")
Back to Journals » International Journal of Nanomedicine » Volume 16
Crucial Role of PLGA Nanoparticles in Mitigating the Amiodarone-Induced Pulmonary Toxicity
Authors Motawea A , Ahmed DAM , El-Mansy AA, Saleh NM
Received 2 April 2021
Accepted for publication 17 June 2021
Published 8 July 2021 Volume 2021:16 Pages 4713—4737
DOI https://doi.org/10.2147/IJN.S314074
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Amira Motawea,1 Dalia Alsaied Moustafa Ahmed,2 Ahmed A El-Mansy,3,4 Noha Mohamed Saleh1
1Pharmaceutics Department, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt; 2Forensic Medicine and Clinical Toxicology Department, Faculty of Medicine, Mansoura University, Mansoura, Egypt; 3Department of Medical Histology and Cell Biology, Faculty of Medicine, Mansoura University, Mansoura, Egypt; 4Department of Histology, Horus University, Dumyat al Jadidah, Egypt
Correspondence: Noha Mohamed Saleh Email [email protected]
Background: Amiodarone (AMD) is a widely used anti-arrhythmic drug, but its administration could be associated with varying degrees of pulmonary toxicity. In attempting to circumvent this issue, AMD-loaded polymeric nanoparticles (AMD-loaded NPs) had been designed.
Materials and Methods: AMD was loaded in NPs by the nanoprecipitation method using two stabilizers: bovine serum albumin and Kolliphor® P 188. The physicochemical properties of the AMD-loaded NPs were determined. Among the prepared NPs, two ones were selected for further investigation of spectral and thermal analysis as well as morphological properties. Additionally, in vitro release patterns were studied and kinetically analyzed at different pH values. In vitro cytotoxicity of an optimized formula (NP4) was quantified using A549 and Hep-2 cell lines. In vivo assessment of the pulmonary toxicity on Sprague Dawley rats via histopathological and immunohistochemical evaluations was applied.
Results: The developed NPs achieved a size not more than 190 nm with an encapsulation efficiency of more than 88%. Satisfactory values of loading capacity and yield were also attained. The spectral and thermal analysis demonstrated homogeneous entrapment of AMD inside the polymeric matrix of NPs. Morphology revealed uniform, core-shell structured, and sphere-shaped particles with a smooth surface. Furthermore, the AMD-loaded NPs exhibited a pH-dependent and diffusion-controlled release over a significant period without an initial burst effect. NP4 demonstrated a superior cytoprotective efficiency by diminishing cell death and significantly increasing the IC50 by more than threefold above the pure AMD. Also, NP4 ameliorated AMD-induced pulmonary damage in rats. Significant downregulation of inflammatory mediators and free radicle production were noticed in the NP4-treated rats.
Conclusion: The AMD-loaded NPs could ameliorate the pulmonary injury induced by the pure drug moieties. Cytoprotective, anti‐fibrotic, anti-inflammatory, and antioxidant properties were presented by the optimized NPs (NP4). Future studies may be built on these findings for diminishing AMD-induced off-target toxicities.
Keywords: amiodarone, pulmonary toxicity, PLGA, nanoparticles, BSA, cytotoxicity
Graphical Abstract:
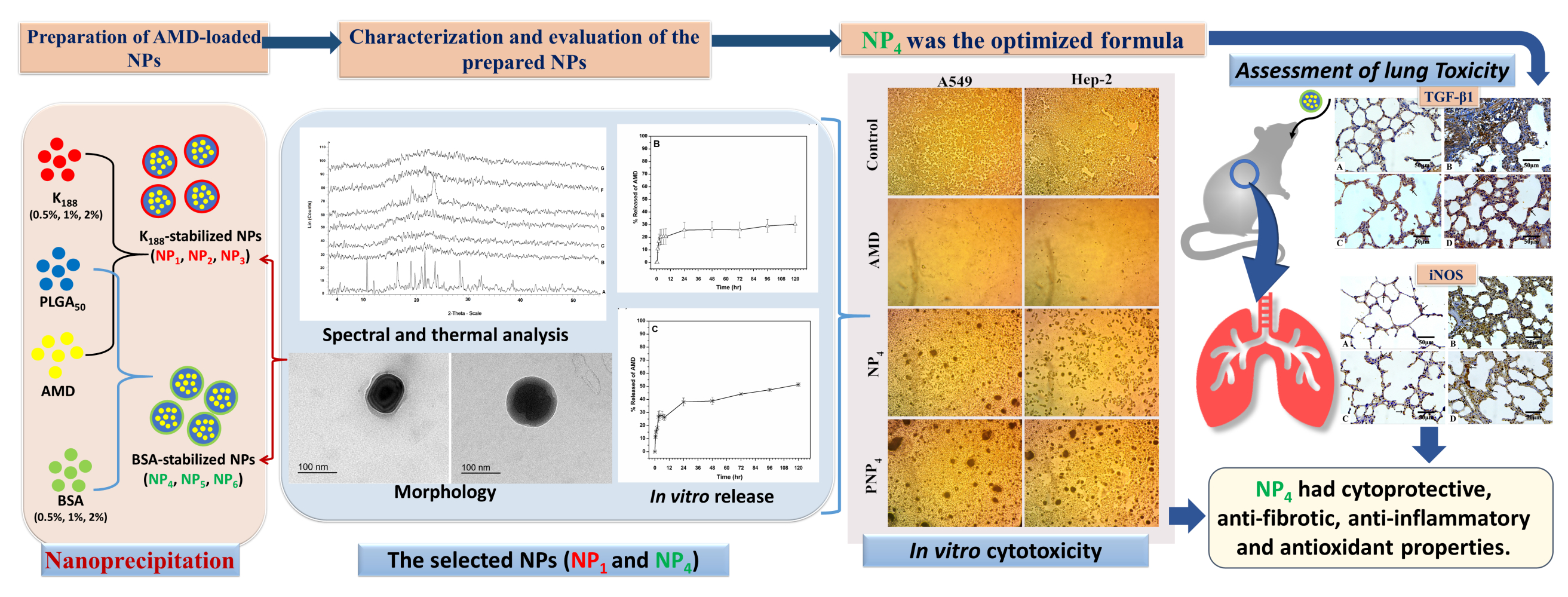
Introduction
Amiodarone (AMD) is an iodinated benzofuran derivative that is used generally in preserving sinus rhythm and controlling ventricular arrhythmias in patients with atrial fibrillation. AMD has a non-competitive adrenergic blocking effect of the inactivated Na+ channels and it reduces the Ca2+ current, outward and inward rectifier K+ current.1 However, low water solubility, high lipophilicity, slow dissolution rate, and poor oral bioavailability of AMD have negatively affected its pharmacokinetic parameters.2,3 AMD is subjected to enterohepatic cycling after its absorption to form an active metabolite (desethyl amiodarone). The implication of this pathway is the prolongation of AMD elimination half-life to about 60 days. Hence, accumulation of AMD takes place with associated severe side effects in off-target organs such as lung, liver, and thyroid gland.4
Pulmonary toxicity is the most dangerous event that leads to respiration malfunction or even death.5,6 Pulmonary toxicities may happen in acute and subacute/chronic patterns. AMD-induced pulmonary toxicity may evolve as chronic interstitial pneumonitis or mimic pulmonary fibrosis if not early diagnosed and treated. Also, AMD toxicity may induce pleural effusions or thickening.5,7,8 Routine pulmonary function testing is not accurate enough to predict or prevent AMD-induced lung toxicity. Hence, the optimal and safe use of AMD in the clinical field requires thorough patient monitoring of lung functions that could endow early pulmonary toxicity.5
To allocate appropriate solutions to these drawbacks, tailored delivery of a drug via nanotechnology-based techniques can be applied. Different systems have been exploited to improve the biodistribution of drugs, enhance the pharmacological effects, control the release of the drug, and limit the expected adverse drug reactions.9–13 Among the different systems, polymeric nanoparticles (NPs) have attracted significant attention over the last few decades as multipurpose means for drug delivery due to their safety and biodegradability.14 The production of NPs is particularly important in combating diseases that require long-term therapy such as heart diseases. Poly(lactic-co-glycolic acid) (PLGA) is the most interesting biodegradable and biocompatible copolymer. It has been used in the preparation of NPs to allow a controlled and targeted delivery to specific areas.15,16 PLGA is an FDA-approved polymer for human use, and hence, several PLGA-based formulations are present in the market. The percentages of 50% lactic acid and 50% glycolic acid is the commonly applied ratio of PLGA in drug delivery. It is worth noting that the biodegradation of PLGA occurs in an aqueous environment by cleavage of the ester bonds in the polymer backbone via bulk erosion. The biodegradation results in the production of monomeric acids (lactic and glycolic acids).17,18 The final products are safe because lactic acid is eliminated from the body via the tricarboxylic acid cycle as carbon dioxide and water. Glycolic acid is eliminated by either direct renal excretion or the tricarboxylic acid cycle.19
For the preparation of NPs, the nanoprecipitation method offers diverse advantages such as simplicity, high drug entrapment, and the reproducibility of the results.20–22 A tensioactive agent (stabilizer) is normally added in the aqueous phase to stabilize the formed NPs by exposing its hydrophobic and hydrophilic regions toward the organic and aqueous phases, respectively. The type and concentration of the stabilizer can significantly influence the properties of NPs.23,24 In our study, AMD-loaded NPs were formulated at varying levels of Kolliphor® P 188 (K188) and bovine serum albumin (BSA) concentrations to illuminate the most efficient characteristics for optimized size, AMD encapsulation, and safety. BSA, one of the most abundant proteins, is safe, non-immunogenic, soluble in water, biodegradable to produce harmless degradation products, and thus an ideal candidate for NPs preparation.25,26 K188 is a nonionic triblock copolymer comprised hydrophilic blocks (polyethylene oxide, PEO) and hydrophobic ones (polypropylene oxide, PPO) in the form of PEO/PPO/PEO. Based on their physiochemical properties and biocompatibility, poloxamer-based systems are promising candidates in drug delivery.27,28 It is crucial to assess the biological and toxicological effects of NPs via in vitro and in vivo testing for a better understanding of NPs-induced responses.29
The present study aimed to develop a nano-carrier of AMD with efficient encapsulation, controllable release, and potential reduction of pulmonary toxic effects. To fulfill this target, AMD-loaded NPs were designed and characterized. Physicochemical characteristics of NPs could be adjusted by varying the formulation parameters. The promising formulations of NPs were subjected for additional evaluation regarding spectral and thermal analysis, morphology, and in vitro release patterns. Further, in vitro and in vivo studies were performed on cell lines and a rat model, respectively, to compare the toxicological profile of encapsulated AMD versus free one.
Materials and Methods
Materials
Amiodarone Hydrochloride (AMD) was obtained kindly from Global Napi Pharmaceuticals, Egypt. Purasorb PDLGA®5010, Mwt 153 kDa (PLGA50) was kindly given by Corbion Biomaterials, Holland. Kolliphor® P 188, Mwt 7680–9510 Da (K188) was obtained from BASF Co., Germany. Bovine serum albumin (BSA) was purchased from MP Biomedical LLC, France. Dimethyl sulfoxide (DMSO) was procured from Sigma-Aldrich Co., Germany. A549 (Human epithelial lung carcinoma) and Hep-2 (Human laryngeal epidermoid carcinoma) were obtained from Nawah Scientific Inc., Cairo, Egypt. Anti-transforming growth factor β1 (TGF-β1) antibody 500-M66 was obtained from Santa Cruz Biotechnology Inc, USA. Anti-inducible nitric oxide synthase (iNOS) Rabbit polyclonal antibody was gained from ABclonal Technology Inc., 500W Cummings Park, Ste. 6500, Woburn, MA 01801, USA. All supplementary chemicals and solvents were of fine analytical grade.
Preparation of AMD-Loaded NPs
AMD-loaded NPs were prepared by the nanoprecipitation method after some modifications.30 At a ratio of 10:1, PLGA50 (2% w/v) and AMD (0.2% w/v) were dissolved in 2 mL of DMSO to obtain a clear solution (organic phase). Then, the organic phase was added dropwise into an aqueous solution of a stabilizer (40 mL) under magnetic stirring (Hot Plate and Magnetic stirrer, Misung Scientific Co., LTD, Korea). Two types of stabilizers; K188 and BSA were used at three concentrations (0.5%, 1%, and 2% w/w). The compositions of the prepared NPs are shown in Table 1. After the complete addition of the organic phase, the obtained dispersion was probe-sonicated (Sonics Vibra cell, Sonic & Materials, USA) using pulsed mode (1 sec On, 2 sec Off) with 750 W Power and 20 kHz Frequency at 100% amplitude for 5 min. To allow complete diffusion of DMSO, the dispersion was magnetically stirred overnight at room temperature. The dispersions were filtered through a filter paper to eliminate macroparticles if they were present. AMD-loaded NPs were separated by centrifugation at 10,000 rpm/25 °C for 1 hour (Benchtop Centrifuge, Sigma Laborzentrifugen, Germany) and pellets were washed once by deionized water (DIW), gathered, and kept at −20°C. AMD-loaded NPs were lyophilized under reduced pressure and at −80 °C (SIM FD8-8T, SIM International, USA). Then, the lyophilized NPs were collected and stored at 4 °C for further characterization. Corresponding AMD-free NPs (PNPs) were fabricated using the above-mentioned procedure without the incorporation of AMD.
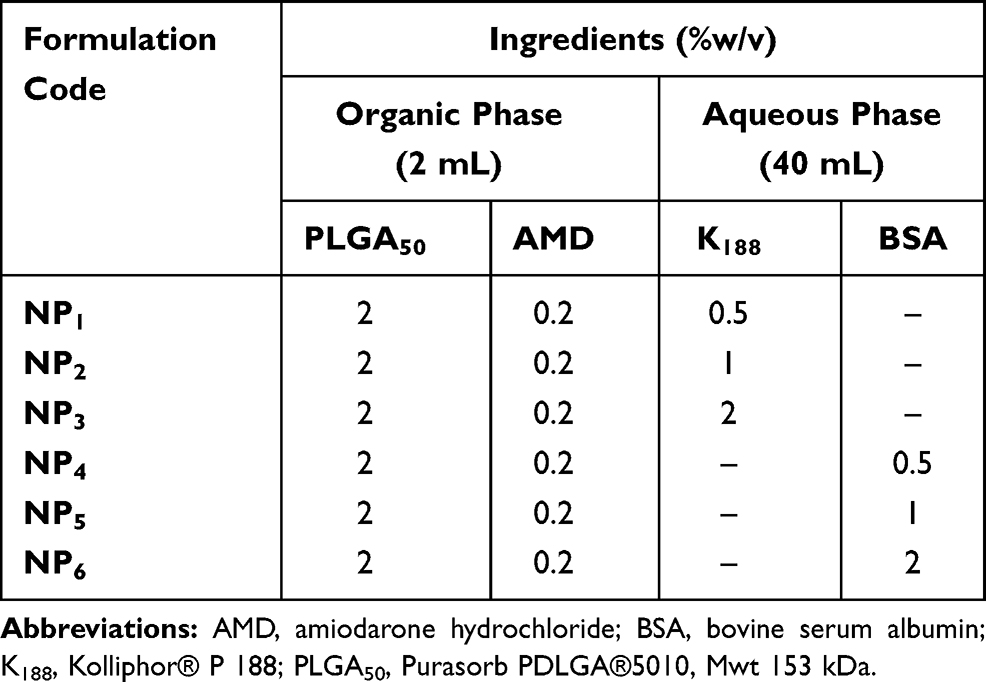 |
Table 1 Compositions of the Prepared NPs |
Characterization of AMD-Loaded NPs
Hydrodynamic Diameter and Surface Charge
Hydrodynamic diameters (PS) and size distribution (PDI) of the prepared NPs after appropriate dilution were analyzed by dynamic light scattering technique (DLS) using Zetasizer (Malvern Instruments Ltd, England). The zeta-potential (ZP) of NPs was also determined using the same apparatus. All measurements were conducted in triplicates and were represented as mean ± SD (n=3).
Encapsulation Efficiency, Loading Capacity, and Yield
Clear supernatants that recovered after the centrifugation, were used for determining the encapsulation efficiency of AMD-loaded NPs (EE%) indirectly. Non-encapsulated (free) AMD was measured in the supernatants spectrophotometrically at 242 nm31 versus supernatants of the corresponding AMD-free NPs as blank (UV-VIS Spectro double beam, Labomed Inc., USA). After lyophilization, the weight of the NPs was determined for the calculation of the loading capacity (LC%) and the yield (Y%). The outcomes of NP1 and NP4 were promising and therefore, they were selected to undergo further investigations (refer to Table 1 for compositions of NP1 and NP4). The following equations were used for the determination of EE%, LC%, and Y%.
