Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Critical role of tumor necrosis factor receptor 1 in the pathogenesis of pulmonary emphysema in mice
Authors Fujita M ![]() , Ouchi H, Ikemage S, Harada E, Matsumoto T, Uchino J, Nakanishi Y, Watanabe K
, Ouchi H, Ikemage S, Harada E, Matsumoto T, Uchino J, Nakanishi Y, Watanabe K
Received 21 March 2016
Accepted for publication 7 May 2016
Published 28 July 2016 Volume 2016:11(1) Pages 1705—1712
DOI https://doi.org/10.2147/COPD.S108919
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Masaki Fujita,1 Hiroshi Ouchi,2 Satoshi Ikegame,2 Eiji Harada,2 Takemasa Matsumoto,1 Junji Uchino,1 Yoichi Nakanishi,2 Kentaro Watanabe1
1Department of Respiratory Medicine, Faculty of Medicine, Fukuoka University, 2Research Institute for Diseases of the Chest, Graduate School of Medical Sciences, Kyushu University, Fukuoka, Japan
Abstract: COPD is a major cause of chronic morbidity and mortality throughout the world. Although tumor necrosis factor-α (TNF-α) has a critical role in the development of COPD, the role of different TNF receptors (TNFRs) in pulmonary emphysema has not been resolved. We aimed to clarify the role of TNFRs in the development of pulmonary emphysema. TNF-α transgenic mice, a murine model of COPD in which the mice spontaneously develop emphysema with a large increase in lung volume and pulmonary hypertension, were crossed with either TNFR1-deficient mice or TNFR2-deficient mice. After 6 months, the gross appearance of the lung, lung histology, and pulmonary and cardiac physiology were determined. In addition, the relationship between apoptosis and emphysema was investigated. Pulmonary emphysema-like changes disappeared with deletion of TNFR1. However, slight improvements were attained with deletion of TNFR2. Apoptotic cells in the interstitium of the lung were observed in TNF-α transgenic mice. The apoptotic signals through TNFR1 appear critical for the pathogenesis of pulmonary emphysema. In contrast, the inflammatory process has a less important role for the development of emphysema.
Keywords: TNF-α, receptor, emphysema, apoptosis
Introduction
COPD is a major cause of chronic morbidity and mortality throughout the world. It is characterized by airflow limitation that is not fully reversible and is an umbrella term for the commonly associated manifestations of pulmonary emphysema, alveoli destruction, and chronic bronchitis. Pulmonary emphysema is characterized by reduced maximal expiratory flow, increased lung volume, and alveolar wall destruction.1,2 The disease has a strong relationship with tobacco smoking, and as many as 2 million people in the US suffer from pulmonary emphysema. However, only 10%–20% of smokers develop clinically significant COPD, and a precise understanding of COPD pathogenesis remains elusive. Moreover, there is no proven clinically effective treatment that promotes recovery from established emphysema,3–6 and despite extensive research, the clinical management of patients with pulmonary emphysema remains mostly supportive.
Despite the suggestion that COPD might be an autoimmune disease,7 the current paradigm for COPD pathogenesis is that a chronic airflow limitation results from an abnormal inflammatory response to inhaled particles and gases in the lung, resulting in alveoli and bronchial inflammation in susceptible smokers. Inflammation in the peripheral airspaces at different stages of disease severity has been investigated.8 Furthermore, a protease–antiprotease imbalance has been shown to be a major contributor to COPD,9 and emphysema development may involve alveolar cell loss through apoptosis.10 Therefore, several important processes appear to contribute to COPD development.11 In addition to these factors, chronic inflammation affects lung morphogenesis and causes several pathological involvements, including COPD.12
Tumor necrosis factor-α (TNF-α) is a pleiotropic cytokine critical for controlling a vast array of immunological responses, including host defense. TNF-α binds to one of the two receptors (TNF receptor [TNFR] 1 and TNFR2), which are found on almost all cell types,13,14 evoking numerous reactions, such as apoptosis, tumor cell lysis in vitro, hemorrhagic necrosis of tumors, shock, tissue damage, bone resorption, fever, T-cell proliferation, dermal necrosis, and insulin resistance.15 We have investigated the relationship between TNF-α and respiratory diseases using TNF-α transgenic (TNF-tg) mice. These mice overexpress TNF-α mainly in the lungs and develop emphysematous changes that are similar to COPD.16,17 There are several lines of clinical evidence suggesting that TNF-α appears to be important in COPD,18–20 with loss of body mass associated with higher levels of serum TNF-α18 and high levels of TNF-α present in sputum from COPD patients.19 Several studies also suggest that TNF-α appears to contribute to COPD pathogenesis in animal studies.8,20 Although TNF-α has a critical role in the development of COPD, the role of different TNFRs in pulmonary emphysema has not been resolved. Recently, D’hulst et al21 reported that TNFR2 played a more significant role in cigarette-induced pulmonary inflammation and pulmonary emphysema. Here, we clarify the role of TNFRs in the development of COPD using TNF-α-overexpressing mice.
