")
Back to Journals » Journal of Inflammation Research » Volume 14
Dexmedetomidine Attenuates Ischemia/Reperfusion-Induced Myocardial Inflammation and Apoptosis Through Inhibiting Endoplasmic Reticulum Stress Signaling
Authors Yang Y , Wang H , Song N, Jiang Y, Zhang J, Meng X , Feng X, Liu H , Peng K , Ji F
Received 18 November 2020
Accepted for publication 16 March 2021
Published 31 March 2021 Volume 2021:14 Pages 1217—1233
DOI https://doi.org/10.2147/JIR.S292263
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Yu-fan Yang,1,* Hui Wang,1,2,* Nan Song,1,* Ya-hui Jiang,1 Jun Zhang,1 Xiao-wen Meng,1 Xiao-mei Feng,3,4 Hong Liu,5 Ke Peng,1 Fu-hai Ji1
1Department of Anesthesiology, First Affiliated Hospital of Soochow University, Suzhou, Jiangsu, People’s Republic of China; 2Department of Anesthesiology, Wuxi People’s Hospital Affiliated to Nanjing Medical University, Wuxi, Jiangsu, People’s Republic of China; 3Department of Anesthesiology, University of Utah Health, Salt Lake City, UT, USA; 4Transitional Residency Program, Intermountain Medical Center, Murray, UT, USA; 5Department of Anesthesiology and Pain Medicine, University of California Davis Health, Sacramento, CA, USA
*These authors contributed equally to this work
Correspondence: Ke Peng; Fu-hai Ji
Department of Anesthesiology, First Affiliated Hospital of Soochow University, 899 Pinghai Road, Suzhou, Jiangsu, 215006, People’s Republic of China
Tel +86-159-6215-5989
; +86-512-6797-2352
Email [email protected]; [email protected]
Background: Endoplasmic reticulum stress (ERS)-mediated myocardial inflammation and apoptosis plays an important role in myocardial ischemia/reperfusion (I/R) injury. Dexmedetomidine has been used clinically with sedative, analgesic, and anti-inflammatory properties. This study aimed to determine the effects of dexmedetomidine pretreatment on inflammation, apoptosis, and the expression of ERS signaling during myocardial I/R injury.
Methods: Rats underwent myocardial ischemia for 30 min and reperfusion for 6 h, and H9c2 cardiomyocytes were subjected to oxygen-glucose deprivation/reoxygenation (OGD/R) injury (OGD for 12 h and reoxygenation for 3 h). Dexmedetomidine was administered prior to myocardial ischemia in rats or ODG in cardiomyocytes. In addition, the α 2-adrenergic receptor antagonist (yohimbine) or the PERK activator (CCT020312) was given prior to dexmedetomidine treatment.
Results: Dexmedetomidine pretreatment decreased serum levels of cardiac troponin I, reduced myocardial infarct size, alleviated histological structure damage, and improved left ventricular function following myocardial I/R injury in rats. In addition, dexmedetomidine pretreatment increased cell viability and reduced cytotoxicity following OGD/R injury in cardiomyocytes. Mechanistically, the cardioprotection offered by dexmedetomidine was mediated via the inhibition of inflammation and apoptosis through downregulating the expression of the ERS signaling pathway, including glucose-regulated protein 78 (GRP78), protein kinase R-like endoplasmic reticulum kinase (PERK), C/EBP homologous protein (CHOP), inositol-requiring protein 1 (IRE1), and activating transcription factor 6 (ATF6). Conversely, the protective effects of dexmedetomidine were diminished by blocking the α 2 adrenergic receptors with yohimbine or promoting PERK phosphorylation with CCT020312.
Conclusion: Dexmedetomidine pretreatment protects the hearts against I/R injury via inhibiting inflammation and apoptosis through downregulation of the ERS signaling pathway. Future clinical studies are needed to confirm the cardioprotective effects of dexmedetomidine in patients at risk of myocardial I/R injury.
Keywords: myocardial ischemia/reperfusion injury, dexmedetomidine, endoplasmic reticulum stress, inflammation, apoptosis, GRP78/PERK/CHOP
Introduction
Myocardial ischemia/reperfusion (I/R) injury is the leading cause of mortality and morbidity in patients with ischemic heart disease.1,2 To date, effective clinical therapy for preventing or treating myocardial I/R injury is still lacking, which is attributable to its complicated and multifactorial pathogenesis. Despite great progress over the decades, the specific mechanism underlying myocardial I/R injury remains elusive. Recent studies showed that multiple mechanisms are involved during the process of myocardial I/R injury, such as inflammation, apoptosis, intracellular calcium overload, endothelial dysfunction, reactive oxygen species, and endoplasmic reticulum stress (ERS).3–8
Under physiological conditions, the endoplasmic reticulum serves many essential roles in cell function, including protein synthesis, protein folding, calcium homoeostasis, and lipid metabolism.8,9 Under a stress condition such as the myocardial I/R process, the endoplasmic reticulum homoeostasis is disrupted, and unfolded or misfolded proteins accumulates, leading to the prolonged unfolded protein response (UPR) during ERS.10–12 The UPR signaling is regulated by three key sensor proteins, namely protein kinase R-like endoplasmic reticulum kinase (PERK), inositol-requiring protein 1 (IRE1), and activating transcription factor 6 (ATF6).13 The activation of PERK upregulates the expression of pro-apoptotic transcription factor C/EBP homologous protein (CHOP).13,14 In addition, glucose-regulated protein 78 (GRP78), an important ERS marker, is the master of UPR in the endoplasmic reticulum.15 The activation of ERS signaling exacerbated inflammation and apoptosis in a variety of stress situations.16–18 A recent study showed that ERS plays a critical role during myocardial I/R injury in rats with diabetes mellitus.19
Dexmedetomidine, a highly selective agonist of α2-adrenergic receptors (α2-ARs), has been used in clinical anesthesia and intensive care unit, exerting sedative, analgesic, and sympatholytic effects.20 In addition, studies suggest that dexmedetomidine protects the hearts against I/R-induced injury, possibly by activating an endothelial nitric oxide synthase/nitric oxide dependent mechanism,21 inhibiting hypoxia-inducible factor 1α (HIF-1α)-induced apoptosis,22 and alleviating intracellular calcium overload through the regulation of 12.6-kd FK506-binding protein/ryanodine receptor 2.23 However, whether dexmedetomidine has an impact on ERS-mediated myocardial I/R injury is still unclear.
Therefore, this study aimed to determine the regulatory mechanism of dexmedetomidine on the ERS signaling during myocardial I/R injury in rats and during oxygen-glucose deprivation/reoxygenation (OGD/R)-induced injury in cardiomyocytes. We hypothesized that pretreatment with dexmedetomidine would exert its cardioprotection via suppressing inflammation and apoptosis through the inhibition of ERS signaling pathway.