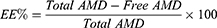
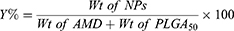
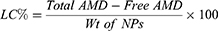
Spectral and Thermal Analysis
Pure AMD, PLGA50, binary physical mixture of AMD and PLGA50 (AMD+PLGA50), AMD-loaded NPs (NP1 and NP4), and corresponding plain NPs (PNP1 and PNP4, respectively) were analyzed by FTIR spectrophotometer equipped with an attenuated total reflectance (ATR) device (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The sample was positioned on the diamond ATR crystal, covered with a glass coverslip, and held with a specialized clamp. The spectra of the tested samples were acquired with a resolution of 4 cm−1 in between 4000 cm−1 and 500 cm−1.
The thermal properties of pure AMD, PLGA50, AMD+PLGA50, AMD-loaded NPs (NP1 and NP4), and corresponding plain NPs (PNP1 and PNP4, respectively) were studied by differential scanning calorimetry (DSC) (Differential scanning calorimeter, Pyris 6 DSC, Perkin Elmer, USA). Approximately 2 mg of each sample was heated in a sealed aluminum pan and an empty pan was used as a reference. In a nitrogen atmosphere of 20 mL/min flow rate, thermograms were recorded in the temperature range of 50–400 °C with a heating rate of 10 °C/min.
XRD was applied to estimate the crystallinity of pure AMD, PLGA50, AMD+PLGA50, AMD-loaded NPs (NP1 and NP4), and corresponding plain NPs (PNP1 and PNP4, respectively) by X-ray diffractometer (Rigaku Denki, Rint-2500VL. Tokyo, Japan). X-ray diffractogram of each sample was recorded using Cu-Kα radiation (λ = 1.542Å) at room temperature in the 2θ ranges from 4° to 55°. The voltage and current of X-ray tubes were 45 kV and 40 mA, respectively, and the chart speed was 10 mm/s.
Morphology
The morphology of the prepared NPs was investigated by using transmission electron microscopy (TEM, JOEL 1010; JEOL Ltd, Tokyo, Japan). Briefly, an aqueous drop of the NPs dispersion was placed on a 200-mesh copper grid coated with carbon and the extra liquid was drawn off by a filter paper. Then, samples were dried at room temperature to be visualized at a voltage of 200 kV.
Determination of AMD Solubility
The solubility of AMD was determined in distilled water and different pH; 1.2 (0.1 N HCl), phosphate buffer at 6.8 (PB6.8), and phosphate buffer at 7.4 (PB7.4). AMD (20 mg) was weighed and placed in a screw-capped tube to which 5 mL of a solvent were added. Each solvent was repeated in triplicates. The tubes were shaken in a thermostated water bath (Grant instrument Cambridge Ltd., England) at 37 °C ± 0.5 °C for 72 h to achieve equilibrium. After that, the tubes were centrifuged at 5000 rpm for 30 min to remove excess undissolved AMD. Supernatants were separated, filtered (OlimPeak, Nylon membrane filters, 0.45 µm, Teknokroma, Spain), diluted, and analyzed spectrophotometrically at 242 nm.
In vitro Release Study and Kinetic Modeling
In vitro release of AMD from the selected NPs was studied using modified Franz cells. The release study was conducted in three media with pH values simulating gastric pH (HCl, pH1.2) for 2 h, intestinal pH (PB6.8) for 6 h, and physiological pH (PB7.4) for up to 5 days. According to the currently published studies on AMD in the literature, the release media with different pH values were used.3,9,32–35 Based on the results of solubility experiments, the volume of the release medium that would be able to achieve the sink condition was placed in the receptor chamber. A pre-equilibrated dialysis cellulose membrane (Mw Cut off 12–14 kDa, Sigma-Aldrich) was mounted between the donor and receptor chambers. AMD (0.3 mg) or equivalent amounts of AMD-loaded NPs were dispersed in 3 mL of distilled water and transferred to each donor chamber. The whole system was maintained at 37 °C ± 0.5 °C and stirred at 100 rpm in a shaking incubator (GFL Gesellschaft für Labortechnik, Burgwedel, Germany). At prearranged intervals, a sample of 2 mL was withdrawn and replenished with a fresh aliquot. The gathered samples were filtered (0.45 μm), appropriately diluted, and spectrophotometrically analyzed at 242 nm. Each experiment was completed in triplicate and the cumulative released concentration of AMD was calculated at each time interval according to the following equation.36
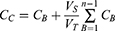
where CC is the calculated concentration, CB is the measured concentration, VS is the volume of the withdrawn sample and VT is the total volume of the release medium. To explain the mechanism of AMD release, release data were kinetically analyzed according to zero-order, first-order,37 and Higuchi diffusion mechanisms.38 For further investigation of release mechanisms, the Korsmeyer–Peppas model was applied to determine the diffusion exponent (n).39 A model to which the release data were best fitted with the maximum correlation coefficient (r2) was the one that described the release of AMD. Based on the outcomes of the above-mentioned experiments, NP4 was selected for further evaluation.
In vitro Study of Cell Toxicity
The cytotoxic effects of pure AMD, NP4, and PNP4 were studied in vitro using A549 (Human epithelial lung carcinoma), and Hep-2 (Human laryngeal epidermoid carcinoma) cell lines. Briefly, the cells were cultured in DMEM media complemented with streptomycin (100 mg/mL), penicillin (100 units/mL), and 10% of heat-inactivated fetal bovine serum in a humidified atmosphere of 5% (v/v) CO2 at 37 °C. Cell viability was assessed by SRB (sulforhodamine B) assay.40,41 Aliquots of 100 µL cell suspension (at a density of 5×103 cells/well) were seeded into 96-well plates and incubated overnight for 24 h. Samples of pure AMD, NP4, and PNP4 were sterilized by UV irradiation before use. Afterward, the cells were treated with additional aliquots of 100 µL media containing pure AMD at concentrations of 0.01, 0.1, 1, 10, and 100 µg/mL, and the cells were incubated for 72 h at 37 °C. AMD-equivalent amounts of NP4 and NP4-equivalent amounts of PNP4 were used to prepare the above concentration series. After that, the media were exchanged with 150 µL of 10% trichloroacetic acid (TCA) for fixation, and the cells were re-incubated for 1 hour at 4°C. The TCA solution was purged, and the cells were rinsed carefully five times with distilled water. SRB solution (0.4% w/v) was added in aliquots of 70 µL to the cells and they were incubated at 25 °C in a dark place for 10 min. After that, the plates were washed with 1% acetic acid thrice and allowed to dry overnight. To dissolve the SRB stain bound to proteins, 150 µL of TRIS (10 mM) was added. Finally, the absorbance intensity was measured spectrophotometrically at 540 nm (A540) using a microplate reader (BMG LABTECH®-FLUO star Omega, Ortenberg, Germany). All the experiments were performed in triplicate. Representative phase-contrast images of cells were captured just before the addition of the SPB reagent (Olympus BX 41 microscope, Olympus, USA). The cytotoxic effect was expressed as the percent cell viability and was used to construct a dose–response curve. Then, the half-maximal inhibitory concentration (IC50, the concentration that killed 50% of cells) in comparison with the untreated (control) cells was calculated from the dose–response curve. The percent cell viability was calculated using the following equation.
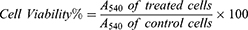
In vivo Evaluation of the Pulmonary Toxicity
Animals
Healthy thirty-two Sprague Dawley male rats (180 and 220 g) aged 6–8 weeks were randomized into four equal groups (n = 8). The procedures were approved by the Institutional Research Board IRB, Faculty of Medicine, Mansoura University (R.20.04.795.R1) in accordance with the “Principles of Laboratory Animal Care” (NIH publication #85-23, revised in 1985).
Formulations and Treatment
The subsequent formulations were tested: AMD suspension (Group I), placebo PNP4 (Group II), and NP4 dispersion (Group III). A control group was involved where animals were received distilled water. The treatment regimen consisted of once-daily oral administration for 6 weeks using a dosage level of 3 mg AMD/kg body weight.13
Specimen Preparation
By the end of the sixth week, the animals were sedated with xylazine (10 mg/kg) and ketamine (50 mg/kg). Then, the lung specimens were gently separated and then fixed in 10% neutral buffered formalin. Then, the specimens were embedded in paraffin blocks and cut into 5 µm sections for the consequent histopathological and immunohistochemical evaluations (IHC).
Histopathological Evaluation
One group of lung sections was deparaffinized and stained routinely with hematoxylin and eosin (H&E). To recognize the extent of fibrosis, the sections were stained with Masson’s trichrome. Five fields from each section were assessed and three sections from each animal were examined. The slides were photographed (at 400× magnification) and scored. The scored elements included alveolar capillary congestion, haemorrhage, infiltration of neutrophils in the vessel wall, and hyaline membrane formation/thickness of the alveolar wall. Each parameter was graded according to a five-point scale as following; 0=minimal (little), 1=mild, 2=moderate, 3=severe, and 4=maximal.42
IHC Evaluation
The second group of sections was subjected to IHC estimation of expression levels of TGF-β1 and iNOS. IHC staining of anti-TGF-β1 antibody was conducted by the streptavidin-biotin technique. Lung sections with 4 μm thickness were deparaffinized and incubated with fresh hydrogen peroxide in methanol (0.3% v/v) for a half-hour at room temperature. Then, the specimens were incubated with an anti-TGF-β1 antibody as the primary antibody. Hematoxylin was applied as a counterstain.43 For anti-iNOS antibody IHC staining, sections were incubated with 10% serum of the animal in which the secondary antibodies were formed for 30 min. Then, they were first incubated with diluted monoclonal anti‐iNOS (1:200) antibodies, in phosphate buffer saline overnight at 4° C, and later with biotinylated secondary antibodies followed by peroxidase‐linked ABC. Controls were performed for background staining by substituting the primary antibody with phosphate buffer saline.44 The sections were examined and photographed. Then, the intensity of the IHC staining was analyzed by ImageJ analysis (NIH, Bethesda, MD, USA) to quantitatively determine levels of TGF-β1 and iNOS expression in different groups.
Statistical Analysis
The results were statistically analyzed using GraphPad Prism version 8 software (San Diego, CA, USA). Data were presented as mean ± SD (standard deviation). Shapiro test was used to test the normality of data. Then, a one-way ANOVA (followed by multiple comparisons of the Tukey’s test) for data expressed in mean ± SD or Kruskal Wallis test (followed by post-hoc Dunn’s) for data expressed as median (range) was applied. The results were considered non-significant when the probability was more than 5% (P > 0.05).
Results and Discussion
Preparation and Characterization of AMD-Loaded NPs
Hydrodynamic Diameter and Surface Charge
In nanotoxicological researches, evaluating the condition of the dispersed NPs has a major consequence for assessing their toxicity.45 Table 2 illustrates the PS, PDI, and ZP of the prepared AMD-loaded NPs. BSA or K188 at different concentrations were added to the dispersion media to consolidate stabilization by prohibiting subsequent agglomeration of the NPs. The drug loading and the size of NPs as functions of the used concentrations of each stabilizer should be considered.46 Our results show that the mean PS of NPs was within the nano-range and ranged from 175.93±1.75 nm to 217.37±3.21 nm.47–50 Noyes and Whitney’s equation indicates that the reduction of PS is considered an efficient method to improve dissolution characteristics of hydrophobic drugs. Furthermore, the great merits of PS of NPs involve decreased uptake by the liver, prolongation of blood circulation time, and enhancement of drug bioavailability.51 PDI is a sign of uniformity and homogeneity of PS where a monodisperse distribution has a PDI of zero. However, a dispersibility with a PDI value in the range of 0.1 to 0.4 was extensively accepted. Our developed NPs exhibited narrow particle size distribution; PDI values were not more than 0.2 (Table 2).
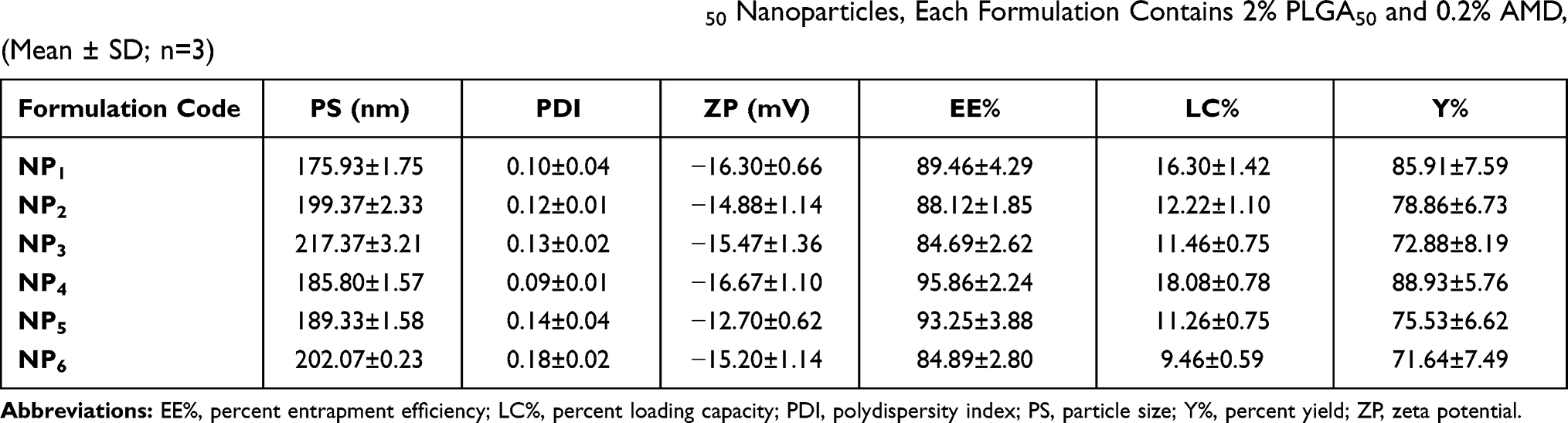 |
Table 2 Physicochemical Properties of AMD-Loaded PLGA50 Nanoparticles, Each Formulation Contains 2% PLGA50 and 0.2% AMD, (Mean ± SD; n=3) |
It could be observed that the increase of K188 or BSA concentration from 0.5% to 2% was accompanied by a growing the PS of NPs (Table 2). Larger PS of NPs that prepared with higher stabilizer concentration could be attributed to the higher viscosity of their aqueous phases. The high viscosity might reduce the net shear stress during emulsification followed by formation of larger nanodroplets and larger NPs after solvent evaporation.52,53 However, larger PS was observed to be accompanied by the formation of agglomerates. Interestingly, no agglomeration or aggregation was noticed during the preparation of NPs using the lowest concentrations of K188 or BSA (NP1 and NP4, respectively).
