Back to Journals » Journal of Inflammation Research » Volume 17
Comprehensive Gene Analysis Reveals Cuproptosis-Related Gene Signature Associated with M2 Macrophage in Staphylococcus aureus-Infected Osteomyelitis
Authors Shi X, Ni H, Tang L, Li M, Wu Y, Xu Y ![]()
Received 19 February 2024
Accepted for publication 8 May 2024
Published 16 May 2024 Volume 2024:17 Pages 3057—3077
DOI https://doi.org/10.2147/JIR.S457414
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Xiangwen Shi,1,2,* Haonan Ni,3,* Linmeng Tang,4,* Mingjun Li,2 Yipeng Wu,2 Yongqing Xu3
1Graduate School, Kunming Medical University, Kunming, People’s Republic of China; 2Laboratory of Yunnan Traumatology and Orthopedics Clinical Medical Center, Yunnan Orthopedics and Sports Rehabilitation Clinical Medical Research Center, Department of Orthopedic Surgery, 920th Hospital of Joint Logistics Support Force of PLA, Kunming, People’s Republic of China; 3First People’s Hospital of Huzhou, the First affiliated Hospital of Huzhou University, Huzhou, People’s Republic of China; 4Department of Radiology, the Second Affiliated Hospital of Kunming Medical University, Kunming, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yongqing Xu, Department of Orthopedic Surgery, 920th Hospital of Joint Logistics Support Force, 212 Daguan Road, Xi Shan District, Kunming, Yunnan, 650031, People’s Republic of China, Email [email protected] Yipeng Wu, Department of Orthopedic Surgery, 920th Hospital of Joint Logistics Support Force, 212 Daguan Road, Xi Shan District, Kunming, Yunnan, 650031, People’s Republic of China, Email [email protected]
Objective: Osteomyelitis is a challenging disease in the field of bone infections, with its immune and molecular regulatory mechanisms still poorly understood. The aim of this study is to explore the value and potential mechanisms of cuproptosis-related genes (CRGs) in Staphylococcus aureus (S. aureus)-infected osteomyelitis from an immunological perspective.
Methods: Initially, three transcriptomic datasets from public databases were integrated and analyzed, and consistent expression of CRGs in S. aureus-infected osteomyelitis was identified. Subsequently, immune infiltration analysis was performed, and M2 macrophage-related CRGs (M2R-CRGs) were further identified. Their potential molecular mechanisms were evaluated using Gene Set Variation Analysis (GSVA) and Gene Set Enrichment Analysis (GSEA). Finally, distinct osteomyelitis subtypes and diagnostic models based on characteristic M2R-CRGs were constructed.
Results: Through correlation analysis with immune cell infiltration, three characteristic M2R-CRGs (SLC31A1, DLD, and MTF1) were identified. Further analysis using unsupervised clustering and immune microenvironment analysis indicated that cluster 1 might activate pro-inflammatory responses, while cluster 2 was shown to exhibit anti-inflammatory effects in osteomyelitis. Compared to Cluster A, Cluster B demonstrated higher levels and a greater diversity of immune cell infiltrations in CRG-related molecular patterns, suggesting a potential anti-inflammatory role in osteomyelitis. A diagnostic model for S. aureus-infected osteomyelitis, based on the three M2R-CRGs, was constructed, exhibiting excellent diagnostic performance and validated with an independent dataset. Significant upregulation in mRNA and protein expression levels of the three M2R-CRGs was observed in rat models of S. aureus-infected osteomyelitis, aligning with bioinformatic results.
Conclusion: The M2R-CRGs (SLC31A1, DLD, and MTF1) may be considered characteristic genes for early diagnosis and personalized immune therapy in patients with S. aureus-infected osteomyelitis.
