Back to Journals » Drug Design, Development and Therapy » Volume 10
Curcumin protects against myocardial infarction-induced cardiac fibrosis via SIRT1 activation in vivo and in vitro
Authors Xiao J, Sheng X, Zhang X, Guo M, Ji X
Received 23 January 2016
Accepted for publication 26 February 2016
Published 29 March 2016 Volume 2016:10 Pages 1267—1277
DOI https://doi.org/10.2147/DDDT.S104925
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Jie Xiao, Xi Sheng, Xinyu Zhang, Mengqi Guo, Xiaoping Ji
Key Laboratory of Cardiovascular Remodeling and Function Research, Chinese Ministry of Education and Chinese Ministry of Health, Qilu Hospital of Shandong University, Jinan, Shandong, People’s Republic of China
Abstract: Curcumin, a polyphenolic compound derived from turmeric, protects against myocardial injury by alleviating oxidative stress, inflammation, apoptosis, and fibrosis. However, the role of curcumin and its mechanism of action on interstitial fibrosis after myocardial infarction (MI) are poorly understood. To clarify, MI was induced by a permanent ligation of the left anterior descending coronary artery in adult mice, and the effects of curcumin were evaluated 4 weeks after the MI event. In vitro, we treated cardiac fibroblasts (CFs) with Ang II, and investigated the anti-fibrotic mechanism of curcumin. Our results showed that curcumin significantly attenuated collagen deposition in vivo and inhibited CF proliferation and migration, and MMP expression. In addition, we found that the down-regulation of SIRT1 after MI was attenuated by curcumin pretreatment, which indicated that the activation of SIRT1 might be involved in the protective action of curcumin. This hypothesis was confirmed by genetic inhibition of SIRT1 (siRNA-SIRT1) in Ang II-treated CFs. Our results provide new insights into the mechanism underlying the anti-fibrotic effects of curcumin in the heart.
Keywords: curcumin, myocardial infarction, angiotensin II, cardiac fibroblasts, fibrosis, SIRT1
Corrigendum for this paper has been published
Introduction
Myocardial infarction (MI) remains the leading cause of morbidity and mortality worldwide, carrying an enormous medical and social burden. Post-MI fibrosis was observed in both infarcted and non-infarcted myocardium. Although fibrosis is essential for normal healing, an excessive level of fibrosis is a poor prognostic factor. Indeed, excessive fibrosis progressively impairs ventricular functions and is associated with increased levels of hospitalization or death related with heart failure.1,2 Cumulative evidence indicates that the renin-angiotensin system is activated after MI. Indeed, Ang II, the central product of the renin-angiotensin system, is involved in the development of myocardial remodeling following MI.3 Ang II induces cardiac fibroblast (CF) proliferation and migration, collagen deposition, and extracellular matrix (ECM) degradation by activating a variety of cell signaling pathways, such as transforming growth factor (TGF)-β and mitogen-activated protein kinase (MAPK) pathways.3–5 Consequently, angiotensin converting enzyme (ACE) inhibitors and Ang II receptor blockers are now well established and widely used treatments for the management of patients with MI. However, the incidence and lethality of heart failure remains very high. Novel pharmacological strategies to inhibit the maladaptive cardiac repair and improve myocardial dysfunction are needed.
Curcumin (Cur), the active component in Curcuma longa, is known to exhibit a variety of beneficial effects, such as anti-inflammation, anti-apoptosis, anti-proliferation, and anti-oxidation.6 The protective effects of Cur on the cardiovascular system have been reported in MI, hypertension, and diabetic cardiomyopathy.7–9 However, while the previous studies about Cur against myocardial injury were mainly focused on its anti-apoptotic and anti-inflammatory effects, the effects of Cur on myocardial fibrosis remain incompletely elucidated. Recently, Cur was proven to attenuate myocardial fibrosis by modulating the expression of the Ang II receptors, AT1 and AT2, in Ang II-treated rats. Additionally, Cur ameliorated collagen deposition in spontaneously hypertensive rats through peroxisome proliferator-activated receptor (PPAR)-gamma activation.8,10 However, the role and underlying mechanisms of Cur against MI-induced myocardial fibrosis remain unclear.
SIRT1, a member of the mammalian sirtuin protein (SIRT1–SIRT7) family, is a conserved nicotinamide adenine dinucleotide (NAD)+-dependent histone deacetylase involved in various biological processes, including gene silencing, DNA repair, cell survival, metabolism, and aging.11 A growing amount of evidence supports the role of SIRT1 in fibrosis in several organs, such as liver, heart, and kidneys.11–13 Recently, the activation of SIRT1 Cur pretreatment was reported to attenuate the mitochondrial oxidative damage induced by myocardial ischemia reperfusion injury.14 Furthermore, Cur-induced SIRT1 activation blocked the neurotoxicity of amyloid-β25–35 in rat cortical neurons.15 However, the question whether Cur could effectively inhibit MI-induced cardiac fibrosis via SIRT1 activation has not been clearly addressed in vivo or in vitro.
Consequently, we hypothesized that SIRT1 activation could mediate the protective effect of Cur against MI-induced myocardial fibrosis. To determine the role and mechanism behind the effects of Cur, we performed a series of experiments, both in vivo and in vitro, to evaluate collagen deposition, CF proliferation and migration, matrix metalloproteinase (MMP)-induced ECM degradation, and SIRT1 expression. Our results revealed that Cur protected against myocardial fibrosis, which was partially mediated by SIRT1 activation.
