Back to Journals » Journal of Hepatocellular Carcinoma » Volume 10
Deciphering the Divergent Gene Expression Landscapes of m6A/m5C/m1A Methylation Regulators in Hepatocellular Carcinoma Through Single-Cell and Bulk RNA Transcriptomic Analysis
Authors Liu HT, Rau CS, Liu YW, Hsieh TM, Huang CY, Chien PC, Lin HP, Wu CJ, Chuang PC, Hsieh CH ![]()
Received 15 November 2023
Accepted for publication 20 December 2023
Published 28 December 2023 Volume 2023:10 Pages 2383—2395
DOI https://doi.org/10.2147/JHC.S448047
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr David Gerber
Hang-Tsung Liu,1,* Cheng-Shyuan Rau,2,* Yueh-Wei Liu,3 Ting-Min Hsieh,1 Chun-Ying Huang,1 Peng-Chen Chien,4 Hui-Ping Lin,4 Chia-Jung Wu,4 Pei-Chin Chuang,5 Ching-Hua Hsieh4
1Department of Trauma Surgery, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, 83301, Taiwan; 2Department of Neurosurgery, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, 83301, Taiwan; 3Department of General Surgery, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, 83301, Taiwan; 4Department of Plastic Surgery, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, 83301, Taiwan; 5Department of Medical Research, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, 83301, Taiwan
*These authors contributed equally to this work
Correspondence: Pei-Chin Chuang; Ching-Hua Hsieh, Tel +886-7-3454746, Email [email protected]; [email protected]
Introduction: RNA modifications mediated by the m6A, m1A, and m5C regulatory genes are crucial for the progression of malignancy. This study aimed to explore the expression of regulator genes for m6A/m5C/m1A methylation at the single-cell level and to validate their expression in cancerous and adjacent para-cancerous liver tissues of adult patients with HCC who underwent tumor resection.
Methods: The bulk sequencing from The Cancer Genome Atlas (TCGA) database and the single-cell RNA sequencing (scRNA-seq) data obtained from the Gene Expression Omnibus (GEO) database were used to identify the dysregulated m6A/m5C/m1A genes for hepatocellular carcinoma (HCC). A real-time polymerase chain reaction (real-time PCR) was used to measure the expression of dysregulated m6A/m5C/m1A genes in collected human HCC tissues and compared with adjacent para-cancerous liver tissues. Immune cell infiltration with these significantly expressed methylation-related genes was evaluated using Timer2.0.
Results: A discrepancy in m6A/m5C/m1A gene expression was observed between bulk sequencing and scRNA-seq. The clustered heatmap of the scRNA-seq-identified dysregulated m6A/m5C/m1A genes in TCGA cohort revealed heterogeneous expression of these methylation regulators within the cancer, whereas their expression in the adjacent liver tissues was more homogeneous. The real-time PCR validated the significant overexpression of DNMT1, NSUN5, TRMT6, IGF2BP1, and IGFBP3, which were identified using scRNA-seq, and IGFBP2, which was identified using bulk sequencing. These dysregulated methylation genes are mainly correlated with the infiltration of natural killer cells.
Discussion: This study suggests that cellular diversity inside tumors contributes to the discrepancy in the expression of methylation regulator genes between traditional bulk sequencing and scRNA-seq. This study identified five regulatory genes that will be the focus of further studies regarding the function of m6A/m5C/m1A in HCC.
Keywords: hepatocellular carcinoma, HCC, methylation, N6-methyladenosine, m6A, N1-methyladenosine, m1A, 5-methylcytidine, m5C, bulk sequencing, single-cell RNA sequencing, scRNA-seq
Introduction
Hepatocellular carcinoma (HCC) is the most common primary liver cancer worldwide, and is distinguished by its rapid development, high invasiveness, and high mortality rate.1 Apart from well-explored epigenetic modifications such as DNA methylation, RNA modifications have emerged as significant contributors to the regulation of gene expression and cellular functions. These post-transcriptional modifications dynamically affect various aspects of RNA metabolism, including RNA processing, export, translation, and stability, and play crucial roles in cancer behaviors.2–8 Research has demonstrated that the development of HCC is governed by intricate genetic and epigenetic mechanisms, in which abnormal RNA modifications play a pivotal role in causing variations in gene expression, thereby influencing the occurrence and progression of HCC.9–15
Methylation involves a complex and dynamically reversible series of structural alterations induced by various enzymes. The discovery of N6-methyladenosine (m6A) has sparked interest in understanding its biological significance.16,17 Research has revealed that m6A plays essential roles in various cellular processes, including RNA splicing and alternative splicing, RNA translation and stability, and regulation of gene expression.18,19 Additionally, the presence of 5-methylcytidine (m5C) is markedly lower than that of m6A, with levels approximately three to ten times lower, accounting for only 0.03–0.1% of cytosines.20,21 Furthermore, N1-methyladenosine (m1A) is 10 times less abundant than m6A.21,22 Dysregulation of modification patterns of m5C23–27 and m1A28–30 in RNA molecules has been linked to cancer development and progression.
