Back to Journals » Journal of Inflammation Research » Volume 14
Ddb1-Cullin4-Associated-Factor 1 in Macrophages Restricts the Staphylococcus aureus-Induced Osteomyelitis
Authors Zong Y, Shan H, Yin F, Ma X, Jiang C, Wang N, Zhou L, Lin Y, Zhou Z, Yu X
Received 18 February 2021
Accepted for publication 12 April 2021
Published 28 April 2021 Volume 2021:14 Pages 1667—1676
DOI https://doi.org/10.2147/JIR.S307316
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Monika Sharma
Yang Zong,1,* Haojie Shan,1,* Fuli Yin,1 Xin Ma,1 Chaolai Jiang,1 Nan Wang,2 Lihui Zhou,3 Yiwei Lin,1 Zubin Zhou,1 Xiaowei Yu1
1Department of Orthopaedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai, 200233, People’s Republic of China; 2Department of Emergency, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, 450052, People’s Republic of China; 3Department of Orthopaedic Surgery, Xiangshan First People’s Hospital, Ningbo, Zhejiang, 315700, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaowei Yu
Department of Orthopaedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai, 200233, People’s Republic of China
Tel + 86-21-3829-7774
Email [email protected]
Introduction: Ddb1-cullin4-associated-factor 1 (DCAF1) is known to regulate protein ubiquitination, while the roles of DCAF1 in osteomyelitis remain unknown. This study aims to investigate the effects of DCAF1 deficiency in macrophages on osteomyelitis and elucidate the molecular mechanism.
Methods: Staphylococcus aureus-induced mouse model of osteomyelitis was established on the DCAF1fl/flLyz2cre/+ and DCAF1fl/flLyz2+/+ (control) mice. Flow cytometry was conducted to analyze the populations of adaptive and innate immune cells. Lipopolysaccharides (LPS)-induced bone marrow-derived macrophages (BMDMs) were established. qRT-PCR and immunoblot analysis were used to determine the levels of inflammation-related biomarkers. ELISA was used to determine the release of inflammatory cytokines including IL-1β, IL-6, and TNF.
Results: The populations of immune cells in the bone marrow and spleen were not affected due to DCAF1 deficiency in macrophages. DCAF1 suppressed inflammatory cytokines in LPS-induced BMDMs. Additionally, DCAF1 deficiency in macrophages induced severe symptoms including less bacterial load in the femur, cortical bone loss, and reactive bone formation. Mechanistic study revealed that DCAF1 deficiency induced p38 hyperactivation.
Discussion: DCAF1 in macrophages suppressed the Staphylococcus aureus-induced mouse model of osteomyelitis.
Keywords: DCAF1, macrophages, Staphylococcus aureus, osteomyelitis, inflammation
Introduction
Osteomyelitis is an inflammatory response in the bone induced by a series of infecting microbial agents including bacteria and fungus.1,2 Osteomyelitis can be caused by a series of factors including open injury, trauma, or surgical infection and can spread into bone marrow, cortex, periosteum, surrounding soft tissues, and vascular channels.1 The incidence of osteomyelitis is around 13 cases per 100,000 children and 90 cases per 100,000 adults.3 Currently, osteomyelitis is still one of the major challenges in orthopedics and becomes problematic partially due to bacterial persistence mechanisms and antibiotic treatment resistance.3,4 Clinical treatment of osteomyelitis focuses on adequate antibiotic coverage and surgical debridement of nonviable tissue.5 However, overuse of antibiotics can induce drug-resistance and impair the innate immune system as well as cause serious adverse effects.6 Therefore, it is important to develop novel treatment method with minimum antibiotic toxicity for the treatment of osteomyelitis. Interestingly, noninvasive therapeutic strategy such as microwave therapy exhibits promising results for the treatment of osteomyelitis.7,8
A series of proposed mechanisms explaining the occurrence and development of osteomyelitis are vascular insufficiency associated infection, contiguous contamination, and hematogenous spread.9,10 Although various infecting microbial agents including bacteria and fungus can cause osteomyelitis, Staphylococcus aureus (S. aureus) is one of the most common bacterial pathogens accounting for 30–80% of bone infection.11 It is a gram-positive bacterium almost present in every organ and is frequently found in the skin, gut mucosa, and upper respiratory tract.12 S. aureus adheres the bone surfaces due to the production of adhesins for bone matrix proteins including bone sialoglycoprotein, collagen, laminin, and fibronectin. In addition, adhesins expressed by S. aureus allow its attachment to cartilage and medical devices surgically implanted in bone.11,13 Besides, S. aureus can invade and survive intracellularly and expresses phenotypic resistance, leading to antimicrobial resistance and the recurrence of osteomyelitis.11,14
When the bone is infected, S. aureus induces an acute inflammatory response, initiating an inflammation cascade. The accumulation of immune cells (T lymphocytes, B lymphocytes, macrophages, and neutrophils) and the release of pro-inflammatory mediators and cytokines eventually lead to bone necrosis, bone resorption, and new bone deposition.10,15,16 Interestingly, the alternation of macrophage phenotype is observed in the patients with chronic osteomyelitis as compared with those with acute joint infection, supporting that macrophages contribute to the inflammatory responses in the osteomyelitis.17
Ddb1-cullin4-associated-factor 1 (DCAF1) is an E3 ubiquitin ligase component associated with some cellular processes including cell survival, cell cycle, and histone modification.18,19 In addition, DCAF1 is essential for DNA replication and is known to modulate the G2/M arrest.20 It is reported that DCAF1 regulates anti-HIV cellular responses in macrophages by inhibiting viral cDNA synthesis.21 To our knowledge, the roles of DCAF1 in osteomyelitis are still unclear. As mentioned, macrophages play important roles in osteomyelitis. Herein, the present study aims to investigate the effects of DCAF1 deficiency in macrophages on osteomyelitis and explore the molecular mechanism.