Materials and methods
Animal model and drug treatment
Eight-week-old C57BL/6J wild-type male mice (weighing 20–25 g) were obtained from Vital River Laboratories (Beijing, People’s Republic of China). The animals were fed regular chow and maintained under standard lighting (12:12 hour, day-night rhythm), temperature (20°C–22°C), and humidity (50%–60%) conditions. After 1 week of acclimatization, the mice were randomly divided into four groups (n=12 each) for treatments, as follows: sham, sham+Cur, MI, and MI+Cur. The sham+Cur and MI+Cur groups received Cur (Sigma-Aldrich Co., St Louis, MO, USA) (100 mg/kg/day orally) dissolved in distilled water (containing a small amount of dimethyl sulfoxide [DMSO] [Sigma-Aldrich Co.] for better dissolution) and administered by gastric gavage. The other two groups received only distilled water containing an equal amount of DMSO. The Cur dosage was chosen according to previous studies.16,17 One week later, the mice in the MI and MI+Cur groups were anesthetized with 2% isoflurane inhalation without ventilation. The skin over the left side of the chest was cut (~ 1.2–1.5 cm). After dissection of the pectoral muscle, a small hole was made at the fourth intercostal space. The heart was smoothly and gently extruded from the hole, and MI was created by permanent left anterior descending coronary artery ligation using a 7-0 suture line. The success of the coronary occlusion was confirmed by pallor and regional-wall motion abnormality of the left ventricle.18 Mice in the sham and sham+Cur groups underwent the same time-matched surgical procedure without the ligation. Mice were euthanized 4 weeks after surgery. All experiments involving animals were performed according to the Animal Management Rules of the Chinese Ministry of Health (document number 55, 2001), and were approved by the Animal Ethics Committee of Qilu Hospital, Shandong University.
Culture of primary CFs
CFs were isolated from 1- to 3-day-old Wistar rats, as described previously.19 Briefly, the ventricular tissue was finely minced and mechanically digested with type II collagenase (120 units/mL; Sigma-Aldrich Co.) using a rotor in a flask. The cell suspensions were plated in a cell culture flask for 90 minutes to separate the fibroblasts and cardiomyocytes. Most CFs adhered to the flask. The cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 4.5 g/L D-glucose, 10% fetal bovine serum (HyClone, Thermo Fisher Scientific, Waltham, MA, USA), penicillin (100 units/mL), and streptomycin (100 μg/mL) in a humidified atmosphere composed of 95% O2–5% CO2. CFs were used at passage 2 or 3.
Cell treatments
Ang II (Sigma-Aldrich Co.) was dissolved in sterile deionized water and Cur was dissolved in DMSO. Both solutions were stored at −20°C. The cells were treated according to four experimental designs. First, the cells were treated with different doses of Ang II (1, 10, and 100 nM) for 48 hours. Second, the cells were treated with 100 nM of Ang II for various times (12, 24, and 48 hours). Third, the cells were pretreated with Cur at different concentrations (5, 10, and 15 μM) for 1 hour. The Ang II groups were pretreated with an equal amount of DMSO and were stimulated with100 nM of Ang II for 24 hours. Fourth, the CFs were transfected with SIRT1-specific siRNA or its negative control (NC) siRNA (GenePharma, Shanghai, People’s Republic of China) according to the manufacturer’s instructions. After 24 hours, the cells were treated with Ang II for 24 hours with or without Cur pretreatment. The siRNA target sequence for SIRT1 was 5′-GGGAUCAAGAGGUUGUUAATTUUAACAACCUCUUGAUCCCTT-3′. The NC siRNA sequence was 5′-ACGUGACACGUUCGGAGAATT-3′.
Histology
The mouse hearts were fixed in 4% formalin for a minimum of 24 hours. The fixed tissues were embedded in paraffin and sectioned (5 μm) for staining. The heart sections were stained with Masson’s trichrome and Picro-Sirius red to evaluate interstitial collagen deposition. For immunohistochemistry, the slides were incubated overnight at room temperature with primary antibodies against collagen I, collagen III, and TGF-β1 (all from Abcam, Cambridge, MA, USA). After washing with phosphate-buffered saline, the slides were incubated with the corresponding secondary antibodies for 1 hour. The results were analyzed using the Image-Pro Plus 6.0 software.
Western blotting analysis
Freshly dissected mouse heart tissue from the peri-infarction border and CFs were harvested. Protein was extracted using cell lysis buffer intended for Western blotting analysis and immunoprecipitation assays (Beyotime Institute of Biotechnology, Shanghai, People’s Republic of China). Equal amounts of protein were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Merck Millipore, Billerica, MA, USA) which were blocked in 5% nonfat milk for 1–2 hours. Then, the blocked membranes were incubated overnight with the primary antibodies against collagen I (Abcam), collagen III (Proteintech Group, Inc., Chicago, USA), TGF-β1, MMP-2, MMP-9 (all Abcam), or SIRT1 (Cell Signaling Technology, Danvers, MA, USA). After washing with Tris-buffered saline and Tween 20, the membranes were incubated with the appropriate secondary antibodies for 1–2 hours at room temperature. The protein bands were detected using an enhanced chemiluminescence detection system and the band density was determined using the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Gelatin zymography
The enzymatic activity of MMPs in CFs was assayed using gelatin zymography. A 10% sodium dodecyl sulfate-polyacrylamide gel containing gelatin at 1 mg/mL was used for electrophoresis. The samples were renatured in washing buffer (50 mM Tris-HCl, 100 mM NaCl, and 2.5% Triton X-100) at room temperature, followed by incubation in a substrate buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM CaCl2, 1 μM ZnCl2) at 37°C for 20 hours. The gel was stained with Coomassie brilliant blue R-250 and destained. The presence of lytic bands from the gelatin digestion indicated MMP-2 (72 kDa) and MMP-9 (92 kDa) activity.