Keywords: osteomyelitis, cuproptosis, Staphylococcus aureus, M2 macrophage, immune infiltration, diagnosis
Graphical Abstract:

Introduction
Osteomyelitis is a prevalent infectious disease in orthopedics, primarily caused by Staphylococcus aureus, which is responsible for 75% of cases.1–3 This condition often develops following trauma, open fractures, or implant-related infections and affects the skin, soft tissues, muscles, and bones.4,5 Challenges in treating osteomyelitis include compromised blood supply and biofilm formation, which hinder the effectiveness of antibiotic treatments both locally and systemically.6,7 These challenges make clinical management difficult, often resulting in prolonged treatment and a high risk of recurrence.8 Consequently, osteomyelitis is not only a significant clinical issue but also imposes considerable economic burdens on patients and healthcare systems.
The clinical diagnosis of osteomyelitis typically involves assessing symptoms, laboratory tests, and radiological exams, but a definitive diagnosis usually requires an invasive bone tissue biopsy.9 In the early stages, the disease may not show symptoms, complicating early detection. During these initial phases, X-ray imaging is often not sensitive enough to detect osteomyelitis.10 Similarly, common inflammatory markers such as C-reactive protein and erythrocyte sedimentation rate lack specificity for osteomyelitis.11,12 Although Magnetic Resonance Imaging (MRI) provides better staging and aids in surgical planning, it mainly achieves higher sensitivity in the later stages of the disease.13 Consequently, the medical community faces a significant challenge in quickly and accurately diagnosing osteomyelitis due to the absence of rapid, straightforward, and specific diagnostic tools.
The development and progression of osteomyelitis infected by Staphylococcus aureus (S. aureus) involve intricate immune responses and inflammatory environments, affecting the balance between osteoblasts and osteoclasts.14 During chronic osteomyelitis, S. aureus can survive within phagocytic cells (macrophages and neutrophils) and non-professional phagocytic cells (such as osteocytes and osteoblasts). Macrophages that harbor intracellular infections are termed “Trojan horse” macrophages, facilitating bacterial dissemination within the body.15,16 Moreover, S. aureus employs microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) and adhesion proteins to anchor itself within bone tissue, which promotes further bacterial attachment.17–19 S. aureus biofilms can actively evade recognition by toll-like receptor 2 (TLR2) and TLR9, thereby biasing macrophage responses towards anti-inflammatory or pro-fibrotic M2 phenotypes.20 The M2 phenotype contributes to a pre-fibrotic response during chronic implant-associated osteomyelitis, ultimately leading to abscess formation.21,22 The Staphylococcal protein A (SpA) plays a crucial role in evading the host’s immune defenses. Additionally, SpA interacts directly with osteoblasts and osteoclasts, altering their function and differentiation, which leads to inflammation.23–25 Kim et al 2015,26 demonstrated that S. aureus activates toll-like receptor 2 (TLR2) in osteoblasts, increasing the production of pro-inflammatory cytokines, thereby causing inflammation and bone damage typical of osteomyelitis. This suggests that targeting the immunological and inflammatory aspects of S. aureus infection could provide valuable therapeutic opportunities for osteomyelitis.
Previous research has shown that S. aureus-infected osteomyelitis is associated with various types of cell death, including apoptosis and pyroptosis.27,28 S. aureus can induce cell death in osteoblasts and reduce their activity, contributing to bone loss through multiple pathways such as pyroptosis, necrosis, apoptosis, and autophagy.29–31 A recent study by Tsvetkov et al 2022,32 identified a new cell death pathway, cuproptosis, characterized by intracellular copper accumulation that triggers lipid peroxidation, mitochondrial enzyme aggregation, and destabilization of iron-sulfur (Fe-S) cluster proteins, leading to cell death. Additionally, Mendelsohn et al 2023,33 observed mitochondrial dysfunction in patients with chronic osteomyelitis, linking it to the accumulation of intracellular reactive oxygen species that induce osteoblast cell death. Interestingly, another study found that using an inhibitor that targets the Programmed cell death 1/Programmed cell death ligand 1 (PD-1/PD-L1) pathway significantly reduced mitochondrial autophagy in F4/80 macrophages in S. aureus-infected osteomyelitis, which in turn decreased bone destruction in affected mice.34 These findings suggest that cuproptosis could have a regulatory role in the pathology of S. aureus-infected osteomyelitis.