Materials and Methods
Ethics
The study protocol was approved by the Animal Ethics Committee of Soochow University, Suzhou, Jiangsu, China (No. 2018–177). This study followed the Guide for the Care of Use of Laboratory Animals (US National Institutes of Health publication No. 85–23, revised in 1996).
Animals
Adult male Sprague-Dawley rats, weighing 280–320 g, were provided by the laboratory animal center of Soochow University. The rats were housed under a standard controlled condition, with room temperature 22–24°C, humidity 50%, 12-h light/dark cycle, 4 rats per cage, and free access to food and water.
Cell Culture
The rat H9c2 cardiomyocytes were obtained from the Shanghai Institute of Life Sciences, Chinese Academy of Sciences, Shanghai, China. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) in an incubator with 95% air and 5% CO2 at 37 °C, as previously described.23 When confluence reached 70–80%, the cells were used for experimental procedures.
Reagents and Antibodies
Dexmedetomidine (Heng Rui Pharmaceutical Co. Ltd., Lianyungang, Jiangsu, China); Trizol (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA); RIPA lysis, phenylmethanesulfonyl fluoride (PMSF), and phosphate buffered saline (PBS) (Beyotime, Shanghai, China); 2,3,5-triphenyltetrazolium chloride (TTC) and dimethyl sulfoxide (DMSO) (Sigma, St. Louis, MO, USA); DMEM (HyClone, Logan, UT, USA); FBS (Biological Industries, Kibbutz Beit-Haemek, Israel); glucose-free DMEM (Gibco, Carlsbad, CA, USA); reverse transcription polymerase chain reaction (RT-PCR) kit (Applied Biological Materials, Richmond, BC, Canada); RT-PCR primers (Sangon, Shanghai, China); anti-GRP78, anti-CHOP, and anti-cleaved caspase 3 (Cell Signaling Technology, Beverly, MA, USA); anti-α2A-AR (Santa Cruz, Dallas, TX, USA); anti-IRE1, anti-phosphorylated-IRE1 (anti-p-IRE1), anti-ATF6, anti-α2B-AR, and anti-caspase 3 (abcam, Cambridge, MA, USA); anti-phosphorylated-PERK (anti-p-PERK) and anti-PERK (ABclonal, Wuhan, Hubei, China); anti-GAPDH (Absin, Shanghai, China); yohimbine (Selleck, Houston, TX, USA); CCT020312 (MedChemExpress, Monmouth Junction, NJ, USA).
Myocardial I/R Injury Model
The rat myocardial I/R injury model was established as previously described.22,24 Rats were anesthetized with intraperitoneal sodium pentobarbital 50 mg/kg. After tracheal intubation, the lungs were ventilated with room air, tidal volume 20 mL/kg, 1:1 ratio, and 70 breaths/min, by using an animal ventilator (R407, RWD, Shenzhen, China). A left thoracotomy was carried out. The left anterior descending artery was occluded for 30 min to induce myocardial ischemia, and then the ligature was removed for myocardial reperfusion. The successful establishment of the model was confirmed by observation of myocardial blanching and S-T segment elevation in an electrocardiography, as previously described.22 Rats in the sham group underwent the same thoracotomy, without the ligation of artery. A heating pad was used to maintain rats’ body temperature at ~37°C throughout the procedure.
Cardiomyocyte OGD/R Injury Model
H9c2 cardiomyocytes were treated with glucose-free DMEM and incubated in a hypoxia chamber with 95% N2 and 5% CO2 at 37 °C, as previously described.23,24 The cells were subjected to OGD for 12 h, followed by reoxygenation for 3 h with the normal DMEM under a normoxia condition.
Randomization and Blinding
The rats or cardiomyocytes were randomly divided into the study groups using an online randomization tool (https://www.sealedenvelope.com/randomisation/). The allocation concealment was ensured with the use of sealed opaque envelopes. The study medications (dexmedetomidine, yohimbine, normal saline, CCT020312, PBS, and DMSO) were prepared by an independent research assistant according to the randomization results. There was no way to distinguish the study medications, because they are all colorless and were kept in identical syringes. The researchers and study outcome observers were blinded to the group allocation until the completion of final analysis.
Experimental Protocol
In part I (Figure 1A), rats were randomly assigned into five groups (n = 8): a sham group and 4 myocardial I/R groups. Rats in the I/R groups underwent myocardial ischemia for 30 min, followed by reperfusion for 0, 2, 6, and 24 h, respectively. Subsequently, rats were assigned into three groups (n = 5): a sham group, a myocardial I/R group (6 h of reperfusion), and a dexmedetomidine pretreatment group (6 μg/kg/h for 10 min followed by 0.7 μg/kg/h for 15 min via the right jugular vein, prior to ischemia). The dosage of dexmedetomidine was based on our previous studies.22,25
 |
Figure 1 Experiment workflow. (A) Part I: rats underwent sham surgery or myocardial ischemia for 30 min and reperfusion for 0, 2, 6, and 24 h, respectively. In addition, rats were treated with dexmedetomidine before myocardial ischemia, followed by reperfusion for 6 h. (B) Part II: rats were treated with dexmedetomidine and/or yohimbine (an α2-adrenergic receptor antagonist) before myocardial I/R process. (C) Part III: rats were treated with dexmedetomidine and/or CCT020312 (a PERK activator) before myocardial I/R process. In addition, survival during 21 days of reperfusion was observed. (D) H9c2 cardiomyocytes were treated with dexmedetomidine and/or CCT020312 before OGD/R injury. Abbreviations: I/R, ischemia/reperfusion; OGD/R, oxygen-glucose deprivation/reoxygenation; Dex, dexmedetomidine; Yoh, yohimbine; CCT, CCT020312. |
In part II (Figure 1B), rats were randomly assigned into five groups (n = 5): a sham group, a myocardial I/R group, a dexmedetomidine pretreatment group (same dosage as in part I), a yohimbine pretreatment group (intravenous yohimbine 12 mg/kg/h for 5 min, followed by 0.5 mg/kg/h for 20 min, prior to ischemia), and a yohimbine prior to dexmedetomidine treatment group. Yohimbine is an antagonist of α2-adrenergic receptors. The dosage of yohimbine were based on our previous studies.22,25
In part III (Figure 1C), rats were randomly assigned into five groups (n = 5): a sham group, a myocardial I/R group, a dexmedetomidine pretreatment group (same dosage as in part I), a CCT020312 pretreatment group (intraperitoneal 2 mg/kg before ischemia), and a CCT020312 prior to dexmedetomidine treatment group. CCT020312 is a highly selective activator of PERK, and its dose was based on our preliminary experiments and previous studies.26,27 In addition, the survival status of rats during 21 days after myocardial I/R injury was observed (n = 18).