Cell proliferation assays
CFs were seeded into 96-well plates for 24 hours, and proliferation was determined using the Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions. Optical density was measured at 450 nm.
Cell migration assay
CF migration assays were performed in Transwell chambers (8 μm pore size; Corning Inc., Corning, NY, USA). Serum-starved CFs (1×105) were seeded into the upper chamber of the migration apparatus. As a chemotactic stimulus, DMEM containing 0.1% fetal bovine serum and supplemented with Ang II alone or with different concentrations of Cur (5, 10, or 15 μM) was placed into the lower chamber for 8 hours. For the SIRT1 knockdown experiments, CFs were transfected in Transwell chambers with the SIRT1-specific siRNA or its NC siRNA according to the manufacturer’s instructions. After 24 hours, CFs were stimulated with Ang II for 8 hours, with or without Cur pretreatment. The results are expressed as the mean number of migrating cells from five random fields (×200) of each Transwell membrane.
Statistical analysis
Results are expressed as the mean ± standard error of the mean. Inter-group comparisons were performed using one-way analysis of variance on results from at least three independent experiments. Differences were considered statistically significant at P<0.05. All statistical analyses were performed using SPSS software version 18.0 (SPSS Inc., Chicago, IL, USA) and Prism 6 software (GraphPad Software Inc., La Jolla, CA, USA).
Results
Cur alleviated interstitial fibrosis after MI
Masson’s trichrome and Picro-Sirius red staining of heart sections revealed greater fibrosis in the interstitial regions of the MI group compared with the sham group (32.87%±1.41% and 5.66%±0.71%, respectively, P<0.05). Cur pretreatment noticeably reduced collagen deposition following MI (23.17%±1.02% versus 32.87%±1.41%, P<0.05), which was confirmed by the immunohistochemical staining results for collagen I, collagen III, and TGF-β1 (Figure 1A–F, P<0.05). Additionally, Cur greatly reduced the infarct size in cardiac cross-sections compared to MI alone (27.84%±2.80% versus 37.11%±2.26%, P<0.05) (Figure 1G, P<0.05).
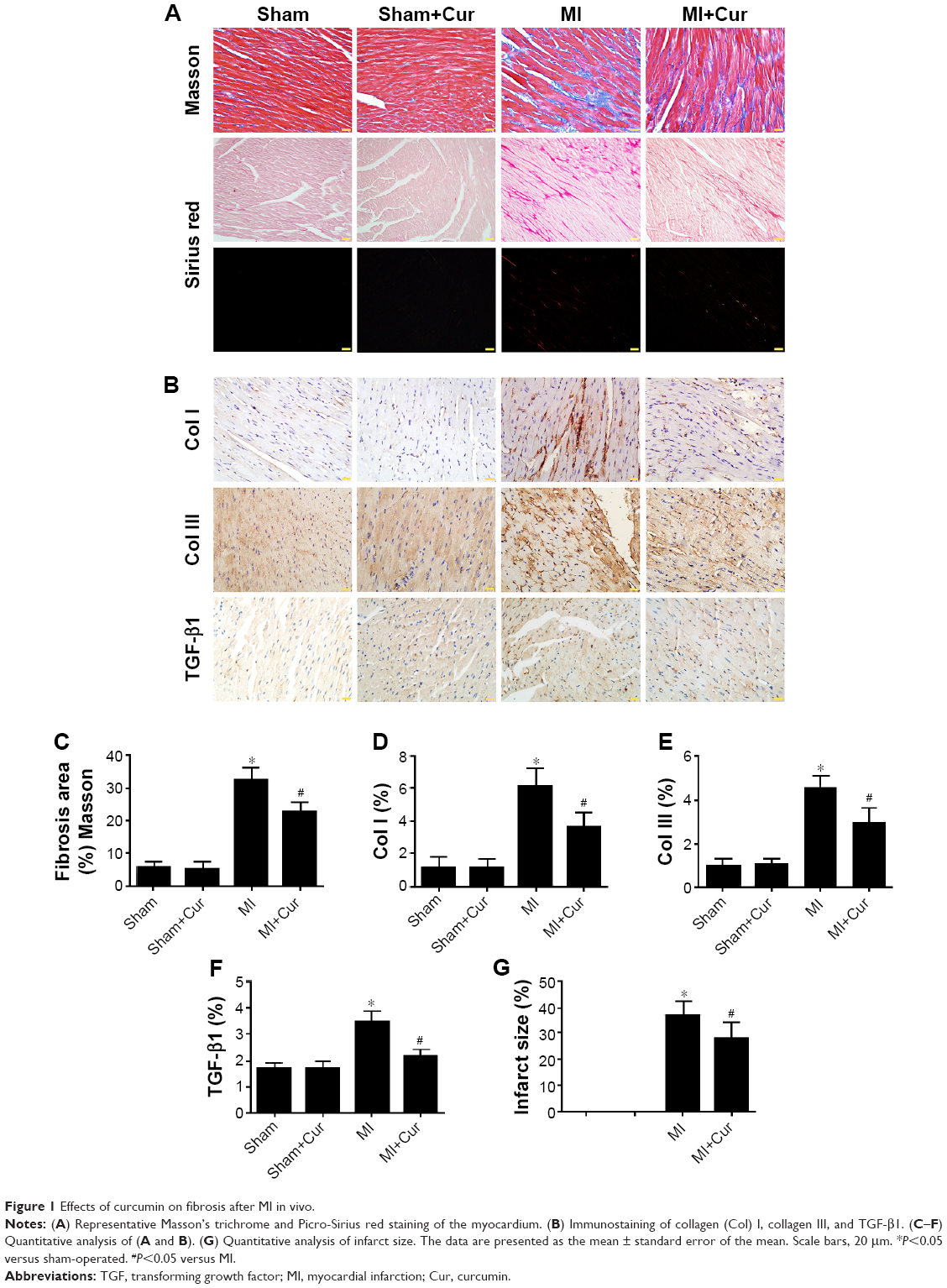 | Figure 1 Effects of curcumin on fibrosis after MI in vivo. |
Cur attenuated the down-regulation of SIRT1 induced by MI in vivo
Four weeks after MI, SIRT1 expression was decreased in the heart tissues, which was attenuated by Cur pretreatment. SIRT1 levels were revealed by Western blotting (Figure 2A and B, P<0.05) and confirmed by immunohistochemistry on cardiac tissue cross-sections (Figure 2C, P<0.05).