In terms of surface charge, the NP charge whether positive or negative is one of the most vital factors affecting the functions of NPs. The ZP of the prepared NPs was consistently negative and in the range of −12.7 to −16.67 mV. The negative ZP values could be attributed to the carboxyl-terminal groups of PLGA50, which were able to ionize in aqueous media.54 It was reported that NPs with negative surface charges had higher kinetic stability than the positively charged NPs. At the same time, the negatively charged NPs had reported exhibiting lower cytotoxic effects than positive ones.55 Also, it could be noticed that the higher ZP value occurred for smaller NPs owing to their higher external specific surface area.56 Such findings indicated the existence of electric repulsion, potential good stability, and accepted dispersion quality of the prepared NPs.45
Moreover, the utilization of BSA as a stabilizing agent was observed to prohibit the coalescence of small droplets completely and provide the kinetic stability of the formed NPs. In this sense, BSA acts as a tensioactive agent by orienting its hydrophobic and hydrophilic regions toward the organic and aqueous phases, respectively.57 It is believed that the stability presented by BSA is owing to a combination of electrostatic repulsion and steric hindrance effects.45 Moreover, poloxamers have nonionic amphiphilic molecules that able to create a sort of steric barrier to stabilize the formed droplets. Nevertheless, this steric repulsion is relatively weaker than the electrostatic repulsion.58–60 The steric repulsion of K188 might be not sufficient to counteract the entropy cost associated with the increase of the surface’s free energy during small droplets formation. In other words, the steric stability of the K188-stabilized droplets is not the same as the dual stability associated with the use of BSA as a stabilizer.28
Encapsulation Efficiency, Loading Capacity, and Yield
EE% is a vital factor in the assessment of drug therapeutic consequences.14 Table 2 documents that AMD was efficiently loaded in the prepared NPs as the values of EE% ranged from 84.69% to 95.86%. Such higher EE% is continuously advantageous to reduce the dose of NPs.61 Similar trends were reported in the literature.9 This efficient entrapment could be ascribed to the extremely strong lipophilicity of PLGA50 as well as AMD. Additionally, the hydrophobic fractions of K188 and BSA interpenetrated into PLGA50 chains to form interconnected networks around the AMD-loaded NPs and thus raised EE% of AMD within their core. It was found that as the percentage of K188 or BSA increased (0.5% to 2% w/w), EE% of AMD within the NPs decreased (Table 2). AMD might diffuse out from the nanodroplets to be solubilized as micelles with increasing stabilizer concentration in the aqueous phase. As more AMD molecules were solubilized in the external aqueous phase, the amount of stabilizer available at the aqueous/organic interface reduced and thus agglomeration of nanodroplets might occur.62,63 The LC% of AMD-loaded NPs as shown in Table 2 varied between 9.46% and 18.08%. The higher values of EE% of NPs might allow an overall their higher loading capacity of AMD. Moreover, the prepared AMD-loaded NPs showed satisfactory Y% values in the range of 71.64% to 88.93% indicating appropriateness of the preparation conditions. Moreover, AMD was successfully encapsulated in NPs up to 95.86 (Table 2). Hence, worthy EE% of AMD might contribute to the reasonable values of Y%.
From the above-mentioned results, it could be concluded that NP1 and NP4 achieved the smallest mean PS of 175.93 and 185.80 nm with the highest EE% of 89.46% and 95.86%, respectively. Moreover, NP1 and NP4 exhibited satisfactory values of ZP, PDI, LC%, and Y%. Hence, subsequent experiments were conducted using these two formulations.
Spectral and Thermal Analysis
ATR-FTIR spectroscopy enables the creation of chemical fingerprints of individual samples to monitor the possibility of interaction between them. The unique molecular vibrations of a system can be allocated to individual components within that system.64 Figure 1 shows the FTIR spectra of pure AMD (A), PLGA50 (B), AMD+PLGA50 (C), NP1 (D), PNP1 (E), NP4 (F), and PNP4 (G). The spectrum of AMD (Figure 1A) revealed the presence of absorption bands between 2850 and 3250 cm−1 that could be attributed to the stretching of aromatic and aliphatic C–H bonds, respectively. The characteristic peaks at 2460 and 2570 cm−1 were ascribed to the tertiary amine. The carbonyl group of di-iodophenyl ketone moiety exhibited a strong absorption band at 1630 cm−1. Peaks at 1249 and 1026 cm−1 were attributed to ether C–O–C stretching and furan ring vibration, respectively. Stretching of the C–I bond could be detected at 613 cm−1.31,35,65 Figure 1B shows the FT-IR spectrum of PLGA50 with characteristic peaks at 1748 cm−1 (C=O group). Also, bending vibration of the aliphatic –CH3 and –CH2 groups appeared at 1453, 1387, and 1271 cm−1. Bands at 1180 and 1084 cm−1 could be correlated to the stretching of the C–O–C link. Absorption peaks between 2850 and 3000 cm−1 were assigned to stretching vibrations of aliphatic C–H.36 The spectrum of AMD+PLGA50 (Figure 1C) illustrates the bands of PLGA50, while AMD’s bands appeared with reduced intensities or even disappeared as a consequence of its dilution by PLGA50. Likewise, the FTIR spectra of PNP1 and PNP4 (Figure 1E and G, respectively) coincide with that of the physical mixture. BSA-related peaks displayed in the spectrum of PNP4 (Figure 1G) at 3289 cm−1 (N–H stretching of amide I), 2945 cm−1 (N–H stretching of amide II), 1647 cm−1 (–NH2 amide I, C=O stretching), 1538 cm−1 (stretching of C–N and N–H bending in amide II), 1396 cm−1 (–CH2) and 1272 cm−1 (stretching of C–N and N–H bending in amide III).66,67 Figure 1D and F show that the characteristic peaks of AMD disappeared in medicated NPs spectra of NP1 and NP, respectively. The absence of AMD characteristic peaks could indicate entrapment of AMD within cores of the medicated NPs instead of its attachment to the NPs surface. Hence, it supposed that the washing step during the preparation of NPs was efficient enough for the removal of surface-attached AMD molecules.68
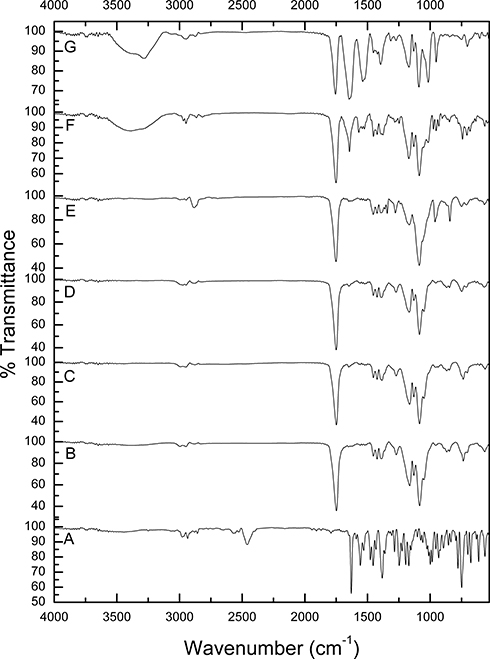 |
Figure 1 FTIR spectra of pure AMD (A), PLGA50 (B), AMD+PLGA50 (C), NP1 (D), PNP1 (E), NP4 (F), and PNP4 (G). |
DSC is an important thermal analysis tool to define any possible change in the physical states of the individual components. Also, the crystalline or amorphous behavior of the AMD-loaded NPs could be checked by DSC.68 Figure 2 displays DSC thermograms of pure AMD (A), PLGA50 (B), AMD+PLGA50 (C), NP1 (D), PNP1 (E), NP4 (F), and PNP4 (G). The crystalline state of pure AMD was demonstrated by a single endothermic peak at 161.63°C which was close to its melting point (Figure 2A).3,35,69 Other peaks at 296.6°C and 317.27°C might be ascribed to AMD decomposition.70 A broad endothermic peak at 275 °C could be observed in the thermogram of PLGA50 (Figure 2B). The absence of a melting point in the PLGA50 thermogram had indicated the amorphous nature of the polymer.71,72 Regarding AMD+PLGA50, the endothermic peak of PLGA50 was noticed at its corresponding position with the absence of those of AMD, owing to the dilution factor (Figure 2C). Remarkably, the thermograms of AMD-loaded NPs (Figure 2D and F) and plain NPs (Figure 2E and G) matched each other regarding the lack of distinctive melting peaks indicating the absence of crystallinity. As well, the vanishing of the AMD peak at 161.63 °C was evident in the thermograms of NP1 and NP4. This finding suggested the homogeneous entrapment of AMD throughout the polymeric matrix of NPs to form amorphous molecular dispersions.68,73,74
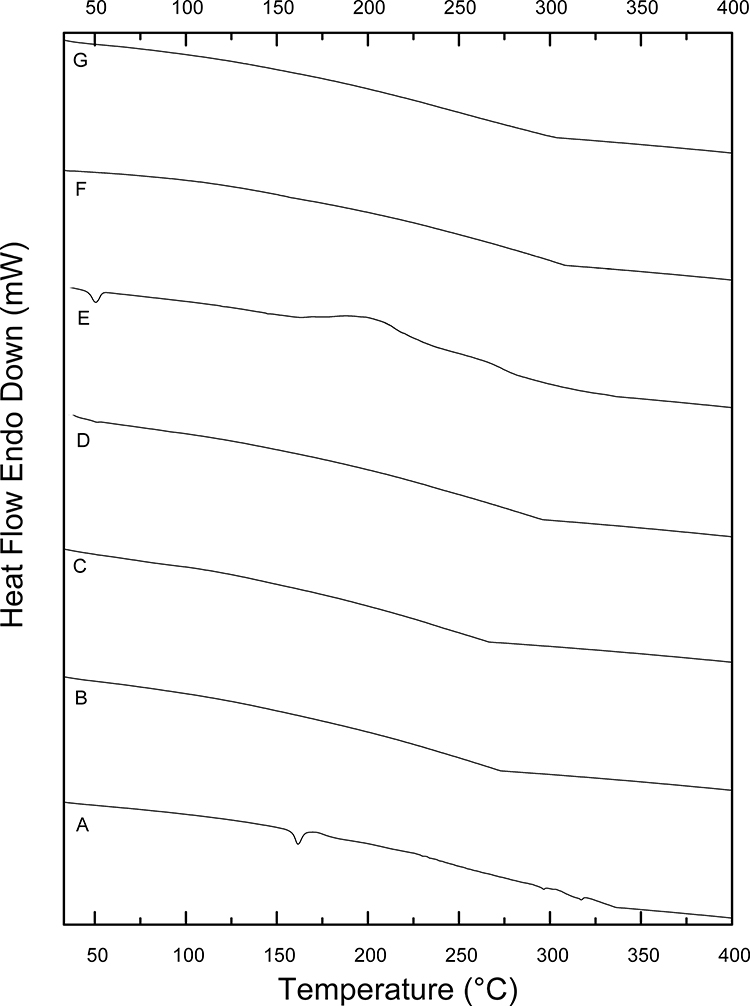 |
Figure 2 DSC thermograms of pure AMD (A), PLGA50 (B), AMD+PLGA50 (C), NP1 (D), PNP1 (E), NP4 (F), and PNP4 (G). |
XRD study is used as an analytical tool for the determination of the crystallinity degree, amorphousness, or bound form.75 Overlaid X-ray diffraction patterns of AMD (A), PLGA50 (B), AMD+PLGA50 (C), AMD-loaded NPs (D and F), and the corresponding PNPs (E and G) are presented in Figure 3. Accordingly, AMD exhibited distinctive sharp peaks at 2θ scattered angles 10.566°, 16.382°, 18.974°, 21.663°, 23.633°, 28.352°, and 38.560° suggesting a crystalline structure and small peaks at different diffraction angles (Figure 3A). Likewise, peaks were noted for PLGA50 and AMD+PLGA50 at lesser intensity (Figure 3B and C). Meanwhile, the diffraction peaks of both plain and AMD-loaded NPs were wider, less sharp, and demonstrated more diffuse X-ray scattering than PLGA50 (Figure 3D, E, and G), possibly because of reduced PS.76 Nonetheless, no characteristic crystalline peaks were noticed in the diffraction pattern for NP4 as depicted in Figure 3F. This implies that AMD was molecularly dispersed throughout the polymeric matrix or was in an amorphous state with simple diffusion.77 Besides, the lack of crystalline structure in NP1 and NP4 might reduce the tendency of the encapsulated drug to be expelled during storage, consequently keeping high drug-loading capacity. These findings confirmed that those formulations may lose their crystalline structure during preparation processes as a consequence of particle size reduction, which produces considerable effects on the solubility and bioavailability of poorly soluble drugs.78
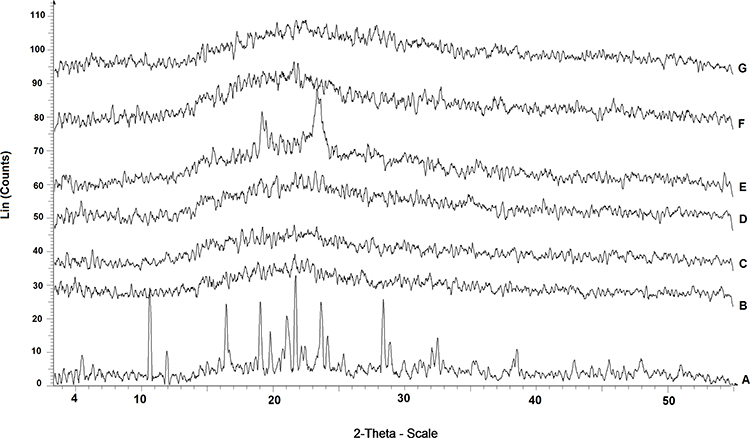 |
Figure 3 X-ray diffraction patterns of AMD (A), PLGA50 (B), AMD+PLGA50 (C), NP1 (D), PNP1 (E), NP4 (F), and PNP4 (G). |
Morphology
Morphological characteristics of NP1 and NP4 were investigated by TEM and shown in Figure 4A and B, respectively. The morphology of K188-stabilised NPs (NP1) shows a high degree of shape variation (Figure 4A). On the other hand, NP4 has a distinctive and uniform spherical shape comprised a dark core with a smooth surface and thick shell. This shell confirmed the existence of BSA layers around the particles. The thickness of the BSA-adsorbed layer can be observed which indicates that BSA sufficiently offered an efficient steric stabilization to the surface of NP4 (Figure 4B). It had been reported that an adsorbed macromolecule layer on the NPs surfaces was principally responsible for steric repulsion and stabilization effect. The magnitudes of these effects are dependent upon the adsorption affinity of macromolecules to the surface as well as their molecular structures.79,80 It was verified that K188 can bind to the PLGA NPs surface via hydrophobic interaction of the PPO moieties, even though the hydrophilic PEO moieties protrude into the surrounding aqueous medium to establish a steric barrier.81 Compared with K188 getting a relatively linear structure,28 protein molecules of BSA have a more compact globular construction after its adsorption onto the hydrophobic surfaces owing to dehydration of protein amino acids. This compact layer could impart longer-range steric repulsive forces on NP surfaces with more uniform assembly.79,82
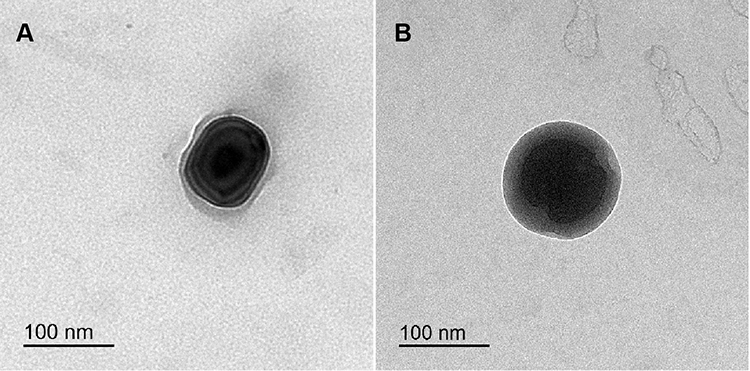 |
Figure 4 TEM images of NP1 (A) and NP4 (B). |
Notably, TEM showed that the PS of the NPs was smaller (<200 nm) than that measured by DLS (Figure 4 and Table 2). This difference could be principally attributed to the fact that the DLS-based measurement of the hydrodynamic radius encloses the double solvation layer. On the other hand, TEM was performed on dried samples of NPs with depletion of surface ions and solvent molecules. Accordingly, the actual diameter assessed by TEM is found generally smaller than that of DLS. Comparable judgments and elucidations have been recognized in the literature for PLGA-based NPs.83
Determination of AMD Solubility
The solubility study was implemented to study the effect of pH on the solubility of AMD. Table 3 shows that the solubility values of AMD were 0.4855, 0.0615, 0.0430, and 0.003 mg/mL in distilled water, pH1.2, PB6.8, and PB7.4, respectively. It was evident that AMD demonstrated the highest solubility in distilled water (pH 5.5) and lower solubilities in the other media. The lower solubility of AMD at the pH values of pH1.2, PB6.8, and PB7.4 might be attributed to the effects of the dissolved anions and cations in these media.84 The fall in solubility at pH1.2 might be clarified by the common ion effect. As the salt form of AMD is hydrochloride and pH1.2 media contained chloride ions too, the degree of dissociation of AMD salt and its solubility diminished by the chloride ion effect.84 By looking at the molecular structure of AMD, the basic tertiary amine side chain was supposed to be ionized in pH1.2 which in turn was responsible for the higher AMD solubility than PB6.8 and PB7.4. The phenomenon of the remarkable drop of solubility in PB6.8 and PB7.4 could be attributed to that in a media with a pH value equal to or higher than pKa of AMD (6.56 ± 0.06), the molecule stays in a unionized form with reduced solubility.32 Based on the obtained results, it was inferred that the solubility of AMD is a pH-dependent one. This phenomenon could be used later to understand the AMD-release pattern in different media.
