Back to Journals » Drug Design, Development and Therapy » Volume 10
Determination of the binding mode for anti-inflammatory natural product xanthohumol with myeloid differentiation protein 2
Authors Fu W, Chen L, Wang Z, Zhao C, Chen G ![]() , Liu X, Dai Y, Cai Y, Li C, Zhou J, Liang G
, Liu X, Dai Y, Cai Y, Li C, Zhou J, Liang G
Received 16 October 2015
Accepted for publication 16 December 2015
Published 27 January 2016 Volume 2016:10 Pages 455—463
DOI https://doi.org/10.2147/DDDT.S98466
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Supplementary video of 50 ns of xanthohumol/MD-2 complex molecular dynamics simulation.
Views: 779
Weitao Fu,1,* Lingfeng Chen,1,* Zhe Wang,1 Chengwei Zhao,1 Gaozhi Chen,1 Xing Liu,1 Yuanrong Dai,2 Yuepiao Cai,1 Chenglong Li,1,3 Jianmin Zhou,1 Guang Liang1
1Chemical Biology Research Center, School of Pharmaceutical Sciences, 2Department of Respiratory Medicine, the Second Affiliated Hospital, Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 3Division of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, Ohio State University, Columbus, OH, USA
*These authors contributed equally to this work
Abstract: It is recognized that myeloid differentiation protein 2 (MD-2), a coreceptor of toll-like receptor 4 (TLR4) for innate immunity, plays an essential role in activation of the lipopolysaccharide signaling pathway. MD-2 is known as a neoteric and suitable therapeutical target. Therefore, there is great interest in the development of a potent MD-2 inhibitor for anti-inflammatory therapeutics. Several studies have reported that xanthohumol (XN), an anti-inflammatory natural product from hops and beer, can block the TLR4 signaling by binding to MD-2 directly. However, the interaction between MD-2 and XN remains unknown. Herein, our work aims at characterizing interactions between MD-2 and XN. Using a combination of experimental and theoretical modeling analysis, we found that XN can embed into the hydrophobic pocket of MD-2 and form two stable hydrogen bonds with residues ARG-90 and TYR-102 of MD-2. Moreover, we confirmed that ARG-90 and TYR-102 were two necessary residues during the recognition process of XN binding to MD-2. Results from this study identified the atomic interactions between the MD-2 and XN, which will contribute to future structural design of novel MD-2-targeting molecules for the treatment of inflammatory diseases.
Keywords: myeloid differentiation 2, xanthohumol, binding mode, inflammation, molecular dynamics simulation
Introduction
Inflammatory responses are triggered by pathogenic bacteria or toxin-mediated injury, which are mediated by pattern receptors expressed by immune cells.1 However, the subsequent production of toxic proinflammatory molecules may result in undesirable outcomes, largely due to collateral tissue damage. Such uncontrolled inflammation plays an important role in the pathogenesis of diseases, such as cardiovascular diseases and sepsis.2–4 For instance, the extensively studied immunostimulatory component of bacteria, lipopolysaccharide (LPS), causes systemic inflammation and sepsis.5–7
LPS-induced inflammation is mediated by the toll-like receptor 4 (TLR4). In this pathway, LPS-binding protein captures LPS, transferring it to CD14 (cluster of differentiation 14)8 for delivery to a coreceptor, myeloid differentiation protein 2 (MD-2).9 TLR4 is a type-I transmembrane protein with a characteristic of horse-shoe/solenoid-shaped ectodomain. MD-2, contains 160 amino acid residues, adopts a β-cup fold with two antiparallel β-sheets that forms a deep hydrophobic pocket for LPS recognition.10,11 LPS directly binds to the hydrophobic pocket in MD-2 protein. Upon LPS binding, the TLR4/MD-2 complex undergoes dimerization and recruits its downstream adaptors.9,10 Two major downstream adaptor molecules of MyD88 and TIR-domain-containing adapter-inducing interferon-β are triggered by the dimerization of two TLR4/MD-2 complexes, which then activates the intracellular proinflammatory cascades.12 In general, MD-2 plays an indispensable role in the activation of the LPS-TLR4 signaling pathway and thus has been confirmed as a suitable target for the therapeutic inhibition of TLR4 signaling.13,14
The study of MD-2, as a target for anti-inflammatory therapy, has drawn great attention and a variety of MD-2 inhibitors have been reported, among which lipid IVa is a popular lipid-like antagonist. Lipid IVa, an underacylated lipid A with four fatty acid chains from Escherichia coli, is an intermediate in the biosynthetic pathway of LPS and can inhibit LPS with four acyl chains invading the hydrophobic pocket of MD-2.15 In the meantime, some other natural and synthetic nonlipid compounds have also been tested. For example, JTT-705 and auranofin form covalent bonds with residue Cys-133 of MD-2, while compounds like taxanes and curcumin form noncovalent interactions embedded into the hydrophobic pocket of MD-2.16–18
Xanthohumol (XN) (Figure 1A) is a prenylated chalcone-type flavonoid of Humulus lupulus L. (Cannabaceae) and it is also used to make commercial flavoring products; XN has a variety of pharmacological functions including anti-inflammation, prevention and treatment of diabetes and antioxidation.19–21 Previous researchers suggested that XN’s anti-inflammatory activities and chemopreventive effects are attributed to blocking LPS binding to MD-2. Peluso et al22 reported that XN can suppress LPS-stimulated inflammatory signaling through direct competitive binding to the hydrophobic cavity of MD-2. However, the binding mechanism of XN in MD-2 pocket are still unclear, preventing effective therapeutic design of XN targeting the MD-2 for the treatment of inflammatory diseases.22 In the present study, we used both computational and experimental methods to explore the interaction between XN and MD-2. Our results indicated that XN can embed into the hydrophobic pocket of MD-2 and form two stable hydrogen bonds with residues ARG-90 and TYR-102.