 | Figure 2 Effects of curcumin on SIRT1 expression after MI in vivo. |
Ang II increased the levels of collagen I, collagen III, and TGF-β1 in CFs
First, we examined the effects of Ang II on collagen deposition and fibrotic TGF-β1 expression in CFs. We tested the Ang II dose-response using concentrations of 1, 10, and 100 nM for 48 hours, and found that the expression of collagen I, collagen III, and TGF-β1 increased gradually in a dose-dependent manner (Figure 3A–C, P<0.05). Indeed, significant increases were detected as early as 12 hours after Ang II incubation, while the maximum effects were observed at 48 hours (Figure 3D–F, P<0.05). However, the effects observed at 24 hours were almost as significant as the effects observed at 48 hours. Therefore, we treated CFs with Ang II (100 nM) for 24 hours for the subsequent experiments.
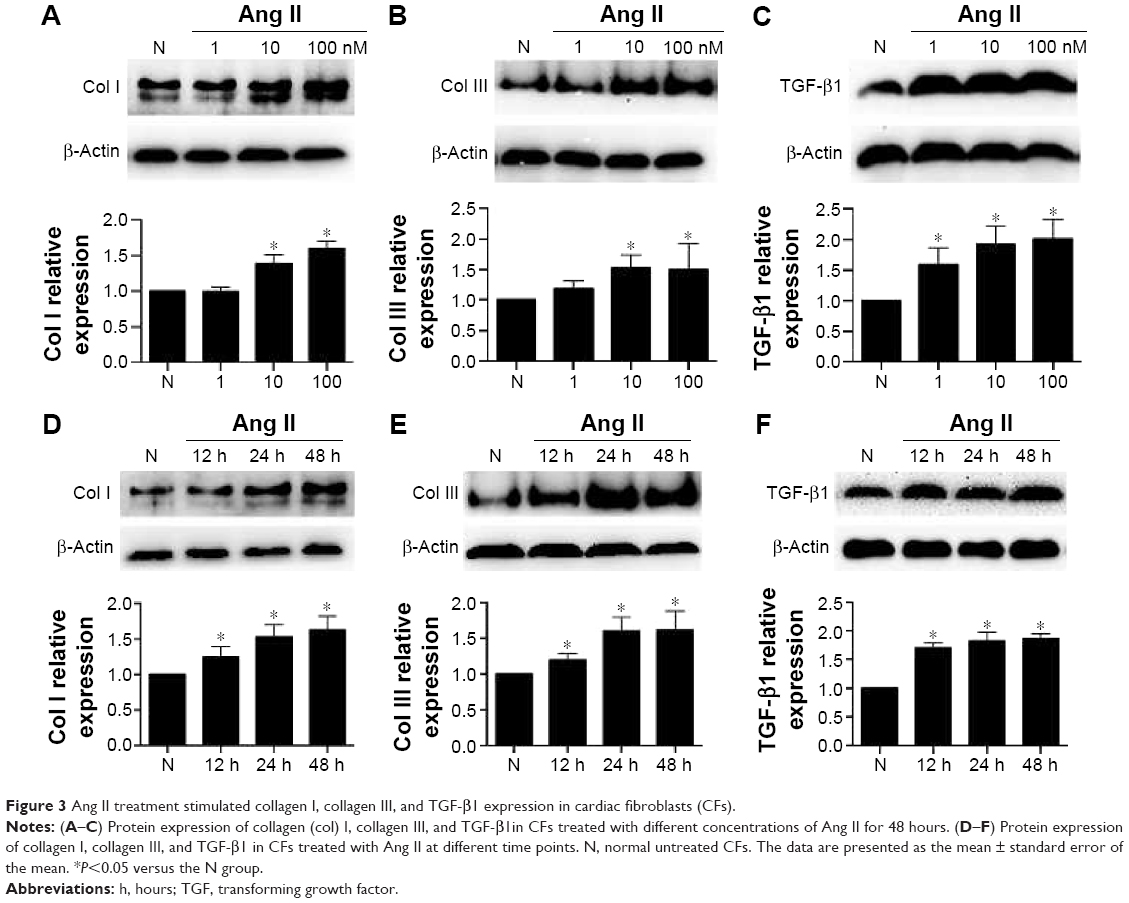 | Figure 3 Ang II treatment stimulated collagen I, collagen III, and TGF-β1 expression in cardiac fibroblasts (CFs). |
Cur increased SIRT1 expression and decreased collagen deposition and ECM degradation
To investigate the protective role of Cur against collagen deposition, we examined the expression of collagen I, collagen III, and TGF-β1 using Western blotting analysis. As shown in Figure 4A, compared with the Ang II-treated group, Cur significantly decreased the expression of related proteins in a dose-dependent manner. In addition, we evaluated the effects of Cur on ECM degradation based on expression and activity of MMP-9 and MMP-2 measured by Western blotting analyses and gelatin zymography. Cur specifically inhibited MMP-2 and MMP-9 expression (Figure 4A and B, P<0.05), as well as MMP-2 activity (Figure 4C and D, P<0.05). However, the activity of MMP-9 was not detected in the medium. Interestingly, we found that Ang II inhibited SIRT1 expression, which was attenuated by Cur pretreatment.
