Back to Journals » ImmunoTargets and Therapy » Volume 15
Development of an Active Chimeric IL13Rα2 ADC for Diffuse Intrinsic Pontine Glioma
Authors Lian X, Allanson VJ, Rasmussen SV ![]() , Chauhan S, Tan GK
, Chauhan S, Tan GK ![]() , Morrow P, Hanson K, Lian EB, Haight AR
, Morrow P, Hanson K, Lian EB, Haight AR ![]() , Hoshino T, Liu X, Berlow NE, Keller C, Lian Y
, Hoshino T, Liu X, Berlow NE, Keller C, Lian Y
Received 13 November 2025
Accepted for publication 14 April 2026
Published 19 May 2026 Volume 2026:15 575767
DOI https://doi.org/10.2147/ITT.S575767
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Michael Shurin
Xiaolei Lian,1– 3 Victoria J Allanson,2 Samuel V Rasmussen,2 Shefali Chauhan,2 Guak-Kim Tan,2 Paige Morrow,2 Kyle Hanson,2 Emily Bing Lian,2 Anthony R Haight,2 Tyuji Hoshino,4 Xianzhi Liu,3 Noah E Berlow,2,* Charles Keller,2,* Yajun Lian1,*
1Department of Neurology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, People’s Republic of China; 2Children’s Cancer Therapy Development Institute, Hillsboro, OR, USA; 3Department of Neurosurgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, 450052, People’s Republic of China; 4Graduate School of Pharmaceutical Sciences, Chiba University, Chiba, Japan
*These authors contributed equally to this work
Correspondence: Yajun Lian, Department of Neurology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, People’s Republic of China, Email [email protected] Charles Keller, Children’s Cancer Therapy Development Institute, Hillsboro, OR, USA, Tel +0801232-8038, Email [email protected]
Introduction: Diffuse intrinsic pontine glioma (DIPG) is a rare pediatric brain tumor with a critical unmet need due to the lack of approved, curative interventions available. The interweaving of malignant cells with normal tissue makes surgical extraction essentially impossible, and radiation provides only transient benefit. The recent ONC201 FDA approval, however, suggests DIPG therapy is tractable. Having identified overexpression of IL13Rα 2 in DIPG tumor tissue versus normal brain tissue, we investigated binding of commercially available IL13Rα 2 monoclonal antibodies. The top candidate antibody was used to generate a chimeric antibody, to which we conjugated deruxtecan to create a preclinical therapeutic candidate.
Methods: We validated the novel antibody–drug conjugate (ADC) in vitro, demonstrating dose-dependent, IL13Rα 2 expression-dependent cell death. We further validated the novel ADC ex ovo in quail xenograft models of DIPG and in vivo in a mouse xenograft model.
Results: The ADC showed tumor reduction in the ex ovo quail embryo model for both IL13Rα 2-high and IL13Rα 2-low DIPG cell models. A proof-of-concept in vivo mouse xenograft experiment demonstrated a reduction in tumor volume beyond antibody treatment alone.
Discussion: The work here represents an important milestone in preclinical development of a novel deruxtecan-based ADC agent for an intractable pediatric brain cancer, concurrent with other ADC agents demonstrating real-world clinical efficacy and gaining approvals in multiple disease indications.
Keywords: diffuse intrinsic pontine glioma, DIPG, chimeric antibody, interleukin-13 receptor alpha, IL13Rα 2, antibody–drug conjugate, ADC
Introduction
Diffuse intrinsic pontine glioma (DIPG), a subtype of diffuse midline glioma (DMG), is a universally lethal central nervous system tumor that afflicts children around the world.1 In the U.S., DIPG occurs in approximately 350 patients per year, representing 20% of all pediatric CNS tumors and 75% of all pediatric brain stem gliomas.2–4 The survival rate among DIPG patients is approximately 30%, 10%, and 1% after one, two, and five years, respectively.5 DIPG originates in the ventral pons region of the midbrain, which controls essential biological functions such as breathing, blood pressure and heart rate. DIPG presents as tumor nodules interwoven among normal tissue, which makes surgical removal impossible in this vital CNS region.6 The only existing clinically accepted treatment is palliative radiation therapy at a total dose of 54–60 Gy over 6 weeks which transiently improves patient neurological function and extends overall survival by 8–12 weeks;7 however, negative short- and medium-term effects of radiation reduces patient quality of life.8 No therapeutic strategy has demonstrated long-term survival benefit compared to standard radiotherapy alone.9 Additionally, the low permeability and active efflux transport of the blood–brain barrier (BBB) hinders most large, hydrophilic chemotherapeutic agents to brain tumor sites.10,11 Thus, new therapeutic approaches and administration strategies are needed urgently.
Convection-Enhanced Delivery (CED) is a drug administration strategy used in CNS malignancies in which a catheter is inserted directly into the brain parenchyma and treatments are infused over a set time interval using a pressure gradient.12 CED was developed to overcome the BBB, for large and/or hydrophilic chemotherapeutic agents. CED delivers anti-tumor compounds in a non-diffusion dependent manner, which can increase local drug concentration while reducing systemic toxicity.13 CED has been explored clinically as a therapeutic administration strategy, supporting further investigation clinical investigation of CED for pediatric brain tumors.14,15
Antibody-drug conjugates (ADCs) are a newer class of oncology therapeutics consisting of a monoclonal antibody (mAb) chemically bound to a cytotoxic “payload” drug. ADCs are designed, in principle, to expand the therapeutic window of cytotoxic drugs by targeting their delivery specifically to cells that express the target cell surface antigen bound by the antibody.16 Designing a novel ADC requires identifying a cell surface antigen target which is differentially overexpressed in tumor cells versus normal tissue, prevalent both in patients and in through the tumor tissue, and which is internalized and trafficked into the lysosomal compartment to release the chemically bound cytotoxic payload.16
ADCs are one of the fastest growing drug classes in oncology as evidenced by FDA approval of thirteen ADCs since 2011, including brentuximab vedotin for relapsed Hodgkin’s lymphoma or relapsed anaplastic large cell lymphoma and enfortumab vedotin for urothelial cancer (the full list of approved ADC therapeutics is provided in Supplementary Table 1; see also https://www.fda.gov/drugs/development-approval-process-drugs/drug-approvals-and-databases). Most of the pivotal, approval-granting trials resulted in longer survival times than cures but have nonetheless motivated numerous additional ADC clinical trials.
In the past decade, interleukin 13 receptor subunit alpha 2 (IL13Rα2) was identified as a promising target for new preclinical research and has been investigated as a clinical target.17 IL13Rα2 has significantly higher expression in numerous cancers compared with non-neoplastic tissue, including head and neck cancer,18 melanoma,19 ovarian cancer,20 AIDS-associated Kaposi’s sarcoma21 and malignant gliomas,22–24 marking IL13Rα2 as a target of therapeutic interest in oncology. Interleukin 13 (IL-13) receptors are composed of two subunits: the functional alpha one subunit (IL13Rα1) and decoyed alpha two subunit (IL13Rα2).25 As a pleiotropic cytokine, IL-13 acts through the IL13Rα1/IL4Rα complex to induce immune activation responses in inflammatory diseases such as asthma and cancer.26,27 IL13Rα2 serves as a decoy signaling receptor to both IL-13 and IL-4, and binds as a monomer with high affinity to IL-13 but not to IL-4.2,27–29 IL13Rα2 has at least 5,000-fold greater affinity for IL-13 than IL13Rα1.25 Despite increased binding affinity, IL13Rα2 is believed to be non-signaling, thereby potentially serving as a decoy receptor to negatively regulate IL-13 and IL-4 signaling.25,29
Importantly, IL13Rα2 can undergo internalization after ligand or antibody binding.27,30,31 Additionally, previous studies suggest IL-13 stimulation of IL13Rα2-expressing DIPG cell lines do not significantly alter proliferation and invasion.2 Our previously published analysis of next-generation sequencing and immunohistochemistry from primary DIPG tumor tissues identified that IL13Rα2 is highly expressed in DIPG tumor samples but essentially absent in normal brain tissue.2,32 Our initial studies investigating the role of IL13Rα2 in proliferation and invasion of DIPG models demonstrate the potential safety of IL13Rα2-targeted treatments in DIPG. Our additional studies using an IL13Rα2-targeting ADC serve as a proof-of-concept of the capability of immunoconjugate agents to eliminate DIPG cells in an IL13Rα2-dependent manner.2 In this report, we further refine the design of, and expand the validation of, a novel IL13Rα2 ADC for DIPG.
Materials and Methods
Cell Culture
A detailed protocol for cell culture is provided in Supplementary Methods 1. Cell Lines RH30 and BJ5TA were obtained from American Type Cell Culture (ATCC). The cell lines SF8628, SJGBM2, VU-DIPG-A, SU-DIPG-IV (DIPG-4), SU-DIPG-VI (DIPG-6), SU-DIPG-XIII (DIPG-13), SU-DIPG-XVII (DIPG-17), and SU-DIPG-XIV (DIPG-24) were obtained through Material Transfer Agreements (MTA) that were subject to Institutional Review board (IRB) approval and protocols.
Luciferase/RFP Lentiviral Transduction
A detailed protocol for luciferase/RFP lentiviral transduction is provided in Supplementary Methods 2.