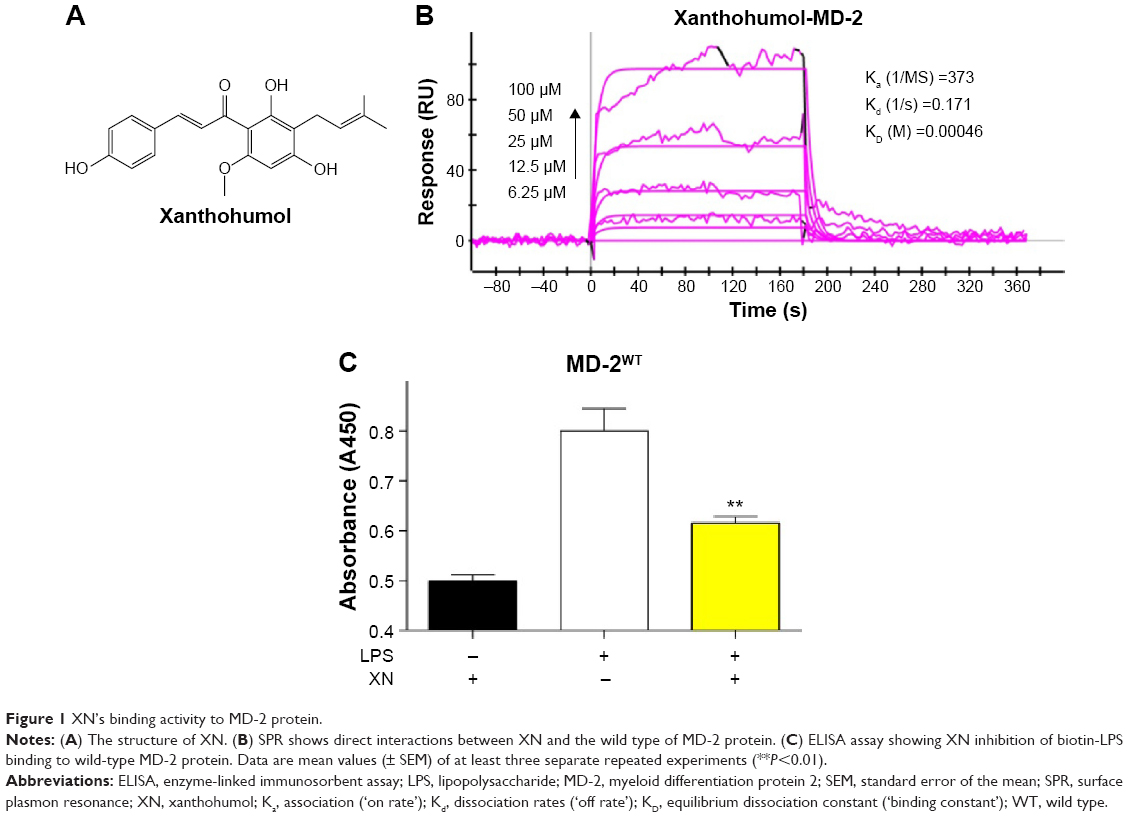 | Figure 1 XN’s binding activity to MD-2 protein. |
Materials and methods
Reagents
The mouse RAW 264.7 macrophages were purchased from ATCC (Manassas, VA, USA). The recombinant human MD-2 (rhMD-2, or rhMD-2 mutant) proteins were purchased from Biowit Technologies (Shenzhen, People’s Republic of China); XN, LPS (from Salmonella typhosa); and the anti-MD-2 antibody from eBioscience (San Diego, CA, USA). No ethical approval was required for the use of these cell lines.
Surface plasmon resonance analysis
The binding affinity of XN to rhMD-2 and rhMD-2 mutants was determined using a ProteOn XPR36 Protein Interaction Array system (Bio-Rad Laboratories, Hercules, CA, USA) with an HTE sensor chip (ProteOn™, #176-5033). Briefly, MD-2 protein (in acetate acid buffer pH 5.5) was loaded to the sensors that were activated with 10 mM NiSO4. The XN samples (at 100, 50, 25, 12.5, and 6.25 μM) were prepared with running buffer (phosphate buffered saline [PBS], 0.1% sodium dodecyl sulfate, 5% dimethyl sulfoxide). Sensor and sample plates were placed on the instrument. The XN samples were then captured in flow cell 1, leaving the second flow cell as a blank. Five concentrations were injected simultaneously at a flow rate of 30 μM/min for 120 seconds of association phase, followed with 120 seconds of dissociation phase at 25°C. The final graphs were obtained by subtracting blank sensorgrams from the duplex or quadruplex sensorgrams. Data were analyzed with ProteOn manager software. KD (equilibrium dissociation constant [‘binding constant’]) was calculated by global fitting of the kinetic data from various concentrations of XN using 1:1 Langmuir binding model.