Several studies have demonstrated the importance of the m6A, m1A, and m5C regulatory genes in RNA modifications31–38 and cancer progression.30,38–40 To regulate methylation, writers modify DNA through various chemical changes, such as methyl group moieties. These molecular decorations can then attract several proteins known as “readers” that identify these moieties. A collection of “erasers” is used to remove the deposited change. Writers, readers, and erasers collaborate to shape the epigenetic landscape.41 A closely grouped subset of regulator genes for m6A/m5C/m1A methylation (hereafter referred to as m6A/m5C/m1A genes) was found to be associated with an unfavorable prognosis and immune microenvironment in HCC. These genes have the potential to serve as innovative tools for evaluating the prognosis of HCC patients31 and aid in the selection of appropriate immunotherapy strategies.32
However, the results of numerous studies on m6A/m5C/m1A genes are based on the analysis of bulk sequencing of HCC samples and do not consider the complex and various compositions of cells in the tumor environment. Therefore, this study aimed to explore the expression of regulator genes for m6A/m5C/m1A methylation at the single-cell level and to validate their expression in cancerous and adjacent para-cancerous liver tissues of adult patients with HCC who underwent tumor resection.
Materials and Methods
The Processing of Bulk RNA-Seq Data from TCGA
This study collected a cohort of 371 HCC tissues and 50 adjacent para-cancerous liver tissues from TCGA database. The purpose of this data collection was to assess the expression levels of m6A/m5C/m1A genes in HCC. Based on previously published literature,5,30,32,37,38,42–44 35 m6A, 16 m5C, and 13 m1A regulator genes were selected (Table S1). The FPKM data were first transformed into transcripts per million and normalized. The analysis of differentially expressed genes (DEGs) for the m6A/m5C/m1A genes between HCC and adjacent para-cancerous liver tissues in TCGA cohort was conducted using the “limma” R package (Table S2). The cutoff thresholds employed for identifying DEGs were |log2 fold change|>1 and a false discovery rate of <0.05.
Source of scRNA-Seq and Data Processing
The scRNA-seq data, obtained from the GEO database (GSE149614, GSE156625, and GSE125449) with 10× scRNA-seq data, was processed using the “Seurat” R package. This involved transforming the 10× scRNA-seq data into Seurat objects and filtering low-quality cells. Quality control was performed by calculating the percentage of mitochondrial or ribosomal genes. To identify highly variable genes, the “FindVariableFeatures” program was used to select the top 2000 genes, while an additional 2000 genes were employed for cell subpopulation identification through PCA. The “SingleR” R package was utilized for the annotation of different cell types. To mitigate any batch effects among between GSE samples and integrate them based on annotations, the canonical correlation analysis implemented as part of Seurat software package was applied. Cell clusters were visualized by utilizing a UMAP map for dimensionality reduction of the data. The cell cycle status was analyzed using the “CellCycleScoring” in the Seurat R package.
Differential Expression of scRNA
The analysis of DEGs for the m6A/m5C/m1A genes between HCC and adjacent para-cancerous liver tissues in the scRNA was conducted using the “FindMarkers” in the Seurat R package. The cut-off thresholds employed for identifying DEGs were |log2 fold change|>1 and an adjusted p<0.05.
Expression of m6A/m5C/m1A Genes in Collected Human Liver Samples
The study included patients with HCC who underwent tumor resection and were aged 20 years or older. Patients with any other cancer, immunocompromised diseases, or autoimmune diseases were excluded. Informed consent was obtained from all patients prior to sample collection. This study included 20 patients with HCC. Tissue specimens (0.5 × 0.5 cm) were collected from both HCC and adjacent para-cancerous liver tissues. Total RNA was extracted from each sample using an RNeasy Mini Kit (Qiagen, Venlo, Netherlands), and RNA quantity and quality were assessed using an SSP-3000 NanoDrop spectrophotometer (Infinigen Biotech, City of Industry, CA, USA). A real-time PCR was used to measure the expression of dysregulated m6A/m5C/m1A genes identified from the scRNA-seq and selected representative m6A/m5C/m1A genes. cDNA was amplified using Power SYBR Green PCR Master Mix with the specific primers listed in Table S3. The expression of methylation-related genes was quantified using the 2-Ct method with normalized cycle threshold values. All results were presented as the mean ± standard error. One-way analysis of variance was conducted for the overall analysis of the differences between group means, followed by a post-hoc Fisher’s least significant difference test. Statistical significance was set at p<0.05. Kaplan–Meier (K–M) survival analysis of significantly expressed methylation-related genes in the real-time PCR was performed using Timer2.0.45
Correlation of Dysregulated m6A/m5C/m1A Genes with Immune Cell Infiltration
Timer2.045 was used to analyze the infiltration relationship between dysregulated m6A/m5C/m1A genes identified by scRNA-seq and specific types of immune cells. A correlation between the dysregulated genes and infiltrated immune cells was considered significant if the absolute value of the correlation coefficient (|Rho|) was >0.1 and p<0.05.
Results
Dysregulated m6A/m5C/m1A Genes Between HCC and Adjacent Para-Cancerous Liver Tissues in the the Cancer Genome Atlas (TCGA) Cohort
From the bulk sequencing of the Cancer Genome Atlas (TCGA) database, 15 upregulated and 6 downregulated m6A genes, 4 upregulated and 2 downregulated m1A genes, and 7 upregulated and 5 downregulated m5C genes were identified in HCC, compared to that in adjacent para-cancerous liver tissues (Figure 1). The clustered heatmap of the dysregulated m6A/m5C/m1A genes revealed their heterogeneous in the adjacent para-cancerous liver tissue compared to that in HCC (Figure 2).
