Back to Journals » OncoTargets and Therapy » Volume 14
Development and Analytical Validation of a Targeted Next-Generation Sequencing Panel to Detect Actionable Mutations for Targeted Therapy
Authors Wang D, Ma K, Deng W ![]() , Li J, Xiang S, Zhang Y
, Li J, Xiang S, Zhang Y ![]() , Fu Y, Dai H, Huang B
, Fu Y, Dai H, Huang B ![]()
Received 27 December 2020
Accepted for publication 18 March 2021
Published 7 April 2021 Volume 2021:14 Pages 2423—2431
DOI https://doi.org/10.2147/OTT.S299381
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
Dandan Wang,1,2,* Kai Ma,3,* Wei Deng,4,* Jingyu Li,1 Shaohua Xiang,1 Yang Zhang,2 Ying Fu,2 Heng Dai,2 Bingding Huang1,2
1College of Big Data and Internet, Shenzhen Technology University, Shenzhen, Guangdong, 518118, People’s Republic of China; 2Department of Research and Development, Sinotech Genomics Inc., Shanghai, 230001, People’s Republic of China; 3Department of Thoracic Surgery, National Cancer Center, Cancer Hospital & Shenzhen Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Shenzhen, Guangdong, 518116, People’s Republic of China; 4Department of General Surgery, Beijing Friendship Hospital, Capital Medical University & National Clinical Research Center for Digestive Disease, Beijing, 100050, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Bingding Huang
College of Big Data and Internet, Shenzhen Technology University, Shenzhen, Guangdong, 518118, People’s Republic of China
Tel +86-755-23256024
Email [email protected]
Background: The ability to rapidly, inexpensively, and accurately identify cancer patients based on actionable genomic mutations in tumour specimens is becoming critically important in routine clinical diagnostics. Targeted panel sequencing is becoming popular because it enables comprehensive and cost-effective diagnosis. However, the implementation of a next-generation sequencing (NGS) assay in clinical settings requires careful analytical validation to demonstrate its ability to detect multiple genomic variants.
Materials and Methods: Here, we developed a custom-targeted NGS panel to identify actionable variants, including single nucleotide variants, insertions, and deletions; copy number variants; and gene fusions, across 73 genes for targeted cancer therapy. We implemented a practical validation strategy with diluted samples and reference standard samples that modelled key determinants of accuracy, including mutant allele frequency, insertion/deletion length, amplitude of copy number, and hotspot gene fusions.
Results: The analytical validation results demonstrated that our panel can identify different types of genomic alterations in these test samples with high levels of accuracy, sensitivity, and reproducibility.
Conclusion: Our panel could be deployed as a routine clinical test to comprehensively detect actionable variants in cancer patients to guide targeted therapy decisions.
Keywords: next-generation sequencing, actionable mutations, targeted therapy
Introduction
With the elucidation of key effects of actionable mutated genes in tumorigenesis and therapy, more and more drugs and therapeutics targeting cancer-associated mutations have been developed, and the identification of such mutations has become a standard of care for cancer treatment. For example, screening for the EGFR somatic mutation is now recommended for non-squamous non-small cell lung carcinomas (NSCLCs).1,2 The national comprehensive cancer network (NCCN) guidelines for NSCLCs strongly endorse broader molecular profiling to identify rare driver mutations for which effective drugs may already be available.3,4 However, the requirement for interrogating hundreds of cancer-related genes simultaneously with a tiny amount of input DNA in one biopsy is still challenging. The existing mature methodologies only detect one type of mutation or a limited number of hotspots. Examples of such methodologies include ddPCR, which only interrogates a single hotspot variant in one array, and fluorescence in situ hybridization (FISH), which tests whether known gene fusion has occurred based on the use of predefined probes.5 Therefore, high-throughput, inexpensive, multiplex biomarker assays for molecular stratification are needed to guide therapeutic choices and support clinical trial design. Although high-coverage whole exome or whole genome sequencing can reliably assess tumour genomics, cost and bioinformatic demands currently restrict their routine clinical implementation to stratify patients for treatment.
In the clinical setting, targeted next-generation sequencing (NGS) is often cost-effective and valuable, as it allows for a more clinically relevant interpretation, robust sequencing coverage of target genes, and faster turnaround time, and can concurrently analyse multiple genomic mutations with a limited bioinformatic analysis burden.6,7 In recent years, a number of clinical molecular laboratories have validated and applied NGS platforms for routine diagnostic screening of solid tumours.8–12 Amplicon-based and hybridization capture methods have been used to enrich target genomic regions. The former method, however, introduces artefacts such as polymerase errors and loss of information about the original molecule count, making it inaccurate in estimating the number of unique input DNA molecules and copy number variations. Recent advancement using unique molecular indexes in the amplicon-based method has greatly reduce the error rate.13–15 Hybridization capture overcomes these drawbacks by pulling down the original DNA molecules directly. Another advantage of hybridization capture is that it is suitable for highly degraded formalin-fixed paraffin-embedded (FFPE) samples since the capture only requires partial overlap of probes with target regions. Consequently, hybridization capture-based panels can provide more accurate estimates of copy number variation and can be used for the detection of known actionable gene fusions.16 However, the implementation of a targeted NGS assay in a clinical setting requires careful analytical validation to demonstrate the assay’s ability to detect multiple genomic mutations.