Enzyme-linked immunosorbent assay
A 96-well microplate was coated overnight at 4°C with antihuman MD-2 antibody in 10 mM Tris–HCl buffer (pH 7.5). The plate was washed with PBS Tween-20 and blocked with 3% bovine serum albumin in high purity water for 1.5 hours at room temperature. rhMD-2 (4 μg/mL) in 10 mM Tris–HCl buffer (pH 7.5) was added to a precoated plate and incubated for 1.5 hours at room temperature. After washing with PBS Tween-20, biotin-labeled LPS (InvivoGen, San Diego, CA, USA) was incubated for 1 hour at room temperature. Compound XN (0.1 or 1 μM) was added to the biotin-LPS before addition to the wells. After further washing streptavidin-conjugated horseradish peroxidase (Beyotime, Shanghai, People’s Republic of China) was added to the wells for 1 hour at room temperature. The horseradish peroxidase activity was determined using TMB substrate solution (eBioscience, San Diego, CA, USA). The optical density of each well was measured at 450 nm. Data were expressed as mean values (± standard deviation) of three separate experiments, each performed in duplication.
Molecular docking
The binding pose of probe XN in MD-2’s binding site was predicted by the software Glide (Schrödinger, Inc., NY, USA). The crystal structure of MD-2 in complex with lipid IVa was downloaded from the protein data bank (PDB code: 2E59) and prepared using the Protein Preparation Wizard in Maestro (Schrödinger, Inc.).15 The process included removal of all nonbonded heteroatoms and water molecules. Hydrogen bonds were added and optimized to the structure. Other preparation steps involved removal of bad contents, optimization of bond lengths and creation of disulfide bonds. The XN was built by using Maestro and converted to a 3D structure from the 2D structure using LigPrep (Schrödinger, Inc.). The resulting structures were saved in Maestro format.
The crystal structure of MD-2 was used to generate grid with van der Waals radius scaling 1.00 and partial charge cut-off at 0.25. The scoring grid was generated by enclosing the residues 30 Å around lipid IVa in the binding site using Receptor Grid Generation (Schrödinger, Inc.), following the standard procedure. The receptor grid file and the prepared XN were docked using Glide standard precision, while the ligand sampling was set to be flexible. Glide score, an empirical docking scoring functions implemented by the Optimized Potentials for Liquid Simulations 2005 force field, was used to infer the affinity and further analyze the binding mode and molecular dynamics (MD) simulations.
MD simulations
MD simulations of XN/MD-2 complex system and apo MD-2 system were carried out, respectively. The initial XN/MD-2 complex came from the top ranked binding pose in the glide docking results. Before MD simulation, XN was extracted from the XN/MD-2 complex and the charge was prepared by gaussian09 and antechamber.23–25 Molecular mechanics parameters from the ff99SB and GAFF force fields were assigned to the protein and the ligand, respectively, using the LEaP module of AMBER (Assisted Model Building with Energy Refinement) 11 software packages.26–29 The MD-2 structure of apo MD-2 system was prepared by Protein Preparation Wizard of Maestro. All the water molecules and the specified ligand were deleted. The two systems were all solvated in a box of TIP3P water molecules with a hydration shell of 10 Å. In addition, an appropriate number of chloride ions were used to neutralize these two systems.
MD simulation was carried out with AMBER 11 software packages and the two systems were using the same protocol.30,31 Prior to the MD productive simulation, we performed an equilibration protocol consisting of an initial minimization of the water box of 1,000 steps, 500 steps for the steepest descent and 500 steps in the conjugate gradient. Then, the TIP3P water box was heated at constant volume until 300 K using a time constant for the heat bath with a coupling time of 100 ps. Equilibration was at 300 K and constant pressure of 100 ps. Before the production of MD, the whole system was equilibrated at 100 ps at a constant pressure of 1 bar and turned on the Langevin temperature scaling with a collision frequency of 2 ps. The last step used 50 ns of MD simulation without any restraints. Furthermore, nonbonded interactions were cut off at 8.0 Å. Periodic boundary conditions were turned on in every step of the whole process.32 Particle mesh Ewald was also performed to deal with the long-range electrostatic interactions under periodic boundary conditions.33 The SHAKE method was used to constrain hydrogen atoms and the time step was set to 2 fs.34,35 The coordinates were saved every 10 ps for the subsequent analysis.
Results and discussion
Determination of the direct binding of XN to MD-2
Previous studies reported that MD-2 is a potential target for XN but the binding mode of XN to MD-2 is still unknown.22 In this study, we performed biochemical and computational experiments to determine the molecular interactions between MD-2 and XN. At the beginning, surface plasmon resonance (SPR) experiments were carried out to assess the binding affinity of XN to MD-2. As shown in Figure 1B, XN bound to the MD-2 protein in a dose-dependent manner with a relatively high affinity with a KD value of 0.00046 M. Furthermore, using a biotin–streptavidin-based enzyme-linked immunosorbent assay (ELISA) system, we found that biotin-tagged LPS (biotin-LPS) bound rhMD-2 previously coated onto plates, while coincubation with XN significantly reduced binding of biotin-LPS with rhMD-2 (Figure 1C). These results suggest that XN has the same binding site as LPS with a higher binding affinity.