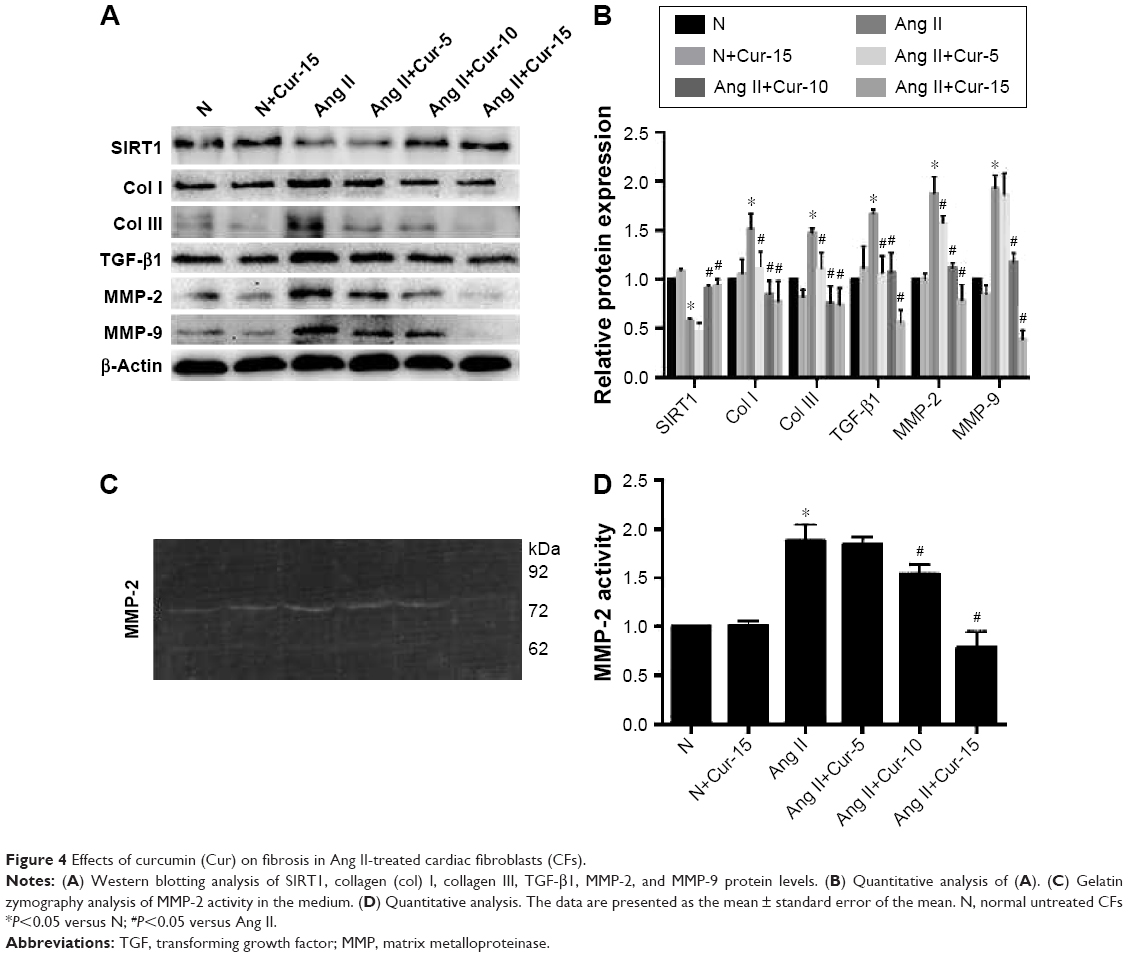 | Figure 4 Effects of curcumin (Cur) on fibrosis in Ang II-treated cardiac fibroblasts (CFs). |
Cur inhibits the proliferation and migration of CFs induced by Ang II
CFs’ proliferation and migration play an important role in myocardial fibrosis. Transwell migration assays were used to evaluate the effects of Cur on CFs’ migration. After 8 hours, we found that Cur strongly inhibited CFs’ migration in a dose-dependent manner (Figure 5A and B, P<0.05). Next, we investigated the CFs’ proliferation using CCK-8 assays. CFs displayed rapid growth over 24 hours after Ang II stimulation, which was delayed by Cur pretreatment, particularly at 15 μM (Figure 5C, P<0.05).