Materials and methods
Animals
SP-C/TNF-tg mice, TNFR1-knockout mice (TNFR1−/−), and TNFR2-knockout mice (TNFR2−/−) on a C57BL/6 background were used in this study. Both TNFR1−/− mice22 and TNFR2−/− mice23 were purchased from Jackson Laboratory. The TNF-tg is heterogeneous, so TNF-tg × TNFR1−/− mice were obtained by initially crossing TNF-tg mice with TNFR1−/− mice. Next, TNF-tg × TNFR1+/- mice crossed with TNFR1−/− mice as confirmed by polymerase chain reaction methods (Figure 1). TNF-tg × TNFR2−/− mice were obtained using similar methods. These mice were bred in an animal facility documented to be free of murine-specific pathogens. Additional detail on the method for making these measurements is provided in an online data supplement.
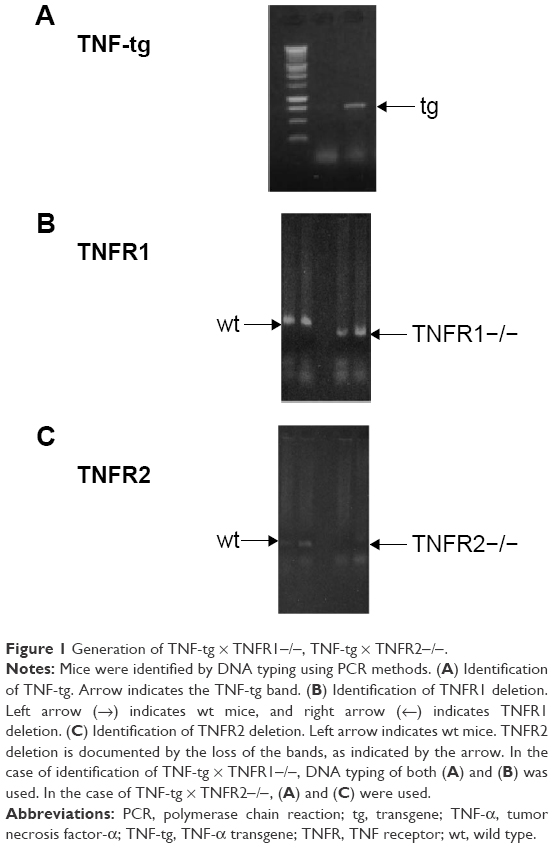 | Figure 1 Generation of TNF-tg × TNFR1−/−, TNF-tg × TNFR2−/−. |
This study was approved by the Institutional Animal Care and Use Committee (IACUC) of Fukuoka University, and followed the Guidelines for Animal Experimentation, Fukuoka University. The IACUC are charged with protecting the safety and welfare of animals used in research at or in conjunction with Fukuoka University.
Pulmonary and cardiovascular physiology
Pulmonary physiological parameters were examined using previously described methods.16,24,25 After measurements, mice were sacrificed by intraperitoneal injection of sodium pentobarbital. Functional residual capacity was measured by saline displacement. The heart was then carefully dissected and divided into the right ventricular (RV) wall and left ventricular (LV) wall with septa (S), and each was weighed separately.