The binding mode of XN to MD-2
Molecular docking was used to analyze the binding mode of XN to MD-2. Docking results showed that XN could be inserted into the large hydrophobic binding pocket of MD-2 and easily occupy a large portion of the LPS binding site (Figure 2A and B), which was consistent with our previous experimental results. In addition, other important interactions to MD-2 were also observed such as hydrogen bonding to residues ARG-90, GLU-92, and TYR-102 (Figure 2C). As with previous study, XN and related prenylated flavonoids had the same interactions with the TYR-102 residue, underscoring the role of TYR-102 in XN’s binding to MD-2. Our results were similar with the previously reported study. Additionally, based on the molecular docking results, we selected a docked complex that interacted with TYR-102 with lowest binding energy.
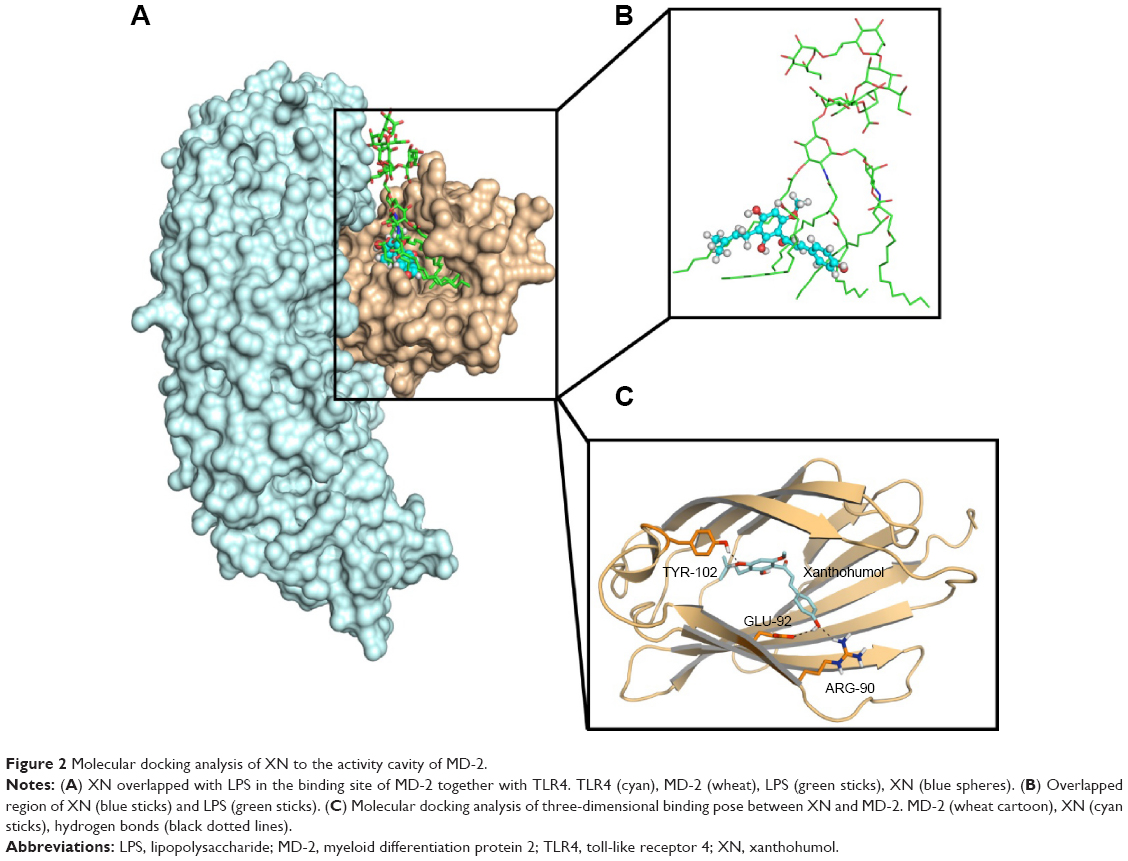 | Figure 2 Molecular docking analysis of XN to the activity cavity of MD-2. |
The stability of the docked complex
The results from the above docking analysis lead us to explore the dynamic behavior of the XN/MD-2 complex. In order to confirm the stability of docked conformation and to probe the function of XN on MD-2, conventional MD simulations on XN/MD-2 complex and apo MD-2 were conducted. For the initial structure as reference, the root-mean square deviation (RMSD) was detected for all Cα atoms in the proteins and all heavy atoms of XN. From our study, the trajectories that we established were more stable after 10 ns, therefore, only the latter part of the trajectories were considered for further analysis. As shown in Figure 3A, after 10 ns of MD simulation, the RMSD values of Cα atoms for MD-2 and XN gradually stabilized to 3.0 and 0.4 Å, respectively. However, the apo MD-2 converged the RMSD values in the system at 3.5 Å after 10 ns simulation (Figure 3B). Therefore, the above results suggest that two systems became stable in the end. Then, we extracted the initial and last snapshots from MD trajectories of two systems to verify the conformation change of the two systems. As depicted in Figure 3C and D, the XN/MD-2 complex should be stable; the docked XN/MD-2 complex in the end snapshot of the MD simulation structure and the initial structure were examined to show that they overlapped with slight positional difference. But the apo MD-2 showed a significant deviation until the end of the simulation, resulting in a backbone RMSD of about 3.5 Å that revealed a greatly changed conformation. What’s more, to quantify the “openness” of the hydrophobic cavity of MD-2, we cited a collective distance (Dtotal) defined as the total distance between the center of mass of MD-2 and to each center of mass of β strand.36 In Figure 4A, the time evolution of Dtotal for apo MD-2 as well as the values of Dtotal became smaller about 5 Å than from the beginning (Video S1). On the contrary, in Figure 4B, values of Dtotal of XN/MD-2 complex increased slightly, which indicate that XN can stabilize the XN/MD-2 complex (Video S2). Garate and Oostenbrink36 also had previously reported the same result with the simulated apo MD-2 and showed that the cavity of MD-2 closes rapidly in the absence of any ligand, indicating a strong hydrophobic nature and the instability of apo MD-2. Therefore XN not only directly binds to MD-2, but also plays a crucial role in the hydrophobic cavity of the MD-2 conformation.