In part IV (Figure 1D), H9c2 cardiomyocytes were randomly assigned into five groups (n = 8): a control group, an OGD/R group (OGD for 12 h and reoxygenation for 3 h), a dexmedetomidine pretreatment group (1 μM dexmedetomidine for 1 h before OGD/R), a CCT020312 pretreatment group (10 μM CCT020312 for 2 h before OGD/R), and a CCT020312 prior to dexmedetomidine treatment group. The dose of dexmedetomidine and CCT020312 in cardiomyocytes were based on previous studies.23,28
Enzyme-Linked Immunosorbent Assay (ELISA)
The commercial ELISA kits were used to measure the serum cardiac troponin I (cTnI) levels (Life diagnostics, Lincoln, NE, USA) and myocardial interleukin (IL)-1β and IL-6 levels (Multi Sciences, Hangzhou, Zhejiang, China). The absorbance values at 450 nm were recorded on a microplate reader (SpectraMax190, Molecular Devices, Sunnyvale, CA, USA). The concentrations of cTnI, IL-1β, and IL-6 were determined by using a standard curve approach.
Myocardial Infract Size
The myocardial infarct size was measured using the Evans blue/TTC staining method, as previously described.22,24 At the end of myocardial reperfusion, the Evans blue staining was performed with re-occlusion of the LAD. The hearts were removed and frozen at −20°C for 30 min, and then the left ventricle was cut into 2-mm thick slices. The slices were stained with 1% TTC at 37°C for 30 min, treated with 4% paraformaldehyde overnight, and imaged by using a CanoScan LiDE 300 scanner (Canon, Tokyo, Japan). The images were analyzed with the Image J software (National Institutes of Health, Bethesda, MD, USA). The area at risk is defined as the myocardial area not stained by Evans blue, and the infarct area is defined as the area not stained by TTC. The myocardial infarct size is expressed as the ratio of the infarct area to the area at risk.
Echocardiography
Echocardiography was conducted using the Vevo 770 system (VisualSonics, Toronto, Canada) at 7 days after myocardial reperfusion, as previously described.29 Briefly, the rats were anesthetized, and M-mode echocardiology were carried out on the parasternal short-axis view and long-axis view of the heart. The left ventricle ejection fraction (EF) was determined as: EF (%) = (EDV − ESV)/EDV (%); EDV = end diastolic volume, and ESV = end systolic volume.
Hematoxylin and Eosin (HE) Staining
The I/R-induced myocardial histopathological changes were visualized using the HE staining. The rat hearts were harvested and then fixed in 4% paraformaldehyde for 48 h. The left ventricular tissues were frozen sectioned into 5-μm thick slices. Next, the slices were stained with hematoxylin/eosin and photographed under a Leica AF6000 microscope (Solms, Hesse, Germany).
Terminal Deoxynucleotidyl Transferase-Mediated Nick End-Labeling (TUNEL)
The myocardial tissues were fixed in 4% formaldehyde for 24 h, embedded in paraffin, and sectioned into 4-μm thick slices. Myocardial apoptosis was tested by using the TUNEL assay with the in situ Cell Death Detection kit-POD (Roche, Basel, Switzerland). The slices were photographed under the Leica AF6000 microscope. For each slice, the TUNEL positive cells (nuclei stained brown) were identified in five random and non-overlapping fields. The apoptosis index was calculated as the ratio of TUNEL positive cells to total number of cells.
Cell Viability and Cytotoxicity Assay
Cell viability was measured using the Cell Counting Kit-8 assay kit (Beyotime, Shanghai, China), and cytotoxicity was assessed using the lactate dehydrogenase (LDH) assay kit (Beyotime, Shanghai, China), as previously described.22,23 The absorbance values at 490 nm were detected using the SpectraMax190 microplate reader.
Western Blotting
The whole protein of rat left ventricular tissues or cardiomyocytes was extracted using the RIPA lysis with PMSF. A bicinchoninic acid reagent kit (Beyotime, Shanghai, China) was used to determine the protein concentration. The proteins were separated in 10% gels using a sodium dodecyl sulphate-polyacrylamide gel electrophoresis and then transferred onto polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% non-fat milk at room temperature for 2 h and then incubated at 4°C overnight with the specific primary antibodies: anti-GRP78 (1:1000), anti-p-PERK (1:1000), anti-PERK (1:1000), anti-CHOP (1:1000), anti-cleaved caspase 3 (1:1000), anti-caspase 3 (1:1000), anti-IRE1 (1:1000), anti-p-IRE1 (1:1000), anti-ATF6 (1:1000), anti-α2A-AR (1:200), anti-α2B-AR (1:2000), and anti-GAPDH (1:3000). Next, the membranes were washed and incubated at room temperature for 2 h with the HRP-conjugated secondary antibodies (1:5000, Santa Cruz, CA, USA). For each sample, three replicates were tested. The bands were detected using an enhanced chemiluminescence kit (NCM, Suzhou, China) under a Tanon 5200 luminescent imaging workstation (Tanon, Shanghai, China). The protein intensity was analyzed with the Image J software and normalized to GAPDH as control.
Quantitative RT-PCR
Total RNA of rat left ventricle was extracted by using the Trizol reagent. Reverse transcription and quantitative real-time PCR were conducted with the RT-PCR kit in a 20 μL reaction volume on a Roche Light Cycler 480 system (Roche, Bedford, MA, USA). For each sample, three replicates were tested. The target gene expression was calculated by using the 2−ΔΔCT method and normalized to GAPDH as control. The specific primer sequences were:
GRP78, 5ʹ-ACACCTGACCGACCGCTGAG-3ʹ (forward), 5ʹ-GCCAACCACCGTGCCTACATC-3ʹ (reverse);
CHOP, 5ʹ-CTGAAGAGAACGAGCGGCTCAAG-3ʹ (forward), 5ʹ-GACAGGTGATGCCAACAGTTC-3ʹ (reverse);
GAPDH, 5ʹ-GGTTGTCTCCTGCGACTTC-3ʹ (forward), 5ʹ-CCTGTTGCTGTACCGTATTCAT-3ʹ (reverse).
Statistical Analysis
Before initiating this study, a sample size calculation was performed using the PASS software (version 11.0.7; NCSS, LCC., Kaysville, UT). According to the results of our preliminary experiment, the serum cTnI level was 10.2 ± 1.1 ng/mL (mean ± standard deviation [SD]) in the myocardial I/R group. We hypothesized that pretreatment with dexmedetomidine would reduce the value of serum cTnI to 7.1 ± 1.1 ng/mL (ie, a relative 30% reduction). To detect such a difference with a power of 80% and a significance level of 0.05, three rats were needed in each group. Considering correction for multiple comparisons, we finally included 5 rats in each group. For the survival analysis, we hypothesized that dexmedetomidine pretreatment would increase the survival rate at 21 days after myocardial I/R injury from 40% to 80%. We estimated that 17 rats were required in each group to detect such a difference in survival with a power of 80% and a significance level of 0.05, and we finally included 18 rats in each group.
An independent statistician performed the statistical analysis using the GraphPad Prism software (version 7.0, GraphPad, San Diego, CA, USA). Variables are shown as mean ± standard deviation and compared by using one-way or two-way analysis of variance followed by Tukey or Dunnett test. Kaplan–Meier plots were used to depict the survival outcome, and the between-group differences were analyzed using hazard ratio (HR) and 95% confidence interval (CI) in the Log rank test. A two-sided P value < 0.05 denotes a statistically significant difference.