Lung histology, morphometry, and Tdt nick-end labeling
Lungs were inflated at 25 cmH2O static pressure by intratracheal instillation of 4% paraformaldehyde in phosphate-buffered saline before 4 μm-thick tissue sections were stained with hematoxylin and eosin. The mean linear intercept, an indicator of air space size, was calculated for each mouse as described previously.16,24 To detect apoptosis, lung specimens were stained using the In Situ Cell Death Detection Kit (Boehringer Mannheim, Indianapolis, IN, USA) according to the manufacturer’s protocol.26 In addition, apoptosis was confirmed by electron microscopy. Lungs were fixed with 2.5% glutaraldehyde for 18 hours. Next, lungs were refixed with 1% OsO4 and divided into small sections and embedded in epon. Finally, epon-embedded sections were cut using a glass knife and observed by electron microscopy.
Bronchoalveolar lavage fluids and TNF-α enzyme-linked immunosorbent assay
The lungs were lavaged five times with 1 mL aliquots of phosphate-buffered saline, and cells in the lavage fluid were counted with a hemocytometer. The level of TNF-α in bronchoalveolar lavage (BAL) fluids was determined using a mouse TNF-α enzyme-linked immunosorbent assay kit (R&D Systems. Inc., Minneapolis, MN, USA) according to the manufacturer’s protocol. For gelatin zymography, 40 μL of BAL fluid was electrophoresed in a sodium dodecyl sulfate polyacrylamide gel containing 1% gelatin under nonreducing conditions as described previously.16
Statistics
The data were expressed as the mean ± standard error. A Mann–Whitney U test was used to compare the two groups. A P-value of <0.05 was considered as significant difference. For comparing mortality, the Kaplan–Meier estimation for survival curves was used. The statistical analyses were performed using a StatView 5.0 (SAS Institute Inc., Cary, NC, USA).
Results
SP-C/TNF-tg overexpresses TNF-α in the lung, which results in chronic inflammation, the destruction of lung tissue and alveolar enlargement, similar to the changes seen in pulmonary emphysema.16 As described previously,16 physiological assessment indicated a decrease in elastic recoil and an increase in lung capacity consistent with pulmonary emphysema. Although these findings were found at high altitude (1,600 m), a similar phenotype has been attained in Fukuoka city, at sea level.27 After establishment of the mice, TNF-α levels in BAL fluids were determined by enzyme-linked immunosorbent assay. Similar levels of TNF-α were produced by all three mice, with higher levels compared with wild-type (wt) mice (wt, 9.69+0.62 pg/mL; TNF-tg, 74.8+2.2 pg/mL; TNF-tg × TNFR1−/−, 84.0+1.8 pg/mL; TNF-tg × TNFR2−/−, 85.8+9.3 pg/mL).
Previous studies have shown that the lungs of TNF-tg mice had a characteristic phenotype: larger with orange-colored pleural surfaces compared with wt mice.16,27 In contrast to these findings, the lungs from TNF-tg × TNFR1−/− mice were similar to wt mice at the macrolevel, while TNF-tg × TNFR2−/− mice exhibited smaller lungs than those from TNF-tg mice but were larger and orange in color when compared with wt mice (Figure 2). The lung weight is consistent with the macrolevel findings of the lungs (Table 1).
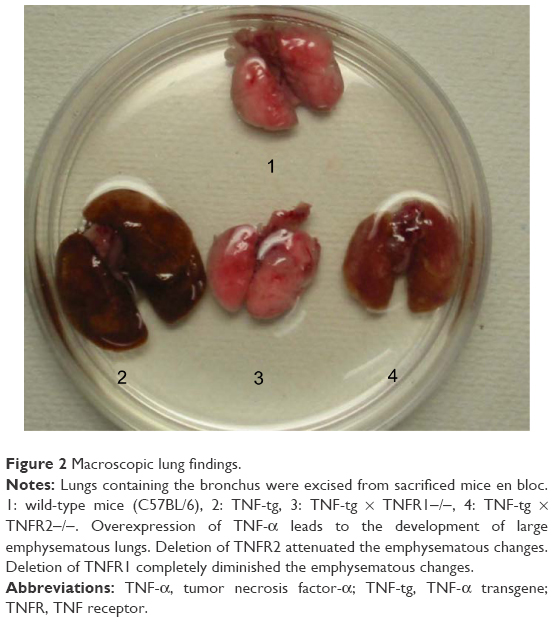 | Figure 2 Macroscopic lung findings. |
 | Table 1 Mice used in this experiment |
Histological analysis of the lungs from TNF-tg mice showed evidence of inflammatory cell (mainly lymphocyte) infiltration and alveolar enlargement, whereas these features were attenuated in the lungs of TNF-tg × TNFR2−/−. Significantly, there was no significant histological difference between wt mice and TNF-tg × TNFR1−/− mice, suggesting that the inflammation was completely abrogated (Figure 3). These histological findings were confirmed by a morphometric study using the mean linear intercept, which showed significant alveolar enlargement in TNF-tg and TNF-tg × TNFR2−/−, compared with wt and TNF-tg × TNFR1−/− (Figure 4A), and TNF-tg compared with TNF-tg × TNFR2−/−.