In this study, we developed a custom 73-gene hybridization-capture-based panel called ActionAll to identify somatic single nucleotide variants (SNVs), insertions/deletions (INDELs), copy number variations (CNVs), and occurrences of gene fusion in one single array without matched normal tissue for cancer patient samples. These targeted genes were selected from different knowledge bases such as OncoKB,17 CIViC18 and Cosmic,19 and relevant literature. Custom biotinylated DNA probes with 120 base pairs (bp) were designed to cover all exons of the 73 targeted genes as well as select hotspot introns of actionable gene fusions. Captured libraries were sequenced on Illumina sequencers. Then, the sequencing reads were processed using an analysis pipeline developed in-house to identify SNVs, short INDELs (<30 bp), CNVs, and gene fusion events. To analytically validate our panel, we assessed the sensitivity and specificity of our panel using a number of diluted samples to detect SNVs and INDELs. Furthermore, we demonstrated the ability of our panel to successfully detect known copy number variants and gene fusions using two selected reference standard samples from Horizon Discovery Inc., UK. Intra- and inter-run replicates were also performed to assess the reproducibility of our panel. The analytical validation results demonstrated that the ActionAll panel can identify different types of variants in tumour samples with high levels of accuracy, sensitivity, and reproducibility. Therefore, we have deployed ActionAll in our laboratory as a routine clinical test to comprehensively detect actionable and other variants in cancer patients to guide targeted therapy decisions and identify the most suitable clinical trials for individual patients.
Materials and Methods
Panel Design
Based on the gene data content of commercial cancer panels, literature, and knowledgebases, we composed a list of 73 genes that have actionable or clinically relevant mutations for targeted therapy in all solid tumours (Supplementary Table 1). The proposed panel targets all the exons corresponding to the canonical transcripts of these 73 selected genes. The hotspot intronic regions of 13 targetable gene fusions are also included in the panel (Supplementary Table 2). The capture probes (120 bp) for this custom panel were ordered from Integrated DNA Technologies Inc., USA. We hypothesised that this panel would be able to detect clinically actionable variants for cancer patient samples with high sequencing coverage at a relatively low cost. We regularly monitor knowledgebases including approved treatment options matched with genomic variants (OncoKB, CIViC and Cosmic, etc.) for new actionable variants. New probes will be designed to cover such variants and be added to this panel after validation.
Samples for Analytical Validation
In contrast to diagnostic assays for a limited number of genomic sites, analytical validation of an NGS-based panel sequencing assay is a complex challenge. A single tumour specimen can harbour multiple types of genomic alterations at any position in the tested regions and at a wide range of allele frequencies (AF). Reference specimens containing all possible somatic alterations in all cancer-related genes do not exist. To validate the accuracy of SNV detection, five HapMap normal cell lines from the 1000 Genomes Project20 were ordered from the Coriell Institute (http://ccr.coriell.org/). Then, five different diluted samples were created with these five normal cell lines with different dilution ratios (Table 1), which leveraged the abundance and population distribution of known germline base-substitution variation to create test specimens that included hundreds of variants across the targeted exons and spanned a range of AFs (5–100%). For INDEL validation, cancer cell line RL952 was purchased from American Type Culture Collection (http://atcc.org) because it has a list of known INDELs (Supplementary Table 3). Additionally, to validate the accuracy of our panel and analysis pipeline, two reference standard samples HD701 and HD753 with confirmed somatic variants (SNV, INDEL, CNV, and fusion) were purchased from Horizon Discovery Inc., UK (https://horizondiscovery.com). We also participated in the external quality assessment (EQA) program by the National Centre for Clinical Laboratories of China (NCCL)21 and, thus, five EQA samples were purchased from NCCL (https://www.nccl.org.cn/) for the assessment of the reproducibility of our panel.