 |
Table 3 Solubility Values of AMD in Different Dissolution Media at 37 °C ± 0.5 °C, (Mean ± SD; n=3) |
In vitro Release
The in vitro release behavior of AMD was expressed in the form of a cumulative percentage released as shown in Figure 5. The release of AMD was investigated at consecutive values of pH; pH1.2, PB6.8, and PB7.4 representing the physiological fate of NPs across the stomach, intestine, and blood, respectively. It has been well recognized that the average residence time in the stomach is 2 h. So, the release of AMD was conducted at pH1.2 for 2 h and then PB6.8 was used to foresee the release in the other parts of the gastrointestinal tract (GIT). Besides, pH7.4 was utilized to observe the possible release patterns upon absorption of the NPs to reach the circulatory system.85,86
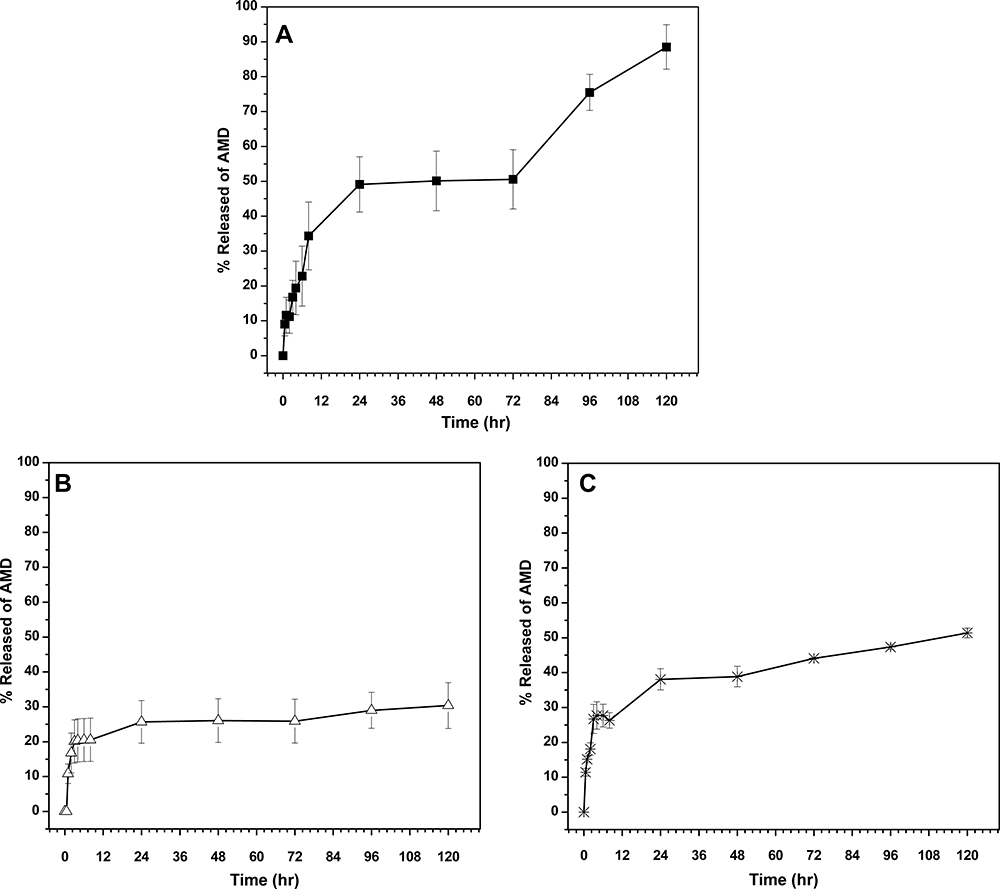 |
Figure 5 The in vitro release profiles of AMD from its suspension (A) and the selected NPs; NP1 (B) and NP4 (C). |
As depicts in Figure 5A, marked fast release rates of 34.3% and 88.5% were exhibited by pure AMD at pH6.8 (up to 8 h) and pH7.4 (up to 5 days), respectively. Conversely, a slow release of AMD from NP1 and NP4 was observed at simulated gastric pH to reach 16.8% and 18.1%, respectively, with no noteworthy burst release (Figure 5B and C). The sustained initial pattern of release may be attributed to that almost all AMD was successively entrapped within the cores of the medicated NPs. This verdict was found to be harmonic with our early discussed findings under the NPs characterization section. If a drug is not efficiently entrapped, the disadvantageous early and premature release will happen.74,87 It has been documented that a compact polymeric coat around a drug can sustain its release.74 Nevertheless, slow degradation of PLGA50 occurs through water invasion to the polymeric matrix followed by hydrolysis of the ester bonds. This process leads to the creation of pores through them and hence the entrapped drug can release.87–89 The drug release is regulated also by its physicochemical properties and hence the observed slow release of pure AMD (13% after 2 h in pH1.2) could be related to its poor aqueous solubility as indicated in Table 3. 11,31 At pH6.8, further release of AMD from NP1 and NP4 continued in the same slow manner to reach 20.5% and 26.3%, respectively, by the end of the eighth hour. After that, higher amounts of AMD (30.4% and 51.4%) were released from the medicated NPs (NP1 and NP4, respectively) at pH7.4.
Considering the acquired results, we concluded that the release patterns of AMD-loaded NPs exhibited controlled delivery manners. However, the percentages of AMD released from NPs at pH6.8 and pH7.4 were higher than the corresponding ones at pH1.2. It could be observed that the release of AMD from NP4 took place in a more likely pH-dependent manner than that of NP1. Poloxamers are nonionic surfactants and can be adsorbed to the NPs surface by hydrophobic interaction in the form of micellar-like aggregates.27,90 Due to the absence of ionizable groups, the poloxamers showed no changes in their aggregation behavior over a wide pH range.91
On the other hand, the adsorption of BSA onto the surface of NPs is driven by hydrophobic, electrostatic interactions, and hydrogen bonding.92 Hence, modification of the BSA charge influences its adsorption capability on the solid surface of NPs. At gastric pH (pH1.2), BSA would be more positively charged due to the value of pH is lower than its isoelectric point (pH 5).93,94 As PLGA50 NPs were found to be negatively charged (the carboxyl-terminal groups), the further attraction between BSA and the surface of NP4 might be enhanced with expected retardation of AMD release in gastric pH. It has been reported that BSA at neutral pH is highly negatively charged indicating that the electrostatic interaction is insignificant. Hence, the adsorption force between BSA and PLGA50 was reduced because it was controlled only by the hydrophobic interaction and hydrogen bonding.92 In such a situation, the incidence chance of AMD release to occur in the intestine increased, which is the preferable site for drug release.
We hypothesized that the release of AMD from NPs might start in the lumen of the gastrointestinal tract. Overall, the average optimal reported size for NP transcytosis in gastrointestinal applications appears to be preferentially ranged from 100 to 500 nm. For NPs larger than 500 nm, they cannot be absorbed as intact particles.95–99 Based on their PS (not more than 190 nm) as stated under Hydrodynamic Diameter and Surface Charge, NPs could transport across the gastrointestinal tract by passive diffusion, reach the systemic circulation, and then release further AMD. Our release finding showed that the AMD-release can occur under the intestinal and physiological conditions. This result was expected to be optimistic because the release in the high surface area of the intestinal environment could support the systemic absorption of AMD. Additionally, it had been reported that orally delivered PLGA-based NPs with a size lower than 500 nm can escape the intestinal wall enzymes that mediate metabolism and the gastrointestinal tract excretion facilitated by P-glycoprotein.100
The in vitro release data were fitted to the kinetic models as listed in Table 4. It was observed and concluded that the Higuchi model gave the highest value of r2 at different pH values. Additionally, the AMD-release kinetic model of NP1 at pH6.8, NP4 at pH1.2, and both medicated NPs at pH7.4 fitted well with Fickian-diffusion (n < 0.5). However, NP1 followed non-Fickian diffusion (anomalous) model (0.5<n< 1) at pH1.2. In general, the Fickian mechanism indicates that the release of the drug is mainly mediated by its diffusion across the polymeric matrix. While non-Fickian one signifies that the release was controlled by both drug diffusion and polymer erosion.
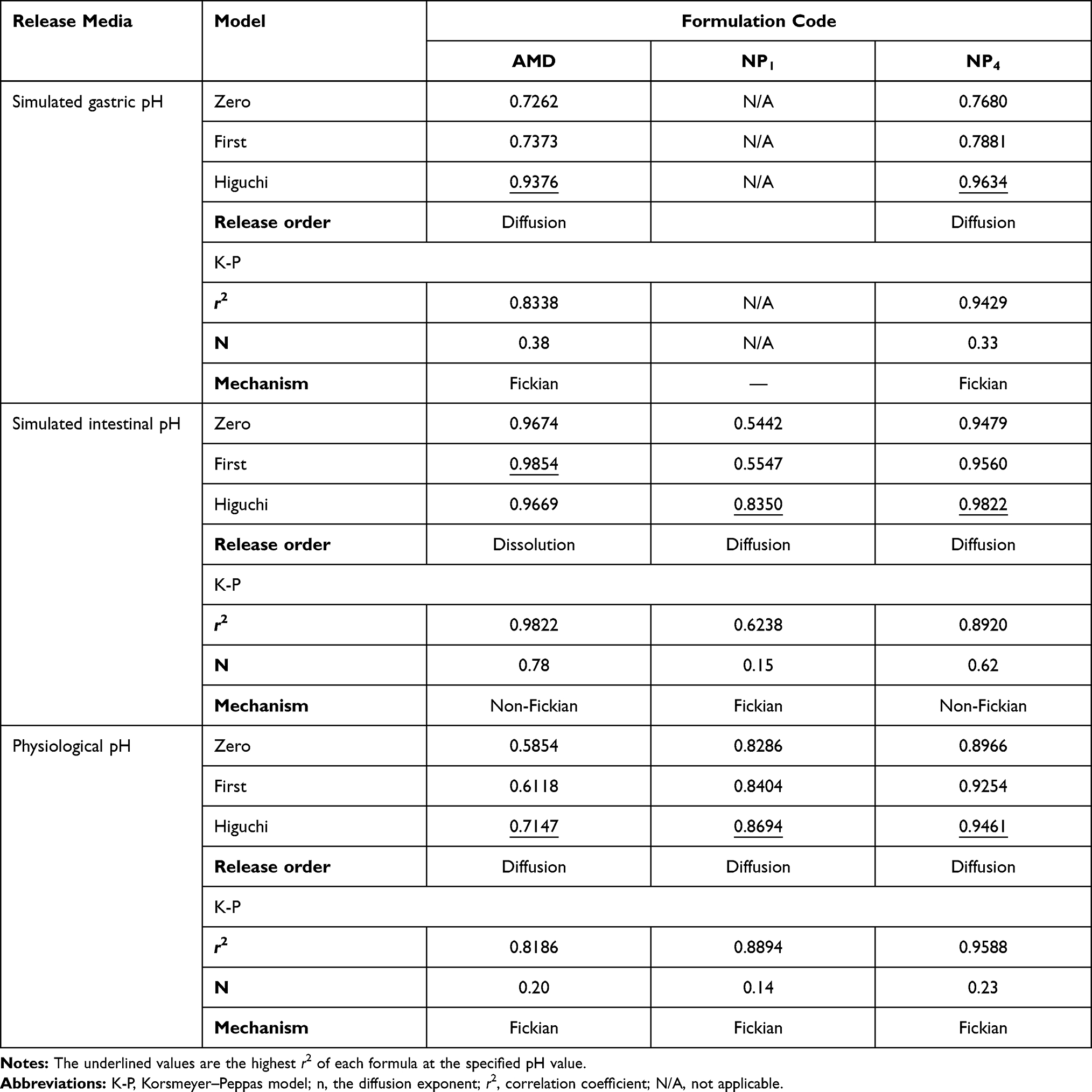 |
Table 4 Release Kinetics of in vitro Release Data |
When we took all the obtained outcomes together, it was concluded that NP4 was a promising delivery system of AMD and hence it deserved to undergo further studies.
In vitro Study of Cell Toxicity
This experiment was conducted to explore the cytotoxicity induced by AMD on cultured cells.7 The impacts of AMD were assessed on the inhibition of cell proliferation using the SRB colorimetric assay. This method is based on the ability of the SRB dye to attach to the basic amino acid residues of proteins. Compared to other assay methods, the SRB one has superior cell line-independent staining accompanied with higher sensitivity and linearity with cell number.101 For toxicity screening, fast-growing robust cell lines are often employed. In consequence of a higher rate of proliferation and metabolic activity of cancer cell lines, they are more resilient to NPs toxicity than normal ones. To discover the precise effects of NPs on specific organs, the cytotoxicity assay must involve cells that represent the exposure route and the targeted organs.102,103 As pulmonary toxicity is the most serious toxic concern of AMD, the potential cytotoxic effects of pure AMD, NP4, and PNP4 were monitored using A549, and Hep-2 cell lines.
The percentages of cell viability are illustrated in Figure 6. Moreover, demonstrative phase-contract images were taken, and they were used to analyze the cellular morphology of the treated cell lines after exposure to the tested formulations for 72 h. Table 5 documents the values of IC50 of NP4, PNP4, and AMD in the studied cell lines. It was evident that free AMD treatment caused a marked decline in cell viability in a dose-dependent fashion. The IC50 values of AMD were 10.49 and 9.5 µg/mL for Hep-2 and A549, respectively (P <0.05). Also, the obtained data have shown that the percentages of cell viability were noticeably enhanced when the cell lines were treated with the AMD-equivalent concentrations of NP4. The values of IC50 significantly increased by more than threefold to reach 47.89 µg/mL, and 30.46 µg/mL for A549, and Hep-2 cell lines, respectively (P <0.0001). The marked increase of IC50 of NP4 indicates a cytoprotective behavior of our developed system. The AMD-free NPs (PNP4) had no noticeable cytotoxic effect on all the treated cells. The harmlessness of PNP4 may be ascribed to the good biocompatibility and biodegradability of PLGA50.
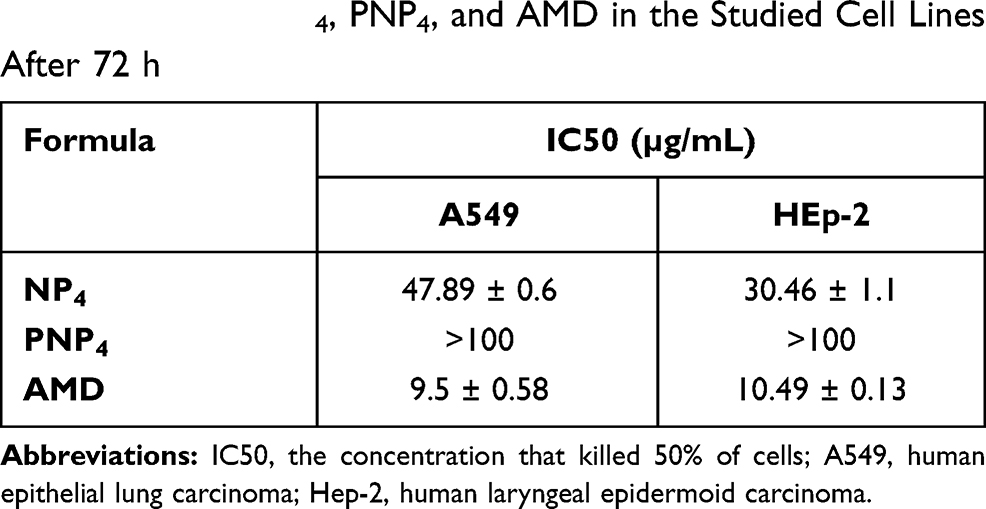 |
Table 5 IC50 of NP4, PNP4, and AMD in the Studied Cell Lines After 72 h |
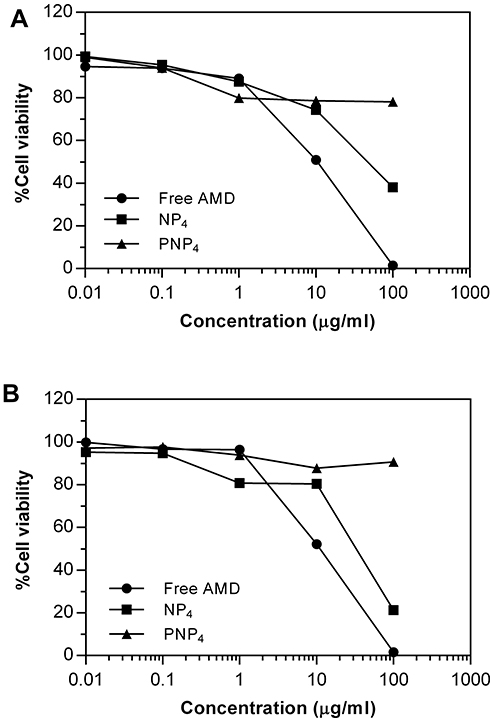 |
Figure 6 Cell viability of A549 (A), and Hep-2 (B) after treatment with pure AMD, NP4, and PNP4 for 72 h. |
The morphological changes of A549 and Hep-2 cell lines are shown in Figure 7. The morphology was analyzed through phase-contrast microscopy after the treatment with 100 µg/mL of pure AMD, NP4, and PNP4 for 72 h. The alterations in the morphology of cells were compared relative to control cells (untreated). Figure 7 depicts the control cells, which maintained their normal morphology with typical viability and high confluency of monolayer cells. The treated cells showed different cellular structures. The free AMD-treated cell lines displayed extensive growth inhibition and offered nearly complete cell death. On the other hand, the NP4- and PNP4-treated cells showed a reduction in cell volume and cell shrinkage. PNP4 caused fewer morphological changes compared to NP4 which exhibited some apoptotic characteristics like losing contact between neighboring cells. Hep-2 was the most sensitive cell line with a marked reduction of cell number (Figure 7). This finding was agreeable with the above-observed pattern of cell viability and IC50 (30.46 µg/mL).