This study represents the first systematic investigation into the differences in cuproptosis-related genes (CRGs) and the immune microenvironment between patients with S. aureus-infected osteomyelitis and healthy controls (Figure 1). From an immunological standpoint, the research focused on M2 macrophage-related CRGs (M2R-CRGs), leading to the development of reliable diagnostic and subclassification models specifically for patients with S. aureus-infected osteomyelitis. These models provide insights into potential mechanisms of immune and inflammatory regulation mediated by the three identified M2R-CRGs in osteomyelitis. Additionally, the study included experimental validation of the expression levels of three M2R-CRGs, further substantiating the findings.
 |
Figure 1 The specific flowchart of this study. |
Materials and Methods
Microarray Data Processing
Gene expression data for the three osteomyelitis datasets were sourced from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). The GSE6269 dataset included 15 osteomyelitis samples and 6 health samples, the GSE16129 dataset included 70 osteomyelitis samples and 29 health samples, and GSE30119 included 39 osteomyelitis samples and 44 health samples. The GSE6269 and GSE16129 datasets were sequenced by the GPL96 platform, and the GSE30119 dataset was sequenced by the GPL6947 platform, and all datasets were included only human species. To verify the consistency of the results, the GSE6269 and GS16129 data sets were combined and used as the training set, and the ComBat function in the “sva” package35 of R software was used to harmonize 85 osteomyelitis and 35 healthy samples, and the separate GSE30119 data set was used as the validation set. ComBat36 is a commonly used method for correcting batch effects in gene expression data. It assumes that most genes do not exhibit differential expression across batches, but allows for some genes to be differentially expressed. Specifically, it is essential to ensure that the data is appropriately normalized before implementing ComBat to minimize the impact of technical biases. Initially, data with higher raw gene expression levels are log-transformed, taking the log2 values. Subsequently, normalized data obtained from the GSE6269 and GSE16129 datasets are used as inputs for the ComBat function. Following batch effect estimation, expression data adjustments, and Empirical Bayes Shrinkage, the unwanted technical variations introduced by batch effects are eliminated.
Based on previous studies,37,38 19 CRGs were extracted as pre-prepared gene sets for further identification and analysis. The details of CRGs were shown in Supplementary Table S1. The gene expression data were output in log2 form for larger values.
Identification of Differentially Expressed (DE)-CRGs
Based on 19 pre-prepared CRGs, the expression of CRGs in each sample in the training set was extracted using the “limma” package39 and the results were output in Supplementary Table S2. To explore the CRGs with significant differences between osteomyelitis samples and healthy control samples, DE-CRGs were screened by differential expression analysis, and a differential heatmap and a boxplot were created using “pheatmap” and “ggpubr” packages to visualize the DE-CRGs. This analysis screened for DE-CRGs using specific criteria: an adjusted P-value of less than 0.05 and an absolute log fold change (FC) greater than 0.585.
Functional Enrichment Analysis
The potential biological functions of DE-CRGs were further explored through enrichment analysis. The enrichment analysis of Gene Ontology (GO) biological process (BP), cellular component (CC), molecular function (MF) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways was performed using the online website (https://hiplot.com.cn), where the top 5 significantly enriched functions and pathways were identified. These were visualized in the form of enrichment network diagrams, and a P<0.05 was considered significant enrichment.
Immune Infiltration Analysis in Osteomyelitis
Given the osteomyelitis-mediated inflammation and immune microenvironment characterized by the infiltration of multiple immune cells such as B cells, T cells, and macrophages, the immune infiltration of each osteomyelitis and control group was scored using the “CIBERSORT” algorithm.40 This comprehensive assessment of the overall immune landscape of osteomyelitis included a comparison of the infiltration abundances of 22 immune cells in osteomyelitis and control samples, which were visualized using histograms and boxplots by the “ggpubr” package. The importance of the anti-inflammatory phenotype of M2 macrophage polarization in osteomyelitis treatment prompted further correlation analysis between DE-CRGs and immune cells. M2 macrophages showed significant correlation with DE-CRGs. Finally, osteomyelitis samples were categorized into high- and low-expression groups based on the median expression of the target genes, to explore the differences in the proportion of M2 macrophages between these groups.