 |
Table 1 Five HapMap Normal Samples and Their Dilution Ratios in Five Diluted Samples for SNV Validation |
Sample Preparation and Targeted Next-Generation Sequencing
Targeted sequencing libraries were constructed using 200 ng of sheared DNA. Briefly, each cell line DNA was sonicated to an average size of around 200 bp using a Covaris S220 focused-ultrasonicator with Covaris microTUBES. Fragmentation was followed by pre-capture library preparation using an Accel-NGS 2S Hyb DNA Library Kit (Swift Biosciences, USA) according to the manufacturer’s protocol. Indexing PCR was performed for a total of six cycles before beads purification. A QC procedure was performed by quantifying purified libraries using the Qubit dsDNA HS Assay Kit (Life Technologies, USA) on Qubit 3.0 (Invitrogen, USA). Libraries yielding less than 500 ng are failed. A quality control check on the average fragment size of the pre-capture libraries was also performed using a 2100 Bioanalyzer (Agilent Technologies, USA). Indexed samples were then pooled at equal quantities (typically 500 ng each for 4–5 samples per pool), and hybridization was performed on each multi-sample pool using custom capture DNA probes (Integrated DNA Technologies Inc., USA) for 16–24 h. The hybridized products were captured by streptavidin-coated beads (Invitrogen, USA) and amplified for 12 PCR cycles with KAPA HiFi HotStart ReadyMix (Roche, Switzerland). Post-capture libraries were quantified by Qubit 3.0, and fragment size was determined by 2100 Bioanalyzer. Captured libraries yielding less than 20 ng or average fragment size less than 250 bp are failed. Sequencing was performed on an Illumina NovaSeq platform (Illumina, USA) using 2 × 150 bp paired end reads.
Data Analysis
First, the quality control and adapter trimming step were performed using fastp22 on raw sequencing data to remove adapter contamination and filter out low-quality reads. Then, the clean reads were mapped to a human reference genome (hg19) using BWA-MEM algorithm version 0.7.16.23 Different QC values such as mapping rate, on target rate and average sequencing depth (minimum depth required: 500X) were calculated to ensure the quality of mapped reads. For variant calling, we used a pipeline developed in-house based on samtools mpileup24 to detect somatic SNVs and INDELs.25,26 Briefly, plenty of candidate SNVs/INDELs were identified in tumour samples with at least three supported reads and the required mapping quality and base quality score. In the calling process, a series of filters were applied on the raw SNV/INDEL calls, including noise estimation from known SNPs, strand bias filtering, and noise filtering from neighbouring regions, to ensure reliable variant detection. In our experience, most false-positive variants originated from alignment errors and repeat regions. These kinds of variants were removed using a blacklist containing common mistakes from a pool of normal samples. Then, by querying in a reference normal sample, which was a mixture of five HapMap normal samples, those mutations present in the reference normal were categorized as germline, and the rest mutations were categorized as somatic. The final high confident variants (SNVs with AF ≥ 5% and small INDELs AF ≥ 2%) were then annotated with UCSC RefSeq gene information, dbSNP,27 1K Genome,20 ExAC,28 GnomAD,28 COSMIC,19 and Clinvar29 using SNPEff.30 Copy number variants (CNVs) were identified using CNVkit31 (CNV gain threshold ≥ 4 copies and CNV loss ≤1 copies). The actionable gene fusion events were identified using BreakID and the validation of fusion genomics breakpoints has been reported previously.32 All the detected variants were manually reviewed using the Integrative Genomics Viewer.33
Analytical Performance
A representative approach was taken to validate SNV detection in the panel. Five HapMap normal samples and those five diluted samples were sequenced individually. In total, 320 single nucleotide polymorphism (SNP) sites in the five HapMap normal samples from the dbSNP27 database (build 147) in the coding regions of the ActionAll panel were examined. SNP sites consistent with a homozygous (AF > 90%) or heterozygous (40% ≤ AF ≤ 60%) state were used in the test set and were classified as private SNP or non-private SNP. Private SNPs were defined as those SNPs present in only one HapMap normal sample, whereas non-private SNPs were present in more than one HapMap normal sample. Both private SNPs and non-private SNPs were treated as confirmed SNVs for validation in the diluted samples. Then, the expected AF for each SNV in the diluted samples was calculated based on the number of alternate alleles present in mixtures with different constituents and different ratios of the constituents. To evaluate the detection capability and limitation of our variant analysis pipeline in detecting SNVs, we calculated the sensitivity and specificity values. For sensitivity calculation, each confirmed SNV was assigned as either true positive (TP) if detected in the diluted sample or false negative (FN) if not detected. Sensitivity was then calculated as TP/(TP+FN). For specificity calculation, each called SNV was classified as TP if a matched SNV was found in the undiluted samples, or as false positive (FP) if not found. Specificity was then calculated as TP/(TP+FP). Moreover, because sequencing depth plays an important role in the accuracy for SNV calling, in silico down-sampling (random selection of subsets of reads) was carried out to examine the performance over a wide coverage range (500X to 1000X) using the same procedure. A similar approach was taken for INDEL validation as well. The accuracy of our panel in detecting all kinds of variants (SNV, INDEL, CNV, and gene fusion) was determined by two reference standard samples with confirmed somatic mutations. To assess the reproducibility and repeatability of our assay, we also performed inter-run and inter-operator reproducibility tests using five EQA samples.