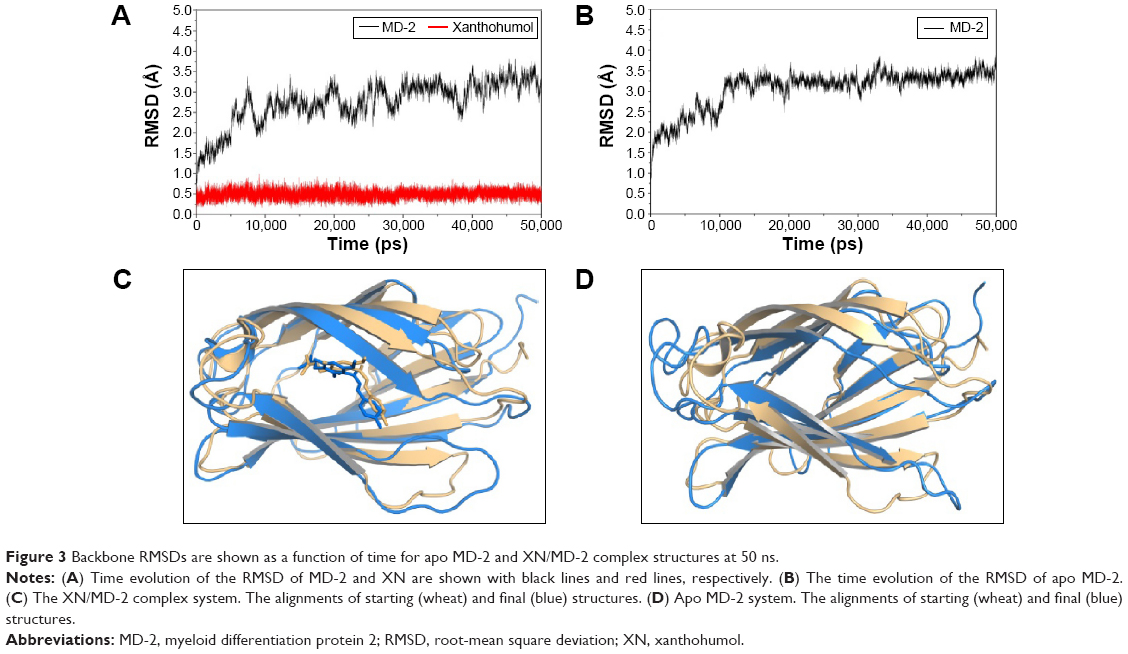 | Figure 3 Backbone RMSDs are shown as a function of time for apo MD-2 and XN/MD-2 complex structures at 50 ns. |
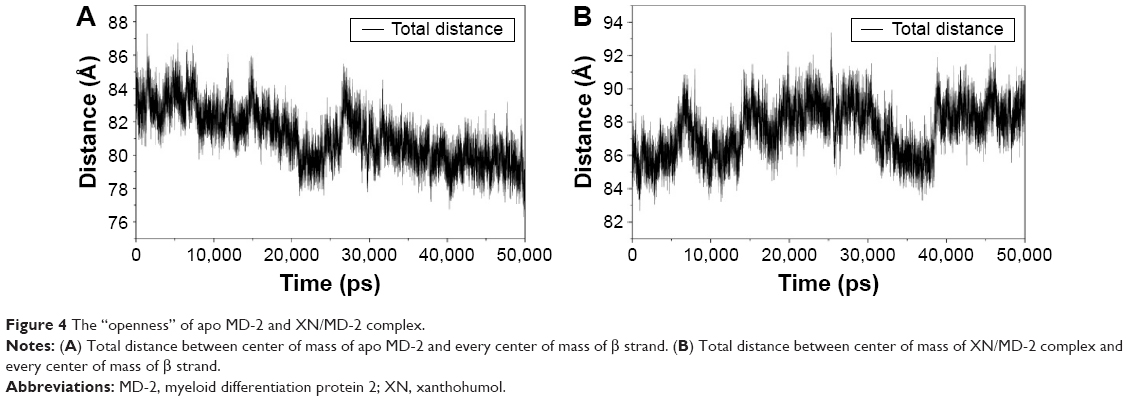 | Figure 4 The “openness” of apo MD-2 and XN/MD-2 complex. |
The main driving force for XN/MD-2 binding is a combination of several strong hydrogen bonds, which plays an important role in the protein-ligand recognition process. An analysis of the hydrogen bonds was then carried out to explore the interactions between MD-2 and XN where the most frequent hydrogen bonding partner was considered. As shown in Figure 5A, the distance between atoms in the docked complex, which form hydrogen bonds, was monitored during the whole MD simulation process and plotted. As a result, vast profiles of hydrogen bonds were observed, which fluctuated from 2.5 to 4.5 Å with an average of 3.0–3.5 Å hydrogen bonds in the XN/MD-2 complex. Regarding the XN/MD-2 complex, amidogens of ARG-90 and the hydroxyl of TYR-120 formed hydrogen bonds with hydroxyl and methoxy groups of XN, respectively, but ARG-90 was more stable than TYR-120 (Figure 5B and C). Moreover, although the distance between the two mentioned hydrogen bonds fluctuated slightly, they stabilized slightly with a distance ~3–3.5 Å in 50 ns of MD simulation. The detailed distances of evolutions in the two hydrogen bonds are illustrated in Figure 5B and C. Lastly, it is unknown as to why the hydrogen bond between GLU-92 and XN disappeared rapidly compared to the docking complex. This may be related to an inadequate amount of attraction.
