Back to Journals » Drug Design, Development and Therapy » Volume 17
Dendritic Cell-Derived Exosomes Driven Drug Co-Delivery Biomimetic Nanosystem for Effective Combination of Malignant Melanoma Immunotherapy and Gene Therapy
Authors Lin J, Huang N, Li M ![]() , Zheng M
, Zheng M ![]() , Wang Z, Zhang X, Gao H, Lao Y, Zhang J, Ding B
, Wang Z, Zhang X, Gao H, Lao Y, Zhang J, Ding B ![]()
Received 1 May 2023
Accepted for publication 4 July 2023
Published 19 July 2023 Volume 2023:17 Pages 2087—2106
DOI https://doi.org/10.2147/DDDT.S414758
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Jiecheng Lin,1,2,* Na Huang,2,* Mingjuan Li,2 Mengyuan Zheng,2 Zhuoxiang Wang,2 Xiaojuan Zhang,2 Huan Gao,2 Yunzhe Lao,2 Jie Zhang,2 Baoyue Ding2
1School of Pharmaceutical Sciences, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, 214122, People’s Republic of China; 2Department of Pharmaceutics, Jiaxing Key Laboratory for Photonanomedicine and Experimental Therapeutics, College of Medicine, Jiaxing University, Jiaxing, 314001, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Baoyue Ding; Jie Zhang, Department of Pharmaceutics, Jiaxing Key Laboratory for Photonanomedicine and Experimental Therapeutics, College of Medicine, Jiaxing University, No. 118 Jiahang Road, Jiaxing, Zhejiang Province, 314001, People’s Republic of China, Tel/Fax +86-0573-83643808, Email [email protected]; [email protected]
Purpose: Malignant melanoma (MM), the most lethal skin cancer, is highly invasive and metastatic. These qualities are related to not only genetic mutations in MM itself but also the interaction of MM cells with the immune system and microenvironment. This study aimed to construct a combined immunotherapy and gene therapy drug delivery system for the effective treatment of MM.
Methods: Mature dendritic cell (mDC) exosomes (mDexos) with immune induction functions were used as carriers. BRAF siRNA (siBRAF) with the ability to silence mutated BRAF in MM was encapsulated in mDexos by electroporation to construct a biomimetic nanosystem for the codelivery of immunotherapy and gene therapy drugs (siBRAF-mDexos) to the MM microenvironment. Then, we investigated the nanosystem’s serum stability and biocompatibility, uptake efficiency in mouse melanoma cells (B16-F10 cells), cytotoxicity against B16-F10 cells and inhibitory effect on BRAF expression. Furthermore, we evaluated its antimelanoma activity and safety in vivo.
Results: SiBRAF-mDexos were nanosized. Compared to siBRAF, siBRAF-mDexos displayed significantly increased serum stability, biocompatibility, uptake efficiency in B16-F10 cells, and cytotoxicity to B16-F10 melanoma cells; they also had a significantly greater inhibitory effect on BRAF expression and induced T-lymphocyte proliferation. Moreover, compared with siBRAF, siBRAF-mDexos showed significantly enhanced anti-MM activity and a high level of safety in vivo.
Conclusion: The study suggests that the siBRAF-mDexo biomimetic drug codelivery system can be used to effectively treat MM, which provides a new strategy for combined gene therapy and immunotherapy for MM.
Keywords: malignant melanoma, dendritic cell exosomes, BRAF siRNA, immunotherapy, gene therapy
Graphical Abstract:

Plain-Language Summary
Malignant melanoma (MM) is the most lethal skin cancer. Recent studies have found that the invasion and metastasis of MM are related not only to MM gene mutation but also to the interaction between MM cells and the immune system and microenvironment. The application of RNAi-based therapeutics to silence the gene mutation at BRAF V600E, which is present in most MM cases, can significantly improve immunotherapy effectiveness and inhibit the invasion and metastasis of MM. In this study, we used mDexos, mature dendritic cell exosomes with immune-inducing function, as vectors to carry BRAF siRNA (siBRAF) that can silence the MM gene mutation to construct a biomimetic nanosystem, siBRAF-mDexos, for combined of MM immunotherapy and gene therapy. The average diameter of siBRAF-mDexos was 128.46 ± 13.34 nm, and the zeta potential was −14.1 ± 1.3 mV. Compared with those of free siBRAF, the serum stability, biocompatibility, cell uptake efficiency and BRAF gene silencing effect of siBRAF-mDexos were significantly improved, and the proliferation of B16-F10 mouse melanoma cells was significantly inhibited. Moreover, compared with siBRAF, siBRAF-mDexos showed significantly enhanced anti-MM activity and a high level of safety in vivo. This study provides a new idea for the use of exosomes combined with immunotherapy and gene therapy for MM.
Introduction
Malignant melanoma (MM) is an aggressive malignant skin cancer with the highest mortality rate among skin cancers. In recent years, the global incidence of MM has been increasing rapidly.1,2 MM tends to invade and metastasize, and is insensitive to chemotherapy and radiotherapy. There are currently no effective treatments for metastatic malignant melanoma (MMM). The prognosis of patients with MM is abysmal, as the 5-year survival rate is 10–15%, and the average median survival time is only 6–9 months.3,4 Research on the pathogenesis of MM has revealed, that MM invasion and metastasis are related to many factors, including gene mutations inherent to MM and the interplay between MM cells and the immune system and microenvironment.5,6 Therefore, immunotherapy and gene therapy have become promising strategies for MMM.
Recently, critical breakthroughs have been made in anti-MM immunotherapy research. Many forms of MM immunotherapy have entered clinical trials and been applied in the clinic. Specific immunotherapy has become the most promising immunotherapeutic strategy for treating MM due to its low toxicity, ability to specifically kill tumour cells, and high long-term efficacy.2,7 However, studies have found that immunotherapy cannot achieve the expected efficacy due to the unique tumour microenvironment (TME) of MM.8–10 The primary method by which tumour cells undergo immune clearance is mediated by cytotoxic T lymphocytes (CTLs), especially through CD8+ T-cell-mediated cytotoxicity (CMC). CD8+ T cells must be activated to initiate an immune response. However, dendritic cells (DCs), the antigen-presenting cells in the MM microenvironment, are primarily immature and lack costimulatory factors that can sufficiently activate T cells, which induces immune anergy. In addition, MM patients with specific gene mutations exhibit a poor response to immunotherapy and are prone to metastasis and recurrence.9,10 Up to 50%~70% of MM patients have the BRAF V600E mutation, which can promote the growth and proliferation of human MM cells and enhance tumour cell invasion and angiogenesis. This mutation is one of the main causes of further invasion and metastasis of MM and treatment failure.11,12 MM with gene mutation is particularly vulnerable to immune blockade, which causes immunotherapy failure. Amini-Adle et al13 conducted a clinical study on PD-1 immunotherapy in patients in whom treatment with the BRAF inhibitor (BRAFi) ipilimumab or chemotherapy failed and analysed the treatment effect of a PD-1 antibody in patients with and without the BRAF V600E mutation. The results indicate that for advanced melanoma, BRAFi therapy failure also results in anti-PD-1 therapy failure. Therefore, to improve the efficacy of immunotherapy against MM, we must improve the MM microenvironment, such as by promoting the maturation of infiltrating DCs in the microenvironment, activating the T-cell immune response, and inhibiting the BRAF V600E mutation.
Mature DC (mDC) exosomes (mDexos) have been found to contain immune-related components, including antigen MHC class I and II molecules (often in complex with antigenic epitopes), costimulatory molecules (such as CD80, CD86, and CD40), intercellular adhesion molecules (such as ICAM-1) and integrins.14 These immune-related components enable mDexos to have some of the functions of mDCs, which can directly act on T cells to induce an immune response.15,16 Therefore, mDexos can be used as an immunotherapeutic drug for MM to activate T cells in the TME of MM in order to exert cytotoxic effects and exert a specific immune response.
The BRAF V600E mutation can be suppressed using gene therapy. RNA interference (RNAi) can inhibit the proliferation of MM cells and reverse the immune tolerance of antigen-presenting cells.17 Sumimoto et al11 used a lentiviral vector to deliver BRAF V600E-specific siRNA into melanoma cells. The siRNA inhibited the production of IL-10, IL-6, and VEGF, thus inhibiting extracellular regulated protein kinase (ERK) phosphorylation and restoring DCs to a mature state. However, siRNA has low cell transfection efficiency and is easily degraded by nucleases in vivo, resulting in poor stability in vivo. Therefore, nanocarriers are needed for encapsulation to obtain higher transfection efficiency and greater stability in vivo.
Exosomes (Exos) from biological sources are cellular vesicles with a diameter of only 30~120 nm. They have been widely used as excellent nanodrug carriers because of the wide range of exosomal sources and because of their high stability, good biocompatibility with the immune system, small particle size for avoiding phagocytosis by macrophages and ability to directly penetrate biological membranes.18,19 In addition, the lipid bilayer membrane structure of Exos can protect the genes within them from degradation and improve their stability in vivo. Moreover, Exos can directly release the encapsulated genes into receptor cells via cell membrane fusion to achieve greater delivery efficiency.20,21 Therefore, Exos can deliver small DNA, siRNA, and miRNA molecules for precise and highly effective gene therapy.
In this study, mDexos with both immune function and drug-loading capacity were selected as carriers, and BRAF siRNA (siBRAF), a targeted gene drug that can effectively inhibit the invasion and metastasis of MM, was encapsulated by electroporation to construct a bionic nanosystem called siBRAF-mDexos. This system was used for specific codelivery of immunotherapy and gene therapy drugs to the MM microenvironment (as shown in Figure 1A). The mDexos in the bionic nanosystem activated T cells in the MM microenvironment and induced an immune response, while siBRAF in the bionic nanosystem interfered with the expression of BRAFV600E and inhibited the growth and metastasis of MM (as shown in Figure 1B). The particle size, zeta potential, serum stability, and haemolytic toxicity of the bionic nanosystem in vitro were all investigated in detail. Moreover, the uptake efficiency and inhibitory effect on cell proliferation of siBRAF-mDexos were examined in B16-F10 cells, and the anti-MM effects and in vivo safety of the system were evaluated using B16-F10 tumour-bearing C57BL/6J mice.