Materials and Methods
Antibody and Reagent
Primary antibodies against phosphorylated p38 (p-p38) (Thr180/Tyr182, #9211), p38 (#9212), p-JNK (Thr180/Tyr185, #9255), and p-IKKα/β (Ser176/180, #2694) were purchased from Cell Signaling Technology (Danvers, MA, United States). Primary antibodies against IKBα (H4, sc-1643), JNK1 (F3, sc-1648), and IKKα (C6, sc-166231) were purchased from Santa Cruz Biotechnology (Dallas, TX, United States). The primary antibody against DCAF1 was purchased from Abcam (ab1766, Shanghai, China). Lipopolysaccharides (LPS, Escherichia coli 055:B5) were purchased from Sigma-Aldrich (St. Louis, MO, United States). TNF was purchased from PepTech Biosciences (Brownsville, TX, United States).
Animals
DCAF1fl/fl (C57BL6J background) and Lyz2-Cre mice (C57BL6J background) were purchased from Jackson Laboratory (Bar Harbor, ME, United States). Myeloid cell conditional knockout mice (DCAF1fl/flLyz2cre/+) were constructed by GemPharmatech (Nanjing, China). DCAF1fl/flLyz2+/+ mice were constructed and used as the control group. The animals were kept at constant temperatures of 23–26°C with 55±5% humidity under sterile conditions. The study was approved by the Ethics Committee of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital. The study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (eighth edition, NIH).
Staphylococcus aureus-Induced Mouse Model of Osteomyelitis
Staphylococcus aureus (S. aureus, ATCC® BAA-2437™) were cultured on a tryptic soy agar plate or tryptic soy broth with or without erythromycin (10 μg/mL). To prepare the concentrated bacterial suspension, the strain was cultured in the Roswell Park Memorial Institute (RPMI) medium containing 1% casamino acids. The mouse model of osteomyelitis was established by the injection of bacterial suspension (2 μL; 106 CFU) into the intramedullary canal. In the mock infection group, the mice were injected with same volume of phosphate-buffered saline (PBS) into the intramedullary canal.
Animal behaviors were observed daily and body weight was recorded every 2 days. The mice were sacrificed when human endpoint criteria were met including 1) the animals are not able to ambulate, eat, or drink; 2) body weight reduces greater than 20% of body weight; and 3) the animals exhibit hunched posture.
If the mice have not reach to the human endpoint criteria, the experimental mice were sacrificed at the indicated time points shown in data, and assessed the reactive bone formation and the cortical bone loss were determined. After 16 days of post-infection, femurs were harvested and fixed in neutral buffered formalin for 48 hours. Bones were scanned using a μCT50. The diaphysis and distal epiphysis of each femur were visualized in the scout-view radiographs. Each imaging scan resulted in 1000 slices (10 mm) of the femur. Three-dimensional volumetric analyses were conducted by contouring transverse image slices. For analysis of cortical bone loss, volume of cortical bone destruction was determined by segmenting the image with a lower threshold of 0 and an upper threshold of 595 mg HA/ccm, sigma 1.3, and support 1. To measure reactive bone formation, an inclusive contour was placed around the outer perimeter of the bone. Bone was segmented from non-mineralized tissues in the VOI with a lower threshold of 400 mg HA/ccm, sigma 1.3, and support 2.
Isolation and Culture of Murine Bone Marrow-Derived Macrophages (BMDMs)
BMDMs were isolated from mice according to a previous report.22 The mice were sacrificed and the tibia and femur bones were collected. After removal of surrounding muscle, the exposed bone marrow was flushed out the ends of the bones using a 25-gauge needle and a 10-mL syringe filled with PBS. Clumps were gently disaggregated and passed through a 70 μm cell strainer. The cell suspension was centrifuged at 250 g for 5 min at room temperature to pellet cells. BMDMs were cultured in the complete Gibco Dulbecco’s Modified Eagle Medium (DMEM) with 10% FBS containing 10 ng/mL macrophage colony-stimulating factor (M-CSF) for 5 days.
Flow Cytometry
Flow cytometry was performed according to previous reports.23,24 We collected the spleen and removed the surrounded fat tissues. Spleen cell suspensions were prepared by using a plastic plunger of syringe combined with a 70-µm cell strainer. In addition, bone marrow was collected and washed with sterile PBS. After that, the bone marrow was suspended in FACS buffer containing 2% FBS to prepare the bone marrow cell suspensions. Cell suspensions were stained with fluorescent-dye conjugated antibodies against CD3, B220, CD4, CD8, Ly6G, CD11b, and F4/80 and analyzed using multicolor flow cytometer. Antibodies were purchased from Abcam or Invitrogen. FlowJo was applied to gate the cells and analyze the cell populations.