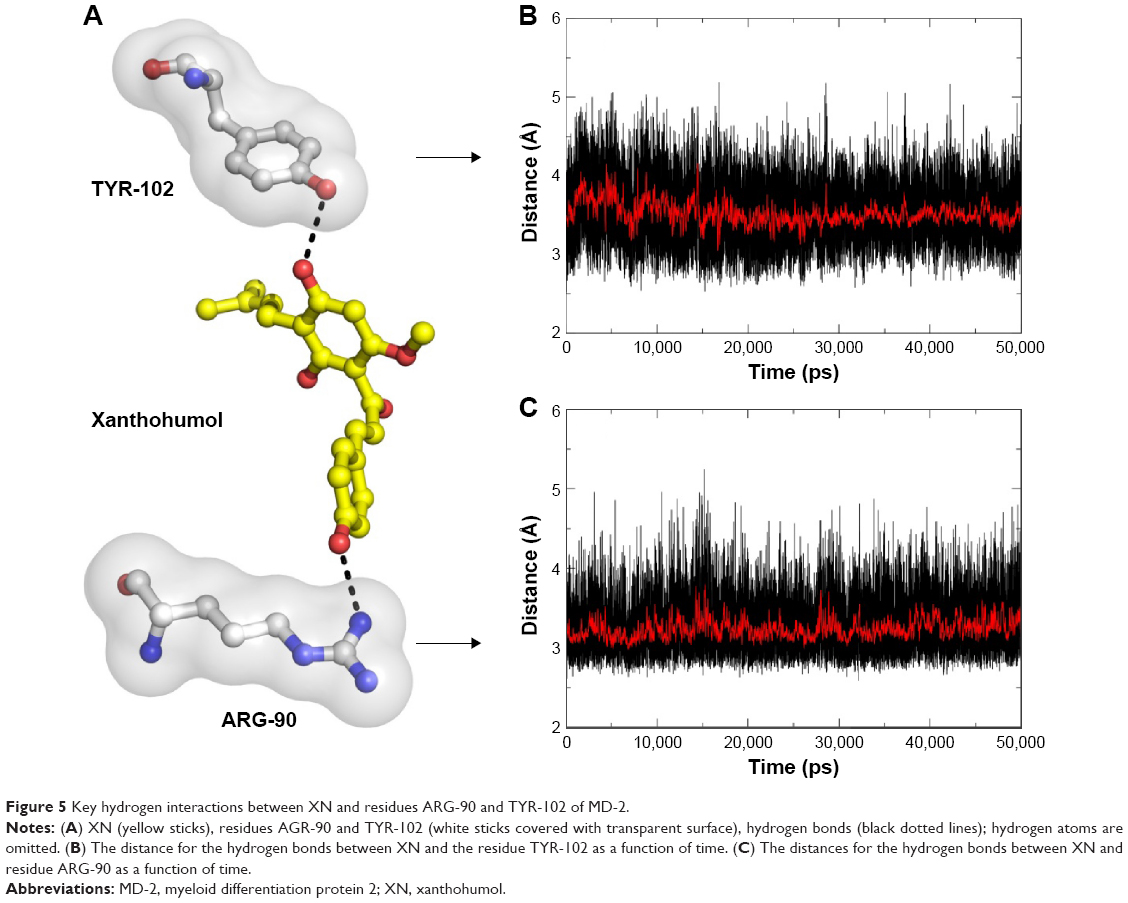 | Figure 5 Key hydrogen interactions between XN and residues ARG-90 and TYR-102 of MD-2. |
The weak interaction between XN and MD-2R90A/Y102A mutant
Based on the computational results in previous studies, we hypothesized that if residues ARG-90 and TYR-102 transform into alanine, the binding affinity may decline. To test our hypothesis, we analyzed the double-mutant protein MD-2R90A/Y102A using SPR to determine whether these two residues would affect the binding affinity of XN to MD-2 (Figure 6A). We found that the KD of XN bound to MD-2R90A/Y102A was 0.939 M, which was >2,000 times higher than that of the wild-type MD-2. In addition, ELISA assay showed that XN was not able to prevent the binding of biotin-LPS with the MD-2R90A/Y102A mutant (Figure 6B). Furthermore, the side chains in the mutation of MD-2 R90A/Y102A were unable to attract hydrogen bonds to XN, indicating that ARG-90 and TYR-102 were the key amino acids on MD-2 that bind XN. These two results further support the notion that the residues ARG-90 and TYR-102 are essential to the recognition process of XN binding to MD-2 proteins.