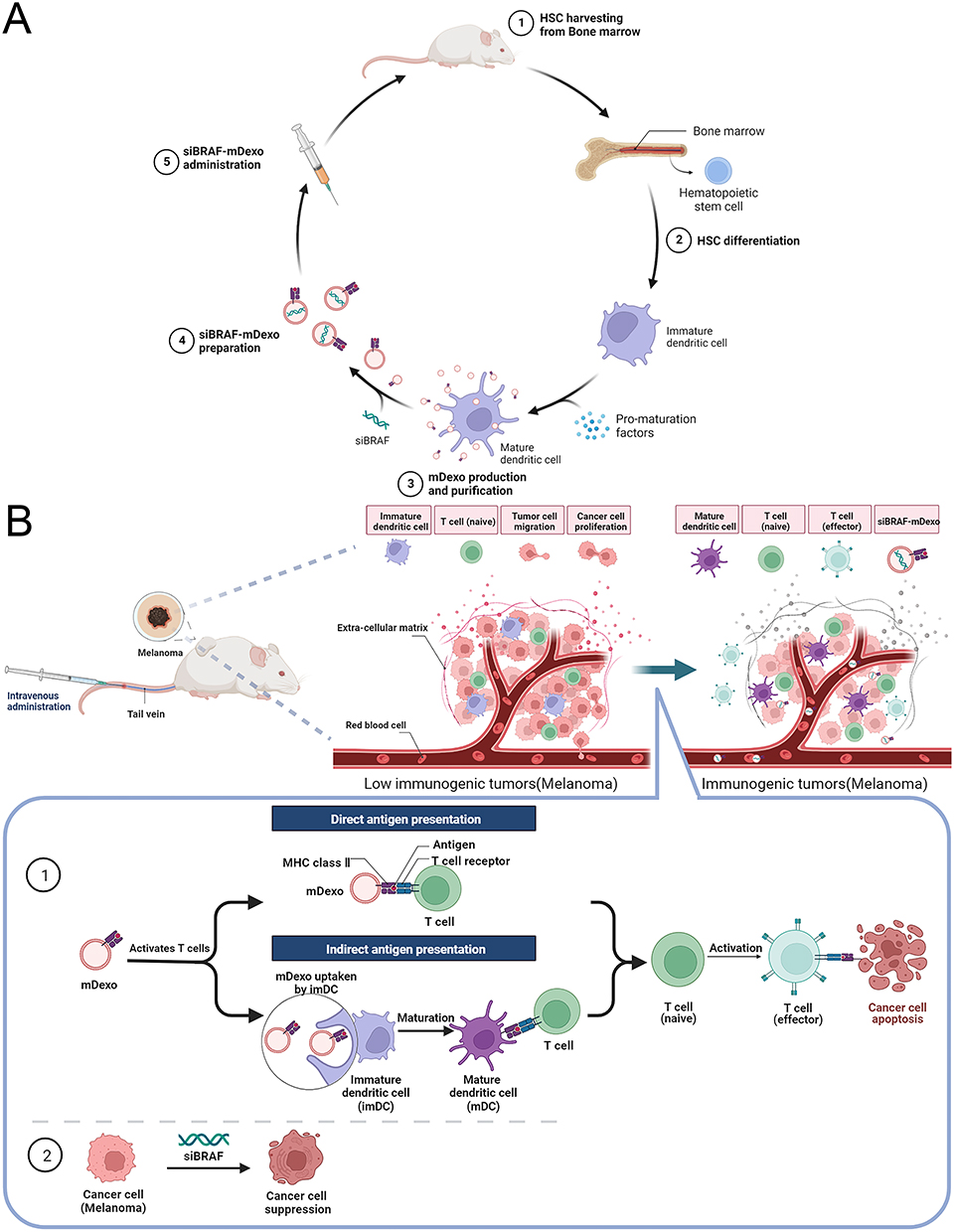 |
Figure 1 Schematics of (A) the construction of the biomimetic nanosystem (siBRAF-mDexos) and (B) the mechanisms of siBRAF-mDexos in MM treatment. |
Materials and Methods
Materials
MojosortTM mouse CD11c nanobeads, PE anti-mouse CD86, APC anti-mouse CD40, PerCP/Cyanine5.5 anti-mouse I-A/I-E, FITC anti-mouse CD11c, and MojosortTM mouse CD8 T-cell isolation kits were purchased from BioLegend Co. (California, USA). Interleukin-4 (IL-4), granulocyte-macrophage colony stimulating factor (GM-CSF), and tumour necrosis factor-α (TNF-α) were purchased from Novoprotein Technology Co. Ltd. (Suzhou, China). Lipopolysaccharide (LPS), radioimmunoprecipitation assay (RIPA) lysis buffer, BCA protein concentration assay kit, 5X SDS‒PAGE sample loading buffer, QuickBlock™ blocking buffer for Western blotting, RNase-free water, InstantViewTM green fluorescent DNA loading buffer (6X), and 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) staining solution were purchased from Beyotime Biotechnology Co. Ltd. (Shanghai, China). Anti-CD9, anti-CD63, and goat anti-rabbit IgG H&L antibodies were purchased from Abcam Co. Ltd. (Shanghai, China). A Cell Counting Kit-8 was supplied by MedChemExpress Biotechnology Co. (Shanghai, China). An anti-B Raf recombinant rabbit monoclonal antibody was supplied by HuaBio Co. Ltd. (Hangzhou, China). ExoQuick-TCTM tissue culture medium Exo precipitation solution was supplied by System Biosciences Biotechnology Co. Ltd. (Beijing, China). Exo-depleted FBS was supplied by Absin Biotechnology Co. Ltd. (Shanghai, China). RPMI 1640 medium, foetal bovine serum (FBS), and penicillin-streptomycin solution were obtained from HyClone™. The following siRNA was synthesized by GenePharma Co. Ltd. (Shanghai, China): siBRAF with sticky ends (5’ labelled with Cy3), sense (5’-3’): agaauggaucuggaucauTT, antisense (5’-3’): augauccagauccaauucuTTCCCAAAAACC. B16-F10 cells were purchased from the BeNa Culture Collection Biotechnology Co. Ltd. (Beijing, China). C57BL/6J mice were provided by Cavens Co. Ltd. (Changzhou, China).
Cell Cultures
B16-F10 cells were cultured in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin‒streptomycin in a 37 °C, 5% CO2 incubator.
mDC Cultures
Bone marrow cells were prepared from the femurs of healthy C57BL/6J mice aged 4–6 weeks for the isolation of mouse haematopoietic stem and progenitor cells (HSPCs). The HSPCs were then seeded into 6-well plates with 3 mL of complete medium containing RPMI 1640 medium with 20% Exo-depleted FBS and 1% penicillin‒streptomycin, supplemented with 20 ng/mL IL-4 and 20 ng/mL GM-CSF for 5 days. Afterwards complete medium containing immature DCs (imDCs) was collected, and the imDCs were then cultured in medium containing 75 ng/mL TNF-α and 1 μg/mL LPS for 2 days to induce their maturation. Finally, mDCs were collected on the 7th day.
Identification of mDCs
Morphological Identification
The mDCs were placed under an inverted microscope (CKX53, Olympus) on days 1, 3, 5 and 7 after inoculation, and were photographed to determine their morphology.
Flow Cytometry
ImDCs and mDCs (1×106) on the 5th day and 7th day prepared as described above were harvested, washed with phosphate-buffered saline (PBS) and centrifuged to discard the supernatant. The cells were resuspended in 100 μL of PBS, and then 5 μL of PE anti-mouse CD86, APC anti-mouse CD40, PerCP/Cyanine 5.5 anti-mouse I-A/I-E, and FITC anti-mouse CD11 was added. Afterwards, the cells were incubated in an ice bath for 15 min, washed three times with PBS, resuspended in 1 mL of PBS, and then loaded onto a flow cytometer (BD FACSVerseTM, BD Bioscience) to determine the expression of the cell-surface specific antigens CD40, CD86 and MHC II.
Isolation of imDexos and mDexos
To isolate imDC exosomes (imDexos) and mDexos, the cell supernatants on the 5th day and 7th day prepared as described above were collected and centrifuged in a gradient (300 × g for 10 min, 1200 × g for 20 min, and 10,000 × g for 30 min) to remove the cell debris and microbubbles in the cell culture medium at 4 °C. The supernatant was collected and filtered with a microporous 0.22-μm filter membrane, and then ExoQuick-TCTM tissue culture medium Exo precipitation solution was added. The sample was mixed well, kept at 4 °C for 12 h, and centrifuged at 1500 × g at 4 °C for 30 min. The supernatant was discarded, and PBS was added to resuspend the precipitate which was then centrifuged at 1500 × g at 4 °C for another 30 min to collect the Dexos. The Dexos were resuspended in PBS and stored at −80 °C for future use.
Identification of Dexos
Dexo Morphology
Dexos were diluted with PBS and fixed with 1% glutaraldehyde. Twenty millilitres of the suspension was dropped onto a Formvar/carbon-coated grid, stained with 3% (w/v) phosphotungstic acid for 5 min, and observed and photographed by a transmission electron microscope (HT7800, HITACHI) at 80 kV.
Particle Size and Zeta Potential of mDexos
MDexos were resuspended in deionized water, and the particle size and zeta potential were measured using a laser particle sizer (Litesizer 500, Anton Paar).
Characteristic Protein Detection
RIPA lysis buffer was added to the Dexos at a 1:1 volume ratio, and the Dexos were lysed on ice for 1 h. The lysate was centrifuged at 10,000 rpm for 15 min at 4 °C and the supernatant was collected. Then, 10 μL of the supernatant (protein concentration of 2.5 mg/mL) was added to 5X SDS‒PAGE sample loading buffer at a ratio of 4:1. The mixture was placed in a metal bath at 95 °C, and incubated for 15 min. The samples were loaded and electrophoresed at a constant voltage of 100 V for 90 min and then a constant current of 200 mA for 120 min to transfer the protein bands to PVDF membranes. The membranes were blocked with QuickBlock™ blocking buffer for 30 min and separately incubated with anti-CD9 and anti-CD63 antibodies overnight. The membranes were then washed 3 times with 1X TBST and incubated for 2 h with a goat anti-rabbit IgG H&L antibody. Finally, a chemiluminescence imaging system was used to analyse the protein bands on the PVDF membrane.
T-Lymphocyte Proliferation Induced by mDexos
Splenic cells were extracted from healthy C57BL/6J mice aged 4–6 weeks. T lymphocytes were isolated from the mouse splenic cells using the MojosortTM mouse CD8 T-Cell isolation kit and inoculated in 96-well plates at a density of 1×104 cells/100 μL. The mDexos and imDexos were diluted with complete medium to different concentrations (0, 0.025, 0.05, 0.1, 0.2, 0.4, 0.8, 1.6, and 3.2 mg/mL) and 100 μL of sample was added to each well separately, with six duplicate wells made in parallel. The plates were incubated at 37 °C and 5% CO2 under saturated humidity for 48 h. Then, 10 mL of Cell Counting Kit-8 reagent was added to each well, and the plates were incubated for 2 h. Finally, the absorbance value at 450 nm was measured using a multipurpose microplate reader (Synergy Neo2, BioTek).