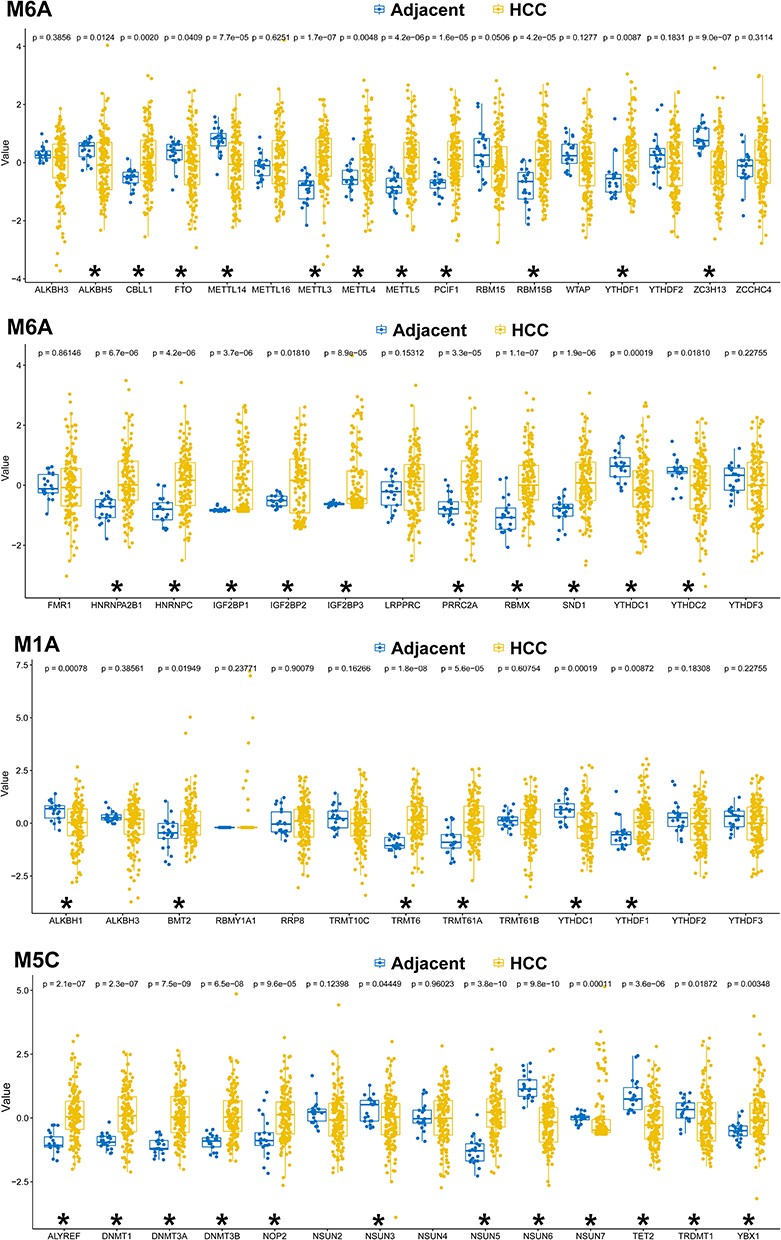 |
Figure 1 Dysregulated m6A/m5C/m1A genes identified from the bulk sequencing of The Cancer Genome Atlas (TCGA) cohort. *Indicates a significant change (p<0.05) between the hepatocellular carcinoma (HCC) and the adjacent para-cancerous liver tissue. |
 |
Figure 2 The clustered heatmap of the dysregulated m6A/m5C/m1A genes in HCC and in adjacent para-cancerous liver tissue. |
Single-Cell Transcriptome Profile of HCC and Adjacent Para-Cancerous Liver Tissue
After quality control filtering, 26,708 unique genes were identified in >73,916 cells. Principal Component Analysis (PCA) was conducted using 2000 variable genes to reduce dimensionality and to classify the cells into 20 clusters. After batch effect correction, Uniform Manifold Approximation and Projection (UMAP) analysis (Figure 3) was used to visualize the results based on HCC vs adjacent para-cancerous liver tissue, Gene Series Accession (GSE) categories in the Gene Expression Omnibus (GEO) database, and cell cycles. Unsupervised dimensionality reduction and graph-based clustering analyses were used to identify nine major cell clusters. The following cell types are described: T-cells, CD8+ T-cells, B-cells, endothelial cells, fibroblasts, hepatocytes, monocytes, macrophages, and NK cells. The association of dysregulated m6A/m5C/m1A genes identified from single-cell RNA sequencing (scRNA-seq) with different cell types is shown in Table 1 and the bubble (Figure 4A) and river (Figure 4B) plots in Figure 4. A summary of the expression of scRNA-seq-identified dysregulated m6A/m5C/m1A genes in bulk sequencing from TCGA cohort is provided in Figure 4C. The clustered heatmap of the dysregulated m6A/m5C/m1A genes in HCC and in adjacent para-cancerous liver tissue according to bulk sequencing from TCGA cohort revealed heterogeneous expression of the m6A/m5C/m1A genes within the cancer when compared to a more homogenous expression of these genes in the adjacent para-cancerous liver tissues.