qRT-PCR
The PCR primers were designed and synthesized by GenScript (Nanjing, China). The sequences of each primer were listed as follows. Il-6 forward: 5ʹ-CAC AGA GGA TAC CAC TCC CAA CA-3ʹ, and reverse: 5ʹ-TCC ACG ATT TCC CAG AGA ACA-3ʹ; Tnf forward: 5ʹ-CAT CTT CTC AAA ATT CGA GTG ACA A-3ʹ, and reverse: 5ʹ- CCA GCT GCT CCT CCA CTT G-3ʹ; Il-1β forward: 5ʹ-AAG CCT CGT GCT GTC GGA CC-3ʹ, and reverse: 5ʹ-TGA GGC CCA AGG CCA CAG GT-3ʹ; Acp5 forward: 5ʹ- GCG ACC ATT GTT AGC CAC ATA CG-3ʹ, and reverse: 5ʹ- CGT TGA TGT CGC ACA GAG GGA T-3ʹ; Calcr forward: 5ʹ-AAG ATG GAC CCT CAT GCC AGT G-3ʹ, and reverse: 5ʹ- CTC GTC GGT AAA CAC AGC CAT G-3ʹ; Rankl forward: 5ʹ- GTG AAG ACA CAC TAC CTG ACT CC-3ʹ, and reverse: 5ʹ-GCC ACA TCC AAC CAT GAG CCT T-3ʹ; Opg forward: 5ʹ- CGG AAA CAG AGA AGC CAC GCA A-3ʹ, and reverse: 5ʹ- TGT CCA CCA AAA CAC TCA GCC-3ʹ;
BMDMs cell suspensions were prepared and homogenized in TRIzol (Invitrogen). After that, an equal volume of pure ethanol was added. After centrifugation, the supernatant was discarded. The column was washed with washing buffer. DNase/RNase-Free water was used to elute the RNA samples. After that, the cDNA library was synthesized by using reverse transcriptase. To analyze the accuracy, the melting curves were observed. The expressions of each target gene were calculated using the 2−ΔΔCq method. The mRNA expression values were normalized with that of the internal control, β-actin.
ELISAs
Cell supernatant was collected from LPS-induced BMDMs at different time points. The release of pro-inflammatory cytokines (IL-1β, IL-6, and TNF) were determined by specific ELISAs (R&D Biosystems).
Immunoblot Analysis
The protein extraction and qualification were performed according to a previous report.25 The BMDMs were collected and incubated with radioimmunoprecipitation assay buffer containing protease inhibitor on ice. Next, the extraction buffer was centrifuged to remove the insoluble components. A bicinchoninic acid protein assay kit was applied to qualify protein concentration. An equal amount of proteins (20 μg) was loaded on the 2–20% precast SDS-PAGE gel followed by transferring to 0.45 μm polyvinylidene fluoride membrane. Membrane blocking was applied by incubating membrane with 5% non-fat milk (room temperature, 2 h). The primary antibodies against IKBα (1:1000), p-IKKα/β (1:1000), IKKα (1:1000), p38 (1:1200), p-p38 (1:1000), JNK (1:1000), p-JNK (1:1000), DCAF1 (1:1000), and β-actin (1:800) were used to incubate with the membrane (4°C, overnight). After that, the membrane was incubated with horseradish peroxidase conjugated-secondary antibodies (room temperature, 2 h). The Biorad Gel Imaging System was applied and the expressions of target proteins were compared to the internal control (β-actin).
Statistical Analysis
SPSS 11.0 was applied to analyze the data. Data were presented as the mean ± the standard error of the mean (SEM). T-test or one- or two-way ANOVA and Student–Newman–Keuls (SNK) test were applied based on the numbers of group. A p-value less than 0.05 was considered as significant difference.
Results
DCAF1 Deficiency Did Not Affect the Development of Immune Cells in Bone Marrow and Spleen
Genotyping analysis was employed by using PCR. The results demonstrated that DCAF1 myeloid cell-conditional KO mice (DCAF1fl/flLyz2cre/+) were successfully constructed (Figure 1A). In addition, immunoblot analysis showed a specific ablation of DCAF1 in the T cells and macrophages of DCAF1fl/flLyz2cre/+ mice (Figure 1B). Next, the populations of adaptive immune cells including B and T lymphocytes were analyzed in the spleen. Interestingly, the populations of B and T lymphocytes showed no significant difference between DCAF1fl/flLyz2cre/+ and DCAF1fl/flLyz2+/+ mice (Figure 1C). Besides, the populations of CD4 and CD8 T lymphocytes also exhibited no significant difference between DCAF1fl/flLyz2cre/+ and DCAF1fl/flLyz2+/+ mice (Figure 1C). These results indicated that DCAF1 deficiency did not affect the development of B and T lymphocytes. Furthermore, our results demonstrated that the populations of CD11b+F4/80+ macrophages and CD11b+Ly6G+ neutrophils exhibited no significant difference between DCAF1fl/flLyz2cre/+ and DCAF1fl/flLyz2+/+ mice, indicating that DCAF1 deficiency did not affect the development of innate immune cells (macrophages and neutrophils, Figure 1D).