Preparation of siBRAF-mDexos
The bionic nanosystem for the codelivery of immunotherapy and gene therapy drugs (siBRAF-mDexos) was prepared by electroporation. The mDexos were dispersed in PBS at a concentration determined by BCA assay, and 2.5 nmol of lyophilized siBRAF (mass of 40 μg) was dissolved in 125 μL of RNase-free water. MDexos and siBRAF at a fixed mass ratio of 7:1 were thoroughly mixed, and then the mixture was transferred to an electroporation cuvette and put into an electroporator (Gene Pulser XcellTM, Bio-Rad). The electroporation parameters were set as follows: voltage 400 V, capacitance 125 μF, resistance ∞ Ω, electrode spacing 4 mm. After electroporation, the samples were transferred to an Eppendorf tube and incubated in an ice bath for 1 h to obtain stable siBRAF-mDexos.
Particle Size and Zeta Potential of siBRAF-mDexos
siBRAF-mDexos were resuspended in deionized water, and the particle size and zeta potential were measured using the laser particle sizer.
Stability in Serum
SiBRAF and siBRAF-mDexos (CsiBRAF: 400 nM) dissolved in RNase-free water were mixed with an equal volume of FBS and then incubated in a constant-temperature water bath at 37 °C. Samples were taken after 0 h, 2 h, 4 h, 6 h, 8 h, 10 h and 12 h of incubation, and RNase inhibitor was added to a final concentration of 2 U/μL. Then, RIPA lysis buffer was added to each sample at a volume ratio of 1:1, and the samples were lysed in an ice bath for 1 h to release undegraded siBRAF. Finally, agarose gel electrophoresis was performed, and fluorescent bands were observed.
In vitro Haemolysis Assay
Sterile fresh rabbit blood was collected and centrifuged at 2500 rpm for 5 min, and the precipitated red blood cells were washed with saline 3–4 times until the supernatant was not red. Then, the obtained red blood cells were resuspended in saline to obtain the final 2% (v/v) RCB suspension. SiBRAF and siBRAF-mDexos were used to prepare a series of solutions at different concentrations (40, 60, 80, 100, 200, 300, and 400 nM) with saline, and 2 mL of each sample solution was incubated with red blood cells (RBCs) for 3 h at room temperature. Moreover, saline and 0.2% Triton X-100 were used as negative and positive controls, respectively. Subsequently, samples were centrifuged at 5000 rpm for 10 min, and 100 µL of supernatant was collected carefully and transferred to a 96-well plate to measure the absorbance at 540 nm by a multipurpose microplate reader. The haemolysis rate was calculated according to the following formula:

Cytotoxicity Assay
To evaluate the cytotoxicity of siBRAF-mDexos in vitro, 1×104 B16-F10 cells per well were seeded in 96-well plates and incubated for 12 h, before treatment with siBRAF and siBRAF-mDexos at different concentrations of 0, 0.3, 1.5, 7.5, 15, 75, 150, 300 and 600 nM. After incubation for 48 h, 10 μL of Cell Counting Kit-8 reagent was added and the cells were further incubated for 2 hours. The absorbance at 450 nm was measured using a multipurpose microplate reader. The viability of untreated cells was set at 100%, and the absorbance of wells with medium and without cells was set as zero.
Cellular Uptake Assay
Flow Cytometry Analysis
Flow cytometry analysis was performed to quantify the cellular uptake of siBRAF-mDexos. B16-F10 cells (1×105 cells per well) were seeded in 12-well plates and incubated for 12 h. The culture medium was then replaced with 1 mL of siBRAF and siBRAF-mDexos (CsiBRAF: 60 nM), incubated for 12 h and washed three times with PBS. Medium without any drug was used as the negative control. Thereafter, the cells were detached using trypsin and washed with ice-cold PBS. Finally, the cells were resuspended in 0.5 mL of PBS, filtered with a 200-mesh cell screen, and then analysed by a flow cytometer for detection.
Laser Confocal Microscopy
To investigate the cellular uptake of siBRAF-mDexos, 1×105 B16-F10 cells per well were seeded onto sterilized microscope cover slips placed in 6-well plates and incubated for 12 h. The cells were then treated with siBRAF and siBRAF-mDexos (CsiBRAF: 60 nM) for 12 h. Untreated cells were used as controls. Then, the cells were washed three times with PBS, fixed with 4% paraformaldehyde for 15 min, and stained with DAPI for 10 min. The samples were captured by a laser confocal microscope (FLUOVIEWTM FV3000, Olympus).
BRAF Protein Expression
To detect BRAF protein expression after treatment with siBRAF-mDexos, 1×105 B16-F10 cells per well were seeded in 6-well plates and cultured for 12 h. The cells were then treated with siBRAF and siBRAF-mDexos (CsiBRAF: 60 nM) for 24 h, washed three times with PBS, transferred to an Eppendorf tube, and centrifuged at 1000 rpm for 5 min at 4 °C. The supernatant was discarded, RIPA lysis buffer was added to the cell pellet, and the cells were lysed in an ice bath for 1 h and centrifuged at 10,000 rpm for 15 min at 4 °C. Then, the supernatant was collected and added to 5X SDS‒PAGE sample loading buffer at a ratio of 1:4 (v/v). The mixture was placed in a metal bath at 95 °C for 15 min and subjected to electrophoresis at a constant voltage of 100 V for 90 min. The proteins were then electrophoresed at a constant current of 200 mA for 120 min to transfer the protein bands to PVDF membranes. After blocking with QuickBlockTM blocking buffer for 30 min, the samples were incubated with a recombinant rabbit monoclonal anti-B Raf antibody overnight and washed three times in 1X TBST before being incubated for 2 h with a goat anti-rabbit IgG H&L antibody. The bands on the PVDF membrane were analysed using a chemiluminescence imaging system (Amersham Imager 680, Cytiva).
In vivo Antitumour Efficacy and Safety
Tumour implantation was performed by injecting 2×105 B16-F10 cells (1.0 ×106 cells/mL, 0.2 mL per mouse) into the back subcutaneous region of each male C57BL/6J mouse aged six weeks. The procedure was carried out in a laminar flow hood using aseptic techniques. The mouse weight and tumour growth after implantation were monitored daily. Mice with a tumour volume (TV) of approximately 100 mm3 were selected for the study. The selected mice were randomly divided into four groups, with six mice in each group. Each group was treated with 0.2 mL of saline, mDexos, siBRAF, or siBRAF-mDexos via tail vein injection. The dose of siBRAF was 1 mg/kg, and that of mDexos was 7 mg/kg per injection based on the mouse body weight. The treatments were administered once every two days, and four treatments were carried out in total. TV and mouse body weight were measured the day after each treatment and monitored every two days. Blood was collected from the retroorbital venous plexus of the mice by capillary tube on the 13th day after administration for subsequent routine blood and liver and kidney function tests. The mice were sacrificed after blood sampling, and the tumours were removed from the skin, photographed, weighed and stored properly for future use. The TV in the mice was calculated as follows:

where a and b are the long and short diameters of the tumour (unit: mm), respectively.
Statistical Analysis
GraphPad Prism 5 software was used for statistical analysis. The data are reported as the mean ± standard deviation (SD). Differences were analysed by one-way analysis of variance and Student’s t-test. A P value <0.05 or <0.01 indicated statistical significance.
Results and Discussion
Identification of mDCs
The mDCs were obtained from murine bone marrow HSPCs. The cell morphology was observed under an inverted microscope, and the results are shown in Figure 2A. The HSPCs were evenly dispersed on the 1st day and began to aggregate into colonies on the 3rd day, and the cells contained fine burr protrusions on their surfaces on the 5th day, identifying them as imDCs. On the 7th day, dendritic or stellate processes were observed on the surfaces of the cells; the cells were at this point considered mDCs. The combination of GM-CSF and IL-4 is a well-established inducer of imDCs. HSPC-derived monocytes are the precursors of DCs and can differentiate into macrophages, osteoclasts and imDCs. The combination of GM-CSF and IL-4 effectively can upregulate the TNF-α converting enzyme and monocyte activity, resulting in the shedding of the ectodomain of the macrophage colony-stimulating factor receptor, inhibiting the phenotypic transformation of monocytes to macrophages or osteoclasts, and promoting the differentiation of monocytes into imDCs.22 Moreover, imDCs can be transformed into mDCs under stimulation with TNF-α and LPS.
 |
Figure 2 Identification of mDCs. (A) Inverted microscopic analysis of morphology, scale bar: 100 μm, magnification: 400X; scale bar: 20 μm, magnification: 1600X. (B) Flow cytometry results. |
The expression rates of the surface-specific antigens CD40, CD86, and MHC II on the imDCs on the 5th day and mDCs on the 7th day after inoculation were measured using flow cytometry. As shown in Figure 2B, the expression rates of MHC II, CD40, and CD86 on the surfaces of imDCs were 0.4%, 18.04%, and 7.93%, respectively, while the expression rates of MHC II, CD40, and CD86 on the surface of mDCs were 43.51%, 95.87%, and 72.26%, respectively. Thus, the expression rates of the three antigens were significantly higher on the surfaces of mDCs than on the surfaces of imDCs. The expression rates of MHC II, CD40, and CD86 on the surfaces of murine DCs treated with TNF-α and LPS were 108 times, 5 times, and 9 times higher than those in the untreated group, respectively. In the immature stage, imDCs strongly express pattern recognition receptors, which can effectively recognize and take up exogenous antigens, and weakly express MHC class II molecules and costimulatory molecules; thus, their ability to present antigens and stimulate the immune response is weak. After antigen uptake or inflammatory stimulation, imDCs begin to migrate under the action of chemokines and gradually mature. The mDCs with strong antigen-presenting ability can activate naive T cells, because they have dendritic processes on their surfaces, low expression of pattern recognition receptors, high expression of MHC class II molecules and the costimulatory molecules CD40 and CD86.23,24 Our results were consistent with previous reports, indicating that this method effectively induced the maturation of DCs.
Identification and Characterization of mDexos
Dexo Morphology
MDexos were isolated from the supernatant of mDCs. The morphology of mDexos was observed by transmission electron microscopy (TEM). As shown in Figure 3A, no significant difference in morphology between imDexos and mDexos was observed, and both types of cells showed a teacup-like morphology, with morphological characteristics typical of Exos.
