apoptosis by blocking GEM-induced NF-κB activation, reducing c-myc, cyclin D1, Bcl-2, and Bcl-xL expression in vitro. In in vivo pancreatic cancer xenograft experiments, the tumor volume was significantly reduced and apoptosis was significantly increased in the combination group.25 Some researchers revealed that the combination of DHA and GEM could significantly reduce the expression of VEGF and MVD and decrease the tumor microvessel density, inhibit tumor neovascularization and proliferation.26 These studies revealed that DHA could elevate the chemotherapeutic effect of GEM mainly by increasing ROS production to inhibit chemoresistance and induce apoptosis. Moreover, DHA might also inhibit tumor neovascularization to enhance the antitumor effect of GEM.
5-FU has no biological activities until it has been converted into 5-fluorouracil deoxynucleotide in vivo, which then combines with thymine synthase (TS) and formyltetrahydrofolate to form a stable triple complex and finally inhibits the activity of TS. So far, inhibition of TS by the active metabolites of 5-FU as the main mechanism for its anti-tumor effects has been well understood.27 Recent studies found that DHA could act synergistically with 5-FU in inhibiting the growth of cancer cells. DHA synergistically inhibited the growth of gastric cancer cells by enhancing the pro-apoptotic effect of 5-FU, which sensitized gastric cancer cells to the 5-FU action by inhibiting Bcl-XL expression and ATF2 phosphorylation and elevating the release of cytochrome C activation of caspase-3.28 Moreover, a combination of DHA and 5-FU significantly promoted gastric cancer cell, SGC7901, apoptosis through inhibiting the expression of SIRT1 and promoting ROS generation as well as phosphorylation of ASK1 and JNK.29 Furthermore, Yao et al showed that DHA effectively reversed the anticancer effect of 5-FU on p53 gene knockout colorectal cancer HCT116 cells (HCT116, TP53-/-) through ROS-mediated apoptosis in DHA treated HCT116 cells.30 The molecular mechanisms might be associated with the up-regulation of Bcl-2 and the down-regulation of Bax. Overall, DHA exerts synergistic anti-tumor effects by inducing apoptosis to enhance the sensitivity of tumor cells to 5-FU.
DHA Promotes the Anti-Cancer Activities of Antibiotic Chemotherapeutic Drugs
An antibiotic chemotherapeutic drug is a kind of antibiotic with a killing effect on tumor cells; it belongs to non-specific cell cycle drugs. Antibiotic chemotherapeutic drugs include two categories according to their different mechanisms. One type is antibiotics, which directly cross-bind to DNA, disrupt its structure and function, and finally prevent its replication, such as mitomycin and bleomycin. The other drug intercalates into the DNA double strand to interfere with its duplication and inhibit RNA synthesis, such as doxorubicin, daunorubicin, and actinomycin D.
Doxorubicin (DOX) is an anti-tumor antibiotic that inhibits the synthesis of RNA and DNA, which has a stronger inhibitory effect on RNA. DOX has a widely anti-tumor spectrum and possesses a killing effect on tumor cells in various cell cycles; it is a cell cycle non-specific drug. P-glycoprotein (P-gp) is a transmembrane glycoprotein with a molecular weight of 170kD. P-gp binds to both drugs and ATP, so that intracellular drugs can be pumped out of the cells, and finally reduces the intracellular drug concentration and develops drug resistance. The higher expression of P-gp on the endothelial cell surface could reduce the penetration of chemotherapy drugs to specific sites. DHA could restrain the expression of P-gp by inhibiting the p53 (R248Q)-ERK1/2-NF-κB signaling pathway, so that liver cancer cells embedding p53 (R248Q) are sensitized to doxorubicin,31 and reverse chemotherapy resistance. DHA elevated the sensitivity of human colon cancer resistant cells HCT8/ADR to DOX trough down-regulating the expression of Bcl-xl and inducing autophagy.32 Another study showed that DHA inhibited the activity of Bcl-2 by combining epirubicin, promoted the release of Beclin 1 and activated Bax to induce apoptosis, which finally led to type I programmed cell death of breast cancer cells. In addition, Beclin 1 initiated excessive autophagy and led to type II apoptosis of breast cancer cells.33 Tai et al showed that the combination of DOX and DHA showed a synergistic effect on HeLa cells through the endogenous apoptosis pathway mediated by caspase-9 and caspase-3.34 Furthermore, in vivo experiments showed that the volume of the tumors significantly reduced after intratumoral injection of DHA combined with DOX, and no obvious toxicity was observed in the liver, spleen, kidney or heart of animals. Another study reported that arginine 8 (R8) modified epirubicin and DHA liposomes, significantly inhibited lung cancer migration by down-regulating the expression of VE-Cad, TGF-β1, MMP-2, and HIF-1a, elevated the concentration of chemotherapy drugs in the selective accumulation of tumor sites, and enhanced the targeted therapy effect.35 Taken together, DHA enhances the anti-tumor effect of DOX on tumor cells by down-regulating P-gp expression and/or promoting apoptosis, inducing autophagy, and inhibiting tumor migration.