 |
Table 1 The Dysregulated m6A/m5C/m1A Genes Identified from the scRNA-Seq in Different Cell Types in the HCC vs the Adjacent Para-Cancerous Liver Tissue |
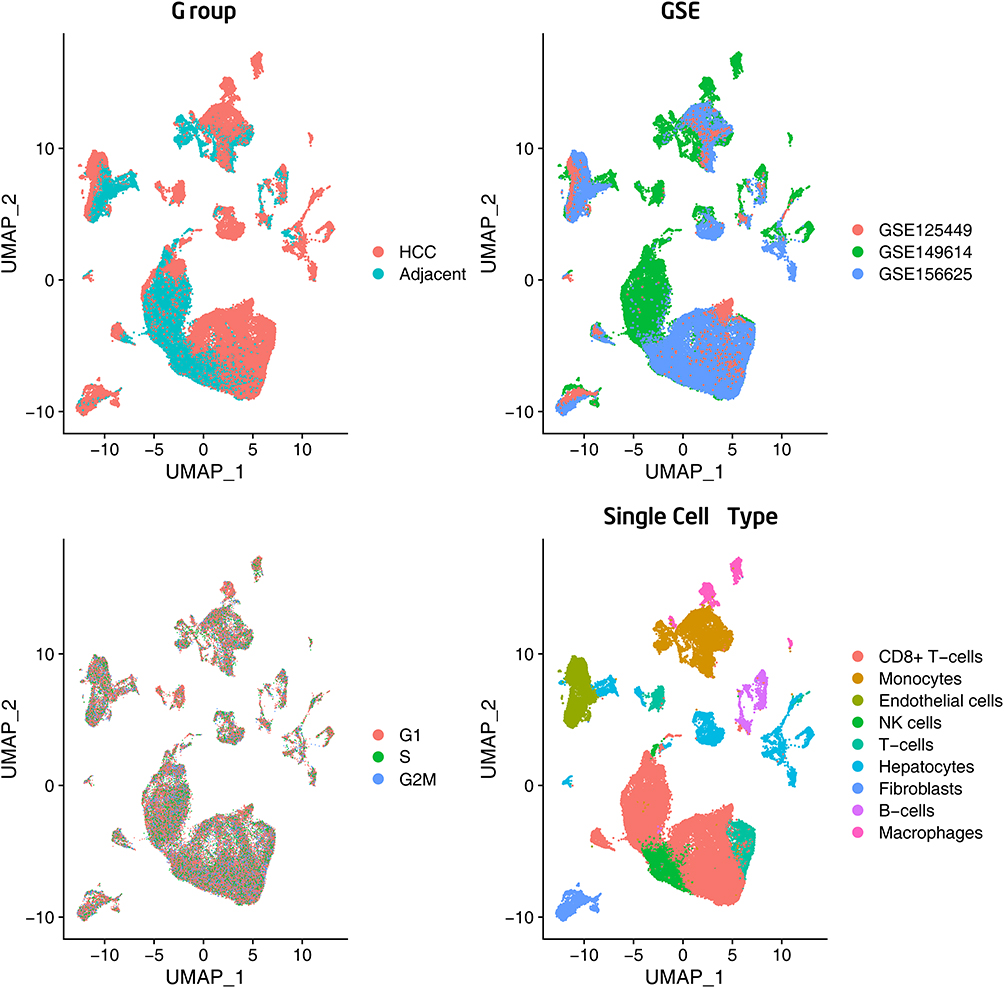 |
Figure 3 Single-cell RNA-sequencing analysis identified based on the HCC vs adjacent para-cancerous liver tissue, GSE categories, cell cycles, and major clusters of the cells. |
 |
Figure 4 (A) The bubble plot conducted to visualize the distribution of m6A/m5C/m1A genes among different cell types. To evaluate the statistical significance of each interaction score, a one-sided Wilcoxon rank-sum test was employed. (B) The river plot shows the association of dysregulated m6A/m5C/m1A genes to different cell types. GSE, Gene Series Accession; UMAP, Uniform Manifold Approximation and Projection; G1, Gap 1; S, Synthesis of DNA; G2 M, Gap 2 and Mitosis. (C) Summary of the expression of scRNA-seq-identified dysregulated m6A/m5C/m1A genes in the bulk sequencing from TCGA cohort. (D) The clustered heatmap of these dysregulated m6A/m5C/m1A genes in HCC and in adjacent para-cancerous liver tissue according to the bulk sequencing from TCGA cohort. *Indicates a significant change (p<0.05) between the hepatocellular carcinoma (HCC) and the adjacent para-cancerous liver tissue. |
The Expression of Methylation-Related Genes Determined Using Real-Time PCR
A real-time polymerase chain reaction (real-time PCR) was used to validate the expression of dysregulated m6A/m5C/m1A genes identified by scRNA-seq and revealed significant overexpression of DNMT1, NSUN5, TRMT6, IGF2BP1, and IGFBP3 (Figure 5A). Although YTHDC1 expression was lower in HCC than in the adjacent para-cancerous liver tissue, as shown by scRNA-seq, the difference was not significant. The upregulation of these genes was consistent with that observed by scRNA-seq. Quantification of the selected representative m6A/m5C/m1A genes from the writers, readers, and erasers categories demonstrated that there was no significant difference in the expression levels of these genes in HCC vs adjacent para-cancerous liver tissue, except that IGF2BP2 expression in HCC was considerably elevated compared to its expression in the adjacent para-cancerous liver tissue (Figure 5B). Kaplan–Meier survival analysis (Figure 5C) through the Timer2.0 website demonstrated that out of the six dysregulated genes studied, IGF2BP3 expression exhibited a significant correlation with the survival of the patients with HCC (hazard ratio [HR]= 1.34, p=0.000226).