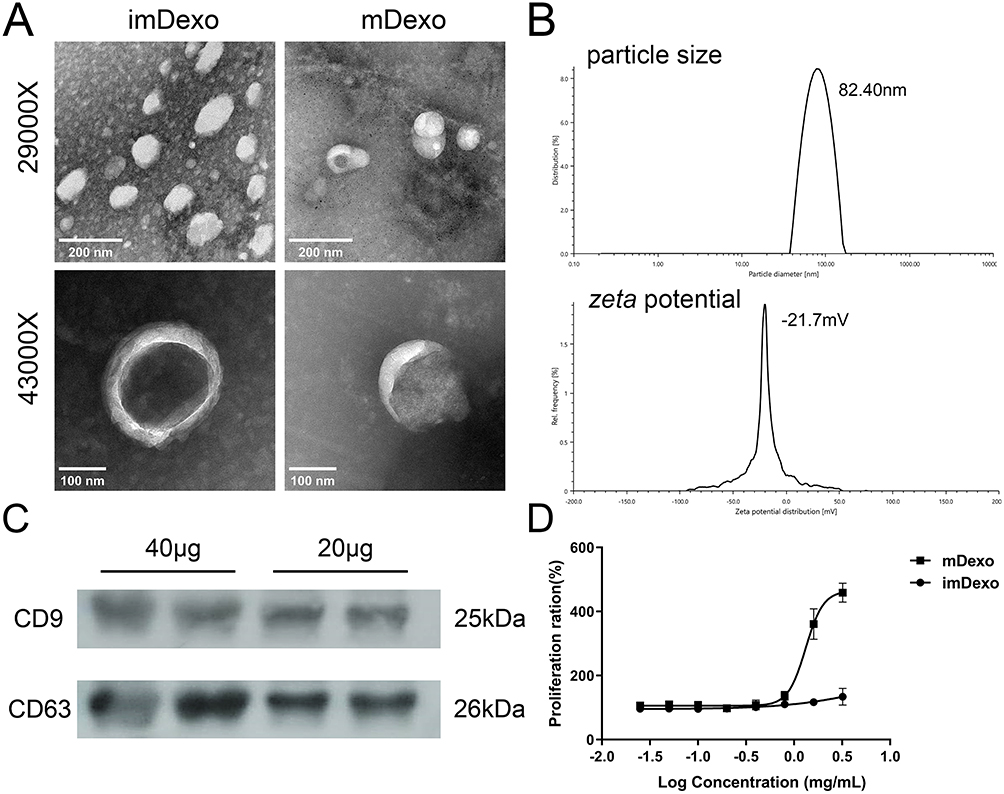 |
Figure 3 Identification and characterization of mDexos. (A) TEM images, scale bars: 200 nm, magnification: 29,000X; scale bars: 100 nm, magnification: 43,000X. (B) Plots of particle size and zeta potential. (C) Western blotting. (D) T-lymphocyte proliferation after 48 h of incubation with different concentrations of Dexos. The data are reported as the mean ± standard deviation (n=6). |
Particle Size and Zeta Potential of mDexos
The particle size and zeta potential of mDexos were investigated with a laser particle sizer. The observed average diameter of the mDexos was 86.37 ± 4.16 nm with the zeta potential of −20.1 ± 1.4 mV (Figure 3B), in accordance with the reported Exo diameter of 30~120 nm with the zeta potential of −10~-50 mV.25,26 The particle size and zeta potential of the extracted mDexos were consistent with the characteristics of Exos.25,26 The mDexos were stable in PBS and were deemed eligible to be used as carriers in the following experiments.
Characteristic Protein Expression
Western blotting was used to detect the protein expression of CD9 and CD63, which are the typical proteins of mDexos. As shown in Figure 3C, mDexos produced obvious bands for CD9 and CD63, indicating that the extracted mDexos contained the CD9 and CD63 proteins.
T-Lymphocyte Proliferation Induced by Dexos
The immune function of mDexos was verified by investigating the ability of mDexos to induce T-lymphocyte proliferation. As shown in Figure 3D, when the concentration was higher than 0.8 mg/mL, the ability of mDexos to induce T-lymphocyte proliferation was much greater than that of imDexos. The ability of the mDexos to induce T-lymphocyte proliferation significantly increased as the concentration increased. The EC50 of imDexos (4.85 ± 0.29 mg/mL) was significantly higher than that of mDexos (1.41 ± 0.08 mg/mL) (P<0.001). These results showed that mDexos were more effective in promoting T-lymphocyte proliferation than imDexos. MDexos contain manya MHC-peptide complexes (MHC class I and II molecules) and costimulatory molecules (such as CD80, CD86, CD40) on their surface. MHC antigen peptide complexes present antigen information to T cells, providing the first signal for T-cell activation, while costimulatory molecules bind to CD28 displayed by T cells become the second signal for T-cell activation. The first and second signals jointly stimulate T-cell proliferation and differentiation, activate CD8+ T cells, and cause these cells to exhibit CMC and a specific immune response.15,16 The above results indicated that the Dexos extracted in this experiment were mDexos and could be used as drug carriers for subsequent melanoma immunotherapy.
Particle Size and Zeta Potential of siBRAF-mDexos
SiBRAF was encapsulated in mDexos by electroporation to construct a biomimetic nanodrug delivery system, named siBRAF-mDexos. The particle size and zeta potential of the siBRAF-mDexos were investigated with a laser particle sizer. As shown in Figure 4, the average diameter of the siBRAF-mDexos was 128.46 ± 13.34 nm, and the zeta potential was −14.1 ± 1.3 mV. Compared with mDexos, the particle size of siBRAF-mDexos were larger, and their zeta potential was slightly lower. The principle of electroporation is to form temporary hydrophilic pores on the exosome membrane by applying an external electric field to increase the permeability of the exosome membrane so that small molecules can pass through the pores and enter the exosome. Since the opening of the temporary pore on the exosomal lipid bilayer membrane is reversible under an appropriate voltage, siBRAF enters the cavity within mDexos and is encapsulated by the lipid bilayer membrane of mDexos after electroporation. Therefore, the structure of each siBRAF-mDexos particle is a lipid bilayer membrane vesicle (mDexo) with siBRAF encapsulated inside (see Figure 1 for the schematic structure of siBRAF-mDexos).
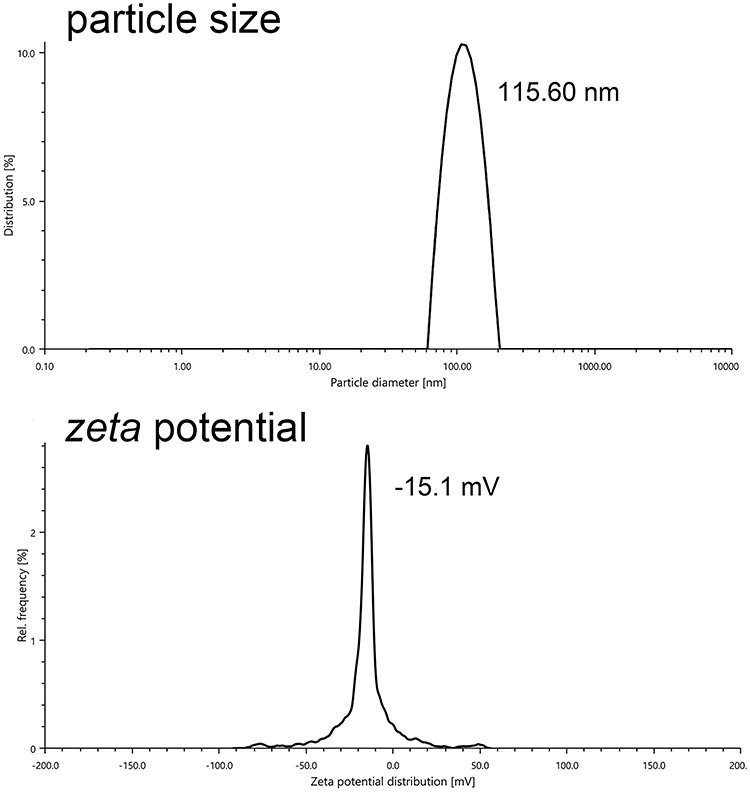 |
Figure 4 Plots of siBRAF-mDexo particle size and zeta potential. |
Serum Stability of siBRAF-mDexos
A major factor that limits the clinical use of siRNA is the instability of naked siRNA in the blood due to ribonuclease degradation.27 In this experiment, the stability of siBRAF and siBRAF-mDexos in serum was investigated by agarose gel electrophoresis. As shown in Figure 5A, the fluorescent band almost completely disappeared after free siBRAF was coincubated with the serum for 4 h, indicating that RNase had completely degraded siRNA in the serum under these conditions. In comparison, the fluorescent band was still clearly visible after siBRAF-mDexos were incubated with serum for 12 h, indicating that encapsulation by mDexos effectively prevented the siRNA from being degraded by RNase in the serum (Figure 5B). Therefore, we predicted that when siBRAF-mDexos are injected into the body, the mDexos will protect siBRAF from being degraded in the blood, thus allowing more siBRAF to reach the melanoma. This prediction laid a foundation for subsequent in vivo experiments.
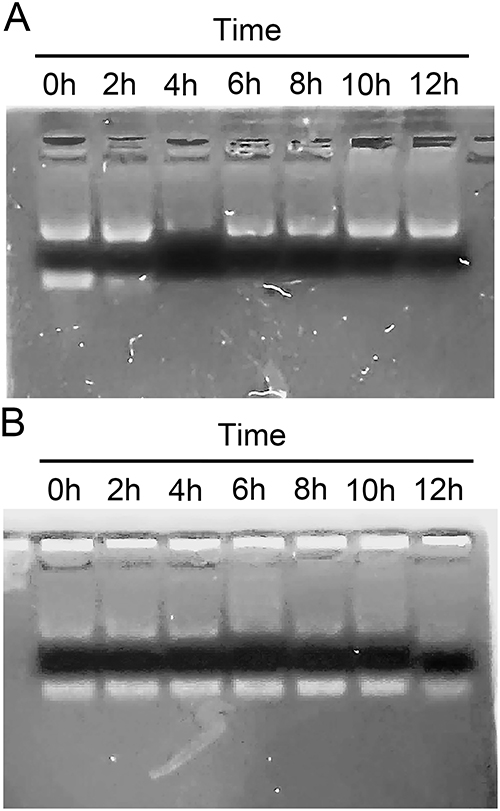 |
Figure 5 Serum stability of siBRAF-mDexos. (A) siBRAF. (B) siBRAF-mDexos. |
Haemolysis of siBRAF-mDexos
A haemolysis assay, which is used to observe whether a tested factor can cause haemolysis and erythrocyte aggregation, should be conducted for injections and other topical preparations that may cause immune or nonimmune haemolysis. As shown in Figure 6, the haemolysis rate of all groups was less than 5%, and the haemolysis rates of the groups administered siBRAF-mDexos at different concentrations were significantly lower than those of the siBRAF treated groups at the same concentrations (P<0.05). These results suggested that the inclusion of mDexos significantly improved the biocompatibility and safety of exogenous siBRAF. The siBRAF in our preparation was encapsulated by mDexos. After entering the bloodstream, the mDexos were in direct contact with blood cells. Because the mDexos were derived from homologous mice and were thus biogenic, while the siBRAF was exogenous, the biocompatibility of biogenic mDexos was higher than that of exogenous siBRAF.