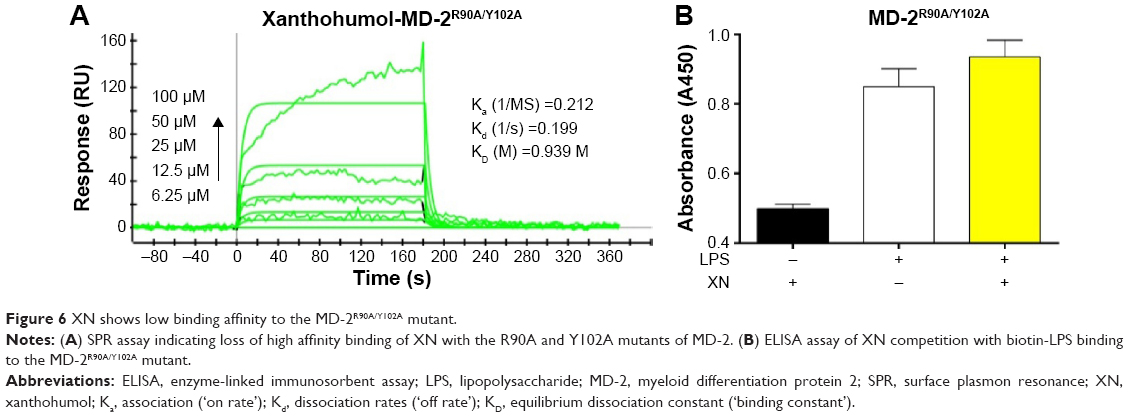 | Figure 6 XN shows low binding affinity to the MD-2R90A/Y102A mutant. |
Conclusion
In this report we described the interaction mode between XN and MD-2 protein. SPR and ELISA experiments were performed to assess the binding affinity and the binding mode of XN to the MD-2 wild-type protein. We applied molecular docking and molecular dynamic simulations to model the dynamic behavior of molecules and macromolecular assemblies at an atomic level. Moreover, the MD-2 site-directed mutants were used to verify the residues of ARG-90 and TYR-102, both of which are crucial for XN to compete with LPS. We found that XN formed two stable hydrogen bonds to ARG-90 and TYR-102 of MD-2, stabilizing the complex. We put forward a strategy to increase hydrophobic property and add hydrogen bond donors or hydrogen bond acceptors for the purpose of forming hydrogen bonds with GLU-92, which was mentioned in the docking study. What’s more, we can retain some pharmacophores and employ scaffold hopping method, known as lead hopping, to discover novel potent MD-2 inhibitors.37,38 In conclusion, by combining theoretical and experimental approaches, we characterized a structure-based drug design of a potential anti-inflammatory inhibitor by targeting the MD-2 protein in the TLR4 signaling pathway.
Acknowledgments
Financial support was provided by the National Natural Science Funding of China (21272179, 81473242, and 81570027), and Zhejiang Provincial Natural Science Foundation (LY14C050004).
Disclosure
The authors report no conflicts of interest in this work.
References
Hecht G. Innate mechanisms of epithelial host defense: spotlight on intestine. Am J Physiol. 1999;277(3 Pt 1):C351–C358. | ||
Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340(2):115–126. | ||
Sacks GP, Studena K, Sargent K, Redman CW. Normal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsis. Am J Obstet Gynecol. 1998;179(1):80–86. | ||
Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96(9):939–949. | ||
Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. | ||
Reiter RJ, Melchiorri D, Sewerynek E, et al. A review of the evidence supporting melatonin’s role as an antioxidant. J Pineal Res. 1995;18(1): 1–11. | ||
Raetz CRH, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. | ||
Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249(4975):1431–1433. | ||
Park SH, Kim ND, Jung JK, Lee CK, Han SB, Kim Y. Myeloid differentiation 2 as a therapeutic target of inflammatory disorders. Pharmacol Ther. 2012;133(3):291–298. | ||
Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458(7242):1191–1195. | ||
Shimazu R, Akashi S, Ogata H, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on toll-like receptor 4. J Exp Med. 1999;189(11):1777–1782. | ||
Zhang H, Tay PN, Cao W, Li W, Lu J. Integrin-nucleated toll-like receptor (TLR) dimerization reveals subcellular targeting of TLRs and distinct mechanisms of TLR4 activation and signaling. FEBS Lett. 2002;532(1–2):171–176. | ||
Christiansen D, Brekke OL, Stenvik J, Lambris JD, Espevik T, Mollnes TE. Differential effect of inhibiting MD-2 and CD14 on LPS-versus whole E. coli bacteria-induced cytokine responses in human blood. Adv Exp Med Biol. 2012; 946:237–251. | ||
Roh E, Lee HS, Kwak JA, et al. MD-2 as the target of nonlipid chalcone in the inhibition of endotoxin LPS-Induced TLR4 activity. J Infect Dis. 2011;203(7):1012–1020. | ||
Ohto U, Fukase K, Miyake K, Satow Y. Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science. 2007;316(5831):1632–1634. | ||
Mancek-Keber M, Gradisar H, Inigo Pestana M, Martinez de Tejada G, Jerala R. Free thiol group of MD-2 as the target for inhibition of the lipopolysaccharide-induced cell activation. J Biol Chem. 2009;284(29): 19493–19500. | ||
Resman N, Gradisar H, Vasl J, Keber MM, Pristovsek P, Jerala R. Taxanes inhibit human TLR4 signaling by binding to MD-2. FEBS Lett. 2008;582(28):3929–3934. | ||