Results and Discussion
Performance of SNV Calling
To simulate a series of SNVs with different AFs, five SNV diluted samples were generated from five HapMap normal samples with different dilution ratios and sequenced using our panel (Table 1). The sequencing reads were downsampled into different depths (500X, 700X, 900X, and 1000X) using samtools (random selection of reads).18 To assess the performance of our approach in detecting SNVs, we calculated the sensitivity and specificity for these five diluted samples at different sequencing depths. The performance results at an AF cut-off of 5% for these diluted samples were summarized in Table 2. At the sequencing depth of 500X, we achieved an average sensitivity of 99.38% (two SNVs not detected) and a specificity of 100% in detecting SNVs (no false positive). At the depths of 700X, 900X and 1000X, all the SNVs with AF ≥ 5% were correctly identified, and there were no false positives. Figure 1 shows the good correlation between detected AF and expected AF for all the detected SNVS. These results demonstrated that our panel and analytical pipeline have excellent performance in detecting SNVs.
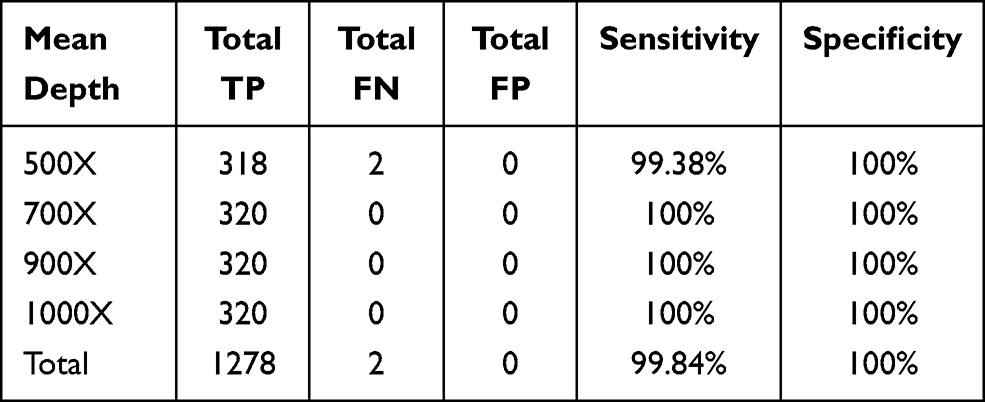 |
Table 2 Performance of SNV Calling at AF ≥5% for Five Diluted Samples at Different Sequencing Depths |
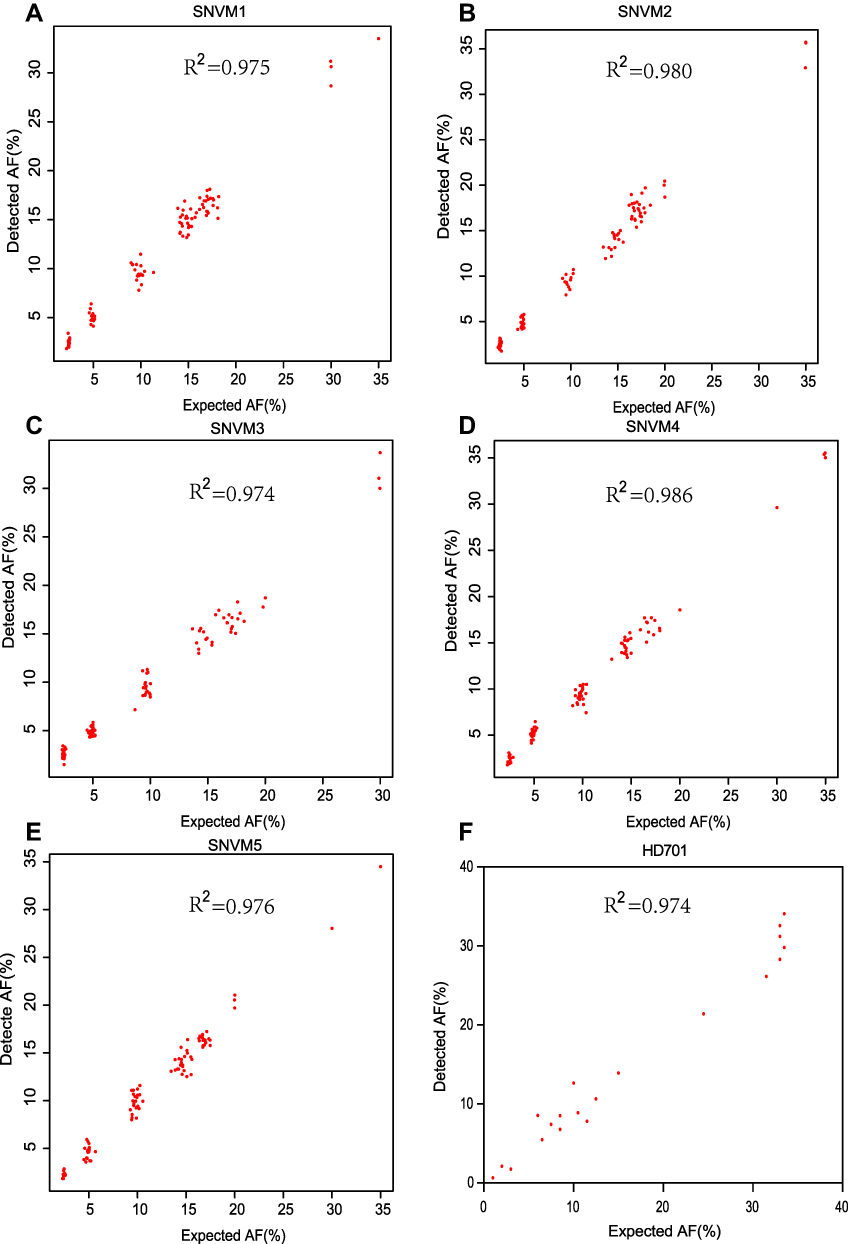 |
Figure 1 Detected AF vs expected AF for five SNV diluted samples (A–E) and the reference standard sample HD701 (F). |
Performance of INDEL Calling
Similar to SNV validation, five INDEL diluted samples were generated from one cancer cell line RL952 and one HapMap normal sample NA18536 to simulate a series of INDELs with different AFs (diluted ratio: 5%, 10%, 20%, 30%, and 40%). These five INDEL diluted samples were sequenced using our panel and also downsampled into different sequencing depths (500X, 700X, 900X, and 1000X). The sensitivity and specificity results at an INDEL AF