Rapamycin (RAPA) is a specific inhibitor of the mTORC1 complex. RAPA indirectly promotes the occurrence of autophagy by inhibiting the mTORC1 complex.36 Studies showed that RAPA significantly increased the expression of caspase-3 in tumor tissues, which promoted the apoptosis of tumor cells.37 RAPA resistance may be related to the negative feedback activation of phosphatidylinositol 3-kinase-protein kinase B (PI3K-AKT) after mTOR suppression.38 A previous study showed that DHA effectively inhibited the expression of PI3K and p-Akt.39 A combination of DHA with RAPA resulted in dual inhibition of the PI3K/AKT signaling pathway, which further prevented the reactivation of PI3K. Thongchot et al showed that DHA induced the expression of autophagy-related genes ATG12, BNIP3, and ULK1 in cholangiocarcinoma and reduced the expression of mTOR that inhibits autophagy.40 Liu et al demonstrated that DHA and RAPA synergistically promote the apoptosis and death of breast cancer MDA-MB-231 cells by up-regulating DAPK and ATG7.41 Collectively, the synergistic anti-tumor effect of DHA combined with RAPA probably works through the PI3K/AKT/mTOR pathway inhibition and apoptosis promotion.
DHA Elevates the Anti-Cancer Activities of Plant-Derived Chemotherapeutic Drugs
Plant-derived chemotherapeutic drugs are composed of alkaloids and natural products, which could inhibit mitosis or enzymes of tumor cells, preventing the synthesis of proteins that are necessary for cell regeneration. The clinical first-line used plant-derived chemotherapeutic drugs are paclitaxel, curcumin, and vincristine.
Paclitaxel (PTX) is an anti-cancer agent derived from the North American Pacific yew tree (Taxus brevifolia) that specifically acts on M-phase cells and belongs to the cell cycle-specific anti-cancer drugs. PTX could promote the polymerization of tubules into stable microtubules, inhibit their depolymerization, damage the network structure of microtubules, and inhibit mitosis of cells. PTX achieved its anti-tumor effect on cervical cancer by inhibiting cancer cell mitosis and affecting the activity of mTOR signaling pathway.42 Huang et al showed that the inhibition rate of breast cancer cell line MCF-7 was significantly higher in cells treated with DHA combined with PTX compared with that treated with DHA or PTX alone.43 Moreover, DHA combined with docetaxel showed a synergistic effect on prostate cancer cells, which prevented or delayed docetaxel resistance to prostate cancer cells.44 To alleviate the side effects of high-dose PTX in patients, researchers synthesized PEGylated PTX and DHA nanoparticles to evaluate its anti-cancer effect in colorectal cancer cells in vitro and in vivo, and the results showed that DHA-PEG-PTX nanosystems (PD@PPD) showed remarkably increased apoptosis in HT-29 cells, as compared to free drug treatment. More importantly, DHA minimized the side effects of PTX and enhanced anti-tumor efficacy.45
Curcumin (CUR) is a yellow pigment extracted from the rhizomes of turmeric and other ginger plants; it is an acidic polyphenol with unsaturated aliphatic and aromatic groups, which shows anti-inflammatory and anti-oxidation pharmacological effects. Studies revealed that curcumin has an anticancer effect by inhibiting cell proliferation, promoting apoptosis, preventing tumor angiogenesis and metastasis, and inducing autophagy.46–50 Zhao et al demonstrated that the combination of DHA and curcumin synergistically up-regulated the expression of miR-124, subsequently reduced the expression of midkine(MK) and increased cytotoxicity, and finally reduced SKOV3 ovarian cancer cell survival by inducing cell cycle arrest and promoting apoptosis.51
DHA Enhances the Anti-Cancer Efficacy of Platinum-Based Chemotherapeutic Drugs
Platinum-based regimens play a vital role in cancer therapy. Platinum inhibits the division of cancer cells by inducing DNA replication disorders. Five kinds of platinum anti-tumor drugs are widely used in cancer patients, including cisplatin (the first generation), carboplatin (the second generation), and oxaliplatin and loplatin (the third generation). One study showed that most cell lines resistant to cisplatin exhibit a phenotype of reduced platinum accumulation, most likely due to reduced cisplatin uptake rather than increased drug efflux.52 Bcl-2 might also be associated with platinum chemoresistance in lung cancer.53 Therefore, elevation of platinum concentration in tumor cells and autophagy induction might enhance the chemotherapeutic effect of platinum. A previous study showed that DHA increased the sensitivity of cisplatin-resistant gastric cancer cell line SGC7901/DDP cells to cisplatin by inducing apoptosis, which was accompanied by P-gp down-regulation.54 Feng et al showed that DHA significantly enhanced the cytotoxic effect of cisplatin-resistant ovarian cancer cells SKOV3/DDP to cisplatin by inducing apoptosis and autophagy.55 Moreover, another study in esophageal squamous cell carcinoma (ESCC) showed that continuous cisplatin treatment activated Shh signaling and induced cancer stem-like properties in ESCC patients, which subsequently resulted in reduction of cisplatin-induced cytotoxicity in KYSE510 cells. DHA could suppress activation of the Shh pathway and attenuate the cancer stem-like traits in ESCC cells and, finally, enhance the sensitivity of cisplatin on ESCC.56 Qin et al also revealed that DHA could enhance the chemo-sensitivity of HCC cells to cisplatin (DDP) and oxaliplatin (OXA).57 The mechanism behind this enhancement was that DHA altered the morphology of HCC cells induced by DDP and OXA and the expression of EMT biomarkers in HCC cells through AKT-Snail signaling pathway, which improved the efficacy of chemotherapy in HCC patients. In addition, Zhang et al reported that DHA combined with carboplatin (CBP) had a stronger inhibitory effect on Lewis lung cancer (LLC) cells.58 DHA sensitized LLC cells to CBP treatment and induced cell cycle arrest by activating the p38-MAPK signal pathway. In short, DHA could reduce platinum resistance in tumor cells by elevating the concentration of platinum in tumor cells and inhibiting the Shh signaling pathway, while enhancing platinum chemosensitivity by cell cycle arrest and apoptosis induction.