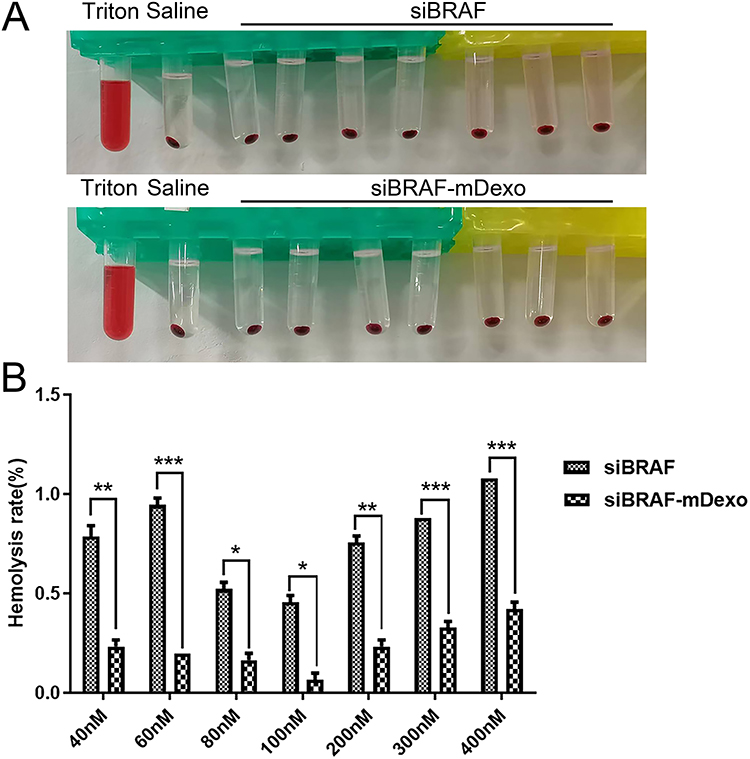 |
Figure 6 Haemolysis of siBRAF and siBRAF-mDexos after 3 h of incubation with RBCs. (A) Photographs of haemolysis test results. (B) Column analysis chart of absorbance at 540 nm wavelength; *P<0.05, **P<0.01, and ***P<0.001 compared with the siBRAF group. The data are reported as the mean ± standard deviation (n=3), one-way ANOVA. |
Cytotoxicity and Cellular Uptake of siBRAF-mDexos
The cell viabilities at different concentrations of siBRAF and siBRAF-mDexos were used to investigate their cytotoxicity in vitro. The results are shown in Figure 7. Both siBRAF and siBRAF-mDexos showed concentration-dependent cytotoxicity in B16-F10 cells. When the concentration of siBRAF-mDexos was higher than 15 nM, the survival rate of B16-F10 cells treated with siBRAF-mDexo was significantly lower than that of the siBRAF group. The IC50 values of siBRAF-mDexos and siBRAF were 56.97 ± 8.64 nM and 254.67 ± 46.89 nM, respectively. The IC50 of siBRAF-mDexos was significantly lower than that of siBRAF (P<0.01). The results indicated that siBRAF-mDexos had a more potent cytotoxic effect on B16-F10 cells than siBRAF.
 |
Figure 7 In vitro cytotoxicity of siBRAF-mDexos at different concentrations against B16-F10 cells for 48 h. The data are reported as the mean ± standard deviation (n=6). |
Flow cytometry and laser confocal microscopy were used to investigate the uptake of siBRAF and siBRAF-mDexos by B16-F10 cells at 12 h. Since the synthesized siBRAF was fluorescently labelled with Cy3, we used the fluorescence intensity of Cy3 to represent the uptake of siRNA. As shown in Figure 8A and B, the uptake rates of free siBRAF and siBRAF-mDexos by B16-F10 cells were 71.09 ±6.65% and 92.83 ±1.54%, respectively. The cellular uptake rate of siBRAF-mDexos was 1.3 times that of free siRNA and significantly higher than that of free siBRAF (P<0.01), indicating that siBRAF encapsulated by mDexos more easily entered B16-F10 cells. As shown in Figure 8C and D, the red fluorescence intensity of Cy3 in the siBRAF-mDexo group was higher than that in the siBRAF group, which was consistent with the flow cytometry results. This finding indicated that more siBRAF in siBRAF-mDexos than free siBRAF was taken up by the B16-F10 cells.
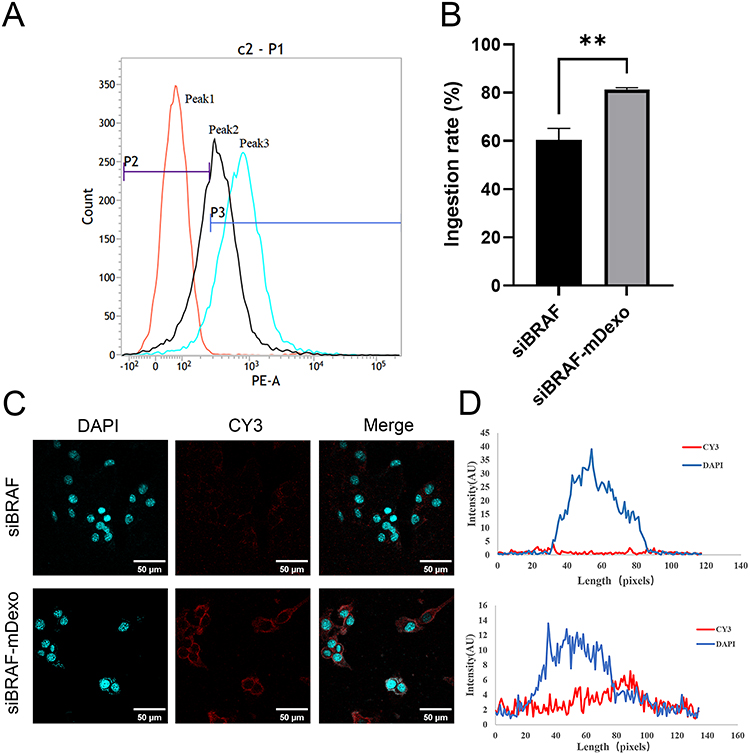 |
Figure 8 Uptake of siBRAF-mDexos in B16-F10 cells over 12 h. (A) One-parameter histogram, peak 1: control, peak 2: siBRAF, peak 3: siBRAF-mDexos. (B) Histogram showing the uptake rate; **P<0.01 compared with the siBRAF group. (C) Fluorescence image, scale bar: 50 μm. (D) Fluorescence colocalization analysis. The data are reported as the mean ± standard deviation (n=3), one-way ANOVA. |
The efficacy of siRNA is limited mainly by the cell membrane, which acts as a barrier. It is difficult for free RNA to pass through the cell membrane, which has a positive surface charge and high lipid content, due to its negative surface charge and high water solubility.28 As the primary medium of cell‒cell communication, Exos can carry siRNA into the cytoplasm via endocytosis and membrane fusion.21 Therefore, the encapsulation of siBRAF in the mDexos increased the siBRAF transfection efficiency into B16-F10 cells and enhanced the killing effect of siBRAF on melanoma. The cytotoxicity of mDexos in B16-F10 cells was also investigated (Supplementary Figure 1). The results of Supplementary experiment 1showed that the mDexos had no significant cytotoxic effect on B16-F10 cells. The cytotoxic effect of the mDexos on B16-F10 cells occurred mainly through secretion of immune-related factors, stimulation of T-cell proliferation and differentiation, and activation of CD8+ T cells, which caused CMC, as confirmed by the results in Figure 3D. However, mDexos did not directly target and kill tumour cells, so they had no cytotoxic effect on B16-F10 cells. Thus, the killing effect of siBRAF-mDexos on B16-F10 cells was produced mainly by siBRAF.
BRAF Protein Expression
The protein expression of BRAF in B16-F10 cells incubated with siBRAF and siBRAF-mDexos for 24 h was investigated by Western blotting. As shown in Figure 9, after B16-F10 cells were incubated with siBRAF and siBRAF-mDexos for 24 h, the BRAF protein bands in the siBRAF-mDexo group were significantly less intense than those in the siBRAF group. Compared with that of the siBRAF group, the greyscale value of the siBRAF-mDexo group was significantly decreased (P<0.05) by 5-fold. Therefore, siBRAF-mDexos exerted an antimelanoma effect by inhibiting the expression of the BRAF protein. In the cellular uptake experiment, compared with free siBRAF, siBRAF-mDexos exhibited a significantly increased uptake efficiency, so more siBRAF was transported into the cytoplasm; this resulted in improved BRAF gene silencing and a stronger killing effect in B16-F10 cells in the siBRAF-mDexo group than in the siBRAF group.