 |
Figure 5 (A) A real-time PCR was performed to measure the expression of dysregulated m6A/m5C/m1A genes identified from the scRNA-seq. (B) A real-time PCR was performed to measure the expression of dysregulated m6A/m5C/m1A genes identified from selected representative dysregulated m6A/m5C/m1A genes from TCGA. (C) Kaplan–Meier (K–M) survival analysis of six dysregulated methylation-related genes using Timer2.0. *Indicates a significant change (p<0.05) between the hepatocellular carcinoma (HCC) and the adjacent para-cancerous liver tissue. |
Correlation of Dysregulated m6A/m5C/m1A Genes with Immune Cell Infiltration
The connection between dysregulated genes involved in the m6A/m5C/m1A processes, as discovered by scRNA-seq, and the infiltration of the corresponding immune cells was examined using Timer2.0.11 The analysis revealed a significant correlation between the five dysregulated genes (ALKBH5, DNMT3A, PPRC2A, WTAP, and YBX1) and the presence of NK cells (Figure 6). The correlation between ALKBH5, DNMT3A, PPRC2A, and NK cell infiltration was negative, whereas the correlation between WTAP and YBX1 was positive. Nevertheless, no statistically significant correlation was found between the expression levels of IGF2BP2 and METTL3 and endothelial cell infiltration. Similarly, the expression levels of RBM15B, YBX1, and ALKBH5 were not significantly correlated with CD8+ T cell infiltration.
 |
Figure 6 Correlation of dysregulated m6A/m5C/m1A genes with immune cell infiltration using Timer2.0. *Indicates a significant change (p<0.05) of the type of infiltrated cells between the hepatocellular carcinoma (HCC) and the adjacent para-cancerous liver tissue. |
Discussion
Overall, our findings revealed a discrepancy in the expression of m6A/m5C/m1A methylation regulator genes between bulk sequencing and scRNA-seq. Cellular diversity inside tumors is a key factor explaining the discrepancy in assessments. The clustered heatmap of the scRNA-seq-identified dysregulated m6A/m5C/m1A genes in TCGA cohort revealed heterogeneous expression of these methylation regulators within the cancer, whereas their expression in the adjacent liver tissues was more homogeneous. The dysregulation of methylation genes from bulk sequencing may not reflect those changes inside the hepatocytes but were derived from other cells in the tumor environment, particularly considering that the expression of some regulator genes was correlated with the immune infiltration of certain immune cells. The expression of regulatory genes for m6A/m5C/m1A methylation at the single-cell level may adhere more to the real situation, considering the complex composition of cells and heterogeneity in the tumor environment deterring the evaluation.
This study found five regulator genes (DNMT1, NSUN5, TRMT6, IGF2BP1, and IGFBP3) to be significantly overexpressed in HCC compared to adjacent para-cancerous liver tissue, and one regular gene (IGF2BP) was found to be significantly overexpressed in the real-time PCR, but not in scRNA-seq. DNMT1 is primarily responsible for the preservation of DNA methylation patterns during replication. It detects hemi-methylated DNA, which occurs when one strand of DNA is methylated but the other is not, and methylates the unmethylated strand to ensure correct methylation inheritance. The upregulation of DNMT1 in HCC leads to aberrant patterns of DNA methylation, characterized by reduced methylation levels across the genome and increased methylation levels in specific genomic regions. Global hypomethylation can potentially induce genomic instability and activate transposable elements, whereas localized hypermethylation of gene promoters can silence tumor suppressor genes that play crucial roles in cell cycle regulation, DNA repair, and apoptosis. DNMT1 overexpression frequently results in the silencing of tumor suppressor genes and the activation of oncogenes, such as c-MET, CDH1, and TERT, thereby increasing tumor development and metastasis.
The main role of NSUN5 is in the post-transcriptional modification of RNA, specifically in the methylation of cytosine at the C5 position of the tRNA. Methylation is crucial for maintaining the stability and functionality of tRNA. tRNA methylation, which is mediated by NSUN5, plays a significant role in ensuring precise and efficient translation by facilitating accurate codon–anticodon interactions and ribosome activity. Abundant NSUN5 expression is observed in HCC.46 In HCC, NSUN5 is associated with poor prognosis and contributes to disease progression.47 However, the exact role of NSUN5 in the development and progression of HCC has not yet been thoroughly investigated.