cut-off of 2% are shown in Table 3. Figure 2 shows the good correlation of detected AF with expected AF for all the detected INDELs. Our INDEL calling algorithm identified all the INDELs correctly except one deletion (BRAF.c1208delC) in the diluted sample INDM1. One possible reason why this deletion was missed might be that its expected AF is relatively low (2.35%), and thus our INDEL calling pipeline could not call it out. Nevertheless, these results demonstrated that our panel and analytical pipeline also have good performance in detecting INDELs at low AF.
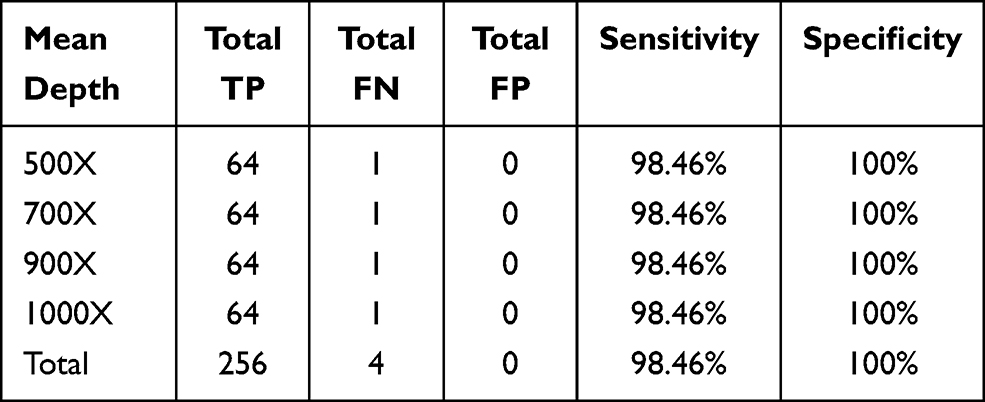 |
Table 3 Performance of INDEL Calling at AF ≥2% for Five Diluted Samples at Different Sequencing Depths |
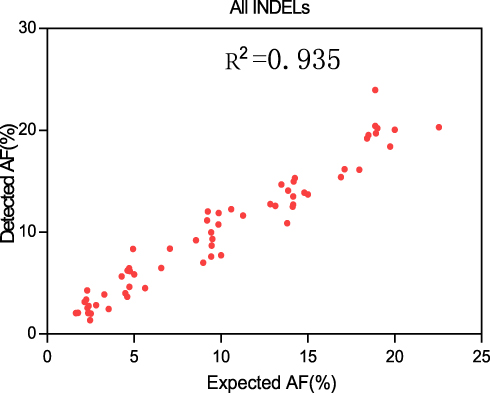 |
Figure 2 Detected AF vs expected AF for five INDEL diluted samples. Please note that the INDELs in these five diluted samples are plotted together. |
Validation Results of Reference Standard Samples
In order to evaluate the detection accuracy of our panel on different variant types (SNV, INDEL, CNV, and gene fusion), two reference standard samples HD701 and HD753 from Horizon Discovery Inc. were sequenced at a mean depth of 500X. HD701 mainly harboured actionable hotspot SNVs and INDELs with different AFs ranging from 1% to 33%, as confirmed by ddPCR assay, whereas HD753 harboured every kind of hotspot variant (SNV, INDEL, CNV, and gene fusion). As shown in Table 4, all the hotspot SNVs and INDELs in HD701 were correctly identified at the mean sequencing depth of 500X, even for those low AF variants in EGFR (L858R: 3%, E746_A750del: 2%, and T790M: 1%). Supplementary Table 4 shows the validation results of HD753 for five hotspot SNVs, six INDELs, two CNVs (MET and MYC), and two gene fusions (SLC34A2-ROS1 and CCDC6-RET) at the sequencing depth of 500X. The results confirmed that different kinds of variants in HD753 were correctly identified using our panel.