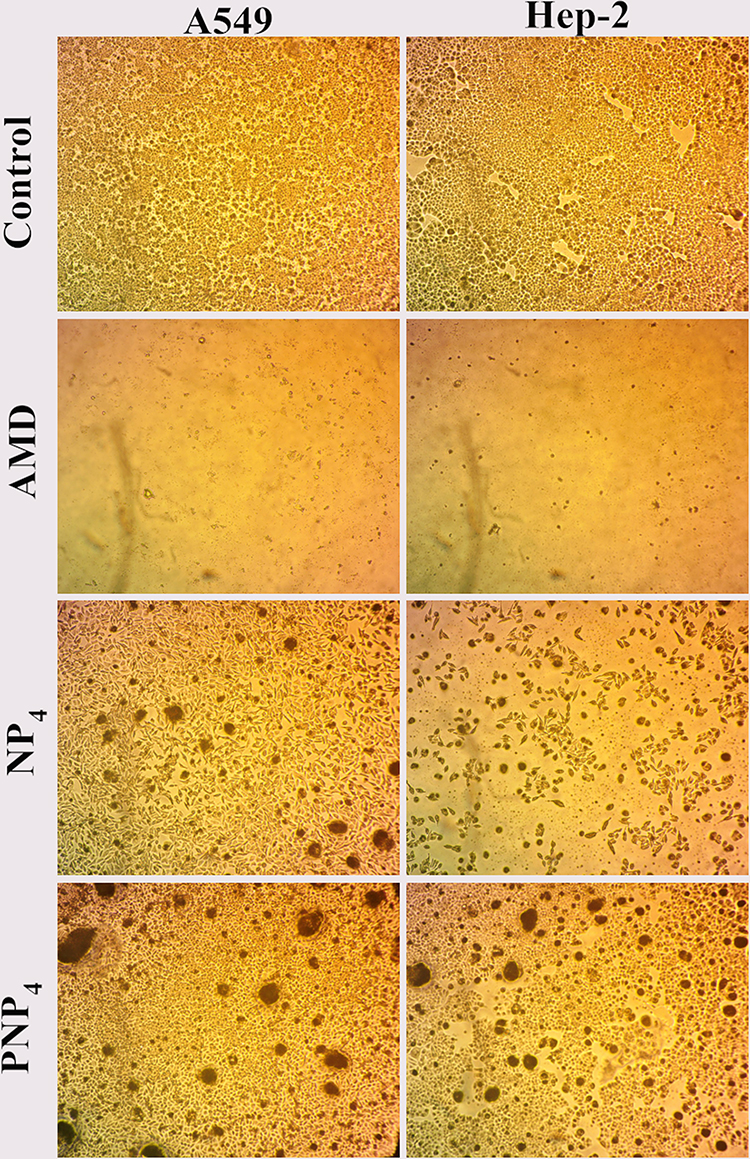 |
Figure 7 The morphological changes of A549, and Hep-2 after treatment with pure AMD, NP4, and PNP4 for 72 h. Notes: Control cells show normal morphology with typical viability, the AMD-treated cells are almost dead with an obvious decline in number, the NP4-treated cells show a reduction in volume and cellular shrinkage, PNP4 caused fewer morphological changes compared to NP4 with some apoptotic characteristics and losing contact between neighboring cells. 100×. |
The AMD-induced cytotoxic effects were clear when the drug was present in its free form (un-capsulated form). In vitro cytotoxicity of AMD was reported by several researchers via different mechanisms.7,104 AMD disrupts the potential of the mitochondrial membrane and decreases the cellular ATP levels in lung cells. Such a mitochondrial dysfunction is a key for the initiation of AMD-induced pulmonary cytotoxicity.105 Also, AMD increases the levels of oxidized glutathione and the reactive oxygen species (ROS) in lung tissues.106 At the same time, the above-mentioned results proved the ability of AMD-loaded NPs to protect lung cells against the cytotoxic properties of free AMD. The cytoprotective effect of NP4 could be attributed to the successful encapsulation of AMD within the core of NP4. Hence, diffusion of AMD out to the media was slow and gradual from the polymeric matrix (see ‘In vitro Release’ section). Such a case offered a brilliant chance to the cells to accommodate their exposure to the diffused amounts of AMD as well as tolerate the associated negative consequences. Similar findings were observed by other investigators.107–109
From the cytotoxic assays and the images of phase contrast microscopy, it could be established that the plain NPs (PNP4) were biosafe and non-toxic. A similar outcome was reported in another study.110 This could be attributed to the biocompatibility of PLGA that was illustrated early in this study. At the same time, encapsulated AMD (NP4) demonstrated a cytoprotective efficiency by diminishing the pure AMD-induced cell death. Hence, NP4 can be used as a novel carrier of AMD. However, further in vivo studies on experimental animals are conducted soon for more investigation of the biosafety of the AMD-loaded NPs.
In vivo Evaluation of the Pulmonary Toxicity
Histopathological Evaluation
Histopathological examination of lung tissues revealed the normal pulmonary architecture of the control group. The alveolar epithelium, consisting of type I and type II pneumocytes, lined up the lung alveoli. The alveoli were separated from each other by thin interalveolar septa (IAS) (Figure 8A). Lung sections of Group I (AMD suspension-treated animals) showed distortion of normal lung architecture with congestion of blood vessels, extravasation of red blood cells (RBCs) into the surrounding structure, and thickening of IAS. Also, peri-bronchial mononuclear cellular infiltration, epithelial shedding from thickened bronchial epithelium, was seen in the thickened IAS of the Group I animals (Figure 8B). Some bronchioles showed the shedding of the epithelial cells into their lumen. These findings were consistent with other scholars who noticed similar changes.8,43,111–115 Both mononuclear cellular infiltration and IAS thickening reduced noticeably in the animals of Group III treated with NP4 dispersion (Figure 8D). Minimal signs of the above elements were observed in the placebo-treated animals (Group II) (Figure 8C).
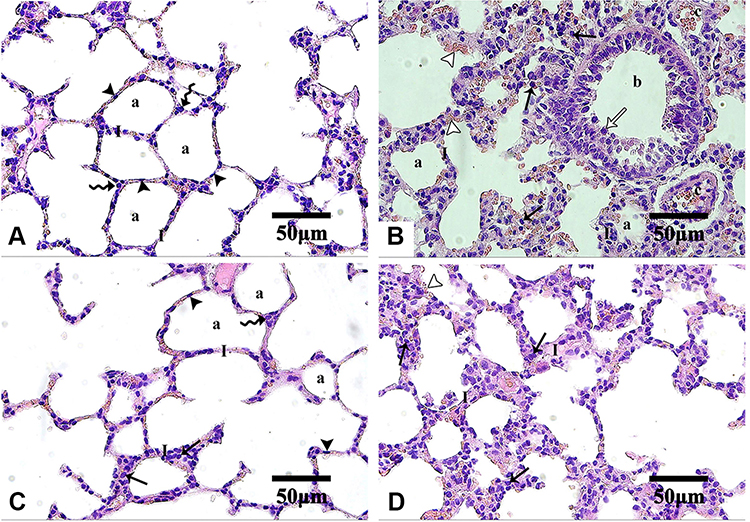 |
Figure 8 Microphotographs of H&E stained-lung sections of the control and the treated groups. Abbreviations: H&E, hematoxylin, and eosin. Notes: Control (A), Group I (B), Group II (C), and Group III (D). In control and Group II, type I pneumocytes (arrowheads) and type II pneumocytes (zigzag arrows) line up alveoli (a) with interalveolar septa (I). Group I shows extravasation of some red blood cells (empty arrowheads) from the congested blood vessels (c). Thickened interalveolar septa were markedly infiltrated by mononuclear cells (arrows) especially around bronchioles (b) which shows thickening of epithelium with the shedding of some cells (empty arrow). Group II shows minimal thickening and cellular infiltration of interalveolar septa. In Group III, cellular infiltration and septal thickening are less marked than Group I. H&E, 400×. |
Lung sections stained with Masson’s trichrome revealed few collagenous fibers in the alveolar wall of both control and Group II (Figure 9A and C, respectively). On the other hand, conspicuous fibrous tissue deposition around bronchioles and in the alveolar wall of the Group I animals were observed (Figure 9B), especially around bronchioles. This agreed with other studies that reported the presence of areas with marked collagenous fibers deposition in the alveolar interstitial tissues and IAS in AMD-treated animals.8,112,113,116,117 Comparatively, the Group III animals exhibited less fibrous tissue deposition (Figure 9D).
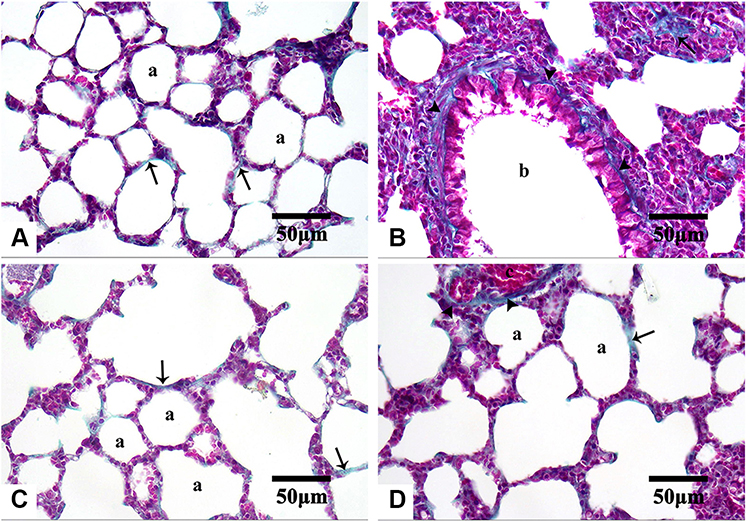 |
Figure 9 Microphotographs of Masson’s trichrome stained-lung sections of the control and the treated groups. Notes: Control (A), Group I (B), Group II (C), and Group III (D). Few collagenous fibers (arrows) are in the alveolar wall (a) of both control and Group II. Noticeable fibrous tissue deposition (arrowheads) looks around bronchioles (b) and in the alveolar wall of Group I. Less amount of fibrous tissue is observed (arrowheads) around congested blood vessels (c) in Group III. Masson’s trichrome, 400×. |
Table 6 shows the scoring results of the lung sections. The obtained findings demonstrated that capillary congestion was significant in both Group I and Group III (P<0.0001 and P<0.05, respectively) and non-significant (P>0.05) in Group II compared to the control group. Significant hemorrhage was discovered in the animals of I and III groups (P<0.001 and P<0.05, respectively) and non-significant hemorrhage (P>0.05) in the Group II animals when related to the control group. Similarly, Group I and Group III exhibited significant infiltration of inflammatory cells (P<0.0001 and P<0.05, respectively) while the Group II animals showed non-significant infiltration (P>0.05) in comparison to the control group. A significant increase in the thickness of IAS was recorded in the Group I animals (P<0.0001) compared to the control group, but the increase in thickness was not significant in the Group II and Group III animals (P>0.05) (Table 6).
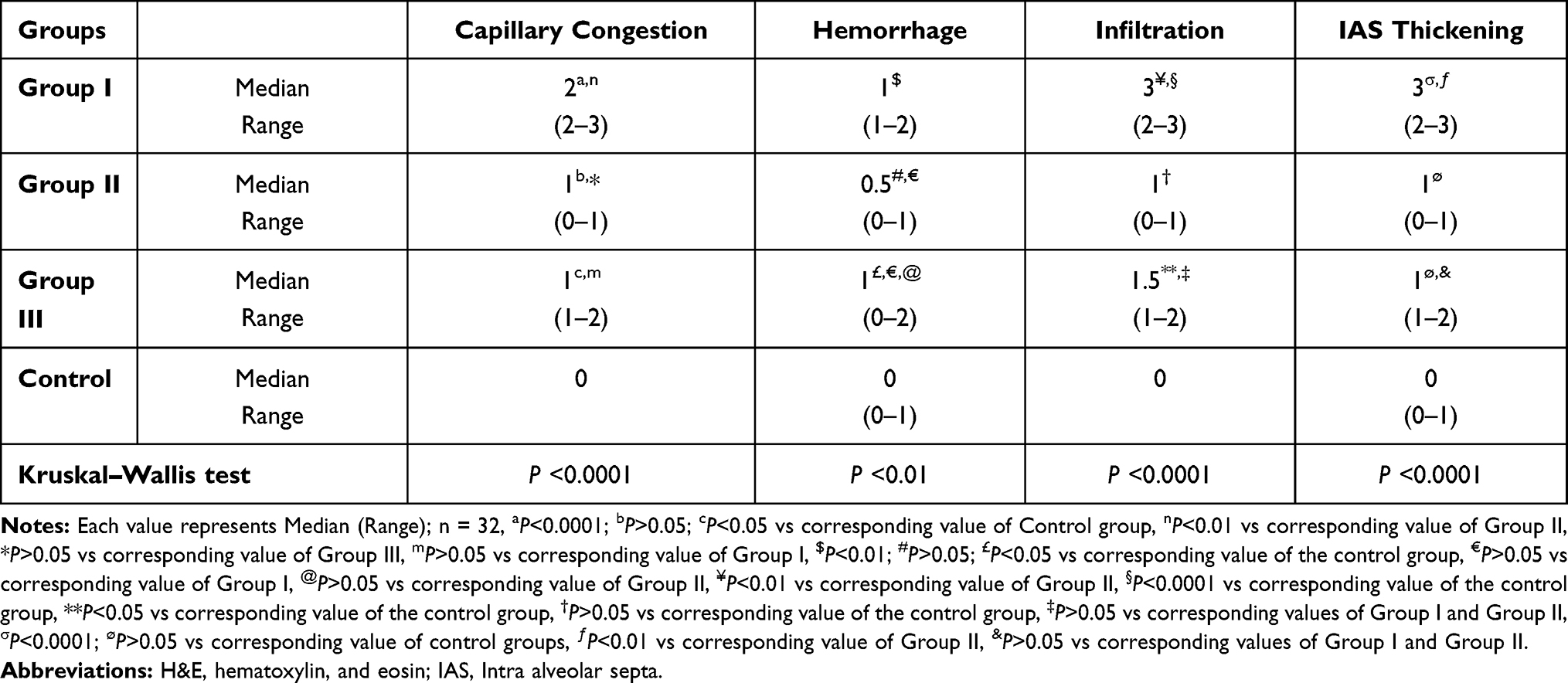 |
Table 6 Scoring the Lung Sections of the Studied Groups |
The above findings indicated that the direct exposure to pure AMD (Group I) resulted in evident toxic effects on the alveolar interstitial tissue and the wall of the pulmonary blood vessels (congestion and extravasation of RBCs) with a marked fibrous tissue deposition, especially around bronchioles. Different scenarios of AMD-induced pulmonary toxicity were reported.118 After AMD administration, pulmonary hypertension and smooth muscle proliferation were developed with consequent deposition of collagen and thickening of the vessel wall. The established vascular modification was characterized by the activation of endothelial cells and the infiltration of inflammatory cells; primarily lymphocytes and macrophages engulfed the extravasated RBCs.113,116,119 A homogeneous interstitial exudate might precede the inflammatory bronchial and vascular cellular infiltration. After that, inappropriate tissue restoration and unsuccessful epithelial regeneration might result in obliteration of bronchiolar lumen and the development of non-specific interstitial pneumonitis.113,120–122 Additionally, AMD was reported to induce pulmonary fibrosis by an unplanned proliferation of fibroblasts followed by their differentiation into myofibroblasts, and massive production of extracellular matrix components (ECM).123–125
To sum up, it was found that the exposure of lung tissues to the free AMD resulted in extreme toxic manifestations. Instead, NP4 ameliorated the AMD-induced pulmonary damage in the treated animals. Such a finding indicated that the incorporation of AMD in the PLGA-based NPs could restrain its toxic influences on the lung tissues. Besides, it was reported that PLGA-based NPs showed an anti‐fibrotic activity against pulmonary fibrosis.126,127
IHC Evaluation
The IHC-stained sections with anti-TGF-β1 antibodies revealed minimal reaction in both the control (Figure 10A) and Group III (NP4 dispersion-treated animals) (Figure 10D) groups. The immunoreactivity was the strongest in the animals treated with pure AMD (Group I) (Figure 10B). The placebo group (PNP4-treated animals) showed the lowest expression of TGF-β1in the lung cells (Figure 10C). The reaction to anti-iNOS antibodies was minimal in both the control (Figure 11A) and Group III (Figure 11D), moderate in Group II (Figure 11C), and strong in Group I (Figure 11B).