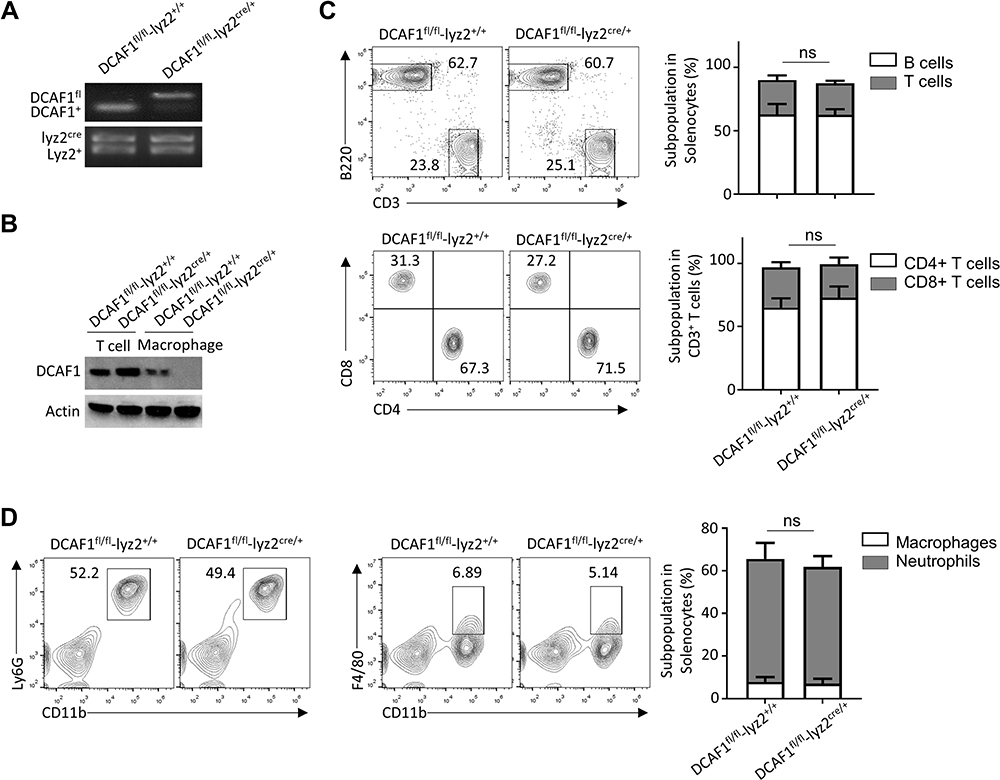 |
Figure 1 The development of immune cells in bone marrow and spleen were not affected due to DCAF1 deficiency in macrophages. (A) The genotype of DCAF1 myeloid cell-conditional KO mice was determined by PCR analysis. (B) The expressions of DCAF1 in the T cells and macrophages of DCAF1fl/fl Lyz2cre/+ mice were determined by Immunoblotting analysis. (C–D) The populations of B lymphocytes, T lymphocytes, macrophages, and neutrophils in the spleen from DCAF1fl/flLyz2+/+ (WT) or DCAF1fl/fl Lyz2cre/+ mice were analyzed by Flow cytometer. Representative FACS plots were shown. In this study, the data were shown as the mean ± SEM. The data were acquired from three independent experiments. The significance of data in (C–D) was calculated by Student’s t-test. ns indicates no statistical significance. |
DCAF1 Suppressed the Levels of Pro-Inflammatory Cytokines in Macrophages
To investigate the effects of DCAF1 on the production of inflammatory cytokines in macrophages, BMDMs isolated from DCAF1fl/flLyz2cre/+ and DCAF1fl/flLyz2+/+ mice were stimulated with LPS. These results demonstrated that the mRNA levels of pro-inflammatory cytokines (Il-1β, Il-6, and Tnf) were significantly elevated in the BMDMs isolated from DCAF1fl/flLyz2cre/+ mice (Figure 2A). Furthermore, the protein levels of pro-inflammatory cytokines (IL-1β, IL-6, and TNF) were also determined by specific ELISAs. Similarly, the results demonstrated that the production of pro-inflammatory cytokines were significantly elevated in the BMDMs from DCAF1fl/flLyz2cre/+ mice as compared with those from DCAF1fl/flLyz2+/+ mice (Figure 2B). Since osteoclasts derived from myeloid progenitors, we further investigated if deletion of DCAF1 affects osteoclast differentiation and activity. The data shown in Figure 2C-D indicated that DCAF1 deficiency had no effect on the osteoclast differentiation. However, DCAF1-deficient in osteoclast also showed an increase in the expressions of multiple inflammatory cytokines (Figure 2E). These results supported that DCAF1 deficiency increased the levels of pro-inflammatory cytokines in macrophages and osteoclast in a transcriptional and post-transcriptional manner.
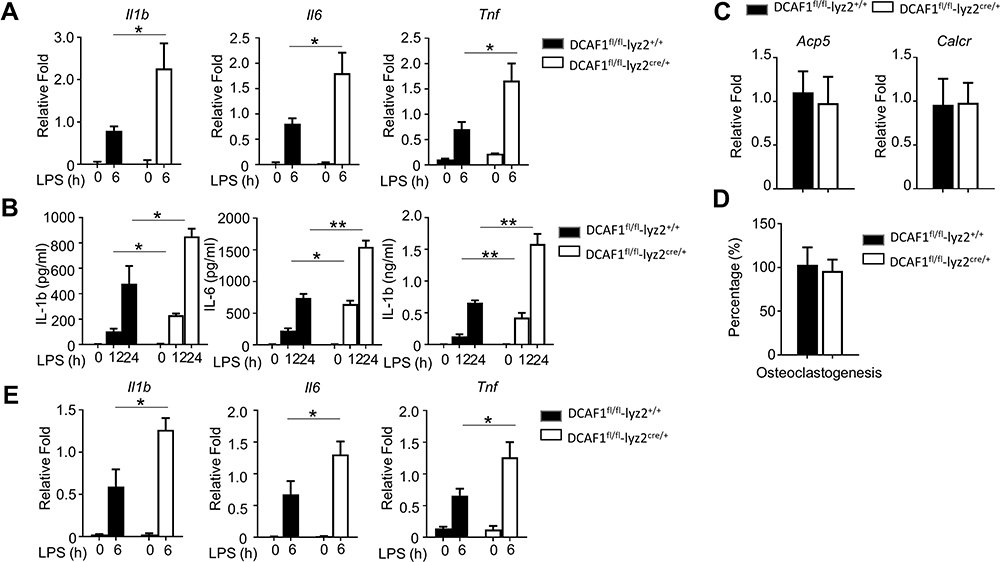 |
Figure 2 Pro-inflammatory cytokines were elevated in macrophages with DCAF deficiency. (A–B) The mRNA and protein levels of pro-inflammatory cytokines in primary BMDMs induced by LPS were determined by qRT-PCR and specific ELISAs, respectively. (C) The expressions of osteoclasts-specific marker genes were detected by qPCR. (D) The efficiency of osteoclast generation from WT and DACF1-deficient BMDMs were measured by TRAP staining. (E) The mRNA levels of pro-inflammatory cytokines in WT and DCAF1-deficient osteoclasts induced by LPS were determined by qRT-PCR. The data were presented as fold change as compared to the internal control. In this study, the data were presented as the mean ± SEM. The significances of differences comparisons between the responses group in the two genotypes were determined by a two-tailed Student’s t-test. Three independent experiments were operated to acquire the results. *P < 0.05; **P < 0.01. |
DCAF1 Deficiency in Macrophages Induced Severe Symptoms in S. aureus-Induced Osteomyelitis
To evaluate the DCAF1 deficiency on inflammation-related symptoms, S. aureus-induced osteomyelitis mouse model was established. Body weight was dramatically reduced after the injection of S. aureus. After 4 days of injection, the body weight began to recover (Figure 3A). The results demonstrated that the body weight loss was more rapidly in DCAF1fl/flLyz2cre/+ mice as compared to the DCAF1fl/flLyz2+/+ mice. Besides, the recovery of body weight was slower in DCAF1fl/flLyz2cre/+ mice as compared to the DCAF1fl/flLyz2+/+ mice.