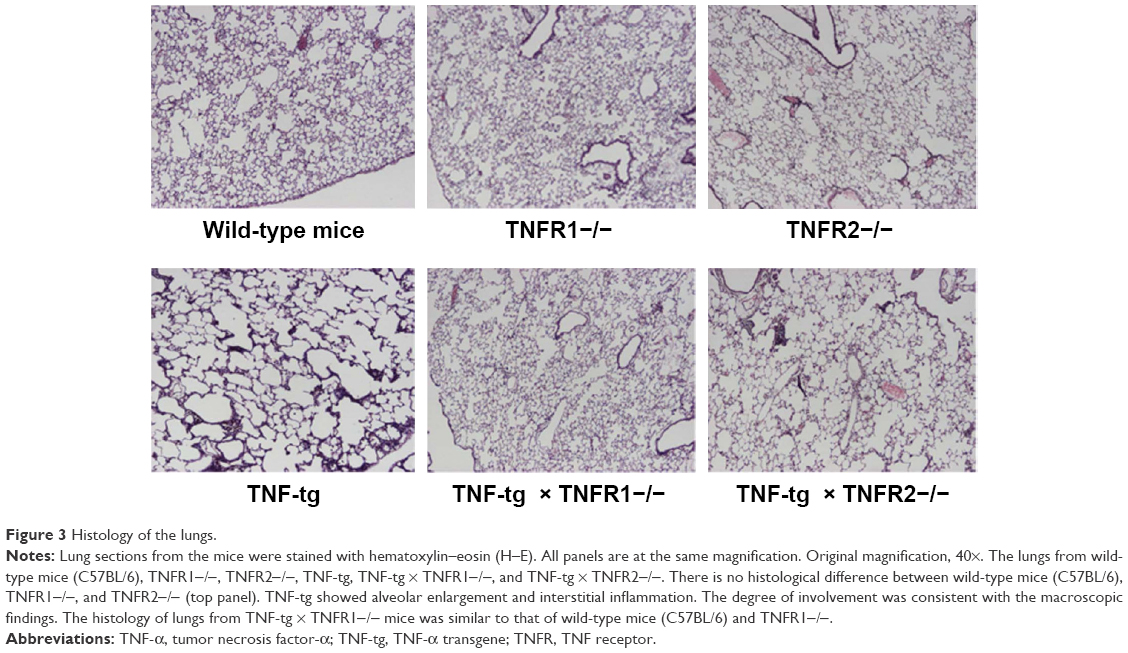 | Figure 3 Histology of the lungs. |
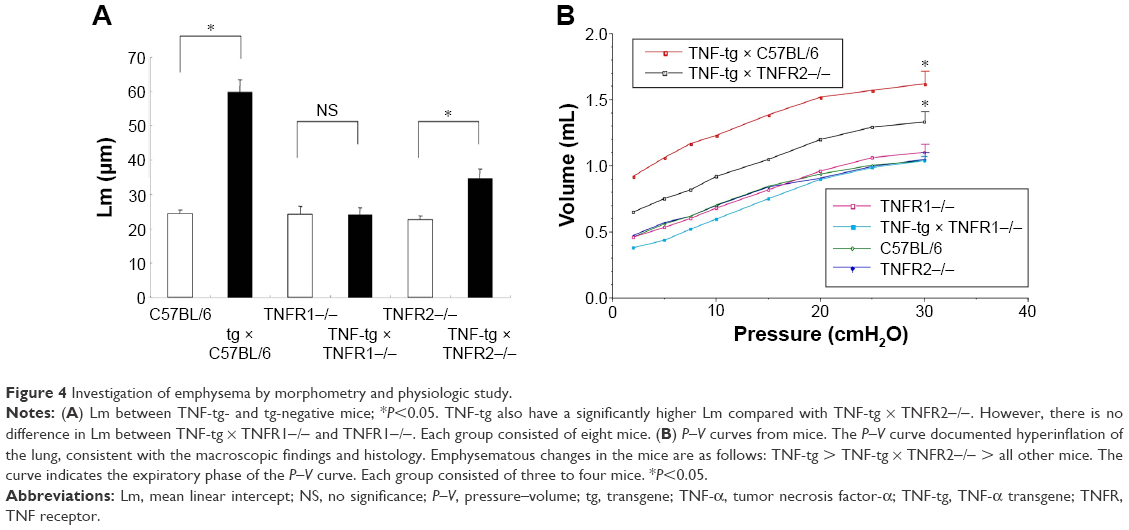 | Figure 4 Investigation of emphysema by morphometry and physiologic study. |
Physiological studies of TNF-tg mice bred in Fukuoka revealed an increase in TLC and functional residual capacity, similar to the phenotype of emphysema but not fibrosis. TNF-tg × TNFR2−/− mice demonstrated an intermediate pressure–volume curve between wt and TNF-tg mice, whereas the pressure–volume curve for TNF-tg × TNFR1−/− mice was similar to that of wt mice (Figure 4B). In the present study, we investigated RV/LV + S and demonstrated that TNF-tg mice also demonstrated RV hypertrophy compared with wt mice. TNF-tg × TNFR2−/− mice showed a slight but not significant increase in RV/LV + S, while the RV/LV + S of TNF-tg × TNFR1−/− was similar to wt mice (Table 1). This indicates that the RV hypertrophy is associated with the involvement of the lung.
Analysis of the cellular infiltrate of the BAL fluids showed that there was an accumulation of neutrophils in TNF-tg mice (~60% macrophages, ~30% neutrophils, and ~10% lymphocytes) but not in TNF-tg × TNFR1−/− mice (~95% macrophages, ~3% neutrophils, and ~2% lymphocytes). Interestingly, TNF-tg × TNFR2−/− mice demonstrated a severe neutrophil accumulation (~20% macrophages, ~75% neutrophils, and ~5% lymphocytes) (Figure 5A). We have previously shown that matrix metalloproteinase (MMP) activation was observed in TNF-tg mice.16 In the present study, a 200 kDa gelatinase was observed in BAL fluids from both TNF-tg and TNF-tg × TNFR2−/− but not in BAL fluids from wt mice or TNF-tg × TNFR1−/− mice (Figure 5B).