Inducible shRNA Lentiviral Transduction
A detailed protocol for inducible shRNA lentiviral transduction is provided in Supplementary Methods 3.
In Vitro Cytotoxic Payload Cell Viability Studies
A detailed protocol for in vitro cytotoxic payload cell viability studies is provided in Supplementary Methods 4.
Immunoblotting
A detailed protocol for immunoblotting is provided in Supplementary Methods 5.
Jess Simple Western Immunoblotting
A detailed protocol for simple western is provided in Supplementary Methods 6.
Live-Cell FACS Antibody Binding Studies
DIPG cells were dissociated with 1 mL 0.05% trypsin (Cat #25300-54, Life Technologies, Grand Island, NY, USA) for 5 mins to obtain single-cell suspensions. 1 mL 0.5% BSA in PBS wash buffer was added to 1.5 mL conical tube to wash cells with a pipette three times, then centrifuged and the supernatant decanted. Cells were counted then resuspended at 106 cells per 100 µL of flow cytometry staining buffer (Cat# FC-001; R&D Systems). one microgram Fc receptor binding inhibitor polyclonal antibody (Cat# 14–9161-73, Thermo Fisher Scientific) was added per 106 cells, gently vortexed and kept on ice for 15 mins. Specific protocols for conjugated and unconjugated antibodies are provided in the Supplementary Methods 7.
Primary antibodies are provided in supplementary materials.
Antibody Surface Plasmon Resonance (SPR) Avidity Studies
The ligand and analyte injections were performed manually in conjunction with the SPR instrument protocol. In these studies, “ligand” is the protein bound to the sensor (not the cognate biological ligand, eg., IL-13 cytokine). The running buffer (buffer 1) was 10 mM HEPES (Cat# H0887-100ML, Sigma-Aldrich) with 200mM NaCl (Cat# S9888-500G, Sigma-Aldrich) as an additive with the pH adjusted to 7.4. The buffer used for loading the ligand (buffer 2) was 10 mM HEPES, with 200 mM NaCl as an additive with the pH adjusted to 7.8. For binding assays, the histidine-tagged IL13Rα2 (Cat# NBP2-23147, Novus Biologicals, Centennial, CO, USA), was immobilized onto the NTA sensor (Cat# SEN-AU-100-10-NTA, Nicoya Lifesciences, Kitchener, ON, Canada) by manually injecting 200 µL of 100 µg/mL13Rα2 protein solution at 20 µL/minute. The running buffer for the immobilization step was buffer 2. The ligand injection was performed three times. Next, the unoccupied area of the NTA sensor was blocked by injecting 150 µL of 100 µg/mL histidine-tagged streptavidin (Cat# ab78833, Abcam, Cambridge, MA, USA) as the blocking protein. Two injections of the blocking protein were performed. Thereafter, the buffer was switched to buffer 1. The analytes, antibodies 2E10 or clone 47, were diluted in buffer 1 to varying concentrations (10, 20, 30, 50, 70 and 100 µg/mL) and 200 µL injections were made at a flow rate of 35 µL/minute. A regeneration step was required after each analyte injection. The regeneration chemical used was 1 mL of glycine-HCl, pH 1.5 (Cat#REG-1.5, Nicoya Lifesciences). All SPR experiments were conducted on a two-channel Nicoya OSPR4.0 instrument at room temperature using different chips at different time points under the same procedures, and the most stable titration curves and Kd values shown in the manuscript.
Antibody VH and VL Sequencing
Sequencing of VH and VL regions was performed by Rapid Novor (Kitchener, ON, N2G 4X8).
Computational Analysis of Avidity and Residue Binding of IL13Rα2 Monoclonal Antibodies
The molecular structures of the clone 2E10 monoclonal antibody and the clone 47 monoclonal antibody were predicted by the AlphaFold2 program through the ColabFold system.33 The model building was limited to the Fab regions, truncated the C-terminal Fc domain of Heavy chains. Both the predicted structures have ordinary Fab forms, and only CDR loops show marked difference between 2E10 and clone 47 antibodies. For residue binding analysis, ZDOCK generated 2,000 predicted binding poses, with additional description available in the Supplementary Methods 8.
Construction of Human-Mouse Chimeric IL13Rα2 ADC (Anti-IL13Rα2::deruxtecan ADC)
A recombinant human-mouse chimeric IL13Rα2 IgG1 antibody (9.44µg/µL) with human Fc derived from murine monoclonal IgG2 2E10 VH and VL sequencing (designated h-2E10) was used to generate the ADC through a maleimide-cysteine linking method using the GGFG-DXd linker payload.
The anti-IL13Rα2::deruxtecan ADC was constructed by WuXi Biologics (Wu Xi City, China). Detailed methods are provided in the Supplementary Methods 9.
In Vitro Cell Viability Studies Using the Human-Mouse Chimeric Anti-IL13Rα2::deruxtecan ADC
Cells in single-cell suspension were plated in a white walled 96-well plate (Cat #136101, Thermo Fisher Scientific) at 150 µL/well growth medium at specific populations in triplicate: SF-8628 at 750 cells/well, DIPG-A, DIPG-4, DIPG-6, DIPG-17 and DIPG-24 at 1500 cells/well. Cells in plates were incubated overnight, then were exposed to unconjugated antibody and human chimeric anti-IL13Rα2::deruxtecan ADC at specific concentrations provided in Supplementary Methods 4. Cells were incubated at 37°C with 5% CO2 after 144 hours, then assayed with CellTiterGlo Luminescent Cell Viability Assay by the manufacturer’s protocol. Luminescence was measured using a BioTek Synergy HT plate reader and IC50 values were calculated using Prism. Each agent-cell line pair was performed in triplicate.
Ex Ovo Anti-Tumor Efficacy Studies Using the Human-Mouse Chimeric Anti-IL13Rα2::deruxtecan ADC
A detailed protocol for ex ovo anti-tumor efficacy studies using ADC is provided in Supplementary Methods 10.
In Vivo Anti-Tumor Efficacy Studies Using the Human-Mouse Chimeric Anti-IL13Rα2::deruxtecan ADC
Six-week-old Nod Scid Gamma (NSG, NOD.Cg-Prkdc<scid> Il2rg<tm1Wjl>/SzJ, Strain: 005557) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). To perform the orthotopic intracranial xenograft of DIPG cells, the bregma of each mouse was identified at a point 1 mm anterior and 2 mm lateral. Each mouse was anesthetized and held in place via mouse adaptor for a stereotaxic surgery device (Cat# 68513, RWD, Shenzhen, Guangzhou, China), and then a hole was made through the skull using a #8 bit in a bone spur drill (Cat# 097852, Fine science tools, Foster City, CA, USA). Next, 500,000 SF-8628 RFP+ Luciferase+ cells in 4 µL PBS were drawn in a 10 μL Hamilton syringe with a 26 G needle. The needle was stereotactically driven 3 mm deep into the brain where 1 µL of cells were injected per min over 3 mins using a digital micropump (Cat# 173128, World Precision Instruments, Sarasota, FL, USA) by a syringe pump controller (Cat# MICRO2T, World Precision Instruments).
Seven days following tumor cell injection, a 10 μL Hamilton syringe with a 33 G needle was drilled through the original skull and 6 μL of 0.317 nmol h-2E10 antibody, 2.536 nmol deruxtecan or 0.317 nmol ADC were injected into the designated intracranial tumor site for 10 mins at a speed of 0.6 μL/min through the digital pump group. The needle stayed in the brain for 5 mins to prevent drug spillage. The solvent for unconjugated antibody and ADC was PBS. The solvent of deruxtecan was 90% corn oil + 10% DMSO. In vivo bioluminescent imaging (BLI) was performed prior to the direct intratumoral injection (IT) of drug or vehicle, and again 7 days later, using 300 µL of 5 mg/mL Cycluc1 Injectable Luciferin Analog (Cat#BIO-545, Medilumine, Montreal, Quebec, Canada) per mouse with an IVIS imaging system (Perkin-Elmer, Waltham, MA) under isoflurane anesthesia. Bioluminescent imaging was performed weekly until day 30 (end of therapy, and sacrifice of first set of animals) and again at day 42 (end of off-treatment observation, sacrifice of second set of animals). All mice were sacrificed at the termination of these studies after long-term observation (maximum 136 days follow-up from time of drug injection). The mice were euthanized using carbon dioxide asphyxiation in accordance with IACUC protocols. The mice were weighed three times a week and observed for behavioral abnormalities of eating, drinking, grooming and locomotion.
All studies in mice were performed after receiving approval from the institutional animal care and use committee (IACUC) at Children’s Cancer Therapy Development Institute and in accordance with relevant guidelines and regulations. The studies were conducted in accordance with ARRIVE guidelines.
Statistics
We used one-way ANOVA analysis to compare luminescence data from different treatment cohorts in the ex ovo studies, and used the Benjamini, Krieger and Yekutieli two-stage linear step-up procedure for post-hoc analysis performed in GraphPad Prism.
We used two-way mixed ANOVA performed in R version 4.2.2 using “anova_test” in the rstatix package through RStudio. Two-way mixed ANOVA was used to analyze luminescence data from the in vivo ADC mouse model study, analyzing all data points up to day 30 (end of therapy) and day 42 (end of off-treatment observation). Significant two-way interactions were analyzed post-hoc using “pairwise_t_test” in the rstatix package, with the Bonferroni method to adjust p-values. The same approach was used to analyze differences in group body weight. Data is presented as mean ± SD.