TRMT6 catalyzes the 2’-O-methylation of the cytosine residue at position 34 within the anticodon loop of specific tRNAs. This modification is essential in ensuring accurate and efficient protein translation.29 Elevated TRMT6 expression has been detected in HCC tissues and is correlated with the staging of Tumor-Node-Metastasis.48 Furthermore, TRMT6-mediated m1A methylation is essential for liver tumorigenesis,48 promotes HCC progression via the PI3K/AKT signaling pathway,49 and functions as a prognostic biomarker of poor clinical outcomes in patients with HCC.50,51 Furthermore, targeting TRMT6 inhibition shows promise as an effective therapeutic approach for HCC.48
Insulin-like growth factor 2 mRNA-binding proteins (IGF2BP1, IGF2BP2, and IGF2BP3) are RNA-binding proteins that are crucially involved in various biological activities encompassing both developmental and pathological contexts. They regulate the stability and translation of crucial regulators involved in cell division and metabolism.52–54 Specifically, IGF2BP1 and IGF2BP3 are widely recognized oncofetal proteins that are predominantly expressed in cancer cells rather than in normal tissues. In cancer, these proteins play crucial roles in initiating and promoting tumor growth.55,56 In patients with HCC, these IGF2BPs are dysregulated, suggesting their involvement in tumor development and progression.57–59 Disrupting the interactions between IGF2BP1, IGF2BP2, IGF2BP3, and IGF1R inhibits HCC growth.60
Overall, this study suggests that the expression of methylation regulator genes in traditional bulk sequencing should be inspected more cautiously because of the associated expression of methylation regulator genes in the complex cellular composition of the tumor environment. Among the many regulator genes, five (DNMT1, NSUN5, TRMT6, IGF2BP1, and IGFBP3), which were identified in the scRNA-seq to be significantly overexpressed in HCC compared to the adjacent para-cancerous liver tissue, should be the focus of further studies regarding the function of m6A/m5C/m1A in HCC. Further evaluation of the impact of these five methylation regulators on the total levels of m6A, m1A, and m5C methylation in the cancer cells would be valuable. Such validation would be valuable in the experiments of cultured cells or hepatoma organoid.
Ethics Statement
The study protocol was approved by the Institutional Review Board of Kaohsiung Chang Gung Memorial Hospital (ref: 202101618B0 and 202201359B0) and adhered to the ethical guidelines outlined in the 1975 Declaration of Helsinki.
Acknowledgments
We appreciate the support provided by the Genomic and Proteomic Core Laboratory of the Department of Medical Research, Kaohsiung Chang Gung Memorial Hospital. Peng-Chen Chien and Ching-Hua Hsieh contributed equally to this work as the corresponding authors. Hang-Tsung Liu and Cheng-Shyuan Rau contributed equally to this work as the first authors.
Funding
This research was funded by the Chang Gung Memorial Hospital (grant numbers CMRPG8N0031 & CMRPG8L1261 to Hang-Tsung Liu).
Disclosure
The authors declare no conflicts of interest.
References
1. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. doi:10.1038/s41572-020-00240-3
2. Boulias K, Greer EL. Biological roles of adenine methylation in RNA. Nat Rev Genet. 2023;24:143–160. doi:10.1038/s41576-022-00534-0
3. Patrasso EA, Raikundalia S, Arango D. Regulation of the epigenome through RNA modifications. Chromosoma. 2023;132:231–246. doi:10.1007/s00412-023-00794-7
4. Su T, Liu J, Zhang N, et al. New insights on the interplays between m(6)A modifications and microRNA or lncRNA in gastrointestinal cancers. Front Cell Dev Biol. 2023;11:1157797. doi:10.3389/fcell.2023.1157797
5. Tang Q, Li L, Wang Y, et al. RNA modifications in cancer. Br J Cancer. 2023;129:204–221. doi:10.1038/s41416-023-02275-1
6. Wang Z, Zhou J, Zhang H, Ge L, Li J, Wang H. RNA m(6) A methylation in cancer. Mol Oncol. 2023;17:195–229. doi:10.1002/1878-0261.13326
7. Wei Y, Li Y, Lu C. Exploring the role of m6A modification in cancer. Proteomics. 2023;23:e2200208. doi:10.1002/pmic.202200208
8. Zhuang H, Yu B, Tao D, et al. The role of m6A methylation in therapy resistance in cancer. Mol Cancer. 2023;22:91. doi:10.1186/s12943-023-01782-2
9. Braghini MR, Lo Re O, Romito I, et al. Epigenetic remodelling in human hepatocellular carcinoma. J Exp Clin Cancer Res. 2022;41:107. doi:10.1186/s13046-022-02297-2
10. Wang S, Gao S, Ye W, Li Y, Luan J, Lv X. The emerging importance role of m6A modification in liver disease. Biomed Pharmacother. 2023;162:114669. doi:10.1016/j.biopha.2023.114669