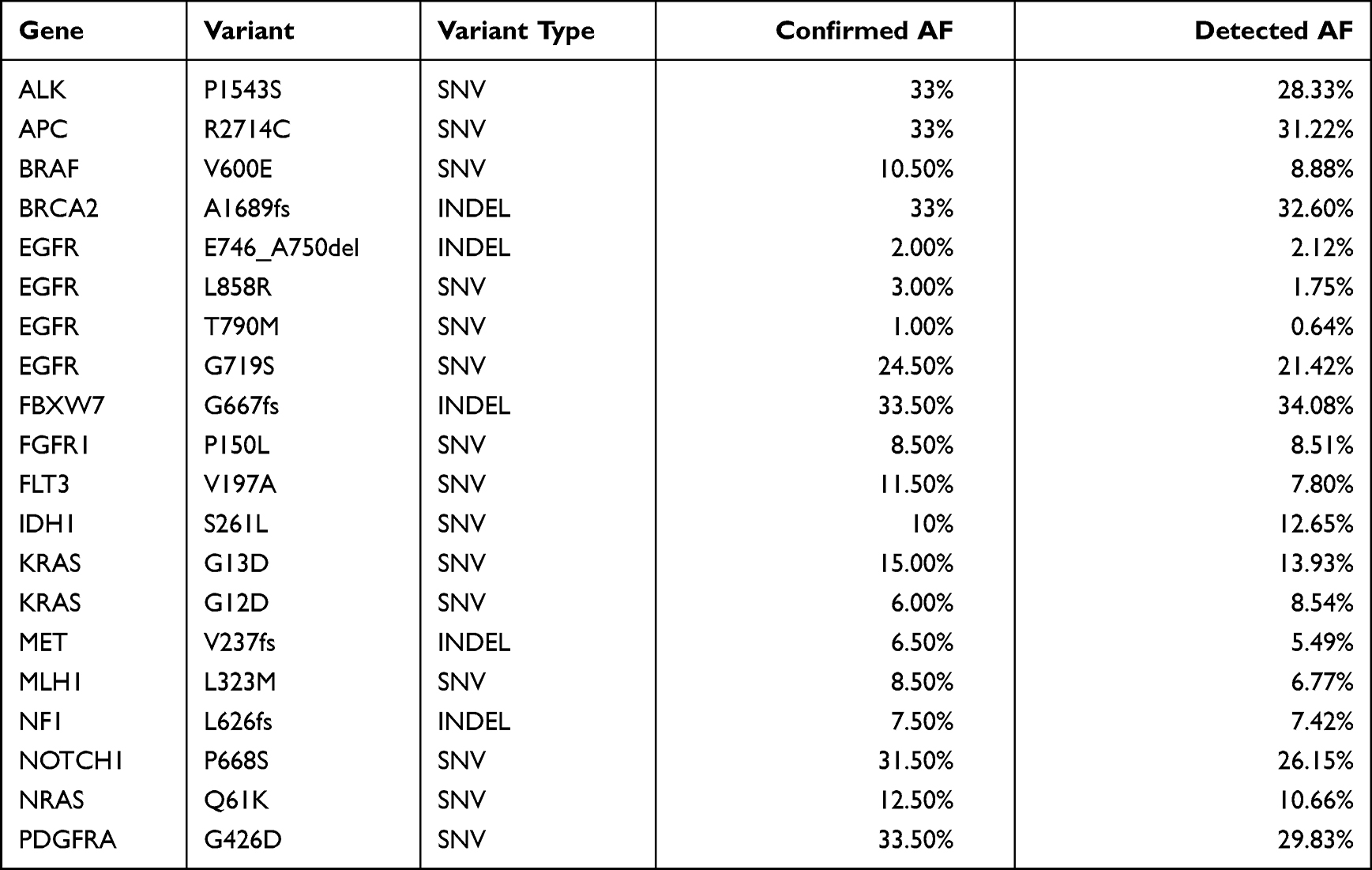 |
Table 4 The Validation Results of Reference Standard Sample HD701 at the Sequencing Depth of 500X for Confirmed Hotspot SNVs and INDELs |
Assessment of Reproducibility
To determine the reproducibility of our assay, we performed intra-run and inter-run reproducibility tests using five clinical EQA samples with confirmed mutations. The intra-run reproducibility replicates were done by three technicians using the same reagents and sequenced in the same lane with different barcodes. The inter-run reproducibility test was performed by the same technician in three different runs. The intra-run and inter-run reproducibility results are summarized in Supplementary Tables 5 and 6. All the confirmed variants were successfully detected in all replicates, with similar variant AFs and at similar levels of sequencing depth. No false-positive variants were detected. The total number of variants called across both intra- and inter-run replicates was generally consistent, which is noteworthy given the differences in sample coverage across replicates. At the same time, the reproducibility of capture probes for our panel was also assessed. Figure 3 shows the normalized mean depths (red) of all targeted regions and their standard deviations (black) for five HapMap normal samples and five clinical EQA samples. All the targeted regions were stably captured across different samples in different runs, which demonstrated that the captured performance of our panel was highly reproducible from sample to sample.