For this study, p < 0.05 was considered statistically significant.
Results
IL13Rα2 Protein Is Differentially Expressed in DIPG Tumor Tissue Versus Normal Brain
To quantify the protein expression of IL13Rα2 in human DIPG tissue samples, normal brain tissue and corresponding DIPG cell models, we conducted Western blotting to compare expression on eight matched human DIPG tumor tissues and normal brain tissues from autopsy material. We also conducted immunoblotting on eight human cell line models (seven DIPG and one GBM) (Supplementary Tables 2–4). Seven of eight DIPG tissues (88%) displayed IL13Rα2 expression which is consistent with previous tissue expression profiling,32 while IL13Rα2 expression was absent in all normal brain tissues (0%). Three of eight DIPG tissue samples (38%) showed strong IL13Rα2 expression. Four cell lines showed strong IL13Rα2 expression (SJ-GBM2, SF-8628, DIPG-4 and DIPG-17), DIPG-6 showed moderate IL13Rα2 expression, and three cell lines (DIPG-A, DIPG-13 and DIPG-24) showed low or absent IL13Rα2 expression (Figure 1a–c, Supplementary Figure 1). Cellular fractionation was conducted on cell lines SJ-GBM2, SF-8628, DIPG-4 and DIPG-17. IL13Rα2 expression was found to be primarily membrane-localized (Supplementary Figure 2), supporting its potential as an accessible target for ADC-based interventions. To verify the specificity of the two antibodies (2E10, clone 47) for IL13Rα2, we stably transduced the IL13Rα2-high DIPG cell line SF-8628 with a doxycycline-inducible lentivirus encoding a shRNA to IL13Rα2 and repeated immunoblots (Figure 1d). The IL13Rα2 expression characterization in cell lines informed subsequent experimental design.
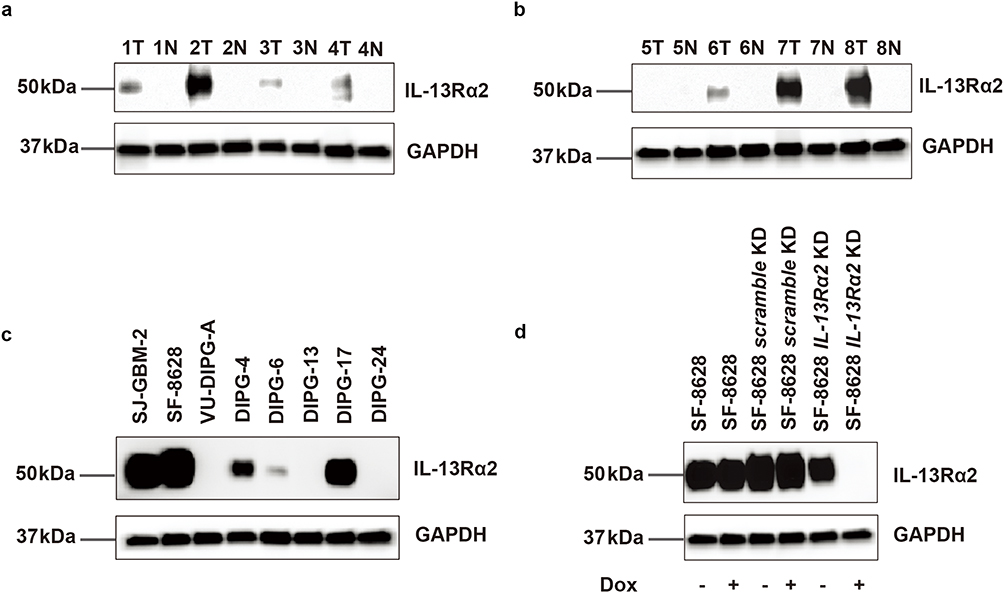 |
Figure 1 IL13Rα2 protein immunoblotting in DIPG. Blots have been cropped to remove space between the protein of interest and the Gapdh control. (a and b) 8 human DIPG tissues versus matched normal brain tissues for IL13Rα2 protein expression. T, tumor tissue, N, normal brain tissue. Numbers are sample numbers. (c) Differential expression of IL13Rα2 protein in 7 DIPG cell line models and 1 GBM cell line model. SJ-GBM-2 is the GBM cell line. (d) Doxycycline-inducible shRNA knockdown of IL13Rα2 in SF-8628 with + representing doxycycline treated and – representing untreated. Uncropped immunoblots are presented in Supplementary Figure 1. |
Topoisomerase Inhibitors are Among the Active Candidate Payloads in DIPG Cell Lines
In clinical trials, DIPG has been unresponsive to chemotherapy agents, including the alkylating agent temozolomide that is a mainstay for other brain tumors.34 The reason for the insensitivity to standard chemotherapy agents has been partially attributed to constraints around drug delivery, which can potentially be overcome by convection-enhanced delivery (CED) should an active agent be developed.35 To this end, we used the six representative DIPG cell lines from the full set described above (SF-8628, DIPG-4, DIPG-6, DIPG-17, DIPG-A, and DIPG-24) to test conventional and unconventional ADC payloads. Because in vitro ADCs require longer exposure to exert effect, we extended drug sensitivity testing up to 6 days (144 hours).
Excluding some resistant DIPG cell lines (DIPG-6 and DIPG-24), the 6-day half maximal inhibitory concentrations (IC50) of exatecan (the chemotherapy from which deruxtecan, Supplementary Figure 3, is derived), thapsigargin, MMAE, DM1, PBD monomer and PBD dimer against DIPG were all less than 10 nM (Supplementary Table 5, Supplementary Figure 4). Data from 3-day studies were also generated (Supplementary Table 6). The set of effective agents along with existing linker chemistry platforms for several of the payloads allowed us to pursue validated payload linker chemistries. PBDs as DNA-binding payloads were excluded because of trends of PBD-containing ADCs being discontinued clinically.36 We also elected not pursue the tubulin inhibitor MMAE as a payload due to conjugation chemistry-associated difficulties around generating ADC products with homogenous drug:antibody ratios (DAR), as ADC heterogeneity is thought have contributed to clinical failure of ADCs for the treatment of GBM.37 MMAF, a hydrophobic analogue of MMAE, has attracted attention because of its safety profile, as MMAE is not extravasated into adjacent tissue. Previous studies have shown that MMAF and MMAE have the same potency after entering cells.38,39 However, depatuxizumab mafodotin failed in Phase III clinical trials for GBM40,41 and belantamab mafodotin failed in phase III clinical trials for multiple myeloma, both of which delivered MMAF as the payload.
The microtubule inhibitor DM1 (mertansine) has the unique chemistry of attaching to the lysine site of the antibody rather than the disulfide bond between the broken antibody chains, which in theory maintains optimal target-binding of the antibody–drug conjugate. Consistent <10 nM activity of DM1 in all cell lines suggests this payload may have applicability in DIPG.
Finally, deruxtecan is a drug-linker form of the clinically investigated chemotherapeutic agent exatecan.42 The dose-limiting adverse effects of exatecan were neutropenia and liver dysfunction in clinical trials.43 Given the 6-day IC50 of 50 nM or less in 5 out of 6 DIPG cell lines, the topoisomerase inhibitor deruxtecan was taken as plausible for the construction of ADC for the treatment of brain tumors. In addition, the ADC synthesized by deruxtecan has a homogenous DAR of 8. Deruxtecan takes all the interchain disulfide bonds broken by the antibody, which makes the deruxtecan-based ADC a homogeneous ADC and avoids the risks potentially associated with heterogeneous ADCs. Although CPT homologous derivatives are the substrate of the P-glycoprotein efflux pump, there may be a risk of drug resistance.44 But a strong trend in the pharmaceutical field related to the anti-HER2 ADC trastuzumab deruxtecan drives us to pursue deruxtecan. Thus, deruxtecan was selected as the ADC toxicity payload-linker for the next series of studies.
Membrane-Bound IL13Rα2 Can Be Recognized in Live Cells by Commercial Monoclonal Antibodies
The next stage in ADC design required selection of an anti-IL13Rα2 antibody capable of recognizing native IL13Rα2 protein on live cells. We studied 3 commercially available antibodies (clone 2E10 from Sigma Aldrich, clone 47 from Biolegend, and clone E7U7B from Cell Signaling Technology) by flow cytometry on DIPG cell lines (SF-8628 and DIPG-24). Each antibody was preincubated with SF-8628 or DIPG-24 cells at the same concentration for 2 hours before performing detection of antibody binding via flow cytometry. Antibody clone 47 had the highest binding affinity to SF-8628 cells (96.6% positive cells), followed by antibody 2E10 (52.8% positive cells) (Figure 2a–d), whereas E7U7B did not bind (data not shown). Antibody clone 47 identified sparsely IL13Rα2 positive cells in cell line DIPG-24 (9.9% positive cells), not otherwise seen in the Western blot (Figure 1c), whereas 2E10 and E7U7B did not bind.