11. Wang Y, Zeng J, Chen W, Fan J, Hylemon PB, Zhou H. Long noncoding RNA H19: a novel oncogene in liver cancer. Noncoding RNA. 2023;9. doi:10.3390/ncrna9020019
12. Xu K, Wu T, Xia P, Chen X, Yuan Y. Alternative splicing: a bridge connecting NAFLD and HCC. Trends Mol Med. 2023. doi:10.1016/j.molmed.2023.07.001
13. Yang L, Tian S, Zheng X, et al. N6-methyladenosine RNA methylation in liver diseases: from mechanism to treatment. J Gastroenterol. 2023;58:718–733. doi:10.1007/s00535-023-02008-4
14. Mao Y, Ding Z, Jiang M, Yuan B, Zhang Y, Zhang X. Circ_0091579 exerts an oncogenic role in hepatocellular carcinoma via mediating miR-136-5p/TRIM27. Biomed J. 2022;45:883–895. doi:10.1016/j.bj.2021.12.009
15. Yu MC, Wu TH, Lee CW, et al. Percentage genome change and chromosome 7q amplification predict sorafenib response in advanced hepatocellular carcinoma. Biomed J. 2021;44:S73–S83. doi:10.1016/j.bj.2020.07.001
16. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi:10.1038/nature11112
17. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi:10.1016/j.cell.2012.05.003
18. Huang J, Yin P. Structural insights into N(6)-methyladenosine (m(6)A) modification in the transcriptome. Genomics Proteomics Bioinf. 2018;16:85–98. doi:10.1016/j.gpb.2018.03.001
19. Yu F, Wei J, Cui X, et al. Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic Acids Res. 2021;49:5779–5797. doi:10.1093/nar/gkab415
20. Huber SM, van Delft P, Mendil L, et al. Formation and abundance of 5-hydroxymethylcytosine in RNA. Chembiochem. 2015;16:752–755. doi:10.1002/cbic.201500013
21. Legrand C, Tuorto F, Hartmann M, et al. Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs. Genome Res. 2017;27:1589–1596. doi:10.1101/gr.210666.116
22. Li X, Xiong X, Zhang M, et al. Base-resolution mapping reveals distinct m(1)A methylome in nuclear- and mitochondrial-encoded transcripts. Mol Cell. 2017;68:993–1005.e1009. doi:10.1016/j.molcel.2017.10.019
23. Cusenza VY, Tameni A, Neri A, Frazzi R. The lncRNA epigenetics: the significance of m6A and m5C lncRNA modifications in cancer. Front Oncol. 2023;13:1063636. doi:10.3389/fonc.2023.1063636
24. Gu X, Ma X, Chen C, et al. Vital roles of m(5)C RNA modification in cancer and immune cell biology. Front Immunol. 2023;14:1207371. doi:10.3389/fimmu.2023.1207371
25. Li M, Tao Z, Zhao Y, et al. 5-methylcytosine RNA methyltransferases and their potential roles in cancer. J Transl Med. 2022;20:214. doi:10.1186/s12967-022-03427-2
26. Song H, Zhang J, Liu B, et al. Biological roles of RNA m(5)C modification and its implications in Cancer immunotherapy. Biomark Res. 2022;10:15. doi:10.1186/s40364-022-00362-8
27. Yu G, Bao J, Zhan M, et al. Comprehensive analysis of m5C methylation regulatory genes and tumor microenvironment in prostate cancer. Front Immunol. 2022;13:914577. doi:10.3389/fimmu.2022.914577
28. Jin H, Huo C, Zhou T, Xie S. m(1)A RNA modification in gene expression regulation. Genes. 2022;13. doi:10.3390/genes13050910
29. Li J, Zhang H, Wang H. N(1)-methyladenosine modification in cancer biology: current status and future perspectives. Comput Struct Biotechnol J. 2022;20:6578–6585. doi:10.1016/j.csbj.2022.11.045
30. Song P, Tayier S, Cai Z, Jia G. RNA methylation in mammalian development and cancer. Cell Biol Toxicol. 2021;37:811–831. doi:10.1007/s10565-021-09627-8
31. Li D, Li K, Zhang W, et al. The m6A/m5C/m1A regulated gene signature predicts the prognosis and correlates with the immune status of hepatocellular carcinoma. Front Immunol. 2022;13:918140. doi:10.3389/fimmu.2022.918140
32. Liu T, Sun L, Li ZZ, et al. The m6A/m5C/m1A regulator genes signature reveals the prognosis and is related with immune microenvironment for hepatocellular carcinoma. BMC Gastroenterol. 2023;23:147. doi:10.1186/s12876-023-02776-6
33. Liu Y, Zhu J, Ding L. Involvement of RNA methylation modification patterns mediated by m7G, m6A, m5C and m1A regulators in immune microenvironment regulation of Sjögren’s syndrome. Cell Signal. 2023;106:110650. doi:10.1016/j.cellsig.2023.110650
34. Mao S, Chen Z, Wu Y, Xiong H, Yuan X. Crosstalk of Eight types of RNA modification regulators defines tumor microenvironments, cancer hallmarks, and prognosis of lung adenocarcinoma. J Oncol. 2022;2022:1285632. doi:10.1155/2022/1285632
35. Qi L, Zhang W, Ren X, et al. Cross-Talk of multiple types of RNA modification regulators uncovers the tumor microenvironment and immune infiltrates in soft tissue sarcoma. Front Immunol. 2022;13:921223. doi:10.3389/fimmu.2022.921223
36. Wang Y, Mao Y, Wang C, et al. RNA methylation-related genes of m6A, m5C, and m1A predict prognosis and immunotherapy response in cervical cancer. Ann Med. 2023;55:2190618. doi:10.1080/07853890.2023.2190618