Results
Elevated mRNA Expression of GRP78 and CHOP During I/R-Induced Myocardial Injury in Rats
Compared to the sham group, the mRNA expression of GRP78 and CHOP in rat myocardium were remarkedly increased in the I/R groups at 0, 2, 6, and 24 h after myocardial reperfusion (Figure 2A and B). By observing the time course of GRP78 and CHOP mRNA expression, ischemia of 30 min followed by reperfusion of 6 h which led to the highest mRNA expression of both genes was selected for the subsequent experiments.
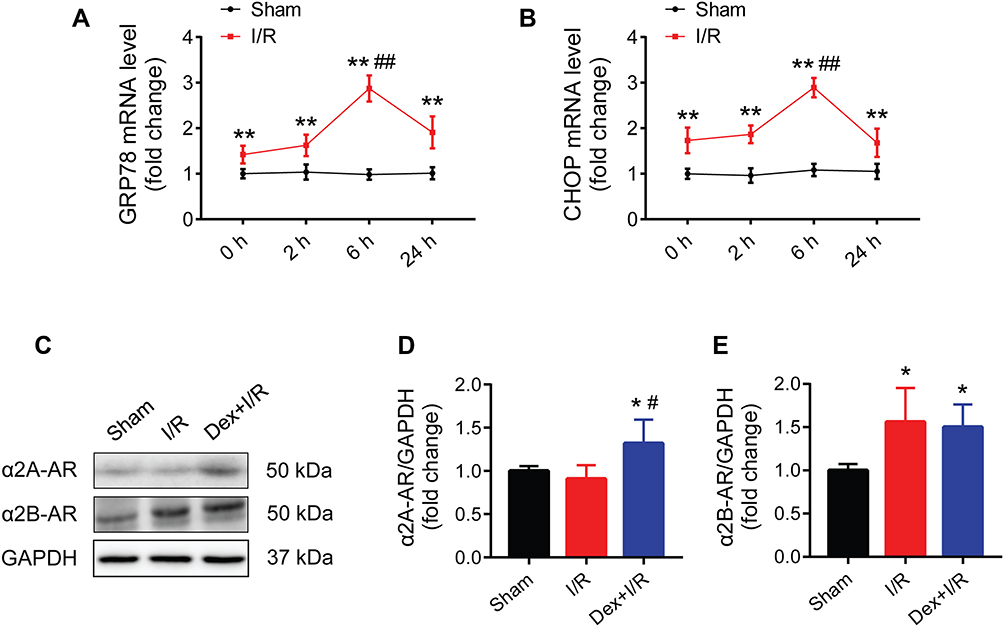 |
Figure 2 Activation of GRP78 and CHOP during myocardial I/R injury in rats, and the expression of two subtypes of α2-ARs in myocardium. (A and B) Time course of myocardial GRP78 and CHOP mRNA expression (n = 8). (C–E) Western blots showing the protein expression of α2A-AR and α2B-AR (n = 5). GAPDH was used as the loading control. *P < 0.05, **P < 0.01 vs the sham group; ##P < 0.01 vs the 0, 2, and 24 h groups (A and B); #P < 0.05 vs the I/R group (D). Abbreviations: I/R, ischemia/reperfusion; Dex, dexmedetomidine; α2-ARs, α2-adrenergic receptors. |
Dexmedetomidine Pretreatment Attenuated I/R-Induced Myocardial Injury in Rats Through the Activation of α2-Adrenergic Receptors
The presence of two subtypes of α2-ARs (α2A-AR and α2B-AR) in rat myocardium were confirmed in the sham, myocardial I/R, and dexmedetomidine pretreatment groups (Figure 2C). The protein expression of α2B-AR increased during myocardial I/R, while both α2A-AR and α2B-AR protein expression were up-regulated in response to dexmedetomidine pretreatment (Figure 2D and E).
The process of myocardial I/R resulted in a significant cTnI release, which was effectively blocked by dexmedetomidine pretreatment (Figure 3A). Remarkable myocardial infract size and histological damage were found following myocardial I/R injury, which was alleviated by dexmedetomidine (Figure 3B–D). In addition, dexmedetomidine pretreatment significantly alleviated I/R-induced left ventricle dysfunction, as evidenced by an increased value of EF (Figure 3E and F). These protective effects of dexmedetomidine were abolished by the addition of yohimbine, while yohimbine alone did not have a significant impact on myocardial I/R injury.
 |
Figure 3 The protective effects of dexmedetomidine against I/R-induced rat myocardial injury and left ventricular dysfunction were blocked by yohimbine. (A) Serum levels of cTnI (n = 5). (B and C) Representative images of myocardial Evans blue/TTC staining and quantitative analysis of infarct size (n = 5). (D) Representative HE staining images of myocardium. Scale bar = 50 μm. (E and F) Echocardiography images and quantitative analysis of left ventricle ejection fraction (n = 5). **P < 0.01 vs the sham group; ##P < 0.01 vs the I/R group; $$P < 0.01 for the comparisons shown. Abbreviations: I/R, ischemia/reperfusion; Dex, dexmedetomidine; Yoh, yohimbine; cTnI, cardiac troponin I; AAR, area at risk; LV, left ventricle; IA, infract area. |
Dexmedetomidine Pretreatment Reduced I/R-Induced Myocardial Inflammation and Apoptosis in Rats Through the Activation of α2-Adrenergic Receptors
Pretreatment with dexmedetomidine notably suppressed I/R-induced myocardial expression of IL-1β and IL-6 (Figure 4A and B). In addition, dexmedetomidine reduced TUNEL apoptosis during myocardial I/R injury (Figure 4C and D). The use of yohimbine abolished the effects of dexmedetomidine. Similarly, yohimbine alone did not alter myocardial I/R-induced inflammation or apoptosis.
 |
Figure 4 The protective effects of dexmedetomidine against I/R-induced rat myocardial inflammation and apoptosis were blocked by yohimbine. (A and B) Myocardial IL-1β and IL-6 levels (n = 5). (C and D) Representative images and quantitative analysis of TUNEL staining in myocardium (n = 5). Scale bar = 200 μm. Arrows indicating the apoptotic cells. **P < 0.01 vs the sham group; #P < 0.05, ##P < 0.01 vs the I/R group; $P < 0.05, $$P < 0.01 for the comparisons shown. Abbreviations: I/R, ischemia/reperfusion; Dex, dexmedetomidine; Yoh, yohimbine. |
Dexmedetomidine Pretreatment Inhibited I/R-Induced Myocardial Expression of GRP78, p-PERK, CHOP, and Cleaved Caspase 3 in Rats Through the Activation of α2-Adrenergic Receptors
The expression of several key factors of ERS signaling including GRP78, PERK, and CHOP were examined. Pretreatment with dexmedetomidine reduced the mRNA expression of GRP78 and CHOP during myocardial I/R injury (Figure 5A and B). Dexmedetomidine also suppressed myocardial I/R-induced phosphorylation of PERK and protein expression of GRP78, CHOP, and cleaved caspase 3 (Figure 5C–G). The addition of yohimbine reversed these effects of dexmedetomidine, while yohimbine alone did not influence the expression of these proteins.