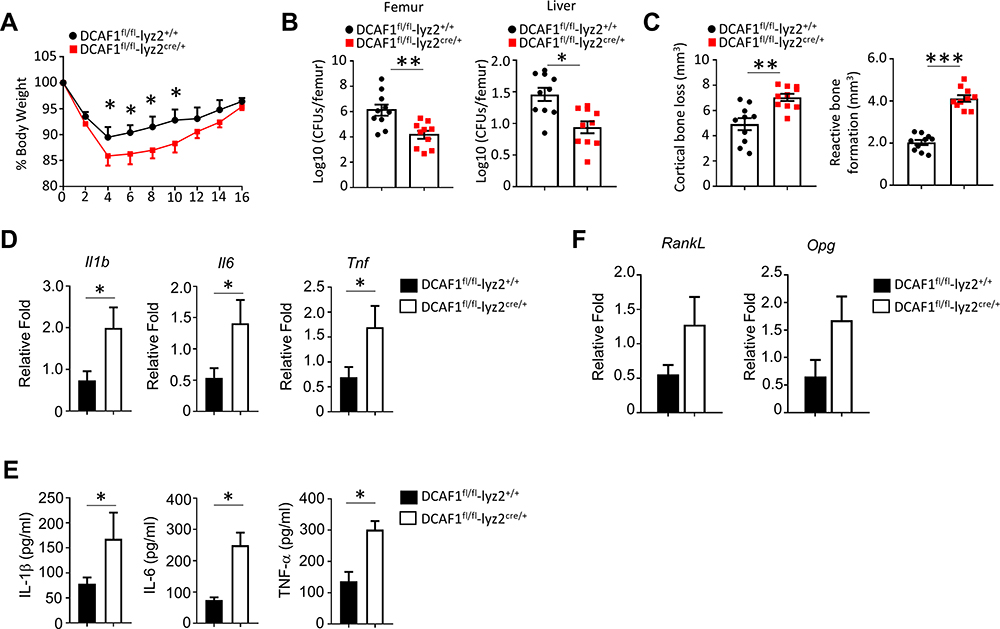 |
Figure 3 DCAF1 deficiency in macrophages induced severe symptoms in S. aureus-induced osteomyelitis. (A) The body weight was monitored every two days after the mice were injected with S. aureus (106 CFU) (n = 10). (B) After the injection of S. aureus, bacterial burdens were enumerated from the infected femurs and liver on day 14 (n=10). (C) The cortical bone loss (mm3) and reactive bone formation (mm3) were determined by μCT three-dimensional analysis. (D) After the injection of S. aureus, inflammatory tissues were collected and the mRNA levels of inflammatory cytokines were determined by qRT-PCR on day 8. In this study, the data were shown as the mean ± SEM. (E) Peripheral blood was collected and the secretion of these inflammatory cytokines were determined by ELISA on day 8. (F) The expressions of RankL and Opg inflammatory tissues were detected by qPCR. Three independent experiments were operated to acquire the results. The significance of data in (A–E) was calculated by Student’s t-test. * P < 0.05; **P < 0.01; ***P < 0.001.. |
Moreover, the results demonstrated that DCAF1fl/flLyz2cre/+ mice exhibited more severe symptoms in S. aureus-induced osteomyelitis, including less bacterial load in the femur, cortical bone loss, and reactive bone formation (Figure 3B and C). However, a significant decrease of S. aureus load in the liver was observed in WT mice (Figure 3B). In addition, the mRNA levels of inflammatory cytokines in the tissues were also determined. The results demonstrated that inflammatory cytokines (Il-1β, Il-6, and Tnf) were significantly elevated in the inflammatory tissues from DCAF1fl/flLyz2cre/+ mice as compared to the DCAF1fl/flLyz2+/+ mice (Figure 3D), which is consistent to the in vitro measurements. Additionally, DCAF1 deficiency also enhanced the expressions of these cytokines in the peripheral blood (Figure 3E), and thereby promoting the expressions of inducers related to osteoclast differentiation. These results indicated that DCAF1 functioned as a negative regulator in the systemic inflammatory response.