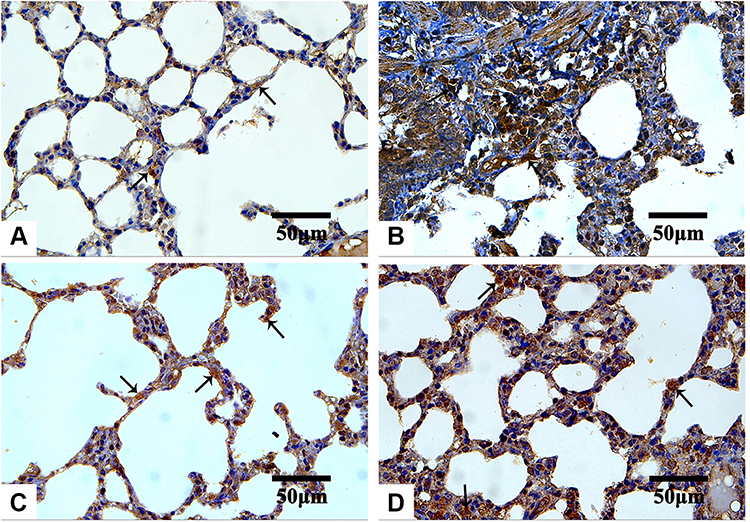 |
Figure 10 Microphotographs IHC estimation of TGF-β1 expression in rat’s lung sections of the control and the treated groups. Abbreviations: IHC, immunohistochemical; TGF-β1, anti-transforming growth factor ß1. Notes: Control (A), Group I (B), Group II (C), and Group III (D). The reaction is minimal in both control and Group III. As indicated by arrows, strong expression is observed in Group I, but it is less marked in Group II. IHC counterstained with hematoxylin, 400×. |
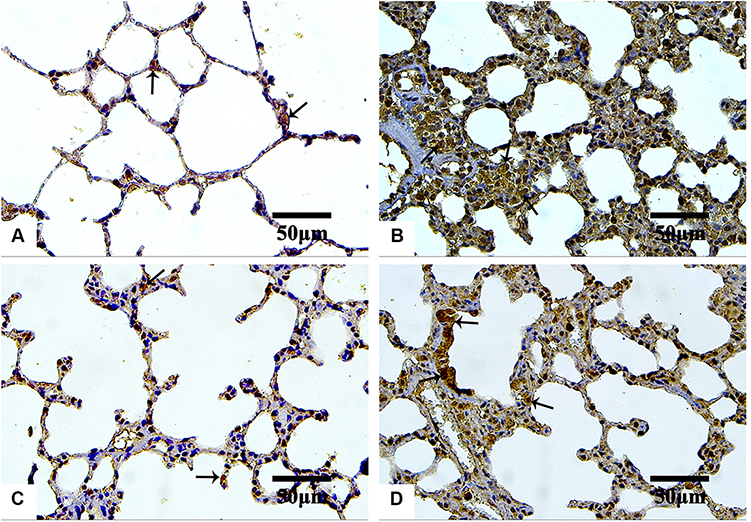 |
Figure 11 Microphotographs IHC estimation of iNOS expression in rat’s lung sections of the control and the treated groups. Abbreviations: IHC, immunohistochemical; iNOS, anti-inducible nitric oxide synthase. Notes: Control (A), Group I (B), Group II (C), and Group III (D). The reaction is minimal in both control and Group III. Minimal reaction as indicated by arrows is seen in both the control and Group III. Strong and moderate expression is observed in Group I and Group II, respectively. IHC counterstained with hematoxylin, 400×. |
Table 7 documents the expression levels of TGF-β1and iNOS in the control and the treated groups. Among the studied groups, a marked increase in the expression levels of both TGF-β1and iNOS (12.34 ± 0.87 and 12.65 ± 0.86, respectively) could be observed in Group I. On the other hand, the placebo group (Group II) presented the lowest expression levels of both TGF-β1and iNOS (3.72 ± 0.33 and 1.96 ± 0.17, respectively). The expression levels of TGF-β1and iNOS were 6.85 ± 0.31 and 3.04 ± 0.26, respectively, in Group III (Table 7). The statistical analysis revealed that treating animals with NP4 dispersion (Group III) and placebo formula (Group II) resulted in a significant reduction in the expression levels of TGF-β1 (P<0.0001) in comparison to the pure AMD treatment (Group I). Additionally, the Group II animals exhibited a much lower expression level of TGF-β1 than the corresponding one of Group III (P<0.0001). Similarly, the expression levels of iNOS were significantly diminished in Group III and Group II in comparison to the corresponding level of Group I (P<0.0001). In comparison to the control group, both Group II and Group III displayed a significant decline in the expression levels of iNOS (P<0.0001).
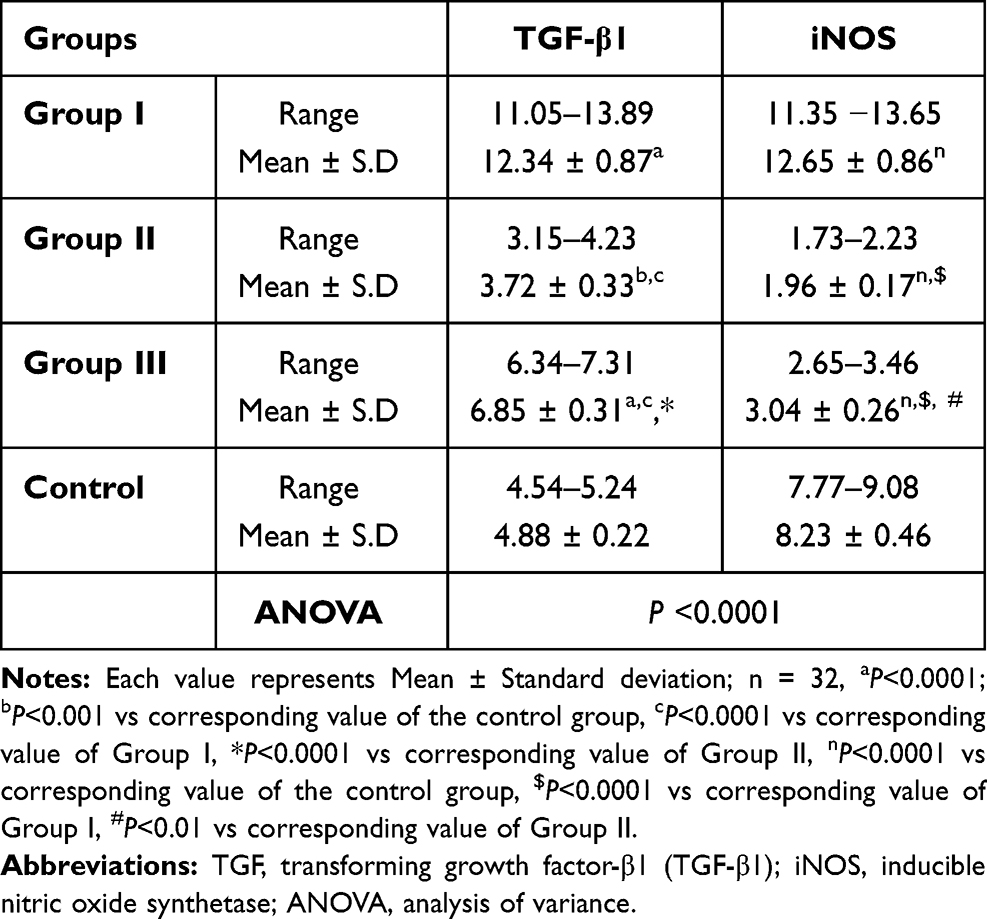 |
Table 7 Expression Levels of TGF-β1and iNOS in the Studied Groups |
In the present study, IHC stained sections revealed a stronger reaction for anti-TGF-β1 antibodies in the pure AMD-treated group. Similar results were obtained by other researchers who reported an elevation of the TGF-β1 expression after AMD administration, accompanied by excessive α-smooth muscle actin (α-SMA) expression.43,114,128 TGF-β1 is a key pro-fibrogenic cytokine that promotes collagen accumulation in numerous aspects. It stimulates the differentiation of fibroblasts into myofibroblasts, enhances collagen production, and reduces collagenase activity.43 TGF-β1 stimulates the production of ROS through the activation of NADPH oxidases and mitochondrial dysfunction.129 Also, TGF-β1 diminishes the expression of the mitochondrial lung superoxide dismutase (SOD) and catalases.130 Furthermore, upregulation of TGF-β1 expression suggested a positive feedback action that increases its production by the activated macrophages.112 It could be concluded that the AMD-induced upregulation of the TGF-β1 expression in lung tissue supports the key role of this cytokine in the establishment and progression of lung fibrosis.
Furthermore, it was reported that the production of reactive nitrogen species (RNS), the alteration of lung prostaglandin, and the overproduction of collagen occurred simultaneously as a result of the exposure of an organ to oxidative stress. RNS involving peroxynitrite (ONOO−), nitrogen dioxide (NO2), and nitric oxide (NO) greatly contribute to respiratory tract pathogenesis. NO is a short-lived, readily diffusible, and potent oxidant created in the lung by the inducible form of iNOS, especially during lung injury or inflammation. The degree of pulmonary fibrosis is strongly correlated with the iNOS levels.109,114 In the current study, the IHC expression of iNOS was extensive in pure AMD-treated animals, suggesting a strong oxidative impact of pure AMD. The overexpression of iNOS after administration of pure AMD was consistent with other previous studies.113,131–133
It was evident from IHC results that treating animals with NP4 dispersion induced significant down-regulation of TGF-β1 and iNOS expression compared to pure AMD treatment. The ameliorative effects of NP4 could be attributed to the role of the biodegradable polymer (PLGA) in controlling the AMD level at the action site (see under ‘In vitro Release’ and ’In vitro Study of Cell Toxicity’ sections). By the gradual release of AMD from NP4 to the lungs, the production of inflammatory mediators and the free radicals in healthy cells might be reduced. Hence, the exaggerated toxicity upon the direct exposure to free AMD could be attenuated. These outcomes are in agreement with other endeavors validating the defensive properties of nanoencapsulation contrasted with the free drug-induced toxicities.109,134,135
In the light of the obtained findings, the present study was concerned with the pulmonary toxicity of the optimized AMD-loaded NPs (NP4) rather than their anti-arrhythmic efficiency. After demonstrating the cytoprotective, anti‐fibrotic, anti-inflammatory, and antioxidant properties of the developed system, it is thought that such a system deserves to undergo further studies. Hence, a separate study has been currently conducted to evaluate the efficiency of NP4 and compare it to that of the pristine AMD. Such a study will be concerned mainly with the efficiency of the AMD-loaded NPs to treat cardiac arrhythmia.
Conclusion
AMD-loaded NPs were successively prepared by the nanoprecipitation method using Kolliphor® P188 or BSA as stabilizers. The optimized formula was prepared using BSA at a concentration of 0.5%. About 96% of AMD could be encapsulated in the PLGA-based matrix of NPs. The developed NPs were able to control the release of AMD without initial drug eruption. Cytoprotective, anti‐fibrotic, anti-inflammatory, and antioxidant properties were presented by the optimized NPs (NP4). Hence, the results substantiate that the polymeric nanoparticles as promising tactics could ameliorate the pulmonary injury induced by the pure drug moieties.
Abbreviations
A549, human epithelial lung carcinoma; AMD, Amiodarone; ATR-FTIR, attenuated total reflectance-Fourier infrared spectroscopy; AV node, the atrioventricular node; BSA, bovine serum albumin; DLS, dynamic light scattering technique; DMSO, dimethyl sulfoxide; DSC, differential scanning calorimetry; EE%, encapsulation efficiency; ECM, extracellular matrix; GIT, gastrointestinal tract; Hep-2, human laryngeal epidermoid carcinoma; PS, Hydrodynamic diameters; IC50, the concentration that killed 50% of cells in comparison with the untreated cells; IAS, interalveolar septa; K188, Kolliphor® P 188; LC%, loading capacity; NO, nitric oxide; NO2, nitrogen dioxide; ONOO−, peroxynitrite; PDI, size distribution; PLGA, poly(lactic-co-glycolic acid); PNPs, AMD-free NPs; PS, particle size; RBCs, red blood cells; RNS, reactive nitrogen species; ROS, reactive oxygen species; SRB, sulforhodamine B assay; α-SMA, α-smooth muscle actin; SOD, superoxide dismutase; TCA, trichloroacetic acid; TEM, transmission electron microscopy; XRD, powder X-ray diffraction; Y%, yield; ZP, zeta potential.
Disclosure
The authors reported no conflicts of interest for this work.
References
1. Ganjehei L, Massumi A, Nazeri A, Razavi M. Pharmacologic management of arrhythmias. Tex Heart Inst J. 2011;38(4):344–349.
2. Mehraein F, Breijo-Marquez FR. A review on amiodarone as an antiarrhythmic drug. In: Abnormal Heart Rhythms. BoD–Books on Demand; 2015:115.