 |
Figure 5 The effects of dexmedetomidine on the expression of ERS signaling during rat myocardial I/R injury were blocked by yohimbine. (A and B) The mRNA expression of myocardial GRP78 and CHOP (n = 5). (C–G) Western blots showing the protein expression of GRP78, p-PERK, PERK, CHOP, cleaved caspase 3, and caspase 3 (n = 5). GAPDH was used as the loading control. *P < 0.05, **P < 0.01 vs the sham group; #P < 0.05, ##P < 0.01 vs the I/R group; $P < 0.05, $$P < 0.01 for the comparisons shown. Abbreviations: I/R, ischemia/reperfusion; Dex, dexmedetomidine; Yoh, yohimbine. |
Activation of PERK Aggravated I/R-Induced Myocardial Injury in Rats and Abolished Dexmedetomidine’s Cardioprotection
To explore the role of PERK in dexmedetomidine pretreatment during myocardial I/R injury, a highly selective PERK activator CCT020312 was used. The use of CCT020312 significantly exacerbated myocardial I/R-induced serum level of cTnI (Figure 6A), myocardial infract size (Figure 6B and C), myocardial histological structure damage (Figure 6D), and left ventricular dysfunction (Figure 6E and F). Moreover, the administration of CCT020312 prior to dexmedetomidine pretreatment reversed the protective effects of dexmedetomidine against myocardial I/R injury.
 |
Figure 6 The PERK activator CCT020312 exacerbated rat myocardial I/R injury and abolished the protective effects of dexmedetomidine. (A) Serum levels of cTnI (n = 5). (B and C) Representative images of Evans blue/TTC staining and quantitative analysis of myocardial infarct size (n = 5). (D) Representative HE staining images of myocardium. Scale bar = 50 μm. (E and F) Echocardiography images and quantitative analysis of left ventricle ejection fraction (n = 5). **P < 0.01 vs the sham group; #P < 0.05, ##P < 0.01 vs the I/R group; $$P < 0.01 for the comparisons shown. Abbreviations: I/R, ischemia/reperfusion; Dex, dexmedetomidine; CCT, CCT020312; cTnI, cardiac troponin I; AAR, area at risk; LV, left ventricle; IA, infract area. |
Activation of PERK Aggravated I/R-Induced Myocardial Inflammation and Apoptosis in Rats and Abolished the Protective Effects of Dexmedetomidine
The PERK activator CCT020312 significantly exacerbated I/R-induced myocardial expression of IL-1β and IL-6 (Figure 7A and B) and myocardial TUNEL apoptosis (Figure 7C and D). The use of CCT020312 diminished the protective effects of dexmedetomidine on myocardial I/R-induced inflammation and apoptosis. In addition, Kaplan–Meier survival analysis showed that the dexmedetomidine group had a significantly higher survival up to 21 days following myocardial I/R injury than the I/R group (77.8% vs 38.9%, HR = 0.28, 95% CI = 0.10 to 0.76, log-rank P = 0.014) (Figure 7E). The addition of CCT020312 prior to dexmedetomidine treatment reduced the survival (55.6% vs 77.8%), but not by a significant amount.
 |
Figure 7 The PERK activator CCT020312 aggravated rat myocardial inflammation and apoptosis, and abolished the protective effects of dexmedetomidine. (A and B) Myocardial mRNA expression of IL-1β and IL-6 (n = 5). (C and D) Representative myocardial images and quantitative analysis of TUNEL staining (n = 5). Scale bar = 200 μm. (E) Kaplan–Meier survival analysis (n = 18). **P < 0.01 vs the sham group; #P < 0.05, ##P < 0.01 vs the I/R group; $$P < 0.01 for the comparisons shown. Abbreviations: I/R, ischemia/reperfusion; Dex, dexmedetomidine; CCT, CCT020312. |
Activation of PERK Blocked the Effects of Dexmedetomidine on CHOP, p-PERK, and Cleaved Caspase 3 During Myocardial I/R Injury in Rats, but Not GRP78, p-IRE1, or ATF6
The PERK activator CCT020312 had little impact on the mRNA expression of GRP78 during myocardial I/R injury, along or with dexmedetomidine (Figure 8A). However, the mRNA expression of CHOP was significantly increased by CCT020312 (Figure 8B). Furthermore, the use of CCT020312 significantly increased I/R-induced protein expression of p-PERK, CHOP, and cleaved caspase 3, but not GRP78, in the presence or absence of dexmedetomidine (Figure 8C–G).
 |
Figure 8 The PERK activator CCT020312 blocked the effects of dexmedetomidine on the expression of ERS signaling during rat myocardial I/R injury. (A and B) The mRNA expression of myocardial GRP78 and CHOP (n = 5). (C–G) Western blots showing the protein expression of GRP78, p-PERK, PERK, CHOP, cleaved caspase 3, and caspase 3 (n = 5). GAPDH was used as the loading control. (H–J) Western blots showing the protein expression of p-IRE1, IRE1, and ATF6 (n = 5). GAPDH was used as the loading control. *P < 0.05, **P < 0.01 vs the sham group; #P < 0.05, ##P < 0.01 vs the I/R group; $P < 0.05, $$P < 0.01 for the comparisons shown. Abbreviations: I/R, ischemia/reperfusion; Dex, dexmedetomidine; CCT, CCT020312. |
In addition, the protein expression of p-IRE1 and ATF6 were significantly increased following myocardial I/R, which was partly blocked by dexmedetomidine pretreatment (Figure 8H–J). The protein expression of p-IRE1 or ATF6 did not change by CCT020312, alone or with dexmedetomidine.
Dexmedetomidine Pretreatment Protected Cardiomyocytes Against OGD/R-Induced Injury Through Inhibiting the ERS Signaling
The process of OGD/R significantly reduced cell viability and increased LDH release in H9c2 cardiomyocytes, which was partially reversed by dexmedetomidine pretreatment (Figure 9A and B). The addition of CCT020312 diminished the protective effects of dexmedetomidine on cell viability and LDH release in cardiomyocytes.