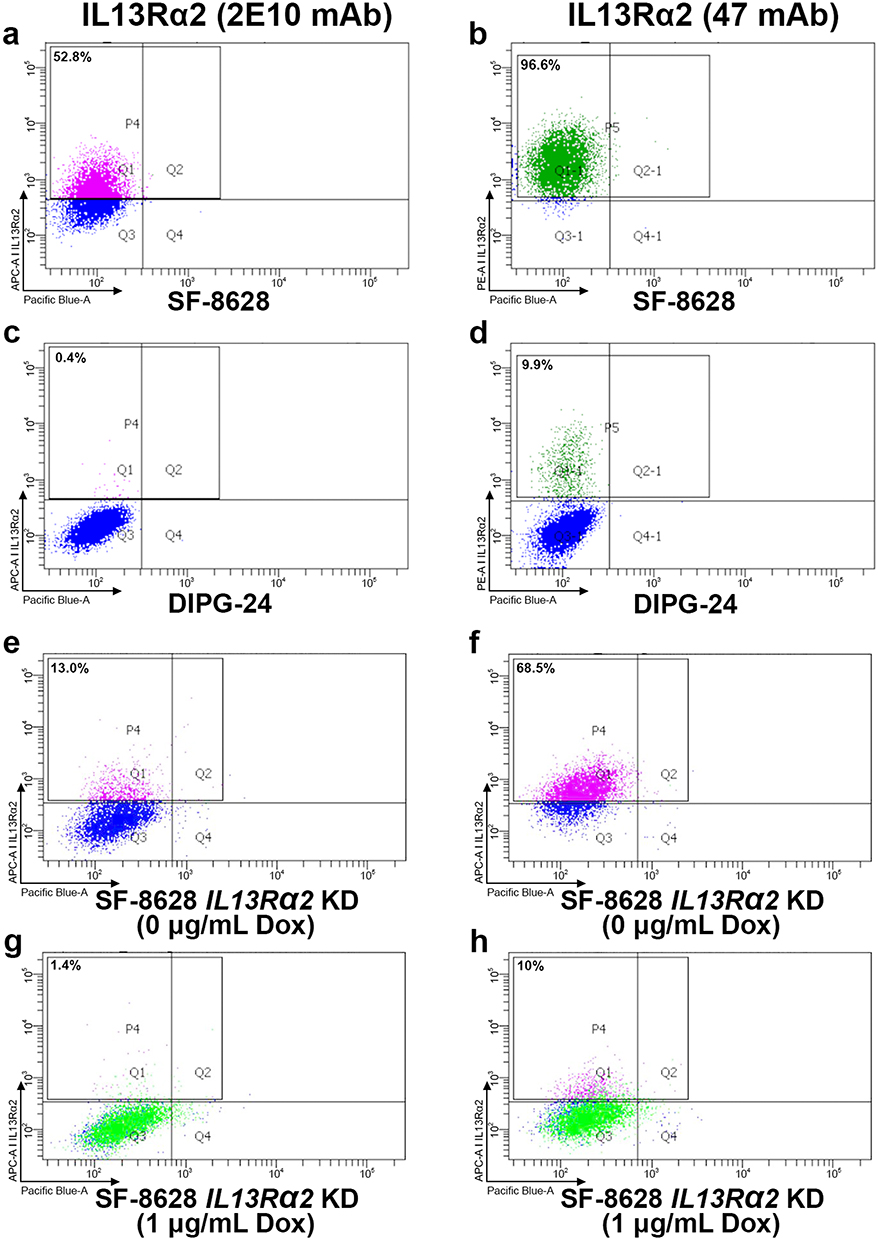 |
Figure 2 FACS validation of antibody binding for DIPG cell lines. Percentage represents the proportion of cells bound by labeled antibody. (a and b) Binding of 2E10 antibody and clone 47 antibodies in cell line SF-8628, respectively. (c) and (d) Binding of 2E10 antibody and clone 47 antibodies in cell line DIPG-24, respectively. (e and f) Antibody binding of 2E10 antibody and clone 47 antibodies, respectively, for cell line SF-8628 in the absence of doxycycline (Dox). (g and h) Diminished antibody binding of 2E10 antibody and clone 47 antibodies, respectively, for cell line SF-8628 in the presence of Dox and IL13Rα2 knockdown (see Figure 1d). nb. The APC-A channel and PE-A channel were used to detect Alexafluor conjugated secondary antibodies, whereas the Pacific Blue-A channel was used for autofluorescence. Gating in the APC-A channel and PE-A channel were done relative to the isotype control for each primary antibody. |
Antibodies 2E10 and Clone 47 Have Specificity for IL13Rα2 in Viable Cells
To verify the specificity of two antibodies (2E10, clone 47) against IL13Rα2 in living cells, we stably transduced the IL13Rα2-high DIPG cell line SF-8628 with a doxycycline-inducible lentivirus encoding a shRNA to IL13Rα2 (the same knockdown construct validated by immunoblotting in Figure 1d). Doxycycline-treated SF-8628 cells or doxycycline-untreated cells were incubated with clone 2E10 and clone 47 antibodies for two hours, then the specificity of IL13Rα2 staining on live cells was detected on FACS (Figure 1d). The percentage of positive stained SF-8628 cells induced by doxycycline (1.4% and 10% for clone 2E10 and clone 47 antibodies, respectively) was reduced compared positive stained SF-8628 cells without doxycycline (13% and 68.5%, respectively), confirming that both antibodies have specificity for IL13Rα2 in living cells (Figure 2e–h).
Computational Analysis of IL13Rα2 Antibody Structure Identifies Differential Avidity in 2E10 and Clone 47 Antibodies
Computational analysis of 2E10 antibody predicted the complementarity-determining region (CDR) loops are compactly folded except for H2 (Figure 3a, Supplementary Figure 5, Supplementary Tables 7–9). The H2 loop has three Tyr residues, two of which are located at the CDR surface, which could strongly contribute to antigen recognition. Two Gln residues at the top of H2 extend outside, which may also interact with the IL13Rα2 antigen (rcsb.org/3d-view/3LB6/1). A side chain of Arg52 on the H2 loop is positioned at the antigen–antigen interface between H2 and H3 loops. A marked characteristic of clone 47 is the lengths of H3 and L1 CDR loops (Figure 3b). Both the H3 and L1 loops extend outside, and Tyr appears at the top of L1 loop. The presence of Tyr in the L1 loop is sometimes observed in antibodies, which effectively increases the binding affinity with antigens. Tyr residues also appear at the top of the H2 and H3 loops. Another characteristic of clone 47 is the presence of five Asp residues in the H2 loop, resulting in a positive charge bias which impacts the specificity of antigen recognition. Binding scores across the proteins are given in Figure 3C.
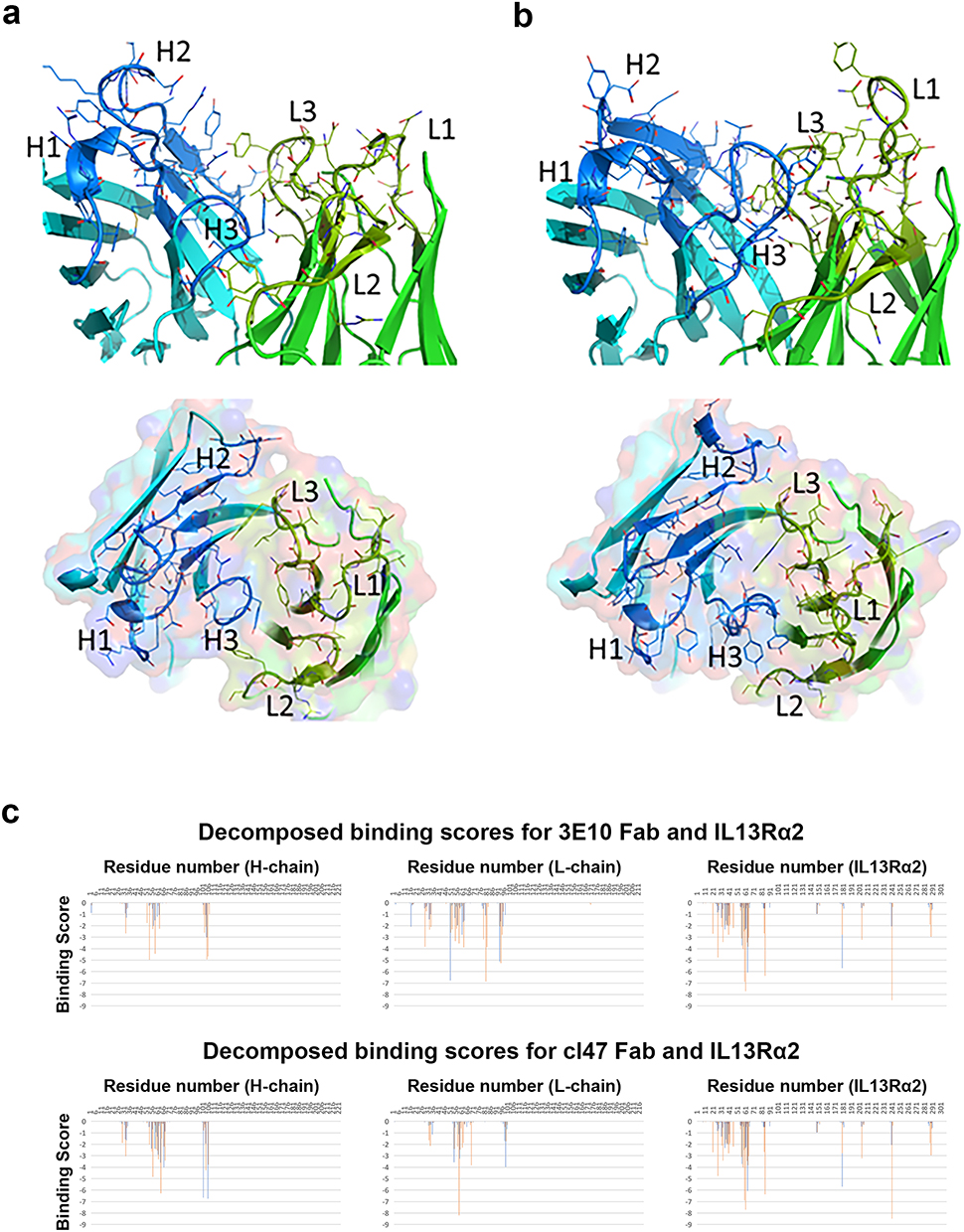 |
Figure 3 Predicted structures of anti-IL13Rα2 antibody molecules. (a) Clone 2E10 predicted structure and (b) clone 47 predicted structure. (upper) Antibody fragment variable region. (lower) Top view of the variable region. The heavy and light chains are depicted in blue and green with the cartoon representation. The side chains of the CDR loops are shown in sticks. The labels, H1, H2, H3, L1, L2, and L3, indicates the positions of the CDR loops. (c) Binding scores at the amino acid level for the heavy and light chains of mAb clone 2E10 (top) and mAb clone 47 (bottom). |
In Surface Plasmon Resonance (SPR) Avidity Testing, Anti-IL13Rα2 Antibody 2E10 Had the Lowest Kd
The affinity of each antibody for IL13Rα2 binding was determined again by SPR. The results shown in Figure 4 demonstrate the Kd of clone 2E10 was higher than clone 47 (Kd = 1.17e−6 M vs 2.04e−5 M, respectively (Figure 4a and b).