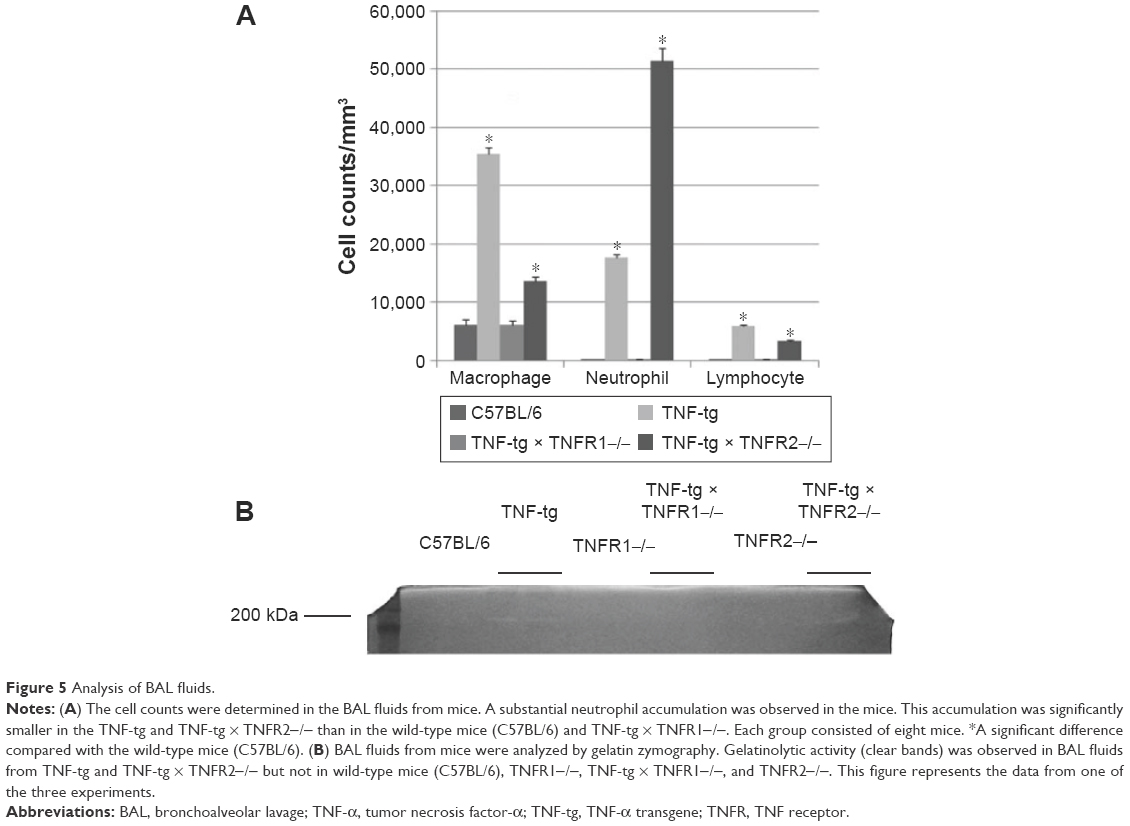 | Figure 5 Analysis of BAL fluids. |
Tdt nick-end labeling staining clearly demonstrated Tdt nick-end labeling-positive signals in the interstitium of the lung in TNF-tg mice, while wt mice and TNF-tg × TNFR1−/− did not show positive signals (Figure 6A and B). Electron microscopy revealed that the apoptotic cells, showing characteristic condensed-nuclear material, were phagocytosed by macrophages (Figure 6C). These data indicated that apoptosis had occurred in TNF-tg mice.
 | Figure 6 Apoptosis in COPD pathogenesis. |
Discussion
TNF-α has been considered to be an important factor for the pathogenesis of pulmonary fibrosis.28,29 Although TNF-tg mice were originally reported as a pulmonary fibrosis model,30 we have demonstrated that TNF-α-overexpressing mice have a phenotype that more closely resembles the physiological features of pulmonary emphysema.16 Others have demonstrated the critical role of TNF-α in the pathogenesis of COPD.17–20 Presently, TNF-α is considered to play a central role in the pathogenesis of COPD. Interferon (IFN)-γ-overexpressing mice demonstrated that chronic inflammation can lead to emphysema,31 similar to TNF-α-overexpressing mice, therefore implying that chronic inflammation is associated with emphysematous changes. However, the role of the TNFR in COPD remains undetermined. Only D’hulst et al21 have reported that TNFR2 plays a certain role in a cigarette smoke-induced emphysema model.
TNF-α binds to two kinds of receptors (TNFR1 and TNFR2), present on virtually all cells throughout the body, and evokes a variety of reactions.13–15 The role of TNF-α and its receptors has been vigorously investigated in the field of infection and immunity. TNFR1 is known to have a major role in host immunity against pathogens,32,33 while the role of TNFR2 remains uncertain. In contrast to TNFR1, TNFR2 does not have a death domain, a signaling element that induces apoptosis.34
In the present study, it is surprising that the offspring from crossing TNF-tg mice with TNFR1-deleted mice (TNF-tg × TNFR1−/−) demonstrated a complete abrogation of the emphysematous changes that are present in TNF-tg mice. In contrast, TNF-tg × TNFR2−/− mice showed only a slight attenuation of the emphysematous changes. Since the TNF-α production was similar in all these mice, the change in lung phenotype could be attributed to the different receptors. It has previously been shown that emphysema was attenuated in TNFR-deficient (both TNFR1 and TNFR2) mice in cigarette smoke-induced emphysema.35 A previous report demonstrated that TNFR2 plays more important roles compared with TNFR1 in emphysema,21 and it is difficult to explain this difference compared with our data. However, it is likely that cigarette smoking induces a more complex array of signaling pathways than TNF-α alone.