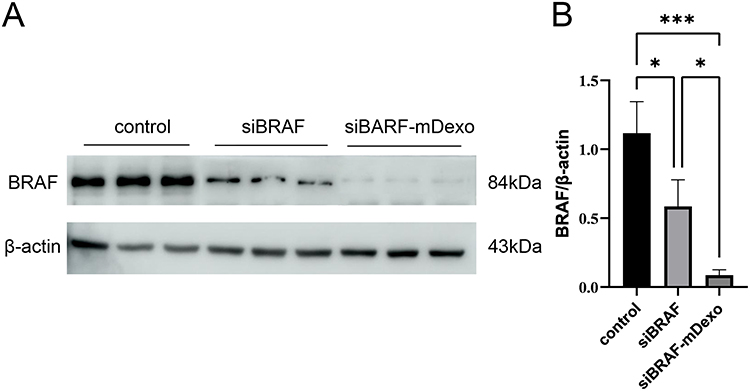 |
Figure 9 BRAF protein expression levels in B16-F10 cells after treatment with siBRAF and siBRAF-mDexos for 24 h. (A) Western blotting, with a sample loading amount of 40 μg. (B) Internal reference-normalized bar chart; *P<0.05 and ***P<0.001. The data are reported as the mean ± standard deviation (n=3), one-way ANOVA. |
The BRAF protein is an activated serine/threonine protein kinase in the MAPK signalling pathway. More than 60% of MM tissues contain BRAF gene mutations, which act as oncogenic drivers that lead to tumour progression and metastasis. Small-molecule inhibitors of BRAF, including vemurafenib, dabrafenib, and encorafenib, have been developed for the clinical treatment of melanoma.29 Compared with standard chemotherapy, small-molecule BRAF inhibitors significantly improve the response rate and survival rate of BRAF-mutant melanoma patients. However, most patients develop apparent drug resistance after long-term use, significantly reducing the therapeutic efficacy of these inhibitors. Therefore, an alternative approach to inhibit BRAF is needed to treat melanoma or slow tumour progression. SiRNA-mediated silencing is a good candidate strategy.30 Unlike traditional small-molecule chemotherapeutic drugs, siRNA can specifically target mRNA in cells and inhibit disease development at the genetic level by silencing the expression of disease-related genes, which does not easily cause drug resistance. However, physiological barriers and in vivo nucleases limit the clinical application of free siRNA. In this paper, mDexos were used as vectors to solve this problem. After incubation with siBRAF-mDexos, the expression of BRAF in B16-F10 cells was significantly reduced. This proved that the siBRAF-mDexos that we prepared had a stronger gene therapy effect than free siBRAF, and suggests that our siBRAF-mDexos can be used for the treatment of melanoma with BRAF gene mutation. Aside from melanoma, other malignancies with BRAF mutations include hairy cell leukaemia, papillary thyroid carcinoma, colorectal cancer, and non-small cell lung cancer; our siBRAF system is also applicable to these cancers. Furthermore, we used exosomes from DCs, rather than exosomes from a specific tumour, so our siBRAF-mDexos can be used in the treatment of a wide range of cancers.
In vivo Safety of siBRAF-mDexos
A C57BL/6J melanoma mouse model was established by subcutaneous injection of B16-F10 cells, and siBRAF, mDexos and siBRAF-mDexos were administered through tail vein injection of mice for treatment. Saline was given as a control to evaluate the in vivo safety and antitumour efficacy of siBRAF-mDexos. The routine blood indicators that we tested were the RBC count, white blood cell (WBC) count, lymphocyte (LYM) count, monocyte (MON) ratio, neutrophil (NEU) count, eosinophil (EOS) count, basophil (BASO) count, haemoglobin (HGB), haematocrit (HCT), mean corpuscular volume (MCV), mean corpuscular haemoglobin (MCH), mean corpuscular haemoglobin concentration (MCHC), RBC distribution width (RDW-CV), platelet (PLT) count and mean platelet volume (MPV). As shown in Table 1, there were no significant differences in these routine blood indicators between the treatment groups and the saline group, indicating that the drugs used did not affect the normal physiology of tumour-bearing mice and supporting the high safety of the drugs in vivo.
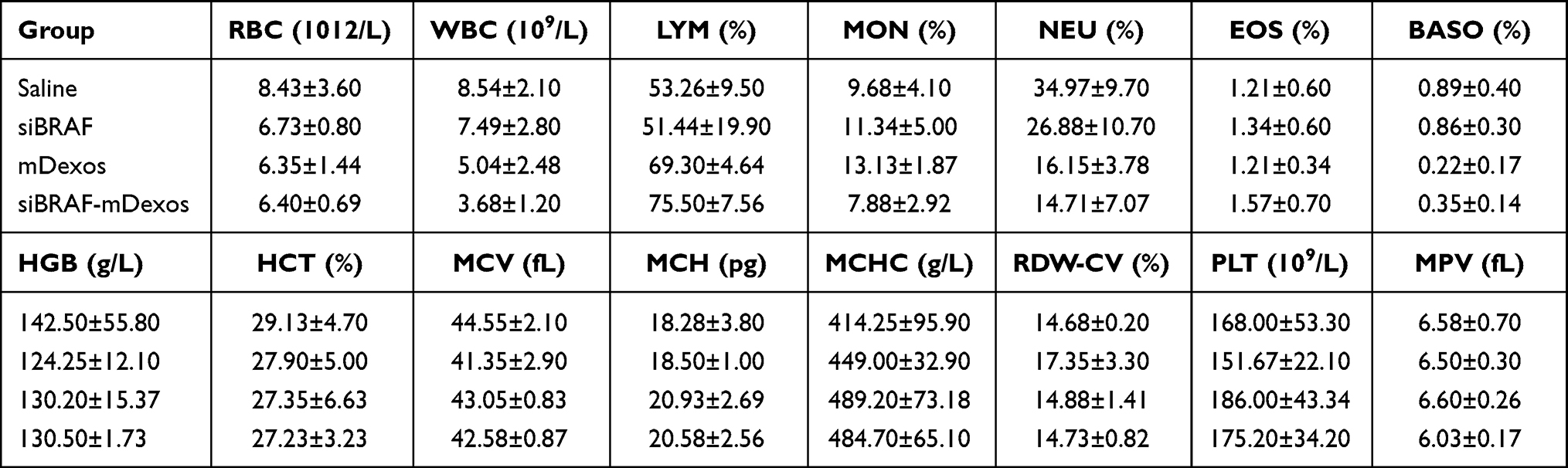 |
Table 1 Blood Parameters of Melanoma Tumour-Bearing Mice. Values are Means ± Standard Deviation (n=6) (Cont) |
As shown in Table 2, the liver function indicators that we measured included aspartate aminotransferase (AST), alanine aminotransferase (ALT), and alkaline phosphatase (ALP). Compared with the non-tumour-bearing mice, the saline-treated tumour-bearing mice exhibited significantly higher AST levels, proving the existence of liver injury in the tumour-bearing mice. In addition, some of the tumour-bearing mice exhibited abnormal swelling or black spots on their liver tissues, suggesting tumour liver metastasis. The values of these three indices did not significantly differ between the saline group and the mDexo group, indicating that mDexos had no apparent hepatotoxicity. The AST values of the siBRAF and siBRAF-mDexo groups were significantly lower than that of the saline group, while there were no significant differences in AST among the siBRAF, siBRAF-mDexo and non-tumour-bearing groups, indicating that siBRAF may have protected the mice from live injury. Many clinical reports have shown that the BRAF V600E mutation is closely related to the metastasis of colorectal cancer to the liver, suggesting that the protective effect of siBRAF in mice was mediated via inhibition of the liver metastasis of melanoma.31–33 Additionally, there were no significant ALT or ALP differences between the three groups and the non-tumour-bearing group, indicating that siBRAF, mDexos and siBRAF-mDexos do not exert hepatotoxicity and that their long-term use will not affect the normal function of the liver.
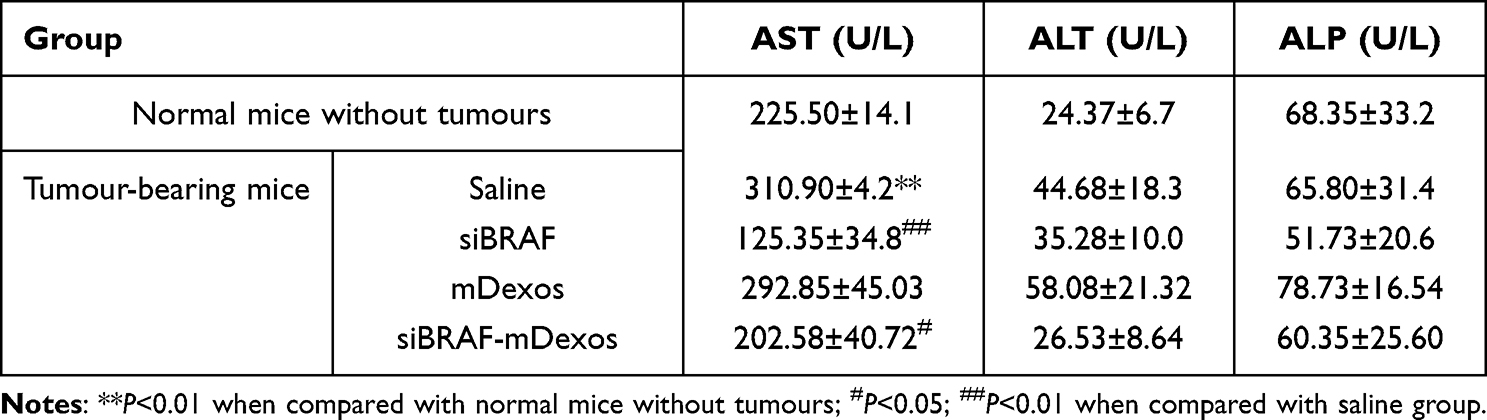 |
Table 2 Liver Indexes of Melanoma Tumour-Bearing Mice. Values are Means ± Standard Deviation (n=6) |
As shown in Table 3, we measured serum creatinine (CREA) and urea nitrogen (UREA) as kidney function indicators. An increase in CREA indicates renal injury, and an increase or decrease in UREA indicates abnormal renal function. There were no significant differences in the two indicators between the mice in the treatment groups and the non-tumour-bearing mice, indicating that these therapeutic drugs do not affect the normal physiological function of the kidneys in mice.