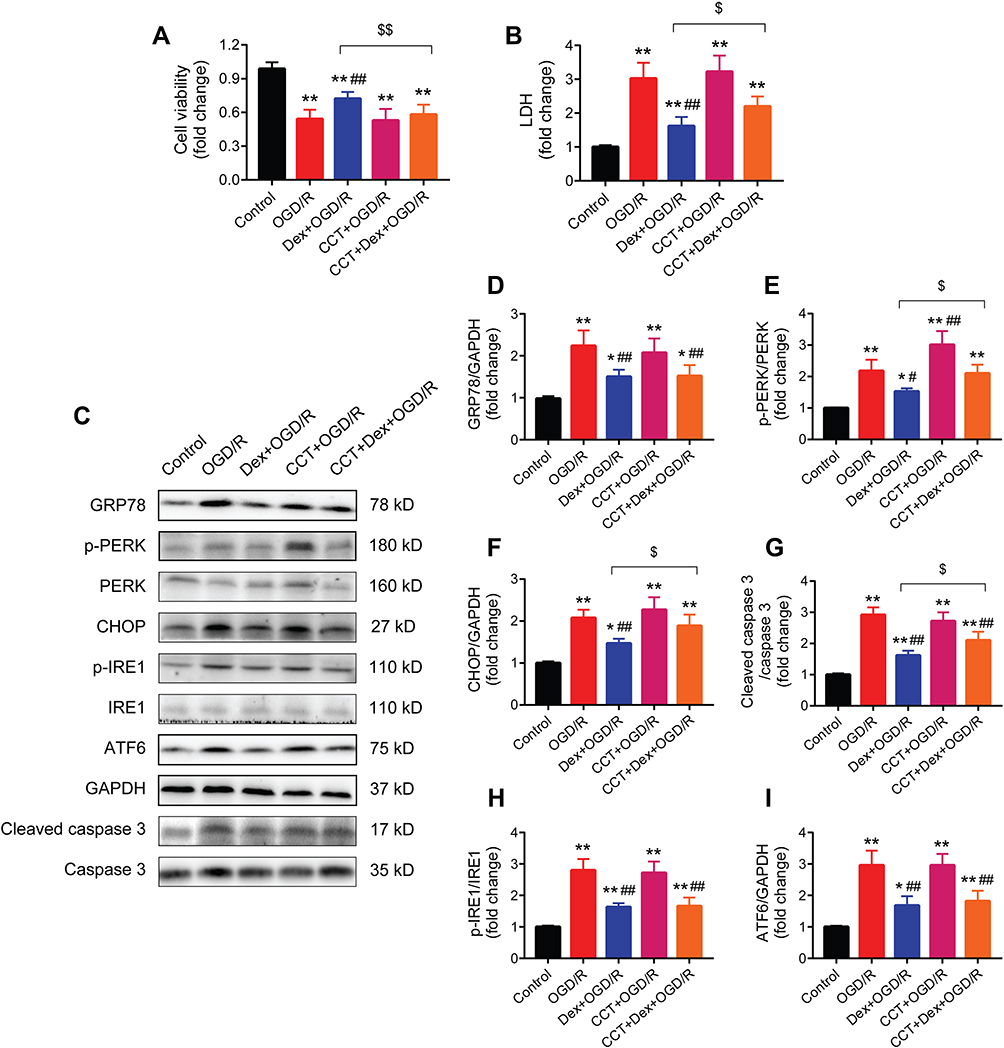 |
Figure 9 The PERK activator CCT020312 blocked the protective effects of dexmedetomidine on cardiomyocytes of OGD/R injury and the expression of ERS signaling. (A and B) Cell viability and LDH assay (n = 5). (C–I) Western blots showing the protein expression of GRP78, p-PERK, PERK, CHOP, p-IRE1, IRE1, ATF6, cleaved caspase 3, and caspase 3 (n = 5). GAPDH was used as the loading control. *P < 0.05, **P < 0.01 vs the control group; #P < 0.05, ##P < 0.01 vs the OGD/R group; $P < 0.05, $$P < 0.01 for the comparisons shown. Abbreviations: OGD/R, oxygen-glucose deprivation/reoxygenation; LDH, lactate dehydrogenase; Dex, dexmedetomidine; CCT, CCT020312. |
The effect of dexmedetomidine pretreatment on the expression of ERS signaling during OGD/R-induced cell injury was also investigated (Figure 9C). Dexmedetomidine suppressed OGD/R-induced activation of GRP78, p-PERK, CHOP, and cleaved caspase 3 (Figure 9D–G). The use of CCT020312 further increased the expression of p-PERK during OGD/R, and blocked the effects of dexmedetomidine on p-PERK, CHOP, and cleaved caspase 3, but not GRP78. In addition, dexmedetomidine significantly reduced OGD/R-induced protein expression of p-IRE1 and ATF6 in H9c2 cardiomyocytes, while CCT020312 had no impact on p-IRE1 or ATF6 protein expression in the presence or absence of dexmedetomidine (Figure 9H and I).
Discussion
This study demonstrates that pretreatment with dexmedetomidine protected the hearts against I/R injury, as reflected by reduced serum cTnI levels, decreased myocardial infarct size and histological structure damage, and improved left ventricle function. In addition, dexmedetomidine pretreatment conferred protective effects at the cellular level. The cardioprotective mechanism of dexmedetomidine was mediated via the inhibition of inflammation and apoptosis through downregulating the expression of ERS signaling pathway. Conversely, the protective effects of dexmedetomidine were diminished by blocking the α2-adrenergic receptors with yohimbine or promoting PERK phosphorylation with CCT020312, and the activation of PERK even aggravated I/R-induced myocardial injury.