DCAF1 Deficiency Induced p38 Hyperactivation
Finally, we explored the underlying mechanisms of DCAF1 on the regulation of inflammation. First, the expressions of NF-κB complex-related components including p-IKKα/β, IKKα/β, IKBα, p-IKBα, and IKKα were determined. Interestingly, the expressions of these components in BMDMs exhibited no significant difference between DCAF1fl/flLyz2cre/+ and DCAF1fl/flLyz2+/+ mice (Figure 4A). However, our results demonstrated that higher levels of p-p38 in the BMDMs from DCAF1fl/flLyz2cre/+ mice (Figure 4B). To confirm the relationship between DCAF1 and p38, LPS was used to induce DCAF1fl/flLyz2cre/+ mice-derived BMDMs with or without p38 inhibitor (Doramapimod; 100 nM). Our results showed that LPS induction dramatically elevated inflammatory cytokines in DCAF1 deficiency conditions, whereas treatment with doramapimod abolished the elevation of pro-inflammatory cytokines (Figure 4C). These results suggested that DCAF1 deficiency induced p38 hyperactivation.
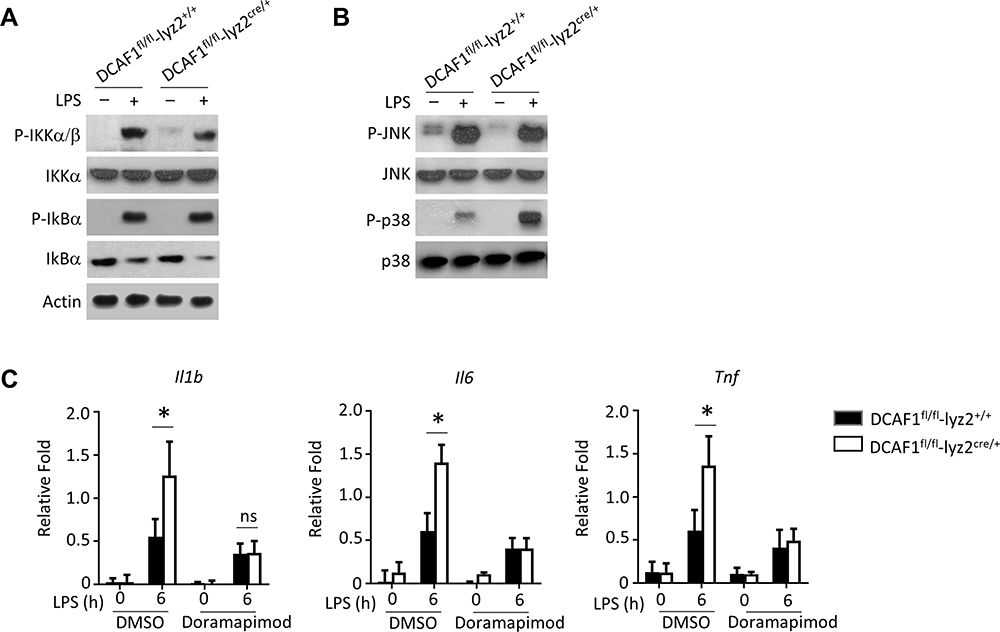 |
Figure 4 DCAF1 deficiency induced p38 hyperactivation. (A–B) BMDMs were isolated from WT or DCAF1fl/fl Lyz2cre/+ mice. Next, LPS was applied to stimulate BMDMs. The phosphorylation and total proteins of NF-kB and MAPKs signal-related markers were determined by Immunoblotting analysis. (C) BMDMs were pre-treated with p38 inhibitor (Doramapimod,100 nM) for 2 h prior to incubating with LPS. Next, the mRNA levels of inflammatory cytokines were determined by qRT-PCR. In this study, the data were presented as the mean ± SEM. The data were acquired from three independent experiments. The significance of data in (C) was calculated by Student’s t-test. *P < 0.05. |
Discussion
In this study, we investigated the effects of DCAF1 deficiency in macrophages on S. aureus-induced mouse model of osteomyelitis. The results demonstrated that DCAF1 deficiency promoted the expressions of inflammatory cytokines in LPS-induced BMDMs and in inflammatory tissues. In addition, DCAF1 deficiency led to severe symptoms including less bacterial load in the femur, cortical bone loss, and reactive bone formation in a mouse model of osteomyelitis. Furthermore, the molecular mechanism study revealed that DCAF1 deficiency was associated with p38 hyperactivation. Taken together, these results supported that DCAF1 in macrophages suppressed the S. aureus-induced osteomyelitis by the regulation of p38 hyperactivation.
DCAF1 is a component of E3 ubiquitin ligase and serine/threonine-protein kinase.19 Many studies demonstrated that DCAF1 is involved in a series of cellular events such as cell survival, cell growth, and cell cycle.19,26 Interestingly, upregulation of DCAF1 is observed in the activated T cells, indicating the important roles of DCAF1 in the T cell’s function.27 Considering the essential roles of DCAF1 for the cells, the effects of DCAF1 deficiency in macrophages on the development of other immune cells were determined in the DCAF1 myeloid cell-conditional KO mice (DCAF1fl/flLyz2cre/+). Interestingly, the results demonstrated that DCAF1 deficiency did not affect the development of innate immune cells (Neutrophils and macrophages) and adaptive immune cells (B and T lymphocytes). These results encouraged us to investigate the effects of DCAF1 deficiency on macrophages-mediated inflammation.