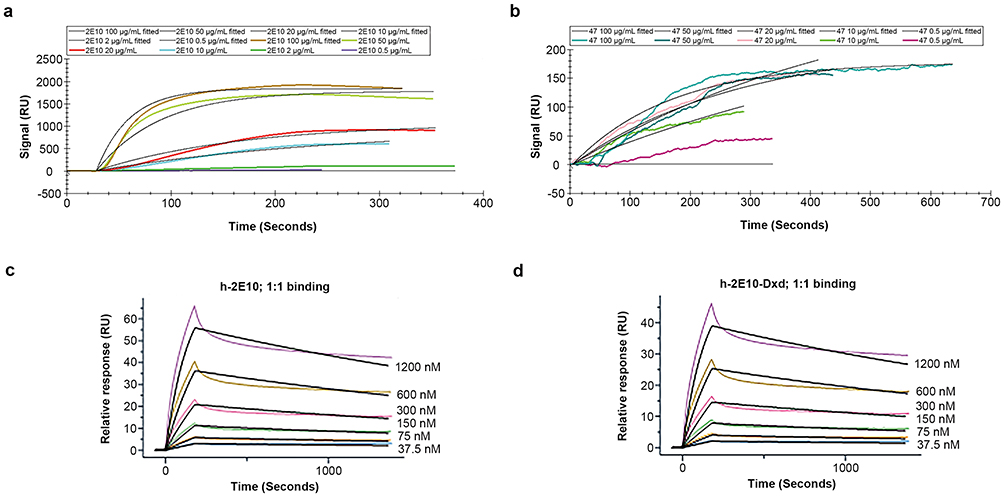 |
Figure 4 Surface plasmon resonance detection of human IL13Rα2 binding to naked antibodies (antibody 2E10, antibody clone 47), the chimeric antibody (h-2E10) and ADC (h-2E10-DXd). (a and b) Analyte concentrations of 2E10 and antibody clone 47 in panels (a and b) are 0.5, 2, 10, 20, 50, and 100 μg/mL. Panels (a and b) use the same NTA sensor chip pre-incubated with ligand-histidine (His)-labeled protein IL13Rα2. (a) Dissociation curve of antibody 2E10 binding ligand in SPR, Kd=1.17e-6 M. (b) Dissociation curve of antibody clone 47-binding ligand in SPR, Kd=2.04e-5 M. (c and d) NTA sensor was preincubated with chimeric antibody h-2E10 (as ligand). (c) Dissociation curve of human-IL13Rα2 protein by SPR after ligand binding, Kd=5.4e-8 M. (d) Dissociation curve of human-IL13Rα2 by SPR on an NTA sensor chip preincubated with ligand (ADC h-2E10-DXd), Kd =5.5e-8 M. |
A Human-Mouse Chimeric Anti-IL13Rα2::deruxtecan ADC Could Be Constructed and Showed Higher Affinity Than the Original Antibody
Having selected clone 2E10 for partial humanization, we humanized the Fc moiety and switched from antibody from the IgG2 to IgG1 isotype. We then used this novel chimeric antibody, designated h-2E10, to construct an antibody–drug conjugate using the deruxtecan payload-linker, designated h-2E10-DXd. The concentrations, molecular weights, and working concentrations of the antibody and ADC were 9.44 μg/μL, 145,000 mol/g, 0.06511 nmol/μL and 8.4 μg/μL, 159,129 mol/g, 0.05283 nmol/μL respectively. For the ADC, the DAR was 7.99, endotoxin level was 0.011 EU/mg and free drug was <0.079% mol/mol. The Kd values of h-2E10 and h-2E10-DXd were 5.41e−8 M and 5.49e−8 M, respectively, which is higher than for the 2E10 monoclonal antibody (Figure 4c and d).
The in Vitro Sensitivity of the Human-Mouse Chimeric Anti-IL13Rα2::deruxtecan ADC Was Consistent with the Expression of IL13Rα2 in DIPG Cell Lines
To demonstrate whether the response to an anti-IL13Rα2 ADC was related to IL13Rα2 expression in DIPG, we examined the IC50 values of the chimeric anti-IL13Rα2::deruxtecan ADC on different DIPG cell lines. The IC50 values for DIPG-4, DIPG-17 of SF-8628 (all high IL13Rα2 expression) were 337 nM, 74 nM and 175 nM, respectively, while the IC50 values of DIPG-A, DIPG-6 and DIPG-24 (with low IL13Rα2 expression) were above 1000 nM (Figures 1c and 5a). These results indicate that the dose-dependent sensitivity to the anti-IL13Rα2 ADC is positively correlated with IL13Rα2 expression. As a control, we examined the effect of unconjugated IL13Rα2 antibody (h-2E10) on cell viability and detected no effect on viability of DIPG cell lines in the same time course (Figure 5b).
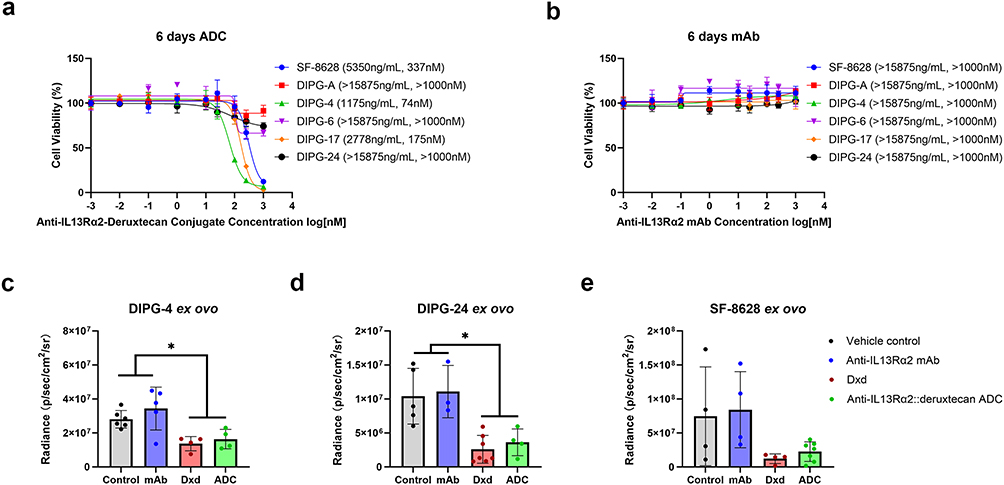 |
Figure 5 Cell growth inhibition by the anti-IL13Rα2::deruxtecan ADC versus h-2E10 mAb in DIPG cell lines (DIPG growth inhibition by the anti-IL13Rα2::deruxtecan ADC versus h-2E10 mAb in DIPG cell lines and by the vehicle control, h-2E10 mAb, DXd and anti-IL13Rα2::deruxtecan ADC in DIPG ex ovo xenograft models) (a) Cell viability of six DIPG cells (SF-8628, DIPG-A, DIPG-4, DIPG-6, DIPG-17 and DIPG-24) incubated with anti-IL13Rα2::deruxtecan ADC for 144 hours. (b) Cell viability of six DIPG cells (SF-8628, DIPG-A, DIPG-4, DIPG-6, DIPG-17 and DIPG-24) incubated with h-2E10 mAb lacking a payload. Values in parentheses are the IC50 values. (c) Tumor viability of DIPG-4 incubated with vehicle control, h-2E10 mAb, DXd and anti-IL13Rα2::deruxtecan ADC for 144 hours in the DIPG ex ovo xenograft model. (d) Tumor viability of SF-8628 incubated with vehicle control, h-2E10 mAb, DXd and anti-IL13Rα2::deruxtecan ADC for 144 hours in the DIPG ex ovo xenograft model. (e) Tumor viability of DIPG-24 incubated with vehicle control, h-2E10 mAb, DXd and anti-IL13Rα2::deruxtecan ADC for 144 hours in the DIPG ex ovo xenograft model. * Represents a statistically difference with a p-value less than 0.05. |
The Ex Ovo Tumor Growth Inhibition of the Human-Mouse Chimeric Anti-IL13Rα2::deruxtecan ADC Could Be Demonstrated in Three DIPG Cell Line-Derived Ex Ovo Xenograft Models
To demonstrate the tumor growth inhibition of the novel ADC, we generated three ex ovo xenograft models using the human DIPG-4, DIPG-24 and SF-8628 DIPG cell lines. The four treatment cohorts per cell line were: vehicle control (PBS), chimeric mAb (h-2E10), DXd and ADC (h-2E10-DXd). After xenograft establishment and then treatment, tumor viability was measured by bioluminescence assay. For the cell line DIPG-4 with high IL13Rα2 expression, the tumor viability of the ADC-treated and payload-treated was significantly lower than that of the h-2E10 mAb group and the vehicle control group (Figure 5c). For the cell line SF-8628 with high IL13Rα2 expression, the ADC and payload treatment resulted in a similar trend in decreased tumor viability but did not reach statistical significance due to variability in the vehicle control and chimeric mAb cohorts (Figure 5d).