DHA Enhances the Anti-Cancer Activities of Targeted Therapies
Targeted therapy is a treatment aimed at identified carcinogenic sites at the cellular and molecular level. The corresponding therapeutic drugs enter the patient, specifically gather at the carcinogenic sites and specifically kill the tumor cells without damaging normal cells. As well-known “biological missiles”, the targeted chemotherapeutic drugs include sorafenib, gemfibrozil, and ABT-263, etc.
Sorafenib (SRF) is an oral multi-kinase inhibitor, which not only inhibits the proliferation of tumor cells by targeting Raf kinase in the Raf/MEK/ERK signaling pathway, but also targets VEGFR2/3 and platelet derivation growth factor receptor β tyrosine kinase to both exert an anti-angiogenic effect and induce apoptosis in liver cancer cells.59 Overexpression of EGFR or ligands in HCC cells leads to sustained activation of EGFR downstream signaling and is related to SRF resistance.60 Zhang et al elucidated that down-regulation of p-ERK may be associated with SRF resistance in liver cancer.61 Shimizu and colleagues found that SRF treatment resulted in autophagosomes accumulation and the activation of autophagic flux in Huh7, HLF, and PLC/PRF/5 cells, which promoted the survival of hepatoma cells by decreasing the efficiency of SRF.62 Therefore, the combination of drugs might reduce the resistance of tumor cells to sofiranib. Hou and colleagues demonstrated that the combination of DHA and SRF could significantly up-regulate APOA1 and MYH11 and down-regulate the expression of GALNT10 to inhibit tumor angiogenesis, migration, and invasion in liver cancer.63 Wang et al also proved that the combination of SRF and DHA had a synergistic anti-cancer effect on HepG2 cells, and low-density lipoprotein-based SRF/DHA lipid nanoparticles (LD-SDN) were more effective in reducing cell survival and improving tumor-targeting effects compared with single administration. The anti-tumor response and delayed tumor growth phenomenon were more significant in LD-SDN-treated xenograft models as well.64 Together, DHA could promote the anti-tumor effect of sorafenib mainly through inhibiting the invasion and migration, promoting the apoptosis, and reducing the resistance of tumor cells to sorafenib.
Gefitinib is a selective epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor that blocks downstream signaling pathways by competing for the ATP binding site of the EGFR tyrosine kinase catalytic region, which inhibits the growth, metastasis and angiogenesis of tumor cells. Data showed that resistance to EGFR-TKIs was associated with impaired EGFR-TKI-mediated apoptosis.65,66 Deng et al found that COX-2 mediated gefitinib resistance by interacting with EGFR and subsequently activating PI3K-AKT signaling in non-small cell lung cancer (NSCLC).67 Jin et al demonstrated that DHA combined with gefitinib induced NCI-H1975 cell cycle arrest at the G2/M phase and apoptosis, and inhibited cell migration and invasion by down-regulation of p-Akt, p-mTOR, p-STAT3, and Bcl-2 and up-regulation of Bax.68 Song et al found that the combination of gemfibrozil and DHA significantly inhibited the proliferation and migration of lung adenocarcinoma cell A549, and arrested the cell cycle at the G0/G1 phase.69 The molecular mechanisms might be associated with the down-regulation of CDK4, cyclin D1, MMP2, and MMP9. Taken together, DHA might increase the anti-tumor effect of gefitinib by suppressing the PI3K/AKT/mTOR pathway and abolishing gemfibrozil resistance by inducing cell cycle arrest, apoptosis, migration, and invasion inhibition.