UPR is an evolutionarily conserved adaptive mechanism that helps cells to restore endoplasmic reticulum homeostasis, involving three key sensor proteins (PERK, IRE1, and ATF6).13 Under mild stress conditions, the UPR process is initiated to protect the cells by eliminating unfolded or misfolded proteins.13 The binding of these proteins to the endoplasmic reticulum chaperone GRP78 represses their activation. Under a situation of prolonged ERS, such as myocardial I/R injury, GRP78 is upregulated and preferentially binds to unfolded or misfolded proteins.13 The decreased inhibitory interactions of GRP78 with the stress sensors allow for phosphorylation of PERK, activation of IRE1, and translocation of ATF6 to Golgi. PERK is a major transducer of ERS, and recent studies showed that the upregulation of PERK/CHOP exacerbated inflammation and apoptosis.16–18,30 Conversely, CHOP-deficient mice exhibited alleviation of myocardial inflammation and apoptosis following myocardial I/R injury.31 These modulators of ERS signaling are recognized as promising targets for research and therapeutic development.8,10
A recent study showed that dexmedetomidine reduced the expression of several ERS-related proteins, inhibited apoptosis, and attenuated myocardial I/R injury in rats with diabetes mellitus.19 Dexmedetomidine also attenuated apoptosis and cell injury in cardiomyocytes subjected to hypoxia/reoxygenation or H2O2 via regulating the ERS signaling.32,33 However, these previous studies failed to detect the expression of key stress sensor protein PERK or its phosphorylation, compromising the evaluation of the effect of dexmedetomidine on ERS signaling. Moreover, these studies did not assess myocardial inflammation which is a major ERS-mediated outcome. In the present study, pretreatment with dexmedetomidine attenuated I/R-induced myocardial inflammation and apoptosis via suppressing the phosphorylation of PERK and IRE1 and inhibiting the expression of GRP78, CHOP, IRE1, and ATF6. Our findings revealed that dexmedetomidine exerted its cardioprotection through the inhibition of the key factors in the ERS signaling pathway. The use of yohimbine abolished the protective effects of dexmedetomidine, suggesting that dexmedetomidine protects the hearts though activating the α2-adrenergic receptors. In addition, the use of CCT020312 diminished dexmedetomidine-mediated cardioprotection, exacerbated I/R-induced myocardial injury, aggravated myocardial inflammation and apoptosis, and activated the expression of p-PERK and CHOP, indicating the essential role PERK/CHOP signaling in dexmedetomidine’s cardioprotection. Besides, recent experimental studies showed that dexmedetomidine alleviated other types of myocardial injury, such as septic heart injury,34,35 doxorubicin cardiotoxicity,36 and liver injury-induced myocardial injury.37 A randomized controlled study also suggested that intraoperative dexmedetomidine infusion reduced myocardial injury in patients with coronary heart disease undergoing non-cardiac surgery.38
We used dexmedetomidine pretreatment before myocardial ischemia for two reasons. First, the time frame of dexmedetomidine pretreatment in this study is clinically relevant. Intravenous dexmedetomidine is administered clinically for sedation or as an anesthetic adjuvant, with a loading dose of 0.5–1 μg/kg before or immediately after anesthesia induction, followed by a continuous infusion of 0.3–0.8 μg/kg/h. The use of dexmedetomidine is expected to reduce perioperative myocardial I/R injury in the clinical settings, which occurs during the surgical procedures or in the early postoperative days. Second, the ERS signaling is activated during the myocardial ischemia phase and continues to show upregulation during the reperfusion phase. We found that the expression of GRP78 and CHOP were increased immediately after myocardial ischemia, suggesting that dexmedetomidine treatment should be used early to inhibit the activation of ERS in its initial phase, other than administered after reperfusion. Regarding the effect of dexmedetomidine post-treatment, a recent study showed that dexmedetomidine administration at the onset of reperfusion inhibited apoptosis and attenuated myocardial I/R injury via targeting the HIF-1α signaling.22 Hence, dexmedetomidine post-treatment also produced cardioprotective effects, but whether a ERS-related mechanism is involved in dexmedetomidine post-treatment during myocardial I/R injury needs further investigations.
This study has some limitations. First, this study determined the regulative mechanism of dexmedetomidine on the GRP78/PERK/CHOP signaling during ERS-mediated myocardial I/R injury. We also measured the expression of IRE1α and ATF6, but we did not further assess their role during dexmedetomidine pretreatment. Second, this study shows the cardioprotection of dexmedetomidine via regulating the ERS signaling pathway. It is reported that dexmedetomidine post-treatment is also cardioprotective,22 but whether dexmedetomidine post-treatment has an impact on the ERS signaling during myocardial I/R injury is unknown. Last, although the cardiac structural development observed in mice and humans are comparable,39 it is still unclear whether dexmedetomidine could reduce or prevent myocardial I/R injury through an ERS-mediated mechanism in humans. While dexmedetomidine has been used clinically for the purpose of sedation, analgesia, and anti-inflammation, future clinical studies are needed to confirm the cardioprotective effects of dexmedetomidine in patients at risk of myocardial I/R injury.
In conclusion, dexmedetomidine pretreatment alleviated myocardial I/R injury via suppressing ERS-mediated inflammation and apoptosis and downregulating the ERS signaling pathway (Figure 10). These findings address a promising effect of dexmedetomidine on ERS-mediated myocardial I/R injury, suggesting that dexmedetomidine may be cardioprotective in high-risk patients.
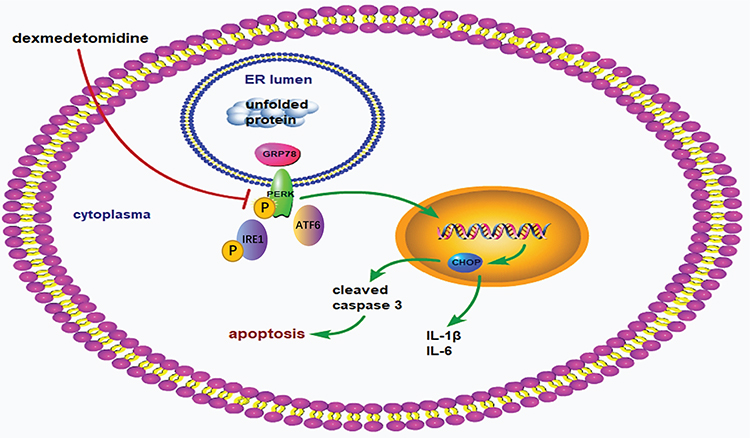 |
Figure 10 Schematic mechanism. The process of myocardial ischemia/reperfusion activates endoplasmic reticulum stress signaling pathway, as reflected by upregulation of GRP78, phosphorylation of PERK and IRE1, and promotion of ATF6. Subsequently, the downstream factor CHOP is activated, leading to increased expression of cleaved caspase 3, IL-1β, and IL-6 in the myocardium. Enhanced myocardial inflammation and apoptosis finally leads to myocardial ischemia/reperfusion injury. Dexmedetomidine pretreatment attenuates myocardial ischemia/reperfusion injury via reducing myocardial inflammation and apoptosis through inhibiting the activation of ERS signaling pathway. |
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357(11):1121–1135. doi:10.1056/NEJMra071667
2. Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123(1):92–100. doi:10.1172/JCI62874
3. Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol. 2010;106(3):360–368. doi:10.1016/j.amjcard.2010.03.032
4. Shi B, Ma M, Zheng Y, Pan Y, Lin X. mTOR and Beclin1: two key autophagy-related molecules and their roles in myocardial ischemia/reperfusion injury. J Cell Physiol. 2019;234(8):12562–12568. doi:10.1002/jcp.28125
5. Zhang T, Zhang Y, Cui M, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22(2):175–182. doi:10.1038/nm.4017
6. Zhang L, Cai S, Cao S, et al. Diazoxide protects against myocardial ischemia/reperfusion injury by moderating ERS via regulation of the miR-10a/IRE1 pathway. Oxid Med Cell Longev. 2020;2020:4957238.