For the cell line DIPG-24 with lower IL13Rα2 expression, the tumor viability of the ADC-treated and payload-treated low IL13Rα2 expression DIPG-24 was significantly lower than that of the h-2E10 mAb group and the vehicle control group, which demonstrated this constructed ADC is sensitive to the low IL13Rα2 expression DIPG tumor (Figure 5e). This result was consistent with significant therapeutic effect of trastuzumab deruxtecan in HER2-Low breast cancer.45
The in Vivo Activity of the Human-Mouse Chimeric Anti-IL13Rα2::deruxtecan ADC Was Confirmed in an IL13Rα2 Expressing Cell Line-Derived Human Orthotopic Xenograft Model
To further validate the therapeutic effect of the constructed ADC, we established an orthotopic xenograft model using the SF-8628 human DIPG cell line (cells growing in vivo Supplementary Figure 6a and injection site is shown in Supplementary Figure 6b). The three treatment arms were the chimeric mAb (h-2E10), deruxtecan and the ADC (h-2E10-DXd). Tumors were engrafted on Day −7, a single dose of the appropriate therapeutic was administered on Day 0, and tumor volume was tracked via luminescent imagine every 4–7 days during the first 30 days and endpoint is Day 136 (Figure 6a). The tumor volume over time of the ADC-treated mice was significantly smaller (p < 0.05) than that of the h-2E10 mAb group at days t = 30 and t = 42 and was similar to that of the payload-linker group that received the same final cytotoxic payload concentration (20 nM) (Figure 6b and c). Tumor volume on other days were recorded, but analysis was only performed on key milestone dates (days t = 30 and t = 42) during the experiment. In all treatment groups, mice had normal ambulation, grooming, eating and drinking throughout the course of the experiment. The juvenile host mice did not lose weight during the study, but in fact mice generally only gained weight with days, except #11 mouse in the deruxtecan group (Figure 6d and Supplementary Table 10). The study was conducted until Day 136, with mice intermittently harvested starting on day t = 30 to recover tumor tissue for immunoblotting; however, too little tumor tissue was present to perform these pharmacodynamic studies. Overall, these results suggest the feasibility of an anti-IL13Rα2 ADC, delivered via intratumoral injection (eg., CED), as a therapeutic option for DIPG.
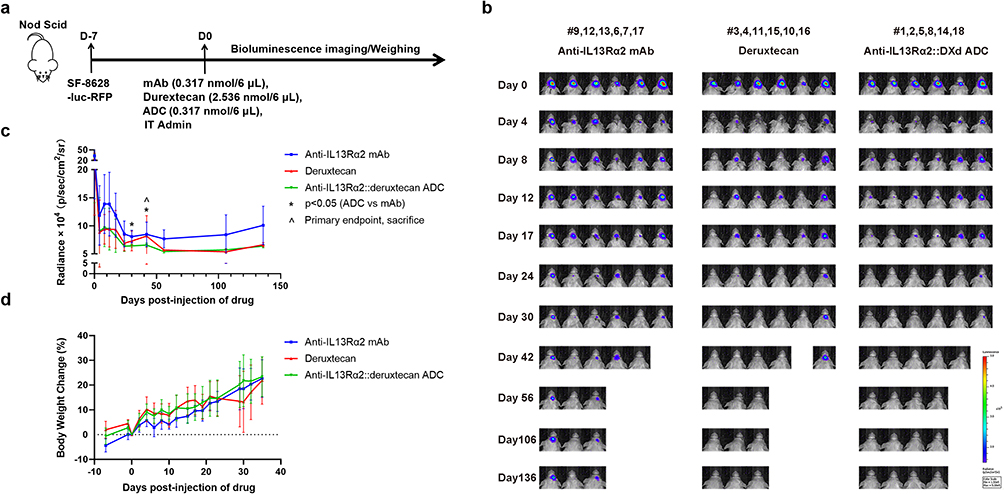 |
Figure 6 The anti-IL13Rα2::deruxtecan ADC in vivo activity in the SF-8628 cell line-derived orthotopic xenograft brain tumor model. (a) 500,000 SF-8628 RFP+ Luciferase+ cells were intracranially injected into Nod Scid mice. After 7 days, mice were divided into 3 groups randomly. One group received 6 μL of 0.317 nmol h-2E10 antibody intratumoral injection, the other group received 6 μL of 2.536 nmol deruxtecan intratumoral injection, the final group was received 6 μL of 0.317 nmol ADC intratumoral injection. (b) Bioluminescence detection of tumor burden in the SF-8628 cell line-derived xenograft model. Color scale: 1.5e5 - 5e5 p/sec/cm2/sr. (c) Quantification of SF-8628 tumor burden bioluminescence intensity in SF-8628 CDX model treated with h-2E10 mAb, deruxtecan, or ADC. The line plot corresponds to quantified radiance values from the images in Figure 5b. Each line represents the mean ± SD tumor load bioluminescence intensity of mice in corresponding group. ^ indicates primary endpoint (t = 42), * = p < 0.05 (ADC vs mAb cohorts) at two analyzed time points (day t = 30, and day t = 42) from two-way mixed ANOVA. (d) Percent change in mean body weight of mice in SF-8628 CDX model treated with h-2E10 mAb, deruxtecan, or ADC. Each line represents the mean ± SD percent weight gain change in the corresponding group. Injection site validation images for mouse studies are given in Supplementary Figure 6. |
Discussion
In our previous published studies, IL13Rα2 is highly expressed in DIPG and absent in normal brain tissues by RNA sequencing and immunohistochemistry, ligand stimulation of IL13Rα2 does not enhance DIPG proliferation or invasion, and a mouse-based anti-IL13Rα2 ADC was effective in the treatment of DIPG in vitro.2,32
The present study further advances the concept of an anti-IL13Rα2 ADC as a treatment for DIPG. We first confirmed that IL13Rα2 was highly expressed at the protein level in DIPG samples but absent in normal brain tissues by immunoblotting. Next, we selected the most plausible payload (DXd) from a clinical point of view activity across DIPG cell lines and acceptance for clinical development. We then determined a monoclonal antibody with affinity and selectivity (clone 2E10) from which we designed a partially humanized (chimeric) antibody and then an anti-IL13Rα2::deruxtecan ADC. Importantly, the chimeric antibody and chimeric ADC demonstrate improved binding properties (Kd values) over the original 2E10 monoclonal antibody, potentially due to higher purity and integrity of intact antibodies resulting from creation of the chimeric products. Finally, the human-mouse chimeric anti-IL13Rα2::deruxtecan ADC was effective and selective across a range of IL13Rα2 cell lines in vitro, three DIPG engrafted quail models ex ovo and a single cell-line derived orthotopic allograft model in vivo using as little as a single intratumor ADC injection.
For the result that the human-mouse chimeric anti-IL13Rα2::deruxtecan ADC is effective in the low (sparse) IL13Rα2 expression cell line DIPG-24 ex ovo xenograft model, but the same treatment was less effective in DIPG-24 in vitro, one explanation could be dilution of Dxd after cleavage from ADC into a large volume of cell culture medium in vitro, lessening the bystander effect. Future studies will investigate whether intratumor heterogeneity of IL13Rα2 expression at the cell membrane exists, and whether IL13Rα2 expression varies with cell cycle phase.
The unexpected tolerability of the payload by intratumoral injection for the orthotopic mouse model bears mention, but using exatecan or its derivatives by CED or other intratumoral approach would need extensive other studies to validate.