37. Wu ZY, Shi ZY. The prognostic value and immune landscapes of m1A/m5C/m6A-associated lncRNA signature in osteosarcoma. Eur Rev Med Pharmacol Sci. 2022;26:5868–5883. doi:10.26355/eurrev_202208_29526
38. Zhao K, Li W, Yang Y, et al. Comprehensive analysis of m(6)A/m(5)C/m(1)A-related gene expression, immune infiltration, and sensitivity of antineoplastic drugs in glioma. Front Immunol. 2022;13:955848. doi:10.3389/fimmu.2022.955848
39. Kong Y, Yu J, Ge S, Fan X. Novel insight into RNA modifications in tumor immunity: promising targets to prevent tumor immune escape. Innovation. 2023;4:100452. doi:10.1016/j.xinn.2023.100452
40. Yang Z, Zhang S, Xia T, et al. RNA modifications meet tumors. Cancer Manag Res. 2022;14:3223–3243. doi:10.2147/cmar.S391067
41. Biswas S, Rao CM. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur J Pharmacol. 2018;837:8–24. doi:10.1016/j.ejphar.2018.08.021
42. Li X, Xiong X, Wang K, et al. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat Chem Biol. 2016;12:311–316. doi:10.1038/nchembio.2040
43. Zhang Q, Liu F, Chen W, et al. The role of RNA m(5)C modification in cancer metastasis. Int J Biol Sci. 2021;17:3369–3380. doi:10.7150/ijbs.61439
44. He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Mol Cancer. 2019;18:176. doi:10.1186/s12943-019-1109-9
45. Li T, Fu J, Zeng Z, et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48:W509–W514. doi:10.1093/nar/gkaa407
46. Sun GF, Ding H. NOP2-mediated m5C methylation of XPD is associated with hepatocellular carcinoma progression. Neoplasma. 2023;70:340–349. doi:10.4149/neo_2023_230110N17
47. Zhang XW, Wu LY, Liu HR, et al. NSUN5 promotes progression and predicts poor prognosis in hepatocellular carcinoma. Oncol Lett. 2022;24:439. doi:10.3892/ol.2022.13559
48. Wang Y, Wang J, Li X, et al. N(1)-methyladenosine methylation in tRNA drives liver tumourigenesis by regulating cholesterol metabolism. Nat Commun. 2021;12:6314. doi:10.1038/s41467-021-26718-6
49. Ye Y, Liu M, Wu F, et al. TRMT6 promotes hepatocellular carcinoma progression through the PI3K/AKT signaling pathway. Eur J Med Res. 2023;28:48. doi:10.1186/s40001-022-00951-1
50. Wang Y, Huang Q, Deng T, Li BH, Ren XQ. Clinical significance of TRMT6 in hepatocellular carcinoma: a bioinformatics-based study. Med Sci Monit. 2019;25:3894–3901. doi:10.12659/msm.913556
51. Zhao Q, Zhang Y, Shao S, Sun Y, Lin Z. Identification of hub genes and biological pathways in hepatocellular carcinoma by integrated bioinformatics analysis. PeerJ. 2021;9:e10594. doi:10.7717/peerj.10594
52. Elcheva IA, Gowda CP, Bogush D, et al. IGF2BP family of RNA-binding proteins regulate innate and adaptive immune responses in cancer cells and tumor microenvironment. Front Immunol. 2023;14:1224516. doi:10.3389/fimmu.2023.1224516
53. Shao W, Zhao H, Zhang S, et al. A pan-cancer landscape of IGF2BPs and their association with prognosis, stemness and tumor immune microenvironment. Front Oncol. 2022;12:1049183. doi:10.3389/fonc.2022.1049183
54. Ramesh-Kumar D, Guil S. The IGF2BP family of RNA binding proteins links epitranscriptomics to cancer. Semin Cancer Biol. 2022;86:18–31. doi:10.1016/j.semcancer.2022.05.009
55. Zhu TY, Hong LL, Ling ZQ. Oncofetal protein IGF2BPs in human cancer: functions, mechanisms and therapeutic potential. Biomark Res. 2023;11:62. doi:10.1186/s40364-023-00499-0
56. Yang J, Qin T, Liu S, Tang H, Liu M, Wang Q. Interaction analysis of miR-1275/IGF2BP1/IGF2BP3 with the susceptibility to hepatocellular carcinoma. Biomarker Med. 2020;14:283–292. doi:10.2217/bmm-2019-0332
57. Liang Y, Chen S, Xie J, et al. Establishment of a prognostic model based on m(6)A regulatory factors and stemness of hepatocellular carcinoma using RNA-seq data and scRNA-seq data. J Cancer Res Clin Oncol. 2023. doi:10.1007/s00432-023-05045-x
58. Czepukojc B, Abuhaliema A, Barghash A, et al. IGF2 mRNA binding protein 2 transgenic mice are more prone to develop a ductular reaction and to progress toward cirrhosis. Front Med. 2019;6:179. doi:10.3389/fmed.2019.00179
59. Waly AA, El-Ekiaby N, Assal RA, et al. Methylation in MIRLET7A3 gene induces the expression of IGF-II and its mRNA binding proteins IGF2BP-2 and 3 in hepatocellular carcinoma. Front Physiol. 2018;9:1918. doi:10.3389/fphys.2018.01918
60. Fawzy IO, Hamza MT, Hosny KA, Esmat G, Abdelaziz AI. Abrogating the interplay between IGF2BP1, 2 and 3 and IGF1R by let-7i arrests hepatocellular carcinoma growth. Growth Factors. 2016;34:42–50. doi:10.3109/08977194.2016.1169532
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.