7. Zeng J, Jin Q, Ruan Y, et al. Inhibition of TGFbeta-activated protein kinase 1 ameliorates myocardial ischaemia/reperfusion injury via endoplasmic reticulum stress suppression. J Cell Mol Med. 2020;24(12):6846–6859. doi:10.1111/jcmm.15340
8. Wang S, Binder P, Fang Q, et al. Endoplasmic reticulum stress in the heart: insights into mechanisms and drug targets. Br J Pharmacol. 2018;175(8):1293–1304. doi:10.1111/bph.13888
9. Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci. 2016;73(1):79–94. doi:10.1007/s00018-015-2052-6
10. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10(1):173–194. doi:10.1146/annurev-pathol-012513-104649
11. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2018;69(2):169–181. doi:10.1016/j.molcel.2017.06.017
12. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi:10.1126/science.1209038
13. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. doi:10.1038/nrm3270
14. Iurlaro R, Munoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016;283(14):2640–2652. doi:10.1111/febs.13598
15. Ibrahim IM, Abdelmalek DH, Elfiky AA. GRP78: a cell’s response to stress. Life Sci. 2019;226:156–163. doi:10.1016/j.lfs.2019.04.022
16. Li Y, Jiang W, Niu Q, et al. eIF2α-CHOP-BCl-2/JNK and IRE1α-XBP1/JNK signaling promote apoptosis and inflammation and support the proliferation of Newcastle disease virus. Cell Death Dis. 2019;10(12):891. doi:10.1038/s41419-019-2128-6
17. Hu H, Tian M, Ding C, Yu S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front Immunol. 2018;9:3083. doi:10.3389/fimmu.2018.03083
18. Guo Q, Li H, Liu J, et al. Tunicamycin aggravates endoplasmic reticulum stress and airway inflammation via PERK-ATF4-CHOP signaling in a murine model of neutrophilic asthma. J Asthma. 2017;54(2):125–133. doi:10.1080/02770903.2016.1205085
19. Li J, Zhao Y, Zhou N, Li L, Li K. Dexmedetomidine attenuates myocardial ischemia-reperfusion injury in diabetes mellitus by inhibiting endoplasmic reticulum stress. J Diabetes Res. 2019;2019:7869318. doi:10.1155/2019/7869318
20. Gerlach AT, Murphy CV, Dasta JF. An updated focused review of dexmedetomidine in adults. Ann Pharmacother. 2009;43(12):2064–2074. doi:10.1345/aph.1M310
21. Riquelme JA, Westermeier F, Hall AR, et al. Dexmedetomidine protects the heart against ischemia-reperfusion injury by an endothelial eNOS/NO dependent mechanism. Pharmacol Res. 2016;103:318–327. doi:10.1016/j.phrs.2015.11.004
22. Peng K, Chen WR, Xia F, et al. Dexmedetomidine post-treatment attenuates cardiac ischaemia/reperfusion injury by inhibiting apoptosis through HIF-1α signalling. J Cell Mol Med. 2020;24(1):850–861. doi:10.1111/jcmm.14795
23. Yuan M, Meng XW, Ma J, et al. Dexmedetomidine protects H9c2 cardiomyocytes against oxygen-glucose deprivation/reoxygenation-induced intracellular calcium overload and apoptosis through regulating FKBP12.6/RyR2 signaling. Drug Des Devel Ther. 2019;13:3137–3149. doi:10.2147/DDDT.S219533
24. Song N, Ma J, Meng XW, et al. Heat shock protein 70 protects the heart from ischemia/reperfusion injury through inhibition of p38 MAPK signaling. Oxid Med Cell Longev. 2020;2020:3908641. doi:10.1155/2020/3908641
25. Zhang JJ, Peng K, Zhang J, Meng XW, Ji FH. Dexmedetomidine preconditioning may attenuate myocardial ischemia/reperfusion injury by down-regulating the HMGB1-TLR4-MyD88-NF-small ka, CyrillicB signaling pathway. PLoS One. 2017;12(2):e0172006. doi:10.1371/journal.pone.0172006
26. Bruch J, Xu H, Rosler TW, et al. PERK activation mitigates tau pathology in vitro and in vivo. EMBO Mol Med. 2017;9(3):371–384. doi:10.15252/emmm.201606664
27. Guo J, Ren R, Sun K, et al. PERK controls bone homeostasis through the regulation of osteoclast differentiation and function. Cell Death Dis. 2020;11(10):847. doi:10.1038/s41419-020-03046-z
28. Stockwell SR, Platt G, Barrie SE, et al. Mechanism-based screen for G1/S checkpoint activators identifies a selective activator of EIF2AK3/PERK signalling. PLoS One. 2012;7(1):e28568. doi:10.1371/journal.pone.0028568
29. Zhang J, Xia F, Zhao H, et al. Dexmedetomidine-induced cardioprotection is mediated by inhibition of high mobility group box-1 and the cholinergic anti-inflammatory pathway in myocardial ischemia-reperfusion injury. PLoS One. 2019;14(7):e0218726. doi:10.1371/journal.pone.0218726
30. Cao L, Chen Y, Zhang Z, Li Y, Zhao P. Endoplasmic reticulum stress-induced NLRP1 inflammasome activation contributes to myocardial ischemia/reperfusion injury. Shock. 2019;51(4):511–518. doi:10.1097/SHK.0000000000001175
31. Miyazaki Y, Kaikita K, Endo M, et al. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):1124–1132. doi:10.1161/ATVBAHA.111.224519
32. Zhu Z, Ling X, Zhou H, Zhang C, Yan W. Dexmedetomidine attenuates cellular injury and apoptosis in H9c2 cardiomyocytes by regulating p-38MAPK and endoplasmic reticulum stress. Drug Des Devel Ther. 2020;14:4231–4243. doi:10.2147/DDDT.S265970
33. Liu XR, Li T, Cao L, et al. Dexmedetomidine attenuates H2O2-induced neonatal rat cardiomyocytes apoptosis through mitochondria- and ER-medicated oxidative stress pathways. Mol Med Rep. 2018;17(5):7258–7264. doi:10.3892/mmr.2018.8751
34. Yu T, Liu D, Gao M, et al. Dexmedetomidine prevents septic myocardial dysfunction in rats via activation of alpha7nAChR and PI3K/Akt- mediated autophagy. Biomed Pharmacother. 2019;120:109231. doi:10.1016/j.biopha.2019.109231
35. Kong W, Kang K, Gao Y, et al. Dexmedetomidine alleviates LPS-induced septic cardiomyopathy via the cholinergic anti-inflammatory pathway in mice. Am J Transl Res. 2017;9(11):5040–5047.
36. Yu JL, Jin Y, Cao XY, Gu HH. Dexmedetomidine alleviates doxorubicin cardiotoxicity by inhibiting mitochondrial reactive oxygen species generation. Hum Cell. 2020;33(1):47–56. doi:10.1007/s13577-019-00282-0
37. Erer D, Ozer A, Arslan M, et al. The protective effects of dexmedetomidine on liver injury-induced myocardial ischemia reperfusion. Bratisl Lek Listy. 2014;115(7):422–426. doi:10.4149/bll_2014_083
38. Xu L, Hu Z, Shen J, McQuillan PM. Does dexmedetomidine have a cardiac protective effect during non-cardiac surgery? A randomised controlled trial. Clin Exp Pharmacol Physiol. 2014;41(11):879–883. doi:10.1111/1440-1681.12296
39. Krishnan A, Samtani R, Dhanantwari P, et al. A detailed comparison of mouse and human cardiac development. Pediatr Res. 2014;76(6):500–507. doi:10.1038/pr.2014.128
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.