ABT-263 (Navitoclax) is a targeted inhibitor against the Bcl-2 family of anti-apoptotic protein members such as Bcl-2, Bcl-xL, and Bcl-w. Many studies have shown that a high concentration of ABT-263 alone can induce tumor cell apoptosis in vivo and in vitro. Researchers found that activation of the endoplasmic reticulum stress response pathway can mediate resistance of uveal melanoma cells to ABT-263.70 Another study showed that the interstitial phenotype of clear cell carcinoma of the ovary (OCCC) cells were resistant to ABT-263, and that pro-apoptotic protein BIM expression was insufficient.71 Mechanistically, EMT-induced transcription factor ZEB1 down-regulated the transcription of BIM by binding to the BIM promoter, and further developed the resistance of OCCC cells to ABT-263. Recently, some studies reported that DHA had a synergistic effect with ABT-263 and induced Bax-dependent apoptosis in NSCLC cells. DHA inhibited the activity of STAT3, down-regulated the expression of Mcl-1and survivin, up-regulated the expression of Bim, and finally worked as a sensitizer of ABT-263 to induce apoptosis of NSCLC cells with EGFR or RAS mutations.72 In leukemia, researchers demonstrated that ABT-263 combined with DHA efficiently improved the treatment of BCR-ABL+B-ALL. DHA inhibited cellular stress response activation, resulting in the down-regulation of Mcl-1, and increased the sensitivity of leukemia cells to ABT-263.73 Hence, DHA might promote ABT-263-induced apoptosis by reversing the resistance of tumor cells to ABT-263 through inhibition of STAT3 and up-regulation of Bim.
DHA Enhances the Efficacy of Radiotherapy
Radiotherapy is a commonly used local and regional treatment for cancer. Studies have shown that the sensitivity of tumor cells to radiotherapy is tightly related to cell cycle status, whereby cells at the G2/M phase are the most sensitive to radiotherapy, followed by cells at the G0/G1 phase; cells at the S phase are insensitive. Moreover, the time and degree of arrest may also alter radiosensitivity.74 Zuo et al showed that DHA has a higher tumor suppressive effect and radiosensitizing effect on lung cancer GLC-82 cells transplanted in nude mice.75 The sensitization effect might be through arresting cell cycle at the G2/M phase and inducing apoptosis. In addition, they also proved that radiotherapy combined with DHA in human lung cancer cells could reduce the proportion of cells at the S phase, increase G0/G1 phase cells, and induce apoptosis. The mechanisms might be through functional retore of p53 and p21 to inhibit Bcl-2 expression to promote GLC-82 cell apoptosis.76 Moreover, PD-L1 expression of NSCLC was positively related with radiation resistance, while DHA could eliminate radiation resistance and synergistically enhance the anti-tumor effect of radiotherapy in NSCLC cells by inhibiting the PD-L1 expression trough inhibiting TGF-β, PI3K/Akt, and STAT3 signaling pathways.77
Clinical Trials of DHA in Cancer Treatment
In addition to in vivo and in vitro studies, two clinical trials of DHA for cancer treatment were identified from Chinadrugtrials.org.cn and Clinicaltrials.gov. The clinical trial in China (registration number: ChiCTR2100041652) investigated dihydroartemisinin combined with chemoradiotherapy in the treatment of locally advanced head and neck squamous cell carcinoma: a multicenter, open-label, randomized control study is at the prospective registration phase. The second trial was also from China (registration number: NCT03402464) and is titled Icotinib Combined with Dihydroartemisinin (DHA) Therapy in Patients with Advanced NSCLC; it is a Phase II study to explore the anti-tumor effect of icotinib combined with DHA on EGFR-mutant lung adenocarcinoma patients. Herein, more clinical trials are needed to further evaluate the clinical application of DHA in cancer treatment.
Conclusions
Although varying amounts of anti-cancer agents are used as therapeutic drugs, drug-resistance remains intractable, which has been perplexing the cancer therapy research community for decades. Recently, researchers found that DHA could reverse drug resistance to chemotherapeutics or enhance anti-cancer activities when combined with some anti-cancer drugs. In this review, we summarized the potential molecular mechanisms or targets of DHA working as a sensitizer in tumor therapy to try to provide useful information for clinicians and patients, although how DHA exactly enhances the anti-cancer activity of tumor cells against chemotherapy, targeted therapy, and radiotherapy remains to be further explored. In addition, in vivo experiments should be applied as models for further target identification in DHA treatment. As a classic first-line anti-malarial drug, the efficacy and safety of DHA has been proven for years, while its role in cancer therapy needs further elucidation. Therefore, more clinical trials are needed to test the efficacy and safety of DHA in cancer patients. In addition, whether DHA could reduce the toxic side effects of anti-tumor drugs or radioactive rays on patients remains unclear. The elucidation of these key issues might be helpful to accelerate the clinical application of DHA as a sensitizer for cancer therapy.
Acknowledgments
The authors thank Nanchong Key Laboratory of Molecular Diagnosis and Therapeutics of Tumor, for providing the research platform of the current study.