DCAF1 is essential for DNA replication and is known to modulate the G2/M arrest.28 It is reported that DCAF1 regulates anti-HIV cellular responses in macrophages by inhibiting viral cDNA synthesis.21 However, the effects of DCAF1 on macrophages-mediated inflammation are still unknown. Herein, we found the elevation of pro-inflammatory cytokines at the transcriptional and post-transcriptional manner in the DCAF1 deficiency BMDMs, supporting the regulatory effects of DCAF1 on inflammatory cytokines in macrophages.
To investigate the effects of DCAF deficiency in macrophages on in vivo inflammation, S. aureus-induced mouse model of osteomyelitis was established. Osteomyelitis is a bone inflammation caused by a series of infecting microbial agents including bacteria and fungus.4 Among them, S. aureus is accounting for 30–80% of bone infection.12 In this study, S. aureus was injected in the DCAF deficiency (DCAF1fl/flLyz2cre/+) and control (DCAF1fl/flLyz2+/+) mice. Interestingly, the results demonstrated that osteomyelitis symptoms including body weight loss, less bacterial load in the femur, cortical bone loss, and reactive bone formation in DCAF deficiency mice. These results supported that DCAF deficiency accelerated the development of osteomyelitis.
When bone infection occurs, S. aureus induces an acute inflammatory response and initiates an inflammation cascade in local inflammatory sites, where many types of immune cells including myeloid and lymphoid cells participates.1,11,15 It is reported that macrophages play important roles in osteomyelitis.29,30 Toll-like receptor is widely expressed in the macrophages and can be activated by the pathogen-associated molecular patterns.29,30 Consequently, the release of pro-inflammatory cytokines (IL-1β, IL-6, and TNF) stimulates osteoclast activation and differentiation and thereby leading to bone resorption.29,30 Herein, we investigated the effects of DCAF deficiency in macrophages on the pro-inflammatory cytokines in inflammatory tissues. The results demonstrated that mRNA levels of pro-inflammatory cytokines were dramatically elevated in the inflammatory tissues, which are consistent with the in vitro measurements. The gram-positive bacterium Staphylococcus aureus is a major pathogen responsible for a variety of diseases including osteomyelitis. Cell wall-associated and secreted proteins and cell wall components such as peptidoglycan and alanylated lipoteichoic acid have been shown to be inflammatory, and contribute to osteomyelitis. Lipoteichoic acid (LTA) has been shown to engage TLR2 and stimulate inflammatory responses. Although LPS and LTA trigger the TLR4 and TLR2 respectively, these two TLRs shared similar downstream signal proteins including MyD88 and TRAF6, which further activated NF-kB and MAPKs.31 Therefore, we believe that LPS stimulation could indicate the potential function of DCAF1 in TLR2 signal pathway even LPS and S. aureus target on different receptors. However, in the further study, S. aureus induced in vitro cell model are warranted to be established.
Finally, we explored the underlying mechanisms of DCAF deficiency on the regulation of macrophage-mediated inflammation. NF-κB is an important transcriptional factor for regulating inflammation.32,33 It is complexed with IκBα in the normal conditions, whereas the phosphorylated IκBα disassociates from NF-κB, leading to the nuclear translocation of p65.33 In this study, the expressions of NF-κB components in BMDMs were not significantly altered in the DCAF deficiency conditions. In addition to the NF-κB signaling pathway, JNK and p38 MAPK pathways also play important roles in the regulation of inflammatory response.34 We then explored the effects of DCAF deficiency on the phosphorylation of JNK and p38. The results demonstrated that a higher level of p-p38 in the BMDMs was observed in the DCAF deficiency conditions. However, treatment with p38 inhibitor (doramapimod) suppressed the elevation of pro-inflammatory cytokines in the BMDMs. Taken together, our data supported the regulatory effects of DCAF1 on inflammation correlated to p38 hyperactivation.
Conclusion
DCAF1 deficiency in macrophages did not affect the development of other immune cells. However, its deficiency promoted inflammatory responses in vitro and in vivo. Consistently, DCAF1 deficiency in macrophages induced severe symptoms in S. aureus-induced mouse model of osteomyelitis. Furthermore, our results supported that DCAF1 regulated macrophage-mediated inflammation and its regulatory effects associated with p38 hyperactivation.
Acknowledgment
This work was funded by a Municipal Human Resources Development Program for Outstanding Leaders in Medical Disciplines in Shanghai (2018BR38) and the National Natural Science Foundation of China (81873993).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lew DP, Waldvogel FA. Osteomyelitis. The Lancet. 2004;364(9431):369–379. doi:10.1016/S0140-6736(04)16727-5
2. Lew DP, Waldvogel FA. Osteomyelitis. N Engl J Med. 1997;336(14):999–1007.
3. Kremers HM, Nwojo ME, Ransom JE, Wood-Wentz CM, Melton III LJ, Huddleston III PM. Trends in the epidemiology of osteomyelitis: a population-based study, 1969 to 2009. J Bone Joint Surg Am. 2015;97(10):837.
4. Brady R, Leid J, Costerton J, Shirtliff M. Osteomyelitis: clinical overview and mechanisms of infection persistence. Clin Microbiol Newsletter. 2006;28(9):65–72.
5. Fang RC, Galiano RD Adjunctive therapies in the treatment of osteomyelitis. Paper presented at: Seminars in plastic surgery. 2009.
6. Gogia JS, Meehan JP, Di Cesare PE, Jamali AA Local antibiotic therapy in osteomyelitis. Paper presented at: Seminars in plastic surgery. 2009.
7. Qiao Y, Liu X, Li B, et al. Treatment of MRSA-infected osteomyelitis using bacterial capturing, magnetically targeted composites with microwave-assisted bacterial killing. Nat Commun. 2020;11(1):1–13.