The main difference between TNFR1 and TNFR2 is the transduction of apoptotic signals, whereby TNFR1 can transduce a death signal.34 There is a large body of evidence describing a relationship between apoptosis and emphysema. One study has also shown that apoptosis in lung tissue is inversely correlated with the surface area.10 The model used a single intratracheal injection of active caspase-3 to induce emphysematous changes.36 This study provides direct evidence that alveolar wall apoptosis is sufficient to cause pulmonary emphysema, even without the accumulation of inflammatory cells. Taking these data into consideration, we hypothesize that apoptosis has a critical role in the pathogenesis of COPD. In the present study, apoptosis has been shown in TNF-tg mice. Another chronic inflammation model using IFN-γ-overexpressing mice also demonstrated the association of apoptosis and emphysematous changes.37 Our data are consistent with the recent hypothesis that apoptosis plays a critical role in the pathogenesis of emphysema.
Although the present data indicated a critical role for TNFR1 in the pathogenesis of COPD, TNFR2−/− mice also demonstrated a slight attenuation in COPD development. Taking this into consideration, the signals through TNFR2 may also play a partial role in COPD pathogenesis. TNF-α induced the expression of MMPs, factors known to be involved in COPD pathogenesis. This activation is thought to be induced by NF-κB,38,39 and it is possible that NF-κB activation through TNFR2 partially contributes to COPD development. We investigated the possible role of MMPs in our model, focusing particularly on gelatinase. We found that TNF-tg mice produced gelatinase, but this was absent in TNF-tg × TNFR−/−. It is clear that many factors, not just TNF-α, contribute to the development of emphysema,34 and it is important to clarify the more precise role of TNFR2 in future studies.
Conclusion
Signals through TNFR1 have critical roles for the pathogenesis in this model. At the present time, there is no effective treatment for pulmonary emphysema, but this study indicated that the blockade of signals through TNFR1 could be a therapeutic target for pulmonary emphysema.
Acknowledgments
The authors appreciate the assistance of Doctor Brian Quinn with language editing. The study was supported by a grant-in-aid for scientific research from the Japan Society for the Promotion of Science, no 14570553 (to MF) and funding from Astra-Zeneca Research Fellow 2003 (to MF).
Disclosure
The authors report no conflicts of interest in this work.
References
Pauwels RA, Buist AS, Calverly PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO global initiative for chronic obstructive lung disease (GOLD) workshop summary. Am J Respir Crit Care Med. 2001;163(5):1256–1276. | ||
The Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) [homepage on the Internet]. 2015. Available from: http://www.goldcopd.org. Accessed January 28, 2016. | ||
Hogg JC. A stimulating treatment for emphysema. Nat Med. 1997;3(6):603–605. | ||
Utz JP, Hubmayr RD, Deschamps C. Lung volume reduction surgery for emphysema: out on a limb without a NETT. Mayo Clin Proc. 1998;73(6):552–566. | ||
Fujita M, Nakanishi Y. The pathogenesis of COPD: lessons learned from in vivo animal models. Med Sci Monit. 2007;13(2): RA19–RA24. | ||
Cornwell WD, Kim V, Song C, Rogers TJ. Pathogenesis of inflammation and repair in advanced COPD. Semin Respir Crit Care Med. 2010;31(3):257–266. | ||
Voelkel N, Taraseviciene-Stewart L. Emphysema: an autoimmune vascular disease? Proc Am Thorac Soc. 2005;2(1):23–25. | ||
Churg A, Dai J, Tai H, Xie C, Wright JL. Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am J Respir Crit Care Med. 2002;166(6):849–854. | ||
Gross P, Pfitzer E, Tolker M, Babayak MA, Kaschak M. Experimental emphysema: its production with papain in normal and silicotic rats. Arch Environ Health. 1965;11:50–58. | ||
Imai K, Mercer BA, Schulman LL, Sonett JR, D’Armiento JM. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J. 2005;25(2):250–258. | ||
Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002–2004. | ||
Petrache I, Natarajan V, Zhen L, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. 2005;11(5):491–498. | ||