Deruxtecan, an ADC drug-linker conjugate, recently received FDA approval for HER2-low breast cancer as part of the ADC trastuzumab deruxtecan.45,46 Deruxtecan consists of the cleavable glycine–glycine–phenylalanine–glycine tetrapeptide-based linker, a self-immolative amino methylene spacer, and a topoisomerase 1 inhibitor payload derived from exatecan, a clinically investigated structural analog of the topoisomerase 1 inhibitor camptothecin (CPT). Four CPT analogues (topotecan, irinotecan, belotecan, and deruxtecan) and the active metabolite of irinotecan (SN-38) have all been utilized in clinically active ADC programs with trastuzumab deruxtecan and Sacituzumab govitecan (SN-38) granted FDA approval.
The CPT derivative topotecan, delivered through CED, was demonstrated to be tolerable and to extend survival time in glioblastoma multiforme (GBM) tumors in small animal models (mice), large animal models (pigs), and human patients.12 Moreover, topotecan is toxic to glioma tumor cells and relatively non-toxic to normal brain tissue in all tested species, resulting in a CPT derivative with a high therapeutic index in GBM.13 However, CPT analog SN-38 is a substrate of P-glycoprotein efflux pump, which presents a risk of drug resistance,44,47 highlighting the importance of careful selection of cytotoxic payload in therapeutic design.
The dogma that ADCs improve selectivity of traditional chemotherapy payloads has recently been challenged, in part because the linker-payload itself has its own unique and sometimes paradoxical toxicity profile.48 Nevertheless, the clinical efficacy of an ADC (fam-trastuzumab deruxtecan-nxki) with next-generation linker technology has dramatically improved outcomes in HER2-low metastatic breast cancer and brought renewed interest in ADCs.49
A potential limitation of this study is that we had no DIPG cell lines available that are IL13Rα2 non-expressing but sensitive to an exatecan-derived payload. As summarized in Supplementary Table 11, cells sensitive to exatecan or its derivatives were sensitive to the anti-IL13Rα2 ADC, while also cells moderately or strongly IL13Rα2 positive were sensitive to the anti-IL13Rα2 ADC. Interestingly, in the ex ovo studies of the payload (DXd) and the anti-IL13Rα2 ADC in DIPG-24 (trace IL13Rα2 protein cell surface expression detected on flow cytometry), DIPG24 was sensitive to both the anti-IL13Rα2 ADC and the positive control DXd. To address this conundrum, further cell lines will be sought that are IL13Rα2 non-expressing but sensitive to an exatecan-derived payload.
From a clinical translation point of view, an ADC would need to be delivered intratumorally to avoid exclusion from the BBB. Fortunately, CED as an intratumoral injection technique is already adopted as a clinically investigated mode of drug delivery.50,51 For example, a clinical trial of topotecan using CED for the treatment of high-grade gliomas is ongoing (NCT02278510). Pre-IND studies to test the suitability of ADC and CED catheter type would need to be conducted prior to finalizing a clinical trial concept.
DIPG is a relatively unique use case amongst ADC therapeutics because the ADC needs to be delivered by CED. However, antibody-based therapeutics have been delivered into DIPG tumors clinically and show a predictable volume of distribution.15 The effectiveness of a CED-delivered antibody-based therapeutic in prospective clinical trials remains to be identified as a benchmark for DIPG.
A limitation of this study is the orthotopic (intracranial) model of DIPG likely required a greater number of tumor cells injected during initial engraftment to develop lethal tumors within the period of observation in the untreated cohort. Rather than repeat intracranial mouse xenograft studies with the chimeric antibody, we deferred such studies until the development of the clinical lead compound, a fully humanized ADC, with which we can perform intracranial mouse xenograft studies using multiple different human DIPG cell lines. Instead, we demonstrated ex ovo activity of our chimeric ADC across a three different cell lines.
From a translational perspective, while a chimeric ADC (brentuximab vedotin) has reached FDA approval, the trend is towards fully humanized antibodies ADCs.52 Creating a fully humanized anti-IL13Rα2 ADC is the next step in drug development, at which time multiple animal models of independent human DIPG tumors will be studied.
In summary, the work presented here is an important preclinical milestone in the development of a new DIPG therapeutic for a patient population that has no life-extending intervention beyond the transient improvement offered by irradiation and represents a blueprint for the development of novel targeted immuno-oncology agents for underserved pediatric patient populations with significant unmet need.
Data Sharing Statement
All data relevant to the manuscript is available in main or supplementary materials.
Ethics Statement
All human tissue samples for primary cell generation were reviewed and approved by the Children’s Cancer Therapy Development Institute’s Institutional Review Board (Advarra, protocol #cc-TDI-IRB-1) and collected through the Cancer Registry for Familial and Sporadic Tumors (CuReFAST) tumor banking study. All patients enrolled in CuReFAST provided informed consent. Patient data and clinical and pathologic information are maintained in a de-identified, encrypted, and secure database.
Acknowledgments
We thank Emma Lashyro, Wonsik Son, Erin Helms and Andrew Woods for technical assistance in this study. We thank Timothy Brown for guidance in study design, as well asMichelle Monte and Suzanne Baker for kindly providing cell lines.
Funding
This work was supported in part by The Lyla Nsouli Foundation, The Matthew Larson Foundation for Pediatric Brain Tumors, and Hero of Children’s Cancer Foundation. Neither foundation was involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit for publication. This work was also funded by the First Affiliated Hospital of Zhengzhou University’s NNSFC matched grant (Grant No: 81771397), as well as impactful fundraising by Ryan McGuire in affiliation with Golf Fights Cancer.
Disclosure
CK has or has had sponsored research agreements with Eli Lilly, Roche-Genentech and Cardiff Oncology as well as research collaborations with Novartis and Nurix and is co-founder of Tio Companies and Therapeutics Ex Ovo. Artisan Biopharma is a wholly-owned subsidiary of cc-TDI. In addition, CK and ARH are non-equity cofounders of Artisan Biopharma which has licensed from Pfizer two anti-IL13ra2 antibodies for ADC development. These antibodies are described in US patent 9,828,428. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Grasso CS, Tang Y, Truffaux N, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. 2015;21(6):555–16. doi:10.1038/nm.3855
2. Lian X, Kats D, Rasmussen S, et al. Design considerations of an IL13Rα2 antibody–drug conjugate for diffuse intrinsic pontine glioma. Acta Neuropathol Commun. 2021;9(1):88. doi:10.1186/s40478-021-01184-9
3. Hoeman CM, Cordero FJ, Hu G, et al. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat Commun. 2019;10(1):1023. doi:10.1038/s41467-019-08823-9
4. Ostrom QT, Cioffi G, Gittleman H, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro Oncol. 2019;21(Suppl Supplement_5):v1–v100. doi:10.1093/neuonc/noz150
5. Dang M, Phillips PC. Pediatric brain tumors. Continuum. 2017;23(6):1727–1757. doi:10.1212/con.0000000000000545
6. Loveson KF, Fillmore HL. Intersection of brain development and paediatric diffuse midline gliomas: potential role of microenvironment in tumour growth. Brain Sci. 2018;8(11):200. doi:10.3390/brainsci8110200
7. Packer RJ, Allen JC, Goldwein JL, et al. Hyperfractionated radiotherapy for children with brainstem gliomas: a pilot study using 7,200 cGy. Ann Neurol. 1990;27(2):167–173. doi:10.1002/ana.410270212
8. Zhou K, Boström M, Ek CJ, et al. Radiation induces progenitor cell death, microglia activation, and blood-brain barrier damage in the juvenile rat cerebellum. Sci Rep. 2017;7(1):46181. doi:10.1038/srep46181
9. Vanan MI, Eisenstat DD. DIPG in children – what can we learn from the past? Front Oncol. 2015;5:237. doi:10.3389/fonc.2015.00237
10. Wang D, Wang C, Wang L, Chen Y. A comprehensive review in improving delivery of small-molecule chemotherapeutic agents overcoming the blood-brain/brain tumor barriers for glioblastoma treatment. Drug Deliv. 2019;26(1):551–565. doi:10.1080/10717544.2019.1616235
11. Warren KE. Diffuse intrinsic pontine glioma: poised for progress. Front Oncol. 2012;2:205. doi:10.3389/fonc.2012.00205
12. Upadhyayula PS, Spinazzi EF, Argenziano MG, Canoll P, Bruce JN. Convection enhanced delivery of topotecan for gliomas: a single-center experience. Pharmaceutics. 2020;13(1):39. doi:10.3390/pharmaceutics13010039
13. Kaiser MG, Parsa AT, Fine RL, Hall JS, Chakrabarti I, Bruce JN. Tissue distribution and antitumor activity of topotecan delivered by intracerebral clysis in a rat glioma model. Neurosurgery. 2000;47(6):1391–1399. doi:10.1097/00006123-200012000-00026
14. Bruce JN, Fine RL, Canoll P, et al. Regression of recurrent malignant gliomas with convection-enhanced delivery of topotecan. Neurosurgery. 2011;69(6):1272–1280. doi:10.1227/NEU.0b013e3182233e24
15. Wembacher-Schroeder E, Kerstein N, Bander ED, Pandit-Taskar N, Thomson R, Souweidane MM. Evaluation of a patient-specific algorithm for predicting distribution for convection-enhanced drug delivery into the brainstem of patients with diffuse intrinsic pontine glioma. J Neurosurg Pediatr. 2021;28(1):34–42. doi:10.3171/2020.11.PEDS20571
16. Drago JZ, Modi S, Chandarlapaty S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat Rev Clin Oncol. 2021;18(6):327–344. doi:10.1038/s41571-021-00470-8
17. Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. 2014;16(10):1304–1312. doi:10.1093/neuonc/nou045
18. Kawakami M, Kawakami K, Kasperbauer JL, et al. Interleukin-13 receptor alpha2 chain in human head and neck cancer serves as a unique diagnostic marker. Clin Cancer Res. 2003;9(17):6381–6388.