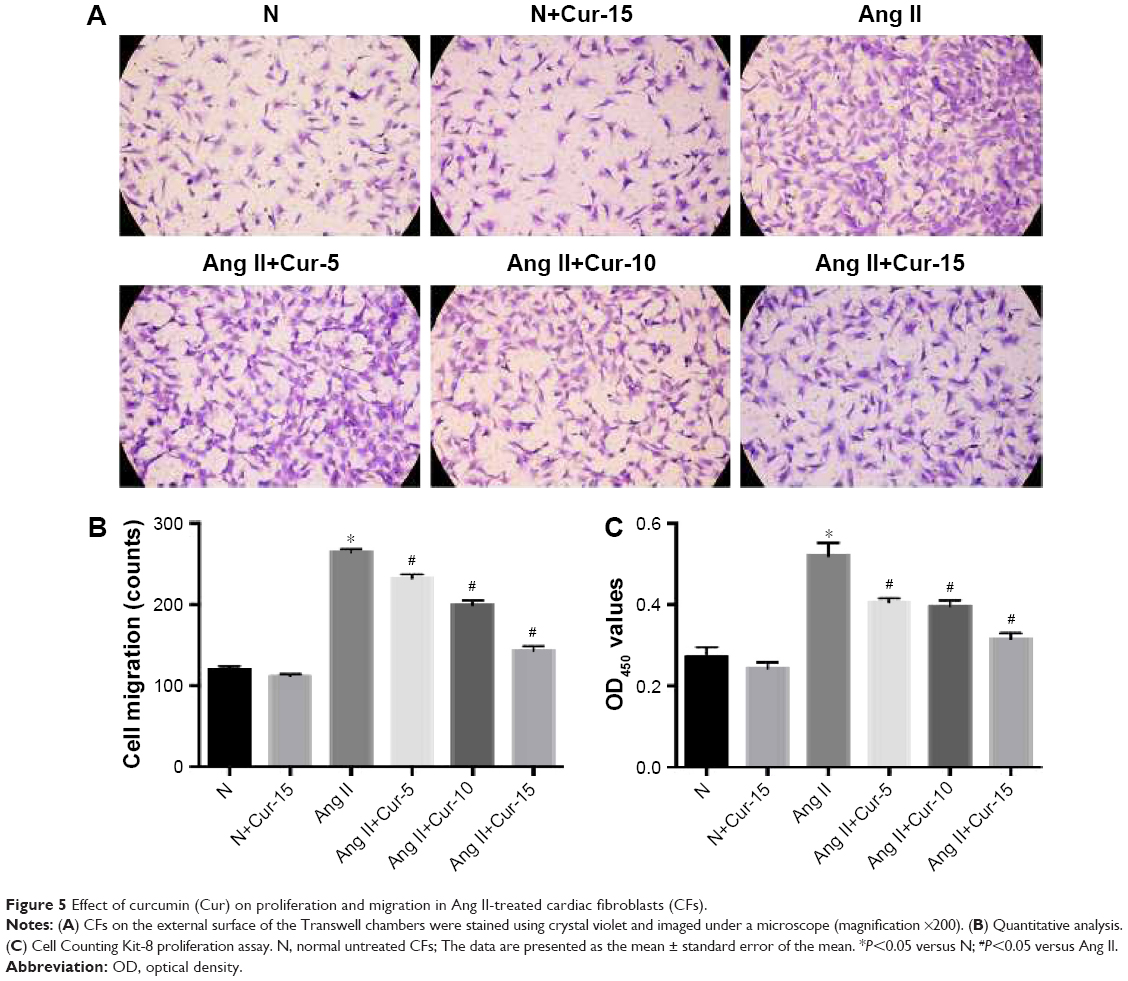 | Figure 5 Effect of curcumin (Cur) on proliferation and migration in Ang II-treated cardiac fibroblasts (CFs). |
SIRT1 siRNA blocks the protective effects of Cur in Ang II-induced myocardial fibrosis
Based on the effects of Cur on SIRT1 expression after Ang II stimulation, we investigated the role of SIRT1 in the protective action of Cur against myocardial fibrosis using SIRT1 siRNA-mediated knockdown. These results showed that SIRT1 knockdown blocked the Cur enhanced SIRT1 expression and increased the expression of collagen I, collagen III, TGF-β1, MMP-2, and MMP-9 (Figure 6A and B, P<0.05). MMP-2 activity was confirmed by zymography (Figure 6C and D, P<0.05). However, MMP-9 activity was not detected in the medium.
 | Figure 6 SIRT1 siRNA attenuated the protective effect of curcumin against collagen deposition and ECM degradation in Ang II-treated cardiac fibroblasts (CFs). |
SIRT1 knockdown alleviated the inhibitory effect of Cur on CFs’ proliferation and migration
Next, we tested the effects of SIRT1 siRNA-mediated knockdown on CFs’ proliferation and migration. The SIRT1 siRNA increased CFs’ migration and proliferation compared to the NC group. The inhibitory effects of Cur on CFs’ migration and proliferation were largely attenuated by SIRT1 siRNA (Figure 7, P<0.05), which confirmed the pivotal role of SIRT1 in the protective effect of Cur against Ang II-induced myocardial fibrosis.