Funding
This research was supported by the Applied Foundational Research Program of Sichuan Province (Grant No.2021YJ0202) and the Science and Technology Support Program of Nanchong (Grant No.16YFZJ0133, 18SXHZ0514, and 18SXHZ0579).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Guo Z. Development of artemisinin antimalarials. Acta Pharm Sin. 2016;51(1):157–164. doi:10.16438/j.0513-4870.2015-1036
3. Li G, Zhou Q, Zhao C, Qu F, Huang L. Current research status of artemisinins. Clin Pharm J. 1998;33(7):3–5. doi:10.3321/j.issn:1001-2494
4. Xu CC, Deng T, Fan MC, Lv WB, Liu JH, Yu BY. Synthesis and in vitro antitumor evaluation of dihydroartemisinin-cinnamic acid ester derivatives. Eur J Med Chem. 2016;107:192–203. doi:10.1016/j.ejmech.2015.11.003
5. Li BN, Bu SX, Sun JY, Guo Y, Lai DM. Artemisinin derivatives inhibit epithelial ovarian cancer cells via autophagy-mediated cell cycle arrest. Acta Biochim Biophys Sin. 2018;50(12):1227–1235. doi:10.1093/abbs/gmy125
6. Xu CH, Liu Y, Xiao LM, et al. Dihydroartemisinin treatment exhibits antitumor effects in glioma cells through induction of apoptosis. Mol Med Rep. 2017;16(6):9528–9532. doi:10.3892/mmr.2017.7832
7. Dong FY, Zhou X, Li CS, et al. Dihydroartemisinin targets VEGFR2 via the NF-κB pathway in endothelial cells to inhibit angiogenesis. Cancer Biol Ther. 2014;15(11):1479–1488. doi:10.4161/15384047.2014.955728
8. Jiang J, Geng GJ, Yu XY, et al. Repurposing the anti-malarial drug dihydroartemisinin suppresses metastasis of non-small-cell lung cancer via inhibiting NF-κB/GLUT1 axis. Oncotarget. 2016;7(52):87271–87283. doi:10.18632/oncotarget.13536
9. Friedman HS, Kerby T, Calvert H. Temozolomide and treatment of malignant glioma. Clin Cancer Res. 2000;6(7):2585–2597.
10. Chen X, Lai M, Tang P, Lai H. Study the influence mechanism of luteolin combined with temozolomide on glioma. Pharm Clin Chin Materia Medica. 2017;33(2):63–66.
11. Choi EJ, Cho BJ, Lee DJ, et al. Enhanced cytotoxic effect of radiation and temozolomide in malignant glioma cells: targeting PI3K-AKT-mTOR signaling, HSP90 and histone deacetylases. BMC Cancer. 2014;14:17. doi:10.1186/1471-2407-14-17
12. Zhang WB, Wang Z, Shu F, et al. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J Biol Chem. 2010;285(52):40461–40471. doi:10.1074/jbc.M110.164046
13. Jiapaer S, Furuta T, Tanaka S, Kitabayashi T, Nakada M. Potential strategies overcoming the temozolomide resistance for glioblastoma. Neurol Med Chir. 2018;58(10):405–421. doi:10.2176/nmc.ra.2018-0141
14. Yan J, Wen J, Chen H, Huang Q, Li X, Mo L. The research on dihydroartemisinin combined with temozolomide inhibits the proliferation and induces apoptosis of glioma cells. Chin J Exp Surg. 2019;36(2):274–276. doi:10.3760/cma.j.issn.1001-9030.2019.02.025
15. Zhang ZS, Wang J, Shen YB, et al. Dihydroartemisinin increases temozolomide efficacy in glioma cells by inducing autophagy. Oncol Lett. 2015;10(1):379–383. doi:10.3892/ol.2015.3183
16. Huang XJ, Li CT, Zhang WP, Lu YB, Fang SH, Wei EQ. Dihydroartemisinin potentiates the cytotoxic effect of temozolomide in rat C6 glioma cells. Pharmacology. 2008;82(1):1–9. doi:10.1159/000125673
17. Lemke D, Pledl HW, Zorn M, et al. Slowing down glioblastoma progression in mice by running or the anti-malarial drug dihydroartemisinin? Induction of oxidative stress in murine glioblastoma therapy. Oncotarget. 2016;7(35):56713–56725. doi:10.18632/oncotarget.10723
18. Nussbaumer S, Bonnabry P, Veuthey JL, Fleury-Souverain S. Analysis of anticancer drugs: a review. Talanta. 2011;85(5):2265–2289. doi:10.1016/j.talanta.2011.08.034
19. Liu C, Liu G. Progress in gemcitabine resistance mechanism of pancreatic cancer cells. People’s Military Surgeon. 2019;62(12):1211–1216.