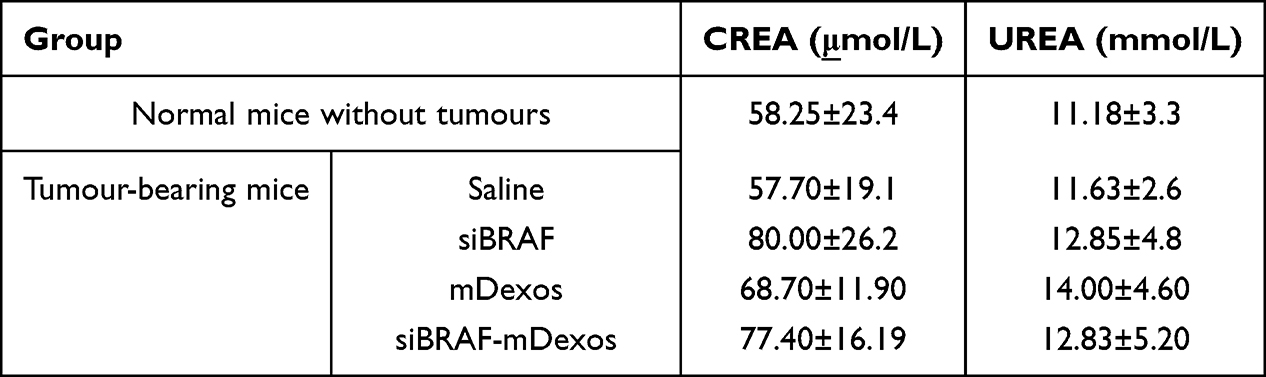 |
Table 3 Kidney Indicators of Melanoma Tumour-Bearing Mice. Values are Means ± Standard Deviation (n=6) |
Mouse weight can be used as an indicator of drug safety. If the weight of a mouse decreased by 20% compared to its initial weight, the animal experiment was concluded, and the mouse was euthanized. As shown in Figure 10A, the weight change curves of the mice showed no significant decreases in the weights of the mice in each group at any point. In addition, there were no significant differences in the weights of the mice between the treatment groups and the saline group at any time point. Compared with the initial weights, the body weights of the mice in each group increased, indicating that the growth of the mice was not affected by tumour growth or the drugs during the experiment. These findings reflected the high safety of the three therapeutic drugs siBRAF, mDexos and siBRAF-mDexos in vivo.
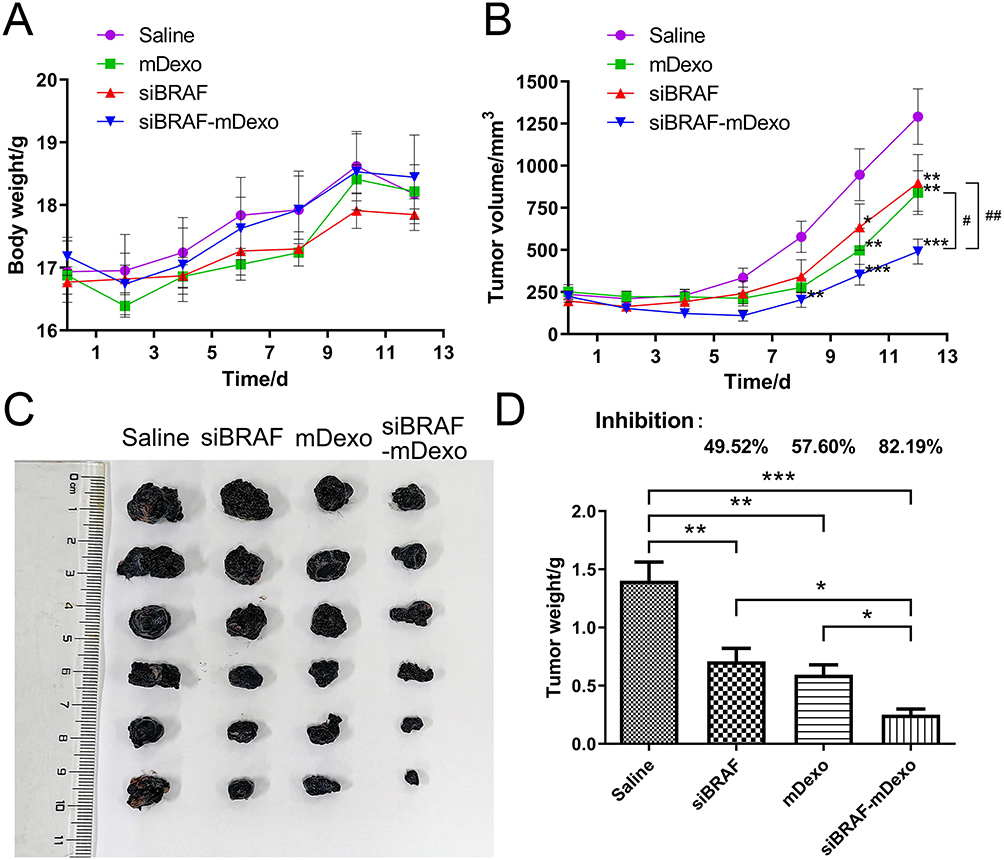 |
Figure 10 Antitumour efficacy of siBRAF-mDexos in vivo. (A) Curves showing the changes in mouse body weight over time. (B) Curves showing the changes in TV over time; *P<0.05, **P<0.01, and ***P<0.001 compared with the saline group; #P<0.05 and ##P<0.01. (C) Photograph of stripped mouse melanoma on day 13. (D) Tumour weight and tumour growth inhibition (TGI) rates; *P<0.05, **P<0.01 and ***P<0.001. The data are reported as the mean ± standard deviation (n=6), one-way ANOVA. |
In vivo Antitumour Efficacy of siBRAF-mDexos
TV Measurements
The tumour growth curve of the mice is shown in Figure 10B. On the 8th day of treatment, the TV of the siBRAF-mDexo group was significantly smaller than that of the saline group (P<0.01). On the 10th day after administration, the TVs of the three treatment groups were significantly smaller than that of the saline group, and there were no significant differences in TV among the treatment groups. On the 12th day of the treatment period, the TVs of the three groups were significantly smaller than that of the saline group, and the TV of the siBRAF-mDexo group was significantly smaller than those of the siBRAF group (P<0.01) and the mDexo group (P<0.05). The photograph of the stripped tumour (Figure 10C) was consistent with the volume change of the tumour growth curve. These results showed that using mDexos to encapsulate siBRAF effectively improved the therapeutic efficacy of siBRAF against melanoma in vivo, which was also consistent with the results of in vitro cellular experiments. Using mDexos to encapsulate siBRAF improved the serum stability of siBRAF and enabled more siBRAF to reach the melanoma during blood circulation. In addition, it enabled more siBRAF to enter melanoma cells via endocytosis and membrane fusion of Exos, which resulted in more effective melanoma treatment.
Tumour Weights and Tumour Growth Inhibition (TGI) Rates
The TGI values were calculated to evaluate the inhibitory effects of the drugs on tumour growth in vivo with the following formula:
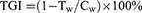
where Tw is the average tumour weight of the treatment group and Cw is the average tumour weight of the control group.
A TGI <40% was invalid, while a TGI ≥40% with a P value <0.05 indicated effective inhibition.
The TGI rates are shown in Figure 10D. The TGI rates of the siBRAF, mDexo, and siBRAF-mDexo groups were 49.52%, 57.60% and 82.19%, respectively. The TGI rate of the siBRAF-mDexo group was the highest, and all of the TGI rates of the treatment groups were higher than that of the saline group (40%), indicating that the three drugs can significantly inhibit melanoma growth.
MDexos function as both immunotherapeutic drugs and carriers. As mDexos are immune-activating drugs, their immunotherapeutic effects are best when the immune system is intact, and the mouse immune system tends to mature after 6 weeks of age; thus, 6-week-old C57BL/6J mice were selected for construction of the melanoma mouse model in the current study.34,35 The TGI rate of the siBRAF-mDexo group was the highest, indicating that siBRAF-mDexos had the strongest antimelanoma activity. However, the TGI rate of the siBRAF group was the lowest, indicating that the in vivo antimelanoma activity of siBRAF was limited, due to the instability of free siRNA in vivo after administration, the ease with which free siRNA is decomposed by nucleases in vivo, and the difficulty with which free siRNA crosses the biofilm barrier into cells to reach the site of action. After 13 days of treatment, the mice were sacrificed, and the tumours were removed from the skin. The tumours had infiltrated the muscle tissue from the subcutaneous layer and connected with the bone, indicating that bone metastasis tended to occur. Some mice in the mDexo group showed tissue infiltration, while those in the siBRAF-mDexo group did not exhibit prominent tissue infiltration of melanoma. This indicates that siBRAF-mDexos may inhibit the invasion and metastasis of melanoma in addition to inhibiting the growth of melanoma.
Abbreviations
MM, Malignant melanoma; DCs, Dendritic cells; Exos, Exosomes; BRAF, V-Raf murine sarcoma viral oncogene homolog B1; siRNA, Small interfering RNA; CCK-8, Cell Counting Kit-8; EC50, Median effect concentration; IC50, Half-maximal inhibitory concentration; RNAi, RNA interference; TME, Tumour microenvironment; CTL, Cytotoxic T lymphocyte; CMC, Cell-mediated cytotoxicity; ORR, Objective response rate; PFS, Progression-free survival; OS, Overall survival; PD-1, Programmed death 1; MHC, Major histocompatibility complex; TLR, Toll-like receptor; ICAM-1, Intercellular cell adhesion molecule-1; IL-12, Interleukin-12; VEGF, Vascular endothelial growth factor; ERK, Extracellular regulated protein kinase; miRNA, MicroRNA; EPR, Enhanced permeability and retention; SPF, Specific pathogen-free; FBS, Foetal bovine serum; GM-CSF, Granulocyte-macrophage colony-stimulating factor; TNF-α, Tumour necrosis factor-α; LPS, Lipopolysaccharide; FITC, Fluorescein isothiocyanate; PE, Phycoerythrin; APC, Allophycocyanin; PBS, Phosphate-buffered saline; RIPA, Radioimmunoprecipitation assay; BCA, Bicinchoninic acid; SDS‒PAGE, SDS–polyacrylamide gel electrophoresis; PVDF, Polyvinylidene fluoride; TBST, Tris-buffered saline with Tween 20; Cy3, Cyanine 3; DAPI, 4’,6-Diamidino-2’-phenylindole; TV, Tumour volume; TGI, Tumour growth inhibition value; TEM, transmission electron microscope; RBC, red blood cell count; WBC, white blood cell count; LYM, lymphocyte count; MON, monocyte ratio; NEU, neutrophil count; EOS, eosinophil count; BASO, basophil count; HGB, haemoglobin; HCT, haematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular haemoglobin; MCHC, mean corpuscular haemoglobin concentration; RDW-CV, red blood cell distribution width; PLT, platelet count; MPV, mean platelet volume; AST, aspartate aminotransferase; ALT, alanine aminotransferase; ALP, alkaline phosphatase; CREA, creatinine; UREA, urea nitrogen.