19. Okamoto H, Yoshimatsu Y, Tomizawa T, et al. Interleukin-13 receptor α2 is a novel marker and potential therapeutic target for human melanoma. Sci Rep. 2019;9(1):1281. doi:10.1038/s41598-019-39018-3
20. Kioi M, Kawakami M, Shimamura T, Husain SR, Puri RK. Interleukin-13 receptor α2 chain. Cancer. 2006;107(6):1407–1418. doi:10.1002/cncr.22134
21. Husain SR, Obiri NI, Gill P, et al. Receptor for interleukin 13 on AIDS-associated Kaposi’s sarcoma cells serves as a new target for a potent Pseudomonas exotoxin-based chimeric toxin protein. Clin Cancer Res. 1997;3(2):151–156.
22. Jarboe JS, Johnson KR, Choi Y, Lonser RR, Park JK. Expression of interleukin-13 receptor α2 in glioblastoma multiforme: implications for targeted therapies. Cancer Res. 2007;67(17):7983–7986. doi:10.1158/0008-5472.Can-07-1493
23. Joshi BH, Plautz GE, Puri RK. Interleukin-13 receptor alpha chain: a novel tumor-associated transmembrane protein in primary explants of human malignant gliomas. Cancer Res. 2000;60(5):1168–1172.
24. Tu M, Wange W, Cai L, Zhu P, Gao Z, Zheng W. IL-13 receptor α2 stimulates human glioma cell growth and metastasis through the Src/PI3K/Akt/mTOR signaling pathway. Tumour Biol. 2016;37(11):14701–14709. doi:10.1007/s13277-016-5346-x
25. Lupardus PJ, Birnbaum ME, Garcia KC. Molecular basis for shared cytokine recognition revealed in the structure of an unusually high affinity complex between IL-13 and IL-13Rα2. Structure. 2010;18(3):332–342. doi:10.1016/j.str.2010.01.003
26. Seyfizadeh N, Seyfizadeh N, Gharibi T, Babaloo Z. Interleukin-13 as an important cytokine: a review on its roles in some human diseases. Acta Microbiol Immunol Hung. 2015;62(4):341–378. doi:10.1556/030.62.2015.4.2
27. Kawakami K, Taguchi J, Murata T, Puri RK. The interleukin-13 receptor α2 chain: an essential component for binding and internalization but not for interleukin-13–induced signal transduction through the STAT6 pathway. Blood. 2001;97(9):2673–2679. doi:10.1182/blood.v97.9.2673
28. David M, Ford D, Bertoglio J, Maizel AL, Pierre J. Induction of the IL-13 receptor α2-chain by IL-4 and IL-13 in human keratinocytes: involvement of STAT6, ERK and p38 MAPK pathways. Oncogene. 2001;20(46):6660–6668. doi:10.1038/sj.onc.1204629
29. Andrews A-L, Nasir T, Bucchieri F, Holloway JW, Holgate ST, Davies DE. IL-13 receptor α 2: a regulator of IL-13 and IL-4 signal transduction in primary human fibroblasts. J Allergy Clin Immunol. 2006;118(4):858–865. doi:10.1016/j.jaci.2006.06.041
30. Pandya H, Gibo DM, Debinski W. Molecular targeting of intracellular compartments specifically in cancer cells. Genes Cancer. 2010;1(5):421–433. doi:10.1177/1947601910375274
31. Debinski W, Dickinson P, Rossmeisl JH, Robertson J, Gibo DM, Lesniak M. New agents for targeting of IL-13RA2 expressed in primary human and canine brain tumors. PLoS One. 2013;8(10):e77719. doi:10.1371/journal.pone.0077719
32. Berlow NE, Svalina MN, Quist MJ, et al. IL-13 receptors as possible therapeutic targets in diffuse intrinsic pontine glioma. PLoS One. 2018;13(4):e0193565. doi:10.1371/journal.pone.0193565
33. Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nature Methods. 2022;19(6):679–682. doi:10.1038/s41592-022-01488-1
34. Cohen KJ, Heideman RL, Zhou T, et al. Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: a report from the Children’s Oncology Group. Neuro Oncol. 2011;13(4):410–416. doi:10.1093/neuonc/noq205
35. Warren KE. Beyond the blood: brain barrier: the importance of central nervous system (CNS) pharmacokinetics for the treatment of CNS tumors, including diffuse intrinsic pontine glioma. Front Oncol. 2018;8:239. doi:10.3389/fonc.2018.00239
36. Hartley JA. Antibody-drug conjugates (ADCs) delivering pyrrolobenzodiazepine (PBD) dimers for cancer therapy. Expert Opin Biol Ther. 2021;21(7):931–943. doi:10.1080/14712598.2020.1776255
37. Anami Y, Otani Y, Xiong W, et al. Homogeneity of antibody-drug conjugates critically impacts the therapeutic efficacy in brain tumors. Cell Rep. 2022;39(8):110839. doi:10.1016/j.celrep.2022.110839
38. Raposo Moreira Dias A, Bodero L, Martins A, et al. Synthesis and biological evaluation of RGD and iso DGR–monomethyl auristatin conjugates targeting integrin α V β 3. ChemMedChem. 2019;14(9):938–942. doi:10.1002/cmdc.201900049
39. Sutherland MSK, Sanderson RJ, Gordon KA, et al. Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-CD30-auristatin conjugates. J Biol Chem. 2006;281(15):10540–10547. doi:10.1074/jbc.M510026200
40. Padovan M, Eoli M, Pellerino A, et al. Depatuxizumab mafodotin (Depatux-M) plus temozolomide in recurrent glioblastoma patients: real-world experience from a multicenter study of italian association of neuro-oncology (AINO). Cancers. 2021;13(11):2773. doi:10.3390/cancers13112773
41. Marin B-M, Porath KA, Jain S, et al. Heterogeneous delivery across the blood-brain barrier limits the efficacy of an EGFR-targeting antibody drug conjugate in glioblastoma. Neuro Oncol. 2021;23(12):2042–2053. doi:10.1093/neuonc/noab133
42. Rowinsky EK, Johnson TR, Geyer CE Jr, et al. DX-8951f, a hexacyclic camptothecin analog, on a daily-times-five schedule: a Phase I and pharmacokinetic study in patients with advanced solid malignancies. J Clin Oncol. 2000;18(17):3151–3163. doi:10.1200/JCO.2000.18.17.3151
43. De Jager R, Cheverton P, Tamanoi K, et al. DX-8951f: summary of phase I clinical trials. Ann NY Acad Sci. 2000;922(1):260–273. doi:10.1111/j.1749-6632.2000.tb07044.x
44. Mattern MR, Hofmann GA, Polsky RM, Funk LR, McCabe FL, Johnson RK. In vitro and in vivo effects of clinically important camptothecin analogues on multidrug-resistant cells. Oncol Res. 1993;5(12):467–474.
45. Hurvitz SA, Hegg R, Chung W-P, et al. Trastuzumab deruxtecan versus trastuzumab emtansine in patients with HER2-positive metastatic breast cancer: updated results from DESTINY-Breast03, a randomised, open-label, Phase 3 trial. Lancet. 2023;401(10371):105–117. doi:10.1016/S0140-6736(22)02420-5
46. Modi S, Jacot W, Yamashita T, et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N Engl J Med. 2022;387(1):9–20. doi:10.1056/NEJMoa2203690
47. Kawabata S, Oka M, Shiozawa K, et al. Breast cancer resistance protein directly confers SN-38 resistance of lung cancer cells. Biochem Biophys Res Commun. 2001;280(5):1216–1223. doi:10.1006/bbrc.2001.4267
48. Colombo R, Rich JR. The therapeutic window of antibody drug conjugates: a dogma in need of revision. Cancer Cell. 2022;40(11):1255–1263. doi:10.1016/j.ccell.2022.09.016
49. Narayan P, Dilawari A, Osgood C, et al. US food and drug administration approval summary: fam-trastuzumab deruxtecan-nxki for human epidermal growth factor receptor 2-low unresectable or metastatic breast cancer. J Clin Oncol. 2023;41(11):2108–2116. doi:10.1200/JCO.22.02447
50. D’Amico RS, Aghi MK, Vogelbaum MA, Bruce JN. Convection-enhanced drug delivery for glioblastoma: a review. J Neurooncol. 2021;151(3):415–427. doi:10.1007/s11060-020-03408-9
51. Mehta AM, Sonabend AM, Bruce JN. Convection-enhanced delivery. Neurotherapeutics. 2017;14(2):358–371. doi:10.1007/s13311-017-0520-4
52. Fu Z, Li S, Han S, Shi C, Zhang Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct Target Ther. 2022;7(1):93. doi:10.1038/s41392-022-00947-7
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.