3. Creteanu A, Pamfil D, Vasile C, et al. Study on the role of the inclusion complexes with 2-hydroxypropyl-β-cyclodextrin for oral administration of amiodarone. Int J Polym Sci. 2019;2019:1–23. doi:10.1155/2019/1695189
4. Van Herendael H, Dorian P. Amiodarone for the treatment and prevention of ventricular fibrillation and ventricular tachycardia. Vasc Health Risk Manag. 2010;6:465–472. doi:10.2147/vhrm.s6611
5. Terzo F, Ricci A, D’Ascanio M, Raffa S, Mariotta S. Amiodarone-induced pulmonary toxicity with an excellent response to treatment: a case report. Respir Med Case Rep. 2020;29:100974. doi:10.1016/j.rmcr.2019.100974
6. Teerakanok J, Tantrachoti P, Chariyawong P, Nugent K. Acute amiodarone pulmonary toxicity after surgical procedures. Am J Med Sci. 2016;352(6):646–651. doi:10.1016/j.amjms.2016.08.013
7. Golli-Bennour EE, Bouslimi A, Zouaoui O, Nouira S, Achour A, Bacha H. Cytotoxicity effects of amiodarone on cultured cells. Exp Toxicol Pathol. 2012;64(5):425–430. doi:10.1016/j.etp.2010.10.008
8. Zickri MB, Fadl SGA, Metwally HG. Comparative study between intravenous and intraperitoneal stem cell therapy in amiodarone induced lung injury in rat. Int J Stem Cells. 2014;7(1):1–11. doi:10.15283/ijsc.2014.7.1.1
9. Lamprecht A, Bouligand Y, Benoit J-P. New lipid nanocapsules exhibit sustained release properties for amiodarone. J Control Release. 2002;84(1–2):59–68. doi:10.1016/S0168-3659(02)00258-4
10. Ahmed MS, Rodell CB, Hulsmans M, et al. A supramolecular nanocarrier for delivery of amiodarone anti-arrhythmic therapy to the heart. Bioconjugate Chem. 2019;30(3):733–740. doi:10.1021/acs.bioconjchem.8b00882
11. Elgart A, Cherniakov I, Aldouby Y, Domb AJ, Hoffman A. improved oral bioavailability of BCS class 2 compounds by Self Nano-Emulsifying Drug Delivery Systems (SNEDDS): the underlying mechanisms for amiodarone and talinolol. Pharm Res. 2013;30(12):3029–3044. doi:10.1007/s11095-013-1063-y
12. Takahama H, Shigematsu H, Asai T, et al. Liposomal amiodarone augments anti-arrhythmic effects and reduces hemodynamic adverse effects in an ischemia/reperfusion rat model. Cardiovasc Drugs Ther. 2013;27(2):125–132. doi:10.1007/s10557-012-6437-6
13. Zhuge Y, Zheng Z-F, Xie M-Q, Li L, Wang F, Gao F. Preparation of liposomal amiodarone and investigation of its cardiomyocyte-targeting ability in cardiac radiofrequency ablation rat model. Int J Nanomedicine. 2016;11:2359–2367. doi:10.2147/IJN.S98815
14. Li J, Kong M, Cheng XJ, et al. Preparation of biocompatible chitosan grafted poly(lactic acid) nanoparticles. Int J Biol Macromol. 2012;51(3):221–227. doi:10.1016/j.ijbiomac.2012.05.011
15. Nadimi AE, Ebrahimipour SY, Afshar EG, et al. Nano-scale drug delivery systems for antiarrhythmic agents. Eur J Med Chem. 2018;157:1153–1163. doi:10.1016/j.ejmech.2018.08.080
16. Júlio A, Caparica R, Costa Lima SA, et al. Ionic liquid-polymer nanoparticle hybrid systems as new tools to deliver poorly soluble drugs. Nanomaterials. 2019;9(8):1148. doi:10.3390/nano9081148
17. Xia Z, Huang Y, Adamopoulos IE, Walpole A, Triffitt JT, Cui Z. Macrophage-mediated biodegradation of poly(DL-lactide-co-glycolide) in vitro. J Biomed Mater Res A. 2006;79A(3):582–590. doi:10.1002/jbm.a.30853
18. Swider E, Koshkina O, Tel J, Cruz LJ, de Vries IJM, Srinivas M. Customizing poly(lactic-co-glycolic acid) particles for biomedical applications. Acta Biomaterialia. 2018;73:38–51. doi:10.1016/j.actbio.2018.04.006
19. Katsuki S, Matoba T, Koga J, Nakano K, Egashira K. Anti-inflammatory nanomedicine for cardiovascular disease. Front Cardiovasc Med. 2017;4. doi:10.3389/fcvm.2017.00087.
20. Hong JS, Srivastava D, Lee I. Fabrication of poly (lactic acid) nano-and microparticles using a nanomixer via nanoprecipitation or emulsion diffusion. J Appl Polym Sci. 2018;135(18):46199. doi:10.1002/app.46199
21. Shakeri F, Shakeri S, Hojjatoleslami M. Preparation and characterization of carvacrol loaded polyhydroxybutyrate nanoparticles by nanoprecipitation and dialysis methods. J Food Sci. 2014;79(4):N697–N705. doi:10.1111/1750-3841.12406
22. Schubert S, Delaney JT, Schubert U. Nanoprecipitation and nanoformulation of polymers: from history to powerful possibilities beyond poly(lactic acid). Soft Matter. 2011;7(5):1581–1588. doi:10.1039/C0SM00862A
23. Cooper DL, Harirforoosh S. Design and optimization of PLGA-based diclofenac loaded nanoparticles. PLoS One. 2014;9(1):e87326. doi:10.1371/journal.pone.0087326
24. Vandervoort J, Ludwig A. Biocompatible stabilizers in the preparation of PLGA nanoparticles: a factorial design study. Int J Pharm. 2002;238(1–2):77–92. doi:10.1016/S0378-5173(02)00058-3
25. Elzoghby AO, Samy WM, Elgindy NA. Albumin-based nanoparticles as potential controlled release drug delivery systems. J Control Release. 2012;157(2):168–182. doi:10.1016/j.jconrel.2011.07.031
26. Li X, He E, Jiang K, Peijnenburg WJGM, Qiu H. The crucial role of a protein corona in determining the aggregation kinetics and colloidal stability of polystyrene nanoplastics. Water Res. 2021;190:116742. doi:10.1016/j.watres.2020.116742
27. Zarrintaj P, Ramsey JD, Samadi A, et al. Poloxamer: a versatile tri-block copolymer for biomedical applications. Acta Biomaterialia. 2020;110:37–67. doi:10.1016/j.actbio.2020.04.028
28. Shaker MA, Elbadawy HM, Al Thagfan SS, Shaker MA. Enhancement of atorvastatin oral bioavailability via encapsulation in polymeric nanoparticles. Int J Pharm. 2021;592:120077. doi:10.1016/j.ijpharm.2020.120077
29. Grijalva M, Vallejo-López MJ, Salazar L, Camacho J, Kumar B. Cytotoxic and antiproliferative effects of nanomaterials on cancer cell lines: a review. Biomed Environ Appl. 2017. doi:10.5772/intechopen.71685
30. Niu X, Zou W, Liu C, Zhang N, Fu C. Modified nanoprecipitation method to fabricate DNA-loaded PLGA nanoparticles. Drug Dev Ind Pharm. 2009;35(11):1375–1383. doi:10.3109/03639040902939221
31. Essa E, Negm M, Eldin E, Maghraby G. Fast disintegrating tablets of amiodarone for intra-oral administration. J App Pharm Sci. 2017;064–072. doi:10.7324/JAPS.2017.70109
32. Rubim AM, Rubenick JB, Gregolin E, Laporta LV, Leitenberg R, Rolim CMB. Amiodarone hydrochloride: enhancement of solubility and dissolution rate by solid dispersion technique. Braz J Pharm Sci. 2015;51(4):957–966. doi:10.1590/S1984-82502015000400021
33. Creţeanu A, Ochiuz L, Vieriu M, Panainte AD, Ţântaru G. In vitro dissolution studies of amiodarone hydrochloride from hydroxy-propyl-β-cyclodextrin/amiodarone inclusion complex formulated into modified-release tablets. Rev Med Chir Soc Med Nat Iasi. 2016;120(3):715–719.
34. Rubim A, Rubenick J, Maurer M, Laporta L, Rolim C. Inclusion complex of amiodarone hydrochloride with cyclodextrins: preparation, characterization and dissolution rate evaluation. Braz J Pharm Sci. 2017;53. doi:10.1590/s2175-97902017000216083.
35. Wang D, Chen G, Ren L. Preparation and characterization of the sulfobutylether-β-cyclodextrin inclusion complex of amiodarone hydrochloride with enhanced oral bioavailability in fasted state. AAPS PharmSciTech. 2017;18(5):1526–1535. doi:10.1208/s12249-016-0646-4
36. Abdel Raheem IA, Abdul Razek A, Elgendy AA, Saleh NM, Shaaban MI, Abd El-Hady FK. Design, evaluation and antimicrobial activity of Egyptian propolis-loaded nanoparticles: intrinsic role as a novel and naturally based root canal nanosealer. Int J Nanomedicine. 2019;14:8379–8398. doi:10.2147/IJN.S219577
37. Martin A, Bustamanate P, Chun AHC. kinetics and drug stability. In: Martin A, editor. Physical Pharmacy.
38. Higuchi T. Mechanism of sustained‐action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52(12):1145–1149. doi:10.1002/jps.2600521210
39. Ritger PL, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Control Release. 1987;5(1):37–42. doi:10.1016/0168-3659(87)90035-6
40. Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82(13):1107–1112. doi:10.1093/jnci/82.13.1107
41. Allam RM, Al-Abd AM, Khedr A, et al. Fingolimod interrupts the cross talk between estrogen metabolism and sphingolipid metabolism within prostate cancer cells. Toxicol Lett. 2018;291:77–85. doi:10.1016/j.toxlet.2018.04.008
42. Jerng J-S, Hsu Y-C, Wu H-D, et al. Role of the renin-angiotensin system in ventilator-induced lung injury: an in vivo study in a rat model. Thorax. 2007;62(6):527–535. doi:10.1136/thx.2006.061945
43. El‐Din AAIS, Allah OMA. Impact of olmesartan medoxomil on amiodarone-induced pulmonary toxicity in rats: focus on transforming growth factor-ß1. Basic Clin Pharmacol Toxicol. 2016;119(1):58–67. doi:10.1111/bcpt.12551
44. Rus A, Peinado MÁ, Castro L, Moral MLD. Lung eNOS and iNOS are reoxygenation time-dependent upregulated after acute hypoxia. Anat Rec. 2010;293(6):1089–1098. doi:10.1002/ar.21141
45. Tantra R, Tompkins J, Quincey P. Characterisation of the de-agglomeration effects of bovine serum albumin on nanoparticles in aqueous suspension. Colloids Surf B Biointerfaces. 2010;75(1):275–281. doi:10.1016/j.colsurfb.2009.08.049
46. Feczkó T, Tóth J, Gyenis J. Comparison of the preparation of PLGA–BSA nano- and microparticles by PVA, poloxamer and PVP. Colloids Surf a Physicochem Eng Asp. 2008;319(1–3):188–195. doi:10.1016/j.colsurfa.2007.07.011
47. Chenthamara D, Subramaniam S, Ramakrishnan SG, et al. Therapeutic efficacy of nanoparticles and routes of administration. Biomater Res. 2019;23(1):20. doi:10.1186/s40824-019-0166-x
48. Luo M-X, Hua S, Shang Q-Y. Application of nanotechnology in drug delivery systems for respiratory diseases (Review). Mol Med Rep. 2021;23(1):1–17. doi:10.3892/mmr.2021.11964
49. Silva-Abreu M, Espinoza LC, Halbaut L, Espina M, García ML, Calpena AC. Comparative study of ex vivo transmucosal permeation of pioglitazone nanoparticles for the treatment of alzheimer’s disease. Polymers. 2018;10(3):316. doi:10.3390/polym10030316
50. Su S, Kang PM. Systemic review of biodegradable nanomaterials in nanomedicine. Nanomaterials. 2020;10(4):656. doi:10.3390/nano10040656
51. Dokoumetzidis A, Macheras P. A century of dissolution research: from Noyes and Whitney to the biopharmaceutics classification system. Int J Pharm. 2006;321(1–2):1–11. doi:10.1016/j.ijpharm.2006.07.011
52. Snehalatha M, Venugopal K, Saha RN. Etoposide-loaded PLGA and PCL nanoparticles I: preparation and effect of formulation variables. Drug Deliv. 2008;15(5):267–275. doi:10.1080/10717540802174662
53. Yadav K, Yadav D, Yadav M, Kumar S. Noscapine loaded PLGA nanoparticles prepared using oil-in-water emulsion solvent evaporation method. J Nanopharm Drug Deliv. 2016;3(1):97–105. doi:10.1166/jnd.2015.1074
54. Wang W, Xi M, Duan X, Wang Y, Kong F. Delivery of baicalein and paclitaxel using self-assembled nanoparticles: synergistic antitumor effect in vitro and in vivo. Int J Nanomed. 2015;10:3737–3750. doi:10.2147/IJN.S80297
55. Shao X-R, Wei X-Q, Song X, et al. Independent effect of polymeric nanoparticle zeta potential/surface charge, on their cytotoxicity and affinity to cells. Cell Prolif. 2015;48(4):465–474. doi:10.1111/cpr.12192
56. Bernal-Chávez SA, Alcalá-Alcalá S, Cerecedo D, Ganem-Rondero A. Platelet lysate-loaded PLGA nanoparticles in a thermo-responsive hydrogel intended for the treatment of wounds. Eur J Pharm Sci. 2020;146:105231. doi:10.1016/j.ejps.2020.105231
57. Betancourt T, Brown B, Brannon-Peppas L. Doxorubicin-loaded PLGA nanoparticles by nanoprecipitation: preparation, characterization and in vitro evaluation. Nanomedicine. 2007;2(2):219–232. doi:10.2217/17435889.2.2.219
58. Qi S, Marchaud D, Craig DQM. An investigation into the mechanism of dissolution rate enhancement of poorly water-soluble drugs from spray chilled gelucire 50/13 microspheres. J Pharm Sci. 2010;99(1):262–274. doi:10.1002/jps.21832
59. Singla P, Chabba S, Mahajan RK. A systematic physicochemical investigation on solubilization and in vitro release of poorly water soluble oxcarbazepine drug in pluronic micelles. Colloids Surf a Physicochem Eng Asp. 2016;504:479–488. doi:10.1016/j.colsurfa.2016.05.043
60. Gossmann R, Langer K, Mulac D. New perspective in the formulation and characterization of Didodecyldimethylammonium Bromide (DMAB) stabilized poly(lactic-co-glycolic acid) (PLGA) nanoparticles. PLoS One. 2015;10(7):e0127532. doi:10.1371/journal.pone.0127532
61. Dhat S, Pund S, Kokare C, Sharma P, Shrivastava B. Risk management and statistical multivariate analysis approach for design and optimization of satranidazole nanoparticles. Eur J Pharm Sci. 2017;96:273–283. doi:10.1016/j.ejps.2016.09.035
62. Manjunath K, Venkateswarlu V, Hussain A. Preparation and characterization of nitrendipine solid lipid nanoparticles. Pharmazie. 2011;66(3):178–186. doi:10.1691/ph.2011.9373
63. Baghbani F, Moztarzadeh F, Mohandesi JA, Yazdian F, Mokhtari-Dizaji M, Hamedi S. Optimization of formulation and process variables for the preparation of novel doxorubicin-loaded sonosensitive nanodroplets. J Clust Sci. 2016;27(5):1519–1536. doi:10.1007/s10876-016-1020-0
64. Keles H, Naylor A, Clegg F, Sammon C. Studying the release of hGH from gamma-irradiated PLGA microparticles using ATR-FTIR imaging. Vib Spectrosc. 2014;71:76–84. doi:10.1016/j.vibspec.2014.01.012
65. Patel A, Shelat P, Lalwani A. Development and optimization of Solid Self Nanoemulsifying Drug Delivery (S-SNEDDS) using D-optimal design for improvement of oral bioavailability of amiodarone hydrochloride. CDD. 2015;12(6):745–760. doi:10.2174/1567201812666150302122501
66. Bronze-Uhle ES, Costa BC, Ximenes VF, Lisboa-Filho PN. Synthetic nanoparticles of bovine serum albumin with entrapped salicylic acid. NSA. 2016;10:11–21. doi:10.2147/NSA.S117018
67. Salehiabar M, Nosrati H, Javani E, et al. Production of biological nanoparticles from bovine serum albumin as controlled release carrier for curcumin delivery. Int J Biol Macromol. 2018;115:83–89. doi:10.1016/j.ijbiomac.2018.04.043
68. Oz UC, Küçüktürkmen B, Devrim B, Saka OM, Bozkir A. Development and optimization of alendronate sodium loaded PLGA nanoparticles by central composite design. Macromol Res. 2019;27(9):857–866. doi:10.1007/s13233-019-7119-z
69. Plomp TA. Analytical profile of amiodarone. In: Analytical Profiles of Drug Substances. Vol. 20. Elsevier; 1991:1–120. doi:10.1016/S0099-5428(08)60529-8
70. Ledeţi A, Vlase G, Vlase T, et al. Solid-state preformulation studies of amiodarone hydrochloride. J Therm Anal Calorim. 2016;126(1):181–187. doi:10.1007/s10973-016-5256-6
71. Mainardes RM, Gremião MPD, Evangelista RC. Thermoanalytical study of praziquantel-loaded PLGA nanoparticles. Rev Bras Cienc Farm. 2006;42(4):523–530. doi:10.1590/S1516-93322006000400007
72. Español L, Larrea A, Andreu V, et al. Dual encapsulation of hydrophobic and hydrophilic drugs in PLGA nanoparticles by a single-step method: drug delivery and cytotoxicity assays. RSC Adv. 2016;6(112):111060–111069. doi:10.1039/C6RA23620K
73. Escalona-Rayo O, Fuentes-Vázquez P, Jardon-Xicotencatl S, García-Tovar CG, Mendoza-Elvira S, Quintanar-Guerrero D. Rapamycin-loaded polysorbate 80-coated PLGA nanoparticles: optimization of formulation variables and in vitro anti-glioma assessment. J Drug Deliv Sci Technol. 2019;52:488–499. doi:10.1016/j.jddst.2019.05.026
74. Venkatesh DN, Baskaran M, Karri VVSR, Mannemala SS, Radhakrishna K, Goti S. Fabrication and in vivo evaluation of Nelfinavir loaded PLGA nanoparticles for enhancing oral bioavailability and therapeutic effect. Saudi Pharm J. 2015;23(6):667–674. doi:10.1016/j.jsps.2015.02.021
75. Gurushankar K, Gohulkumar M, Prasad NR, Krishnakumar N. Synthesis, characterization and in vitro anti-cancer evaluation of hesperetin-loaded nanoparticles in human oral carcinoma (KB) cells. Adv Nat Sci. 2013;5(1):015006. doi:10.1088/2043-6262/5/1/015006
76. Sodeifian G, Sajadian SA. Utilization of ultrasonic-assisted RESOLV (US-RESOLV) with polymeric stabilizers for production of amiodarone hydrochloride nanoparticles: optimization of the process parameters. Chem Eng Res Des. 2019;142:268–284. doi:10.1016/j.cherd.2018.12.020
77. Rahman HS, Rasedee A, How CW, et al. Zerumbone-loaded nanostructured lipid carriers: preparation, characterization, and antileukemic effect. Int J Nanomed. 2013;8:2769–2781. doi:10.2147/IJN.S45313
78. Ali SH, Sulaiman GM, Al-Halbosiy MMF, Jabir MS, Hameed AH. Fabrication of hesperidin nanoparticles loaded by poly lactic co-Glycolic acid for improved therapeutic efficiency and cytotoxicity. Artif Cells, Nanomed Biotechnol. 2019;47(1):378–394. doi:10.1080/21691401.2018.1559175
79. Chen C, Wei J, Li J, Duan Z, Huang W. Influence of macromolecules on aggregation kinetics of diesel soot nanoparticles in aquatic environments. Environ Pollut. 2019;252:1892–1901. doi:10.1016/j.envpol.2019.06.012
80. Louie M, Tilton D, Lowry V. Critical review: impacts of macromolecular coatings on critical physicochemical processes controlling environmental fate of nanomaterials. Environ Sci. 2016;3(2):283–310. doi:10.1039/C5EN00104H
81. Han SK, Kim RS, Lee JH, Tae G, Cho SH, Yuk SH. Core-shell nanoparticles for drug delivery and molecular imaging. In: Kumar CSSR editor. Nanotechnologies for the Life Sciences. Wiley-VCH Verlag GmbH & Co. KGaA; 2007:143–188. doi:10.1002/9783527610419.ntls0110.