Summary
We successfully constructed a biomimetic nanosystem for the codelivery of immunotherapy and gene therapy drugs to the MM microenvironment using mDexos which can activate T-lymphocyte proliferation as carriers to encapsulate siBRAF. The siBRAF-mDexos had a diameter of 128.46 ± 13.34 nm. Compared with those of siBRAF, the serum stability, biocompatibility, uptake efficiency in B16-F10 cells, cytotoxicity to B16-F10 melanoma cells, and inhibitory effect on BRAF protein expression of siBRAF-mDexos were significantly increased. SiBRAF-mDexos also induced T-lymphocyte proliferation. The siBRAF-mDexos showed high antimelanoma efficacy, effectively inhibited melanoma growth and were very safe in vitro and in vivo. In conclusion, the siBRAF-mDexos bionic drug-delivery system is a promising strategy for MM combination therapy. In the future, we will study the effects of siBRAF-mDexos on other cancers with BRAF gene mutations, such as hairy cell leukaemia, papillary thyroid carcinoma, colorectal cancer and non-small cell lung cancer. Our findings will provide more theoretical guidance for wider application of siBRAF-mDexos.
Data Sharing Statement
All data associated with this study are present in the paper and the Supplementary Materials.
Ethics Approval and Informed Consent
The animal research involved in this study was approved by the Laboratory Animal Ethics Committee of College of Medicine, Jiaxing University (Ethical No. JUMC2023-076) and performed in accordance with the Guideline for Ethical Review of Animal Welfare (No. GB/T 35892-2018).
Consent for Publication
All named authors agreed to submit the manuscript for publication.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (81872220 and 81703437), Basic Public Welfare Research Project of Zhejiang Province (LGF18H160034, LGD20H300001, LGF20H300012 and LGD21H300001), Key Research and Development and Transformation project of Qinghai Province (2021-SF-C20), Science and Technology Bureau of Jiaxing (2019AY32009 and 2019AY32012), National College Students’ Innovation and Entrepreneurship Training Program (202110354017), Tumour Nanotargeting and TCM Technology Innovation Team (Key Science and Technology Innovation Team of Jiaxing, 2018), Jiaxing Key Laboratory of Oncological Photodynamic Therapy and Targeted Drug Research, and “Innovative Jiaxing • Excellent Talent Support Program”-Top Talents in Technological Innovation.
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Ruan W, Zhai Y, Yu K, Wu C, Xu Y. Coated microneedles mediated intradermal delivery of octaarginine/BRAF siRNA nanocomplexes for anti-melanoma treatment. Int J Pharm. 2018;553(1–2):298–309. doi:10.1016/j.ijpharm.2018.10.043
2. Hao MZ, Zhou WY, Du XL, et al. Novel anti-melanoma treatment: focus on immunotherapy. Chin J Cancer. 2014;33(9):458–465. doi:10.5732/cjc.014.10118
3. Anestopoulos I, Kyriakou S, Tragkola V, et al. Targeting the epigenome in malignant melanoma: facts, challenges and therapeutic promises. Pharmacol Ther. 2022;240:108301. doi:10.1016/j.pharmthera.2022.108301
4. Li C, Han X. Co-delivery of dacarbazine and All-Trans Retinoic Acid (ATRA) using lipid nanoformulations for synergistic antitumor efficacy against malignant melanoma. Nanoscale Res Lett. 2020;15(1):113. doi:10.1186/s11671-020-3293-3
5. Fattore L, Sacconi A, Mancini R, Ciliberto G. MicroRNA-driven deregulation of cytokine expression helps development of drug resistance in metastatic melanoma. Cytokine Growth Factor Rev. 2017;36:39–48. doi:10.1016/j.cytogfr.2017.05.003
6. Schreibelt G, Bol KF, Westdorp H, et al. Effective clinical responses in metastatic melanoma patients after vaccination with primary myeloid dendritic cells. Clin Cancer Res. 2016;22(9):2155–2166. doi:10.1158/1078-0432.CCR-15-2205
7. Kozakova L, Vondrova L, Stejskal K, et al. The melanoma-associated antigen 1 (MAGEA1) protein stimulates the E3 ubiquitin-ligase activity of TRIM31 within a TRIM31-MAGEA1-NSE4 complex. Cell Cycle. 2015;14(6):920–930. doi:10.1080/15384101.2014.1000112
8. Polak ME, Borthwick NJ, Gabriel FG, et al. Mechanisms of local immunosuppression in cutaneous melanoma. Br J Cancer. 2007;96(12):1879–1887. doi:10.1038/sj.bjc.6603763
9. Dewitte H, Verbeke R, Breckpot K, De Smedt SC, Lentacker I. Nanoparticle design to induce tumor immunity and challenge the suppressive tumor microenvironment. Nano Today. 2014;9(6):743–758. doi:10.1016/j.nantod.2014.10.001
10. Tel J, Aarntzen EH, Baba T, et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res. 2013;73(3):1063–1075. doi:10.1158/0008-5472.CAN-12-2583
11. Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203(7):1651–1656. doi:10.1084/jem.20051848
12. Stachyra-Strawa P, Ciesielka M, Janiszewski M, Grzybowska-Szatkowska L. The role of immunotherapy and molecular-targeted therapy in the treatment of melanoma (Review). Oncol Rep. 2021;46(2):158. doi:10.3892/or.2021.8109
13. Amini-Adle M, Khanafer N, Le-bouar M, Duru G, Dalle S, Thomas L. Ineffective anti PD-1 therapy after BRAF inhibitor failure in advanced melanoma. BMC Cancer. 2018;18(1):705. doi:10.1186/s12885-018-4618-9
14. Viaud S, Ploix S, Lapierre V, et al. Updated technology to produce highly immunogenic dendritic cell-derived exosomes of clinical grade: a critical role of interferon-gamma. J Immunother. 2011;34(1):65–75. doi:10.1097/CJI.0b013e3181fe535b
15. Swaika A, Hammond WA, Joseph RW. Current state of anti-PD-L1 and anti-PD-1 agents in cancer therapy. Mol Immunol. 2015;67(2 Pt A):4–17. doi:10.1016/j.molimm.2015.02.009
16. Kleffel S, Posch C, Barthel SR, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162(6):1242–1256. doi:10.1016/j.cell.2015.08.052
17. Klein S, Mauch C, Brinker K, et al. Tumor infiltrating lymphocyte clusters are associated with response to immune checkpoint inhibition in BRAF V600(E/K) mutated malignant melanomas. Sci Rep. 2021;11(1):1834. doi:10.1038/s41598-021-81330-4
18. Kourembanas S. Exosomes: vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu Rev Physiol. 2015;77(1):13–27. doi:10.1146/annurev-physiol-021014-071641
19. Yanez-Mo M, Siljander PR, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4(1):27066. doi:10.3402/jev.v4.27066
20. den Boorn JG V, Schlee M, Coch C, Hartmann G. SiRNA delivery with exosome nanoparticles. Nat Biotechnol. 2011;29(4):325–326. doi:10.1038/nbt.1830
21. El Andaloussi S, Lakhal S, Mager I, Wood MJ. Exosomes for targeted siRNA delivery across biological barriers. Adv Drug Deliv Rev. 2013;65(3):391–397. doi:10.1016/j.addr.2012.08.008
22. Hiasa M, Abe M, Nakano A, et al. GM-CSF and IL-4 induce dendritic cell differentiation and disrupt osteoclastogenesis through M-CSF receptor shedding by up-regulation of TNF-alpha converting enzyme (TACE). Blood. 2009;114(20):4517–4526. doi:10.1182/blood-2009-04-215020
23. Lu Z, Chang W, Meng S, et al. Mesenchymal stem cells induce dendritic cell immune tolerance via paracrine hepatocyte growth factor to alleviate acute lung injury. Stem Cell Res Ther. 2019;10(1):372. doi:10.1186/s13287-019-1488-2
24. Li H, Wang M, Huang B, et al. Theranostic near-infrared-IIb emitting nanoprobes for promoting immunogenic radiotherapy and abscopal effects against cancer metastasis. Nat Commun. 2021;12(1):7149. doi:10.1038/s41467-021-27485-0
25. Rupert DLM, Claudio V, Lasser C, Bally M. Methods for the physical characterization and quantification of extracellular vesicles in biological samples. Biochim Biophys Acta Gen Subj. 2017;1861(1 Pt A):3164–3179. doi:10.1016/j.bbagen.2016.07.028
26. Jalaludin I, Lubman DM, Kim J. A guide to mass spectrometric analysis of extracellular vesicle proteins for biomarker discovery. Mass Spectrom Rev. 2023;42(2):844–872. doi:10.1002/mas.21749
27. Tuttolomondo M, Ditzel HJ. Evaluation of siRNA stability and interaction with serum components using an agarose gel-based single-molecule FRET labeling method. Methods Mol Biol. 2021;2282:43–56.
28. Faruqu FN, Xu L, Al-Jamal KT. Preparation of exosomes for siRNA delivery to cancer cells. J Vis Exp. 2018;(142). doi:10.3791/58814
29. Proietti I, Skroza N, Bernardini N, et al. Mechanisms of acquired BRAF inhibitor resistance in melanoma: a systematic review. Cancers. 2020;12(10):2801. doi:10.3390/cancers12102801
30. Siu KS, Chen D, Zheng X, et al. Non-covalently functionalized single-walled carbon nanotube for topical siRNA delivery into melanoma. Biomaterials. 2014;35(10):3435–3442. doi:10.1016/j.biomaterials.2013.12.079
31. Margonis GA, Buettner S, Andreatos N, et al. Association of BRAF mutations with survival and recurrence in surgically treated patients with metastatic colorectal liver cancer. JAMA Surg. 2018;153(7):e180996. doi:10.1001/jamasurg.2018.0996
32. Margonis GA, Boerner T, Bachet JB, et al. Demystifying BRAF mutation status in colorectal liver metastases: a multi-institutional, collaborative approach to 6 open clinical questions. Ann Surg. 2022. doi:10.1097/SLA.0000000000005771
33. Bachet JB, Moreno-Lopez N, Viganò L, et al. What is the prognostic impact of BRAF mutation in patients undergoing resection of colorectal liver metastases? Results of nationwide intergroup (ACHBT, French, AGEO) cohort of 249 patients. J Clin Oncol. 2018;36(15_suppl):3554. doi:10.1200/JCO.2018.36.15_suppl.3554
34. Bilkei-Gorzo A, Albayram O, Draffehn A, et al. A chronic low dose of Δ9-tetrahydrocannabinol (THC) restores cognitive function in old mice. Nat Med. 2017;23(6):782–787. doi:10.1038/nm.4311
35. Smithey MJ, Venturi V, Davenport MP, et al. Lifelong CMV infection improves immune defense in old mice by broadening the mobilized TCR repertoire against third-party infection. Proc Natl Acad Sci U S A. 2018;115(29):E6817–E6825. doi:10.1073/pnas.1719451115
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.