8. Wei S, Qiao Y, Wu Z, et al. Na+ inserted metal-organic framework for rapid therapy of bacteria-infected osteomyelitis through microwave strengthened Fenton reaction and thermal effects. Nano Today. 2021;37:101090.
9. Hofstee MI, Muthukrishnan G, Atkins GJ, et al. Current concepts of osteomyelitis: from pathological mechanisms to advanced research methods. Am J Pathol. 2020.
10. Masters EA, Trombetta RP, de Mesy Bentley KL, et al. Evolving concepts in bone infection: redefining “biofilm”,“acute vs. chronic osteomyelitis”,“the immune proteome” and “local antibiotic therapy”. Bone Res. 2019;7(1):1–18.
11. Olson ME, Horswill AR. Staphylococcus aureus osteomyelitis: bad to the bone. Cell Host Microbe. 2013;13(6):629–631.
12. Chambers HF. The changing epidemiology of Staphylococcus aureus? Emerg Infect Dis. 2001;7(2):178.
13. Jensen A, Espersen F, Skinhøj P, Rosdahl V, Frimodt-Møller N. Increasing frequency of vertebral osteomyelitis following Staphylococcus aureus bacteraemia in Denmark 1980–1990. J Infect. 1997;34(2):113–118.
14. Nelson DR, Buxton TB, Luu QN, Rissing JP. The promotional effect of bone wax on experimental Staphylococcus aureus osteomyelitis. J Thorac Cardiovasc Surg. 1990;99(6):977–980.
15. Mal S, Berendt A, Peacock S. Staphylococcus aureus bone and joint infection. J Infect. 2002;44(3):143–151.
16. Fu C, Shi R. Osteoclast biology in bone resorption: a review. STEMedicine. 2020;1(4):e57.
17. Peters K, Koberg K, Rosendahl T, Schmutzler W, Zwadlo-Klarwasser G. Alteration in the pattern of macrophage subtypes in chronic osteomyelitis compared with acute joint infection. Int Orthop. 1995;19(3):162–166.
18. Romani B, Cohen ÉA. Lentivirus Vpr and Vpx accessory proteins usurp the cullin4–DDB1 (DCAF1) E3 ubiquitin ligase. Curr Opin Virol. 2012;2(6):755–763.
19. Schabla NM, Mondal K, Swanson PC. DCAF1 (VprBP): emerging physiological roles for a unique dual-service E3 ubiquitin ligase substrate receptor. J Mol Cell Biol. 2019;11(9):725–735.
20. McCall CM, de Marval PLM, Chastain PD, et al. Human immunodeficiency virus type 1 Vpr-binding protein VprBP, a WD40 protein associated with the DDB1-CUL4 E3 ubiquitin ligase, is essential for DNA replication and embryonic development. Mol Cell Biol. 2008;28(18):5621–5633.
21. Hrecka K, Hao C, Gierszewska M, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474(7353):658–661.
22. Francke A, Herold J, Weinert S, Strasser RH, Braun-Dullaeus RC. Generation of mature murine monocytes from heterogeneous bone marrow and description of their properties. J Histochem Cytochem. 2011;59(9):813–825.
23. Liu C, Ma H, Slitt AL, Seeram NP. Inhibitory effect of cannabidiol on the activation of NLRP3 inflammasome is associated with its modulation of the P2X7 receptor in human monocytes. J Nat Prod. 2020.
24. Shang W, Jiang Y, Boettcher M, et al. Genome-wide CRISPR screen identifies FAM49B as a key regulator of actin dynamics and T cell activation. Proc Natl Acad Sci. 2018;115(17):E4051–E4060.
25. Liu C, Shen Y-J, Tu Q-B, et al. Pedunculoside, a novel triterpene saponin extracted from Ilex rotunda, ameliorates high-fat diet induced hyperlipidemia in rats. Biomed Pharmacother. 2018;101:608–616.
26. Yu C, Ji S-Y, Sha -Q-Q, Sun Q-Y, Fan H-Y. CRL4–DCAF1 ubiquitin E3 ligase directs protein phosphatase 2A degradation to control oocyte meiotic maturation. Nat Commun. 2015;6(1):1–11.
27. Guo Z, Kong Q, Liu C, et al. DCAF1 controls T-cell function via p53-dependent and-independent mechanisms. Nat Commun. 2016;7(1):1–13.
28. Le Rouzic E, Belaïdouni N, Estrabaud E, et al. HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle. 2007;6(2):182–188.
29. Wang Y, Lin Y, Cheng C, et al. NF-κB/TWIST1 mediates migration and phagocytosis of macrophages in the mice model of implant-associated Staphylococcus aureus osteomyelitis. Front Microbiol. 2020;11:1301.
30. Dapunt U, Maurer S, Giese T, Gaida MM, Hänsch GM. The macrophage inflammatory proteins MIP1 (CCL3) and MIP2 (CXCL2) in implant-associated osteomyelitis: linking inflammation to bone degradation. Mediators Inflamm. 2014;2014.
31. Mukherjee S, Karmakar S, Babu SP. TLR2 and TLR4 mediated host immune responses in major infectious diseases: a review. Braz J Infect Dis. 2016;20(2):193–204.
32. Sun W, Liu C, Zhang Y, et al. Ilexgenin A, a novel pentacyclic triterpenoid extracted from Aquifoliaceae shows reduction of LPS-induced peritonitis in mice. Eur J Pharmacol. 2017;797:94–105.
33. Tak PP, Firestein GS. NF-κB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7–11.
34. Huang G, Shi LZ, Chi H. Regulation of JNK and p38 MAPK in the immune system: signal integration, propagation and termination. Cytokine. 2009;48(3):161–169.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.