20. Nakano T, Saiki Y, Kudo C, et al. Acquisition of chemoresistance to gemcitabine is induced by a loss-of-function missense mutation of DCK. Biochem Biophys Res Commun. 2015;464(4):1084–1089. doi:10.1016/j.bbrc.2015.07.080
21. Yang SC, Zhang DW, Shen N, Wang GY, Tang ZH, Chen XS. Dihydroartemisinin increases gemcitabine therapeutic efficacy in ovarian cancer by inducing reactive oxygen species. J Cell Biochem. 2019;120(1):634–644. doi:10.1002/jcb.27421
22. Zhao CB, Gao WJ, Chen TS. Synergistic induction of apoptosis in A549 cells by dihydroartemisinin and gemcitabine. Apoptosis. 2014;19(4):668–681. doi:10.1007/s10495-013-0953-0
23. Zhao CB, Qin GQ, Gao WJ, et al. Potent proapoptotic actions of dihydroartemisinin in gemcitabine-resistant A549 cells. Cell Signal. 2014;26(10):2223–2233. doi:10.1016/j.cellsig.2014.07.001
24. Wang SJ, Gao Y, Chen H, et al. Dihydroartemisinin inactivates NF-kappaB and potentiates the anti-tumor effect of gemcitabine on pancreatic cancer both in vitro and in vivo. Cancer Lett. 2010;293(1):99–108. doi:10.1016/j.canlet.2010.01.001
25. Wang S, Sun B, Pan S, et al. Experimental study of the function and mechanism combining dihydroartemisinin and gemcitabine in treating pancreatic cancer. Chin J Surg. 2010;48(7):530–534. doi:10.3760/cma.j.issn.0529-5815.2010.07.013
26. Shao Z, Xiang S, Zhu J, Zou J. Experimental study of dihydroartemisinin combined with gemcitabine in the treatment of cholangiocarcinoma. Shenzhen J Integr Tradit Chin West Med. 2016;26(22):4–6. doi:10.16458/j.cnki.1007-0893.2016.22.002
27. Baba H, Teramoto K, Kawamura T, Mori A, Imamura M, Arii S. Dihydropyrimidine dehydrogenase and thymidylate synthase activities in hepatocellular carcinomas and in diseased livers. Cancer Chemother Pharmacol. 2003;52(6):469–476. doi:10.1007/s00280-003-0695-8
28. Dai ZR, Mao QF. Dihydroartemisinin increases sensitivity of gastric cancer cells to 5 -fluorouracil through suppression of ATF2 phosphorylation. Zhejiang JITCW M. 2018;28(5):363–365, 369.
29. Wan B, Cao HB, Yu GH. Dihydroartemisinin enhances antitumor effect of 5-fluorouracil against gastric cancer by down-regulating SIRT1 expression. Chin J Pathophysiol. 2017;33(12):2195–2201.
30. Yao ZH, Bhandari A, Wang YH, et al. Dihydroartemisinin potentiates antitumor activity of 5-fluorouracil against a resistant colorectal cancer cell line. Biochem Biophys Res Commun. 2018;501(3):636–642. doi:10.1016/j.bbrc.2018.05.026
31. Yang Y, He JX, Chen J, et al. Dihydroartemisinin sensitizes mutant p53 (r248q)-expressing hepatocellular carcinoma cells to doxorubicin by inhibiting p-gp expression. Biomed Res Int. 2019;2019:8207056. doi:10.1155/2019/8207056
32. Kang XJ, Wang HY, Peng HG, et al. Codelivery of dihydroartemisinin and doxorubicin in mannosylated liposomes for drug-resistant colon cancer therapy. Acta Pharmacol Sin. 2017;38(6):885–896. doi:10.1038/aps.2017.10
33. Hu YJ, Zhang JY, Luo Q, et al. Nanostructured dihydroartemisinin plus epirubicin liposomes enhance treatment efficacy of breast cancer by inducing autophagy and apoptosis. Nanomaterials. 2018;8(10):804. doi:10.3390/nano8100804
34. Tai X, Cai XB, Zhang Z, Wei R. In vitro inhibition of tumor cell viability by combined dihydroartemisinin and doxorubicin treatment, and the underlying mechanism. Oncol Lett. 2016;12(5):3701–3706. doi:10.3892/ol.2016.5187
35. Liu JJ, Tang W, Fu M, et al. Development of R8 modified epirubicin–dihydroartemisinin liposomes for treatment of non-small-cell lung cancer. Artif Cells Nanomed Biotechnol. 2019;47(1):1947–1960. doi:10.1080/21691401.2019.1615932
36. Wong PM, Feng Y, Wang JR, Shi R, Jiang XJ. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun. 2015;6:8048. doi:10.1038/ncomms9048
37. Wang LM, Wang RL. Effect of rapamycin (RAPA) on the growth of lung cancer and its mechanism in mice with A549. Int J Clin Exp Pathol. 2015;8(8):9208–9213.