82. Huangfu X, Jiang J, Ma J, Liu Y, Yang J. Aggregation kinetics of manganese dioxide colloids in aqueous solution: influence of humic substances and biomacromolecules. Environ Sci Technol. 2013;47(18):10285–10292. doi:10.1021/es4003247
83. Courant T, Roullin VG, Cadiou C, et al. Development and physicochemical characterization of copper complexes-loaded PLGA nanoparticles. Int J Pharm. 2009;379(2):226–234. doi:10.1016/j.ijpharm.2009.03.036
84. Plöger GF, Hofsäss MA, Dressman JB. Solubility determination of active pharmaceutical ingredients which have been recently added to the list of essential medicines in the context of the biopharmaceutics classification system–biowaiver. J Pharm Sci. 2018;107(6):1478–1488. doi:10.1016/j.xphs.2018.01.025
85. Krishnaiah YSR, Bhaskar Reddy PR, Satyanarayana V, Karthikeyan RS. Studies on the development of oral colon targeted drug delivery systems for metronidazole in the treatment of amoebiasis. Int J Pharm. 2002;236(1–2):43–55. doi:10.1016/S0378-5173(02)00006-6
86. Li Y, Bi Y, Xi Y, Li L. Enhancement on oral absorption of paclitaxel by multifunctional pluronic micelles. J Drug Target. 2013;21(2):188–199. doi:10.3109/1061186X.2012.737001
87. Loureiro JA, Gomes B, Fricker G, Coelho MAN, Rocha S, Pereira MC. Cellular uptake of PLGA nanoparticles targeted with anti-amyloid and anti-transferrin receptor antibodies for Alzheimer’s disease treatment. Colloids Surf B Biointerfaces. 2016;145:8–13. doi:10.1016/j.colsurfb.2016.04.041
88. Göpferich A. Mechanisms of polymer degradation and erosion. Biomaterials. 1996;17(2):103–114. doi:10.1016/0142-9612(96)85755-3
89. Frasco MF, Almeida GM, Santos‐Silva F, Pereira MC, Coelho MAN. Transferrin surface-modified PLGA nanoparticles-mediated delivery of a proteasome inhibitor to human pancreatic cancer cells. J Biomed Mater Res A. 2015;103(4):1476–1484. doi:10.1002/jbm.a.35286
90. Santander-Ortega MJ, Jódar-Reyes AB, Csaba N, Bastos-González D, Ortega-Vinuesa JL. Colloidal stability of Pluronic F68-coated PLGA nanoparticles: a variety of stabilisation mechanisms. J Colloid Interface Sci. 2006;302(2):522–529. doi:10.1016/j.jcis.2006.07.031
91. Alexander S, de Vos WM, Castle TC, Cosgrove T, Prescott SW. Growth and shrinkage of pluronic micelles by uptake and release of flurbiprofen: variation of pH. Langmuir. 2012;28(16):6539–6545. doi:10.1021/la204262w
92. Kunda NK, Alfagih IM, Dennison SR, et al. Bovine serum albumin adsorbed PGA-co-PDL nanocarriers for vaccine delivery via dry powder inhalation. Pharm Res. 2015;32(4):1341–1353. doi:10.1007/s11095-014-1538-5
93. Spada G, Gavini E, Sorrenti M, et al. Studies of technological parameters influencing the protein-polymeric nanoparticles adsorption process for transmucosal administration. Curr Nanosci. 2012;8(6):819–829. doi:10.2174/157341312803989079
94. Pavlov G, Hsu JT. The pH, temperature, and protein structure effect on β-lactoglobulin A and B separation in anion-exchange chromatography. AiChE J. 2018;64(6):1928–1937. doi:10.1002/aic.16077
95. Cao S, Xu S, Wang H, et al. Nanoparticles: oral delivery for protein and peptide drugs. AAPS PharmSciTech. 2019;20(5):190. doi:10.1208/s12249-019-1325-z
96. Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20(2):101–124. doi:10.1038/s41573-020-0090-8
97. Chen H, Zheng Y, Tian G, et al. Oral delivery of DMAB-modified docetaxel-loaded PLGA-TPGS nanoparticles for cancer chemotherapy. Nanoscale Res Lett. 2010. doi:10.1007/s11671-010-9741-8
98. Murugan K, Choonara YE, Kumar P, Bijukumar D, Du Toit LC, Pillay V. Parameters and characteristics governing cellular internalization and trans-barrier trafficking of nanostructures. Int J Nanomed. 2015;10:2191–2206. doi:10.2147/IJN.S75615
99. Jiang L, Li X, Liu L, Zhang Q. Thiolated chitosan-modified PLA-PCL-TPGS nanoparticles for oral chemotherapy of lung cancer. Nanoscale Res Lett. 2013;8(1):66. doi:10.1186/1556-276X-8-66
100. Yin Win K, Feng -S-S. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26(15):2713–2722. doi:10.1016/j.biomaterials.2004.07.050
101. Vajrabhaya L, Korsuwannawong S. Cytotoxicity evaluation of a Thai herb using tetrazolium (MTT) and sulforhodamine B (SRB) assays. J Anal Sci Technol. 2018;9(1):15. doi:10.1186/s40543-018-0146-0
102. Buckberry LD. Cytotoxicity testing using cell lines. In: Jenkins N editor. Animal Cell Biotechnology: Methods and Protocols. Methods in BiotechnologyTM. Humana Press; 1999:239–252. doi:10.1385/0-89603-547-6:239.
103. Kong B, Seog JH, Graham LM, Lee SB. Experimental considerations on the cytotoxicity of nanoparticles. Nanomedicine. 2011;6(5):929–941. doi:10.2217/nnm.11.77
104. Trohman RG, Sharma PS, McAninch EA, Bianco AC. Amiodarone and thyroid physiology, pathophysiology, diagnosis and management. Trends Cardiovasc Med. 2019;29(5):285–295. doi:10.1016/j.tcm.2018.09.005
105. Card JW, Racz WJ, Brien JF, Massey TE. Attenuation of amiodarone-induced pulmonary fibrosis by vitamin E is associated with suppression of transforming growth factor-β1 gene expression but not prevention of mitochondrial dysfunction. J Pharmacol Exp Ther. 2003;304(1):277–283. doi:10.1124/jpet.102.043208
106. Nicolescu AC, Comeau JL, Hill BC, et al. Aryl radical involvement in amiodarone-induced pulmonary toxicity: investigation of protection by spin-trapping nitrones. Toxicol Appl Pharmacol. 2007;220(1):60–71. doi:10.1016/j.taap.2006.12.031
107. Meng Q, Wang A, Hua H, et al. Intranasal delivery of Huperzine A to the brain using lactoferrin-conjugated N-trimethylated chitosan surface-modified PLGA nanoparticles for treatment of Alzheimer’s disease. Int J Nanomedicine. 2018;13:705–718. doi:10.2147/IJN.S151474
108. Tukulula M, Hayeshi R, Fonteh P, et al. Curdlan-conjugated PLGA nanoparticles possess macrophage stimulant activity and drug delivery capabilities. Pharm Res. 2015;32(9):2713–2726. doi:10.1007/s11095-015-1655-9
109. Saghir SAM, Al-Gabri NA, Khafaga AF, et al. Thymoquinone-PLGA-PVA nanoparticles ameliorate bleomycin-induced pulmonary fibrosis in rats via regulation of inflammatory cytokines and iNOS signaling. Animals. 2019;9(11):951–967. doi:10.3390/ani9110951
110. Aravind A, Varghese SH, Veeranarayanan S, et al. Aptamer-labeled PLGA nanoparticles for targeting cancer cells. Cancer Nano. 2012;3(1–6):1–12. doi:10.1007/s12645-011-0024-6
111. Mahdy AA. The possible ameliorative effect of selenium and vitamins combination against amiodarone-induced alveolar damage in albino rat: histological and immunohistochemical study. J Am Sci. 2014;10(4s):61–71.
112. Chung WH, Bennett BM, Racz WJ, Brien JF, Massey TE. Induction of c-jun and TGF-β1 in Fischer 344 rats during amiodarone-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2001;281(5):L1180–L1188. doi:10.1152/ajplung.2001.281.5.L1180
113. Gawad FAE-R, Rizk AA-E-E, Fouad M. Amiodarone-induced lung toxicity and the protective role of Vitamin E in adult male albino rat. Eur J Anat. 2018;22(4):332–333.
114. Radwan SM, Ghoneim D, Salem M, et al. Adipose tissue–derived mesenchymal stem cells protect against amiodarone-induced lung injury in rats. Appl Biochem Biotechnol. 2020;191(3):1027–1041. doi:10.1007/s12010-020-03227-8
115. Gumuser G, Vural K, Varol T, et al. Assessment of lung toxicity caused by bleomycin and amiodarone by Tc-99m HMPAO lung scintigraphy in rats. Ann Nucl Med. 2013;27(7):592–599. doi:10.1007/s12149-013-0722-8
116. Aboul-Fotouh GI, Zickri MB, Metwally HG, Ibrahim IR, Kamar SS, Sakr W. Therapeutic effect of adipose derived stem cells versus atorvastatin on amiodarone induced lung injury in male rat. Int J Stem Cells. 2015;8(2):170–180. doi:10.15283/ijsc.2015.8.2.170
117. Gado AM, Aldahmash BA. Protective effect of L-carnitine against amiodarone-induced lung toxicity in rats. Internet J Toxicol. 2013;10(1):1–9.
118. Shimizu S, Gabazza EC, Taguchi O, et al. Activated protein C inhibits the expression of platelet-derived growth factor in the lung. Am J Respir Crit Care Med. 2003;167(10):1416–1426. doi:10.1164/rccm.200206-515OC
119. Sood BG, Wykes S, Landa M, De Jesus L, Rabah R. Expression of eNOS in the lungs of neonates with pulmonary hypertension. Exp Mol Pathol. 2011;90(1):9–12. doi:10.1016/j.yexmp.2010.11.003
120. Abo Gazia MM, El-Kordy AM. Attenuation of amiodarone-induced pulmonary fibrosis by vitamin E is associated with suppression of transforming growth factor-β1 gene expression but not prevention of mitochondrial dysfunction. Life Sci J. 2012;9(2):73–85.
121. Larsen BT, Vaszar LT, Colby TV, Tazelaar HD. Lymphoid hyperplasia and eosinophilic pneumonia as histologic manifestations of amiodarone-induced lung toxicity. Am J Surg Pathol. 2012;36(4):509–516. doi:10.1097/PAS.0b013e318243fd9a
122. Barker AF, Bergeron A, Rom WN, Hertz MI. Obliterative bronchiolitis. N Engl J Med. 2014;370(19):1820–1828. doi:10.1056/NEJMra1204664
123. Desai O, Winkler J, Minasyan M, Herzog EL. The role of immune and inflammatory cells in idiopathic pulmonary fibrosis. Front Med. 2018;5. doi:10.3389/fmed.2018.00043.
124. Malaviya R, Kipen HM, Businaro R, Laskin JD, Laskin DL. Pulmonary toxicants and fibrosis: innate and adaptive immune mechanisms. Toxicol Appl Pharmacol. 2020;409:115272. doi:10.1016/j.taap.2020.115272
125. Robert S, Gicquel T, Victoni T, et al. Involvement of matrix metalloproteinases (MMPs) and inflammasome pathway in molecular mechanisms of fibrosis. Biosci Rep. 2016;36:e00360. doi:10.1042/BSR20160107
126. Trivedi R, Redente EF, Thakur A, Riches DWH, Kompella UB. Local delivery of biodegradable pirfenidone nanoparticles ameliorates bleomycin-induced pulmonary fibrosis in mice. Nanotechnology. 2012;23(50):505101. doi:10.1088/0957-4484/23/50/505101
127. Pintado‐Berninches L, Montes‐Worboys A, Manguan‐García C, et al. GSE4-loaded nanoparticles a potential therapy for lung fibrosis that enhances pneumocyte growth, reduces apoptosis and DNA damage. FASEB J. 2021;35(3):e21422. doi:10.1096/fj.202001160RR
128. Shroff A, Mamalis A, Jagdeo J. Oxidative Stress and Skin Fibrosis. Curr Pathobiol Rep. 2014;2(4):257–267. doi:10.1007/s40139-014-0062-y
129. Sturrock A, Cahill B, Norman K, et al. Transforming growth factor-β1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;290(4):L661–L673. doi:10.1152/ajplung.00269.2005
130. Herrera B, Murillo MM, Álvarez-barrientos A, Beltrán J, Fernández M, Fabregat I. Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-β in fetal rat hepatocytes. Free Radic Biol Med. 2004;36(1):16–26. doi:10.1016/j.freeradbiomed.2003.09.020
131. Pullamsetti SS, Savai R, Dumitrascu R, et al. The role of dimethylarginine dimethylaminohydrolase in idiopathic pulmonary fibrosis. Sci Transl Med. 2011;3(87):87ra53–87ra53. doi:10.1126/scitranslmed.3001725
132. Naura AS, Zerfaoui M, Kim H, et al. Requirement for inducible nitric oxide synthase in chronic allergen exposure-induced pulmonary fibrosis but not inflammation. J Immunol. 2010;185(5):3076–3085. doi:10.4049/jimmunol.0904214
133. Kalayarasan S, Sriram N, Sudhandiran G. Diallyl sulfide attenuates bleomycin-induced pulmonary fibrosis: critical role of iNOS, NF-κB, TNF-α and IL-1β. Life Sci. 2008;82(23–24):1142–1153. doi:10.1016/j.lfs.2008.03.018
134. Al-Nemrawi NK, Alshraiedeh NH, Zayed AL, Altaani BM. Low molecular weight chitosan-coated PLGA nanoparticles for pulmonary delivery of tobramycin for cystic fibrosis. Pharmaceuticals. 2018;11(1):28. doi:10.3390/ph11010028
135. Motawea A, Ahmed DAM, Eladl AS, El-Mansy AAE-R, Saleh NM. Appraisal of amiodarone-loaded PLGA nanoparticles for prospective safety and toxicity in a rat model. Life Sci. 2021;274:119344. doi:10.1016/j.lfs.2021.119344
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.