 | Figure 7 SIRT1 siRNA blocked the protective effect of curcumin against migration and proliferation in Ang II-treated cardiac fibroblasts (CFs). |
Discussion
The present study was designed to investigate the potential molecular mechanism involved in the protective effect of Cur against MI-induced myocardial fibrosis. In vivo, Cur decreased the collagen volume fraction and down-regulated the expression of molecular markers of fibrosis, such as collagen I, collagen III, and fibrotic TGF-β1. In vitro, we showed that 100 nM of Ang II significantly promoted collagen deposition, ECM degradation, and CFs’ proliferation and migration. Cur pretreatment inhibited fibrosis in CFs by blunting the aforementioned process. Furthermore, SIRT1 was found to be involved in the protective effect of Cur against MI, and its role was confirmed by SIRT1 siRNA-mediated knockdown.
Myocardial fibrosis is defined as a progressive ECM accumulation phenomenon. Fibrosis following MI is defined as bilateral. Indeed, while appropriate collagen synthesis is essential for injury healing, excessive collagen deposition increases the diastolic stiffness and impairs relaxation, ultimately promoting left ventricular dysfunction and heart failure.20 Collagen deposition and ECM degradation are the main processes involved in the development of fibrosis. Collagen I and III are the predominant fibrillar components of the ECM and are normally designated as fibrosis markers. ECM regulatory proteins, such as MMPs, and cytokines, such as TGF-β1, directly affect ECM turnover. TGF-β1 was reported to influence cell proliferation, migration, and ECM homoeostasis.21,22 Abnormal MMPs’ activity results in ECM disruption, newly formed ECM deposition, and CFs’ proliferation and migration, ultimately leading to fibrosis. MMP-2 was reported to stimulate collagen I expression in a time-dependent manner, while MMP-9 deletion was shown to attenuate collagen accumulation after experimental MI.23 In our study, increased deposition of collagen I, collagen III, and TGF-β1 was observed in the border zone of heart tissue sections of mice after MI compared to sham-operated mice, which was attenuated by Cur pretreatment. For in vitro studies, we used Ang II-stimulated CFs to mimic the fibrotic process after MI, according to a previous study.24 Our results showed that Ang II increased collagen I and collagen III deposition, and stimulated MMP-2 and MMP-9 expression. These effects were inhibited by Cur pretreatment. However, compared to a previous study, MMP-9 activity was not detected by gelatin zymography, which suggested that MMP-9 might have a less important role in Ang II-treated CFs than MMP-2.5 Migration and proliferation were also reduced in Ang II-treated CFs after Cur pretreatment.
SIRT1, a member of the class III group of histone deacetylases, regulates numerous cellular processes such as stress responses, cell survival, development, and aging, by deacetylating target proteins.25 The potential role of SIRT1 in organ fibrosis has been described in several organs, including the liver, heart, and kidneys. Several groups have reported the important roles of SIRT1 in cardiovascular diseases, especially its anti-oxidative stress effects.26,27 However, whether SIRT1 is involved in the Cur-mediated protection against MI-induced fibrosis remains unclear. In the present study, we found that SIRT1 expression decreased after MI, both in vivo and in vitro, and that Cur pretreatment attenuated the SIRT1 down-regulation. To confirm the role of SIRT1 in the anti-fibrotic process, CFs were transfected with SIRT1 and control siRNAs, prior to Cur and Ang II treatments. Our results show that SIRT1 knockdown increased collagen deposition and inhibited the expression and activity of MMPs. Moreover, we found that SIRT1 knockdown inhibited the anti-proliferative and anti-migratory effects of Cur after Ang II stimulation, similar to a previous study in smooth muscle cells exposed to high glucose.28 These results suggest that the SIRT1 pathway plays an important role in the anti-fibrotic effects of Cur in MI.
Conclusion
In summary, Cur attenuated cardiac fibrosis following MI by regulating collagen deposition, ECM degradation, and CFs’ proliferation and migration. The protective effects of Cur were attributed to SIRT1 activation. The present study provides new insights into the mechanism of the anti-fibrotic effects of Cur in the heart. Therefore, Cur could be tested as an auxiliary therapeutic agent, along with classic treatments, to improve the prognosis in patients with myocardial fibrosis after MI.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81070087), the National 973 Basic Research Program of China (2012CB722406), and the Natural Science Foundation of Shandong Province (ZR2010HM063).