Gradisar H, Keber MM, Pristovsek P, Jerala R. MD-2 as the target of curcumin in the inhibition of response to LPS. J Leukoc Biol. 2007;82(4):968–974. | ||
Zhang XL, Zhang YD, Wang T, Guo HY, Liu QM, Su HX. Evaluation on antioxidant effect of xanthohumol by different antioxidant capacity analytical methods. J Chemistry. 2014;2014:1–6. | ||
Kiyofuji A, Yui K, Takahashi K, Osada K. Effects of xanthohumol-rich hop extract on the differentiation of preadipocytes. J Oleo Sci. 2014;63(6):593–597. | ||
Cho YC, Kim HJ, Kim YJ, et al. Differential anti-inflammatory pathway by xanthohumol in IFN-gamma and LPS-activated macrophages. Int Immunopharmacol. 2008;8(4):567–573. | ||
Peluso MR, Miranda CL, Hobbs DJ, Proteau RR, Stevens JF. Xanthohumol and related prenylated flavonoids inhibit inflammatory cytokine production in LPS-activated THP-1 monocytes: structure-activity relationships and in silico binding to myeloid differentiation protein-2 (MD-2). Planta Med. 2010;76(14):1536–1543. | ||
Burger SK, Lacasse M, Verstraelen T, Drewry J, Gunning P, Ayers PW. Automated parametrization of AMBER force field terms from vibrational analysis with a focus on functionalizing dinuclear zinc(II) scaffolds. J Chem Theory Comput. 2012;8(2):554–562. | ||
Khavrutskii IV, Legler PM, Friedlander AM, Wallqvist A. A reaction path study of the catalysis and inhibition of the Bacillus anthracis CapD gamma-glutamyl transpeptidase. Biochemistry. 2014;53(44):6954–6967. | ||
Muddana HS, Sapra NV, Fenley AT, Gilson MK. The SAMPL4 hydration challenge: evaluation of partial charge sets with explicit-water molecular dynamics simulations. J Comput Aided Mol Des. 2014;28(3):277–287. | ||
Beauchamp KA, Lin YS, Das R, Pande VS. Are protein force fields getting better? a systematic benchmark on 524 diverse NMR measurements. J Chem Theory Comput. 2012;8(4):1409–1414. | ||
Wickstrom L, Okur A, Simmerling C. Evaluating the performance of the ff99SB force field based on NMR scalar coupling data. Biophys J. 2009;97(3):853–856. | ||
Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins. 2006;65(3):712–725. | ||
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004; 25(9):1157–1174. | ||
Case DA, Cheatham TE, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. | ||
Salomon-Ferrer R, Case DA, Walker RC. An overview of the Amber biomolecular simulation package. Wiley Interdisciplinary Rev: Comput Mol Sci. 2013;3(2):198–210. | ||
Munoz-Garcia JC, Corzana F, de Paz JL, Angulo J, Nieto PM. Conformations of the iduronate ring in short heparin fragments described by time-averaged distance restrained molecular dynamics. Glycobiology. 2013;23(11):1220–1229. | ||
Sagui C, Darden TA. Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu Rev Biophys Biomol Struct. 1999; 28:155–179. | ||
Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J Chem Theory Comput. 2008;4(1):116–122. | ||
Hess B, Bekker H, Berendsen HJC, Fraaije J. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–1472. | ||
Garate JA, Oostenbrink C. Lipid a from lipopolysaccharide recognition: structure, dynamics and cooperativity by molecular dynamics simulations. Proteins. 2013;81(4):658–674. | ||
Martin YC, Muchmore S. Beyond QSAR: lead hopping to different structures. QSAR Combinator Sci. 2009;28(8):797–801. | ||
Schneider G, Schneider P, Renner S. Scaffold-Hopping: how far can you jump? QSAR Combinator Sci. 2006;25(12):1162–1171. |
Supplementary materials
Video S1 50 ns of apo MD-2 protein molecular dynamics simulation.
Note: The cavity of MD-2 closes rapidly in the absence of any ligand.
Abbreviation: MD-2, myeloid differentiation protein 2.
Video S2 50 ns of xanthohumol/MD-2 complex molecular dynamics simulation.
Notes: Xanthohumol was stable in the hydrophobic cavity of MD-2. MD-2 and xanthohumol were shown as cartoon and sticks, respectively. The sticks of upper left and lower right corner were TYR-102 and ARG-90.
Abbreviation: MD-2, myeloid differentiation protein 2.
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.