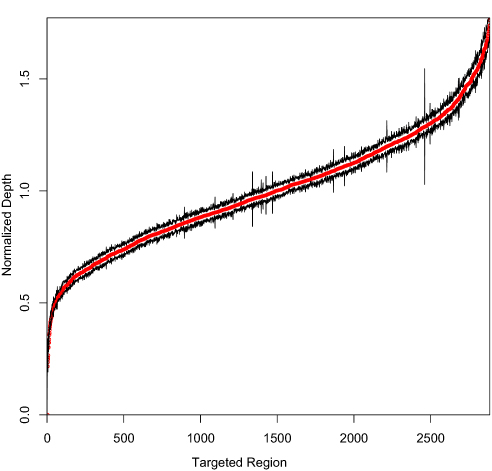 |
Figure 3 The normalized mean depths for all the targeted regions for different validation samples. |
Discussion
Here we developed a custom panel called ActionAll to screen all exons of 73 genes for clinically relevant mutations in all solid tumours as well as hotspot introns of actionable gene fusions. The ActionAll panel was designed to be an efficient, scalable, and cost-effective comprehensive NGS panel. The selection of genes and oncogenic fusions was based on different databases, such as OncoKB, CIViC and COSMIC, and relevant literature. The primary objective of this study was to validate the ability of this ActionAll panel to detect different kinds of variants such as SNV, INDEL, CNV and gene fusion.
In this work, we presents an analytical validation study and performance metrics for the ActionAll panel using a pool of diluted samples and reference standard samples for clinical application. The analytical validation results demonstrated that the ActionAll panel can identify different types of variants (SNV, INDEL, CNV, and gene fusion) in tumour samples with high levels of accuracy, sensitivity, and reproducibility. However, one drawback of our panel is that it prioritizes only well-characterized fusion events and is by design difficult to identify novel oncogenic fusion events from DNA. Recently, a targeted panel that interrogates both RNA and DNA in a single-tube, dual-template assay using anchor multiplexed PCR was deployed clinically.34 However, RNA-Seq sequencing is not suitable in our DNA panel because it is based on hybrid-capture probes. To overcome this drawback, new probes covering novel fusions breakpoints have to be designed and added into our panel after essential validation. Nevertheless, the potential of our panel for routine clinical application will be of great importance for physicians, providing them with a powerful tool to diagnose tumours, monitor tumour dynamics, and evaluate patient responses to targeted therapy. This study affirms the feasibility and clinical utility of targeted sequencing in the oncology arena and provides a foundation for genomics-based stratification in guiding treatment decisions of cancer patients.
Abbreviations
AF, allele frequency; CNV, copy number variant; EQA, external quality assessment; FFPE, formalin-fixed paraffin-embedded; FISH, fluorescence in situ hybridization; INDEL, insertion and deletion; NCCN, national comprehensive cancer network; NGS, next-generation sequencing; NSCLC, non-small cell lung carcinomas; SNP, single nucleotide polymorphism; SNV, single nucleotide variant.
Data Sharing Statement
All data relevant to the study are included in the article or uploaded as Supplementary materials.
Ethics Approval
We confirm that all experiments were performed according to the protocols approved by the institutional review board of the Chinese Academy of Medical Sciences and Peking Union Medical College. All methods were carried out in accordance with relevant guidelines and regulations. Cell line samples used in this analytical validation were purchased commercially from different sources.
Acknowledgment
This work was financed by the Project of Educational Commission of Guangdong Province of China (No. 2020KZDXZ1215). We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
DW, YZ and BH are former employees of SinoTech Genomics, Inc. YF is currently an employee of SinoTech Genomics, Inc. HD is co-founder of SinoTech Genomics, Inc. All other authors declare no competing interests.