Author contributions
All authors contributed toward data analysis, drafting, and revising the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Schelbert EB, Piehler KM, Zareba KM, et al. Myocardial fibrosis quantified by extracellular volume is associated with subsequent hospitalization for heart failure, death, or both across the spectrum of ejection fraction and heart failure stage. J Am Heart Assoc. 2015;4(12). pii: e002613. | ||
Fan GP, Wang W, Zhao H, et al. Pharmacological inhibition of focal adhesion kinase attenuates cardiac fibrosis in mice cardiac fibroblast and post-myocardial-infarction models. Cell Physiol Biochem. 2015;37(2):515–526. | ||
Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292(1):C82–C97. | ||
Hashikata T, Yamaoka-Tojo M, Namba S, et al. Rivaroxaban inhibits Angiotensin II-induced activation in cultured mouse cardiac fibroblasts through the modulation of NF-kappaB pathway. Int Heart J. 2015;56(5):544–550. | ||
Li R, Xiao J, Qing X, et al. Sp1 mediates a therapeutic role of MiR-7a/b in Angiotensin II-induced cardiac fibrosis via mechanism involving the TGF-beta and MAPKs pathways in cardiac fibroblasts. PloS One. 2015;10(4):e0125513. | ||
Duan W, Yang Y, Yan J, et al. The effects of curcumin post-treatment against myocardial ischemia and reperfusion by activation of the JAK2/STAT3 signaling pathway. Basic Res Cardiol. 2012;107(3):263. | ||
Yu W, Wu J, Cai F, et al. Curcumin alleviates diabetic cardiomyopathy in experimental diabetic rats. PloS One. 2012;7(12):e52013. | ||
Meng Z, Yu XH, Chen J, Li L, Li S. Curcumin attenuates cardiac fibrosis in spontaneously hypertensive rats through PPAR-gamma activation. Acta Pharmacol Sin. 2014;35(10):1247–1256. | ||
Wang NP, Wang ZF, Tootle S, Philip T, Zhao ZQ. Curcumin promotes cardiac repair and ameliorates cardiac dysfunction following myocardial infarction. Br J Pharmacol. 2012;167(7):1550–1562. | ||
Pang XF, Zhang LH, Bai F, et al. Attenuation of myocardial fibrosis with curcumin is mediated by modulating expression of angiotensin II AT1/AT2 receptors and ACE2 in rats. Drug Des Devel Ther. 2015;9:6043–6054. | ||
Hsu CP, Zhai P, Yamamoto T, et al. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 2010;122(21):2170–2182. | ||
Wu Y, Liu X, Zhou Q, et al. Silent information regulator 1 (SIRT1) ameliorates liver fibrosis via promoting activated stellate cell apoptosis and reversion. Toxicol Appl Pharmacol. 2015;289(2):163–176. | ||
Rizk SM, El-Maraghy SA, Nassar NN. A novel role for SIRT-1 in L-arginine protection against STZ induced myocardial fibrosis in rats. PloS One. 2014;9(12):e114560. | ||
Yang Y, Duan W, Lin Y, et al. SIRT1 activation by curcumin pretreatment attenuates mitochondrial oxidative damage induced by myocardial ischemia reperfusion injury. Free Radic Biol Med. 2013;65:667–679. | ||
Sun Q, Jia N, Wang W, Jin H, Xu J, Hu H. Activation of SIRT1 by curcumin blocks the neurotoxicity of amyloid-beta25-35 in rat cortical neurons. Biochem Biophys Res Commun. 2014;448(1):89–94. | ||
Zeng C, Zhong P, Zhao Y, et al. Curcumin protects hearts from FFA-induced injury by activating Nrf2 and inactivating NF-kappaB both in vitro and in vivo. J Mol Cell Cardiol. 2015;79:1–12. | ||
Sunagawa Y, Sono S, Katanasaka Y, et al. Optimal dose-setting study of curcumin for improvement of left ventricular systolic function after myocardial infarction in rats. J Pharmacol Sci. 2014;126(4):329–336. | ||
Gao E, Lei YH, Shang X, et al. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010;107(12):1445–1453. | ||
Wang WK, Wang B, Lu QH, et al. Inhibition of high-mobility group box 1 improves myocardial fibrosis and dysfunction in diabetic cardiomyopathy. Int J Cardiol. 2014;172(1):202–212. | ||
Bao MW, Cai Z, Zhang XJ, et al. Dickkopf-3 protects against cardiac dysfunction and ventricular remodelling following myocardial infarction. Basic Res Cardiol. 2015;110(3):25. | ||
Ahamed J, Terry H, Choi ME, Laurence J. Transforming growth factor-beta1-mediated cardiac fibrosis: potential role in HIV and HIV/ART-linked cardiovascular disease. Aids. 2016;30(4):535–542. | ||
Zhang M, Pan X, Zou Q, et al. Notch3 Ameliorates cardiac fibrosis after myocardial infarction by inhibiting the TGF-beta1/smad3 pathway. Cardiovasc Toxicol. Epub 2015 Oct 20. | ||
Ducharme A, Frantz S, Aikawa M, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106(1):55–62. | ||
Sui X, Wei H, Wang D. Novel mechanism of cardiac protection by valsartan: synergetic roles of TGF-beta1 and HIF-1alpha in Ang II-mediated fibrosis after myocardial infarction. J Cell Mol Med. 2015;19(8):1773–1782. | ||
Horio Y, Hayashi T, Kuno A, Kunimoto R. Cellular and molecular effects of sirtuins in health and disease. Clin Sci (Lond). 2011;121(5):191–203. | ||
Samuel SM, Thirunavukkarasu M, Penumathsa SV, Paul D, Maulik N. Akt/FOXO3a/SIRT1-mediated cardioprotection by n-tyrosol against ischemic stress in rat in vivo model of myocardial infarction: switching gears toward survival and longevity. J Agric Food Chem. 2008;56(20):9692–9698. | ||
Alcendor RR, Gao S, Zhai P, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100(10):1512–1521. | ||
Xu J, Li L, Yun HF, Han YS. MiR-138 promotes smooth muscle cells proliferation and migration in db/db mice through down-regulation of SIRT1. Biochem Biophys Res Commun. 2015;463(4):1159–1164. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.