38. Chen K, Lv S. Resistant mechanism about rapamycin and solution. J Int Oncol. 2014;41(10):740–743. doi:10.3760/cma.j.issn.1673-422X.2014.10.007
39. Zhong X, Wang A, Wang H, Feng J, Zheng B, Shi H. DHA-inhibited proliferation through the PTEN/PI3K/Akt pathway in gastric cancer SGC7901 cells. Chin J Clin Oncol. 2013;4:190–194. doi:10.3969/j.issn.1000-8179.2013.04.003
40. Thongchot S, Vidoni C, Ferraresi A, et al. Dihydroartemisinin induces apoptosis and autophagy-dependent cell death in cholangiocarcinoma through a DAPK1-BECLIN1 pathway. Mol Carcinog. 2018;57(12):1735–1750. doi:10.1002/mc.22893
41. Liu QJ, Zhou XY, Li C, Zhang XM, Li CL. Rapamycin promotes the anticancer action of dihydroartemisinin in breast cancer MDA-MB-231 cells by regulating expression of Atg7 and DAPK. Oncol Lett. 2018;15(4):5781–5786. doi:10.3892/ol.2018.8013
42. Chen LP, Wang L, Shen HB, Lin H, Li D. Anthelminthic drug niclosamide sensitizes the responsiveness of cervical cancer cells to paclitaxel via oxidative stress-mediated mTOR inhibition. Biochem Biophys Res Commun. 2017;484(2):416–421. doi:10.1016/j.bbrc.2017.01.140
43. Huang D, Li L, Na X. Inhibitory effect of dihydroartemisinin combined with paclitaxel on proliferation of breast cancer cells. J Front Med. 2018;8(14):160. doi:10.3969/j.issn.2095-1752.2018.14.125
44. Paccez J, Duncan K, Sekar D, et al. Dihydroartemisinin inhibits prostate cancer via JARID2/miR-7/miR-34a-dependent downregulation of Axl. Oncogenesis. 2019;8(3):14. doi:10.1038/s41389-019-0122-6
45. Dai Phung C, Le TG, Nguyen VH, et al. PEGylated-paclitaxel and dihydroartemisinin nanoparticles for simultaneously delivering paclitaxel and dihydroartemisinin to colorectal cancer. Pharm Res. 2020;37(7):129. doi:10.1007/s11095-020-02819-7
46. Guan F, Ding YM, Zhang YM, Zhou Y, Li MG, Wang CH. Curcumin suppresses proliferation and migration of MDA-MB-231 breast cancer cells through autophagy-dependent Akt degradation. PLoS One. 2016;11(1):e0146553. doi:10.1371/journal.pone.0146553
47. Khan MA, Zafaryab M, Mehdi SH, Ahmad I, Rizvi MM. Characterization and anti-proliferative activity of curcumin loaded chitosan nanoparticles in cervical cancer. Int J Biol Macromol. 2016;93(Pt.A):242–253. doi:10.1016/j.ijbiomac.2016.08.050
48. Gabriel S, Felipe TL, Viviane S, et al. Curcumin analog CH-5 suppresses the proliferation, migration, and invasion of the human gastric cancer cell line HGC-27. Molecules. 2018;23(2):279. doi:10.3390/molecules23020279
49. Gaikwad D, Shewale R, Patil V, Mali D, Gaikwad U, Jadhav N. Enhancement in in vitro anti-angiogenesis activity and cytotoxicity in lung cancer cell by pectin-PVP based curcumin particulates. Int J Biol Macromol. 2017;104(Pt.A):656–664. doi:10.1016/j.ijbiomac.2017.05.170
50. Starok M, Preira P, Vayssade M, Haupt K, Salomé L, Rossi C. EGFR inhibition by curcumin in cancer cells: a dual mode of action. Biomacromolecules. 2015;16(5):1634–1642. doi:10.1021/acs.biomac.5b00229
51. Zhao JJ, Pan YC, Li XJ, et al. Dihydroartemisinin and curcumin synergistically induce apoptosis in SKOV3 cells via upregulation of MiR-124 targeting midkine. Cell Physiol Biochem. 2017;43(2):589–601. doi:10.1159/000480531
52. Ohmichi M, Hayakawa J, Tasaka K, Kurachi H, Murata Y. Mechanisms of platinum drug resistance. Trends Pharmacol Sci. 2005;26(3):113–116. doi:10.1016/j.tips.2005.01.002
53. Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16(2):99–109. doi:10.1038/nrc.2015.17
54. Zhang SY, Feng R, Yuan F, et al. The therapeutic effects of dihydroartemisinin on cisplatin-resistant gastric cancer cells. Curr Pharm Biotechnol. 2021;22. doi:10.2174/1389201022666210217114825
55. Feng X, Li L, Jiang H, Jiang KP, Jin Y, Zheng JH. Dihydroartemisinin potentiates the anticancer effect of cisplatin via mTOR inhibition in cisplatin-resistant ovarian cancer cells: involvement of apoptosis and autophagy. Biochem Biophys Res Commun. 2014;444(3):376–381. doi:10.1016/j.bbrc.2014.01.053
56. Cui W, Fang TT, Duan ZH, et al. Dihydroartemisinin sensitizes esophageal squamous cell carcinoma to cisplatin by inhibiting sonic hedgehog signaling. Front Cell Dev Biol. 2020;8:596788. doi:10.3389/fcell.2020.596788
57. Qin Y, Yang G, Li M, et al. Dihydroartemisinin inhibits EMT induced by platinum-based drugs via Akt-Snail pathway. Oncotarget. 2017;8(61):103815–103827. doi:10.18632/oncotarget.21793
58. Zhang BC, Zhang ZM, Wang J, et al. Dihydroartemisinin sensitizes Lewis lung carcinoma cells to carboplatin therapy via p38 mitogen-activated protein kinase activation. Oncol Lett. 2018;15(5):7531–7536. doi:10.3892/ol.2018.8276
59. Feng M, Ding J, Liu S, Tian J, Sun BWL, Guan WX. The effect of gender on endoplasmic reticulum-stress (ER-stress) and apoptosis signals in patients with hepatocellular carcinoma. J Jiangsu Univ. 2017;27(05):432–436.