References
1. Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. Arch Pathol Lab Med. 2013;137:828–860. doi:10.5858/arpa.2012-0720-OA
2. Kerr KM, Bubendorf L, Edelman MJ, et al. Second ESMO consensus conference on lung cancer: pathology and molecular biomarkers for non-small-cell lung cancer. Ann Oncol. 2014;25(9):1681–1690. doi:10.1093/annonc/mdu145
3. Ettinger DS, Aisner DL, Wood DE, et al. NCCN guidelines insights: non-small cell lung cancer, Version 5.2018. J Natl Compr Canc Netw. 2018;16:807–821. doi:10.6004/jnccn.2018.0062
4. D’Haene N, Le Mercier M, De Neve N, et al. Clinical validation of targeted next generation sequencing for colon and lung cancers. PLoS One. 2015;10(9):e0138245. doi:10.1371/journal.pone.0138245
5. Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–1031. doi:10.1038/nbt.2696
6. Jennings LJ, Arcila ME, Corless C, et al. Guidelines for validation of next-generation sequencing-based oncology panels: a joint consensus recommendation of the Association for Molecular Pathology and College of American Pathologists. J Mol Diagn. 2017;19:341–365. doi:10.1016/j.jmoldx.2017.01.011
7. Froyen G, Broekmans A, Hillen F, et al. Validation and application of a custom-designed targeted next-generation sequencing panel for the diagnostic mutational profiling of solid tumors. PLoS One. 2016;11(4):e0154038. doi:10.1371/journal.pone.0154038
8. Cottrell CE, Al-Kateb H, Bredemeyer AJ, et al. Validation of a next-generation sequencing assay for clinical molecular oncology. J Mol Diagn. 2014;16(1):89–105. doi:10.1016/j.jmoldx.2013.10.002
9. Pritchard CC, Salipante SJ, Koehler K, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16(1):56–67. doi:10.1016/j.jmoldx.2013.08.004
10. Singh RR, Patel KP, Routbort MJ, et al. Clinical massively parallel next-generation sequencing analysis of 409 cancer-related genes for mutations and copy number variations in solid tumours. Br J Cancer. 2014;111(10):2014–2023. doi:10.1038/bjc.2014.518
11. Wang SR, Malik S, Tan IB, et al. Technical validation of a next-generation sequencing assay for detecting actionable mutations in patients with gastrointestinal cancer. J Mol Diagn. 2016;18(3):416–424. doi:10.1016/j.jmoldx.2016.01.006
12. Garcia EP, Minkovsky A, Jia Y, et al. Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch Pathol Lab Med. 2017;141(6):751–758. doi:10.5858/arpa.2016-0527-OA
13. Au CH, Ho DN, Kwong A, Chan TL, Ma ESK. BAMClipper: removing primers from alignments to minimize false-negative mutations in amplicon next-generation sequencing. Sci Rep. 2017;7:1567. doi:10.1038/s41598-017-01703-6
14. Lee B, Moon T, Yoon S, Weissman T. DUDE-Seq: fast, flexible, and robust denoising for targeted amplicon sequencing. PLoS One. 2017;12:e0181463. doi:10.1371/journal.pone.0181463
15. Pope BJ, Hammet F, Nguyen-Dumont T, Park DJ. Hi-Plex for simple, accurate, and cost-effective amplicon-based targeted DNA sequencing. Methods Mol Biol. 2018;1712:53–70.
16. Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251–264. doi:10.1016/j.jmoldx.2014.12.006
17. Chakravarty D, Gao J, Phillips SM, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017;2017.
18. Griffith M, Spies NC, Krysiak K, et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet. 2017;49(2):170–174. doi:10.1038/ng.3774
19. Forbes SA, Beare D, Boutselakis H, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(D1):D777–D783. doi:10.1093/nar/gkw1121
20. Auton A, Brooks LD, Durbin RM, et al; Genomes Project C. A global reference for human genetic variation. Nature. 2015;526:68–74.
21. Zhang R, Ding J, Han Y, et al. The reliable assurance of detecting somatic mutations in cancer-related genes by next-generation sequencing: the results of external quality assessment in China. Oncotarget. 2016;7(36):58500–58515. doi:10.18632/oncotarget.11306
22. Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–i90. doi:10.1093/bioinformatics/bty560
23. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
24. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi:10.1093/bioinformatics/btp352
25. Jones DTW, Jäger N, Kool M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488(7409):100. doi:10.1038/nature11284
26. Northcott PA, Buchhalter I, Morrissy AS, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547(7663):311–317. doi:10.1038/nature22973
27. Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi:10.1093/nar/29.1.308
28. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi:10.1038/nature19057
29. Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D7. doi:10.1093/nar/gkx1153
30. Cingolani P, Platts A, Wang le L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: sNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6(2):80–92. doi:10.4161/fly.19695
31. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12(4):e1004873. doi:10.1371/journal.pcbi.1004873
32. Jin L, Lai J, Zhang Y, et al. BreakID: genomics breakpoints identification to detect gene fusion events using discordant pairs and split reads. Bioinformatics. 2019;35:2859–2861. doi:10.1093/bioinformatics/bty1070
33. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–192. doi:10.1093/bib/bbs017
34. Song Z, Xu C, He Y, et al. Simultaneous detection of gene fusions and base mutations in cancer tissue biopsies by sequencing dual nucleic acid templates in unified reaction. Clin Chem. 2020;66(1):178–187. doi:10.1373/clinchem.2019.308833
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.