Hehlgans T, Mannel DN. The TNF-TNF receptor system. Biol Chem. 2002;383(10):1581–1585. | ||
Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104(4):487–501. | ||
Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. N Engl J Med. 1996;334(26):1717–1725. | ||
Fujita M, Shannon JM, Irvin CG, et al. Overexpression of tumor necrosis factor-a produces an increase in lung volumes and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2001;280(1):L39–L49. | ||
Fujita M, Mason RJ, Cool C, Shannon JM, Hara N, Fagan KA. Pulmonary hypertension in TNF-alpha-overexpressing mice is associated with decreased VEGF gene expression. J Appl Physiol (1985). 2002;93(6):2162–2170. | ||
Di Francia M, Barbier D, Mege JL, Orehek J. Tumor necrosis factor-alpha levels and weight loss in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1994;150(5 pt 1):1453–1455. | ||
Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 1996;153(2):530–534. | ||
Sulkowska M, Sulkowski S, Terlikowski S, Nowak HF. Tumor necrosis factor-alpha induces emphysema-like pulmonary tissue rebuilding. Changes in type II alveolar epithelial cells. Pol J Pathol. 1997;48(3):179–188. | ||
D’hulst AI, Bracke KR, Maes T, et al. Role of tumour necrosis factor-alpha receptor p75 in cigarette smoke-induced pulmonary inflammation and emphysema. Eur Respir J. 2006;28(1):102–112. | ||
Rothe J, Lesslauer W, Lotscher H, et al. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 1993;364(6440):798–802. | ||
Erickson SL, de Sauvage FJ, Kikly K, et al. Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature. 1994;372(6506):560–563. | ||
Fujita M, Ye Q, Ouchi H, et al. Retinoic acid fails to reverse emphysema in adult mouse models. Thorax. 2004;59(3):224–230. | ||
Irvin CG, Tu YP, Sheller JR, Funk CD. 5-lipoxygenase products are necessary for ovalbumin-induced airway responsiveness in mice. Am J Phsyiol. 1997;272(6 pt 1):L1053–L1058. | ||
Fujita M, Kuwano K, Kunitake R, et al. Endothelial cell apoptosis in lipopolysaccharide-induced lung injury in mice. Int Arch Allergy Immunol. 1998;117(3):202–208. | ||
Fujita M, Ikegame S, Ye Q, et al. Attenuation of pulmonary hypertension, but not emphysematous change, by breeding emphysema model mice at sea level. Cytokine. 2008;41(3):286–292. | ||
Piguet PF, Collart MA, Grau GE, Kapanci Y, Vassalli P. Tumor necrosis factor/cachectin plays a key role in bleomycin-induced pneumopathy and fibrosis. J Exp Med. 1989;170(3):655–663. | ||
Piguet PF, Collart MA, Grau GE, Sappino AP, Vassalli P. Requirement of tumour necrosis factor for development of silica-induced pulmonary fibrosis. Nature. 1990;344(6263):245–247. | ||
Miyazaki Y, Araki K, Vesin C, et al. Expression of a tumor necrosis factor-alpha transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J Clin Invest. 1995;96(1):250–259. | ||
Wang Z, Zheng T, Zhu Z, et al. Interferon gamma induction of pulmonary emphysema in the adult murine lung. J Exp Med. 2000;192(11):1587–1600. | ||
Black CM, Bermudez LE, Young LS, Remington JS. Co-infection of macrophages modulates interferon gamma and tumor necrosis factor-induced activation against intracellular pathogens. J Exp Med. 1990;172(3):977–980. | ||
Pfeffer K, Matsuyama T, Kundig TM, et al. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73(3):457–467. | ||
Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281(5381):1305–1308. | ||
Churg A, Wang RD, Tai H, Wang X, Xie C, Wright JL. Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 2004;170(5):492–498. | ||
Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am J Respir Cell Mol Biol. 2003;28(5):555–562. | ||
Zheng T, Kang MJ, Crothers K, et al. Role of cathepsin S-dependent epithelial cell apoptosis in IFN-gamma-induced alveolar remodeling and pulmonary emphysema. J Immunol. 2005;174(12):8106–8115. | ||
Hozumi A, Nishimura Y, Nishiuma T, Kotani Y, Yokoyama M. Induction of MMP-9 in normal human bronchial epithelial cells by TNF-alpha via NF-kappa B-mediated pathway. Am J Physiol Lung Cell Mol Physiol. 2001;281(6):L1444–L1452. | ||
Serandour AL, Loyer P, Garnier D, et al. TNF alpha-mediated extracellular matrix remodeling is required for multiple division cycles in rat hepatocytes. Hepatology. 2005;41(3):478–486. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.