60. Ezzoukhry Z, Louandre C, Trécherel E, et al. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int J Cancer. 2012;131(12):2961–2969. doi:10.1002/ijc.27604
61. Zhang Z, Zhou XY, Shen HJ, Wang DX, Wang YH. Phosphorylated ERK is a potential predictor of sensitivity to sorafenib when treating hepatocellular carcinoma: evidence from an in vitro study. BMC Med. 2009;7:41. doi:10.1186/1741-7015-7-41
62. Shimizu S, Takehara T, Hikita H, et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J Cancer. 2012;131(3):548–557. doi:10.1002/ijc.26374
63. Hou CY, Guo DQ, Yu X, Wang SY, Liu TH. TMT-based proteomics analysis of the anti-hepatocellular carcinoma effect of combined dihydroartemisinin and sorafenib. Biomed Pharmacother. 2020;126:109862. doi:10.1016/j.biopha.2020.109862
64. Wang ZF, Duan XX, Lv YH, Zhao YF. Low density lipoprotein receptor (LDLR)-targeted lipid nanoparticles for the delivery of sorafenib and Dihydroartemisinin in liver cancers. Life Sci. 2019;239:117013. doi:10.1016/j.lfs.2019.117013
65. Kiyota M, Kuroda J, Yamamoto-Sugitani M, et al. FTY720 induces apoptosis of chronic myelogenous leukemia cells via dual activation of BIM and BID and overcomes various types of resistance to tyrosine kinase inhibitors. Apoptosis. 2013;18(11):1437–1446. doi:10.1007/s10495-013-0882-y
66. Ng KP, Hillmer AM, Chuah CT, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med. 2012;18(4):521–528. doi:10.1038/nm.2713
67. Deng QF, Fang QY, Ji XX, Zhou SW. Cyclooxygenase-2 mediates gefitinib resistance in non-small cell lung cancer through the EGFR/PI3K/AKT axis. J Cancer. 2020;11(12):3667–3674. doi:10.7150/jca.42850
68. Jin H, Jiang AY, Wang H, Cao Y, Wu Y, Jiang XF. Dihydroartemisinin and gefitinib synergistically inhibit NSCLC cell growth and promote apoptosis via the Akt/mTOR/STAT3 pathway. Mol Med Rep. 2017;16(3):3475–3481. doi:10.3892/mmr.2017.6989
69. Song F, Luo M, Fan N, Zhou J, Li Y. Combination of dihydroartemisinin and gefitinib inhibits cell cycle and migration of lung adenocarcinoma cells in vitro. Armed Police Med. 2019;30(03):228–232.
70. Bellini L, Strub T, Habel N, et al. Endoplasmic reticulum stress mediates resistance to BCL-2 inhibitor in uveal melanoma cells. Cell Death Discov. 2020;6:22. doi:10.1038/s41420-020-0259-2
71. Inoue-Yamauchi A, Oda H. EMT-inducing transcription factor ZEB1-associated resistance to the BCL-2/BCL-X L inhibitor is overcome by BIM upregulation in ovarian clear cell carcinoma cells. Biochem Biophys Res Commun. 2020;526(3):612–617. doi:10.1016/j.bbrc.2020.03.139
72. Yan XH, Li PF, Zhan YH, et al. Dihydroartemisinin suppresses STAT3 signaling and Mcl-1 and Survivin expression to potentiate ABT-263-induced apoptosis in non-small cell lung cancer cells harboring EGFR or RAS mutation. Biochem Pharmacol. 2018;150:72–85. doi:10.1016/j.bcp.2018.01.031
73. Budhraja A, Turnis ME, Churchman ML, et al. Modulation of navitoclax sensitivity by dihydroartemisinin-mediated MCL-1 repression in BCR-ABL+ B-lineage acute lymphoblastic leukemia. Clin Cancer Res. 2017;23(24):7558–7568. doi:10.1158/1078-0432.CCR-17-1231
74. Kempf H, Hatzikirou H, Bleicher M, Meyer-Hermann M. In silico analysis of cell cycle synchronisation effects in radiotherapy of tumour spheroids. PLoS Comput Biol. 2013;9(11):e1003295. doi:10.1371/journal.pcbi.1003295.
75. Zuo Z, Wang J, Wang S, Xin Y, Zhang H. The sensitizing effects of combined treatment with dihydroartemisinin and irradiation on human lung cancer in nude mice. J Modern Oncol. 2013;21(12):2687–2691. doi:10.3969/j.issn.1672-4992.2013.12.13
76. Zuo Z, Wang S, Jiang L, Xin Y, Li W. Effect of dihydroartemisinin combined irradiation on the apoptosis of human lung cancer GLC-82 cells and its mechanism study. Chin J Integr Tradt Chin West Med. 2014;34(10):1220–1224.
77. Zhang H, Zhou F, Wang YY, et al. Eliminating radiation resistance of non-small cell lung cancer by dihydroartemisinin through abrogating immunity escaping and promoting radiation sensitivity by inhibiting PD-L1 expression. Front Oncol. 2020;10:595466. doi:10.3389/fonc.2020.595466
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.