Single-Gene GSEA (ssGSEA) and Single-Gene ssGSVA (ssGSVA)
GSEA analysis is a gene function annotation and analysis method for determining whether a gene set or representative gene collection is significantly enriched under a given biological condition.41 GSVA reflects changes in gene function through the degree of expression variation in a gene set and is a method for assessing changes in pathway enrichment in a sample population.42 To understand the biological functions and metabolic pathways of key DE-CRGs, ssGSEA and ssGSVA were used to further understand the molecular mechanisms. P<0.05 indicated that the pathway was considered significantly enriched.
PPI Network Analysis
To explore genes in osteomyelitis with potential functional and physical interactions with DE-CRGs, a protein PPI network for DE-CRGs was constructed separately using the STRING online website (https://cn.string-db.org/).43 After obtaining the nodes and lines of the PPI network, Cytoscape software (version 3.9.1)44 was used to visualize the hub genes and sort them by the size of the degree centrality (DC). To construct the PPI networks of DE-CRGs, the low confidence value was set to 0.15.
Construction of Transcription Factors (TF) and miRNAs Engage with M2R-CRGs
To further explore the potential molecular mechanisms of M2R-CRGs, TF-miRNAs cohesive regulatory networks associated with hub CRGs were explored and constructed through the NetworkAnalyst online website (https://www.networkanalyst.ca/). Specifically, regulatory networks of hub CRGs interlinked with miRNAs were constructed based on the RegNetwork database (https://regnetworkweb.org/), and the cohesive regulatory networks of hub CRGs and TFs were constructed by ChIP-seq data from the ENCODE database (https://www.encodeproject.org/). Additionally, the GeneMANIA database (http://genemania.org/) was used to further explore the interactions between hub CRGs and construct a gene-gene interaction network based on physical interaction, co-expression, and predication.
Protein-Drug and Gene-Disease Interactions Prediction for M2R-CRGs
Protein and drug target-related information was collected from the DrugBank database (Version 5.0) database (https://go.drugbank.com/), and information on the association of hub CRGs with human diseases was collected from the DisGeNET database (https://www.disgenet.org/), and the acquired interaction networks were visualized by Cytoscape. All the data involved species are human.
Identification of M2R-CRG Patterns
Based on the key M2R-CRG, unsupervised clustering analysis can classify osteomyelitis samples into distinct patterns, potentially aiding in the stratification of osteomyelitis diagnosis and treatment. ConsensusClusterPlus package45 of R software was utilized to perform consistent clustering analysis on osteomyelitis samples, taking into account different characteristics. Clustering parameters were set as follows: maxK=9, reps=50, pItem=0.8, pFeature=1. The most appropriate number of clusters was determined by integrating the results from the consistency score, consistency heatmap and cumulative distribution function (CDF). To ascertain significant differences between the patterns of M2R-CRG with varying characteristics, principal component analysis (PCA) was conducted on the samples both prior to and following clustering.46 Subsequently, the differences of three M2R-CRGs and immune cell infiltration across various CRG clusters were further investigated.
Additionally, DEGs were selected based on various CRG clusters, and osteomyelitis samples were subsequently divided into different gene clusters. The parameter settings mirrored those used for the CRG clusters. The expression differences of M2R-CRGs among these gene clusters were then explored further. PCA was employed to assess the CRG score of each sample, and the consistency of CRG scores between the CRG cluster and gene cluster was compared. Finally, the expression differences in genes from the interleukin family, collagen family, and bone morphogenetic protein-related genes between the two clusters (CRG cluster and gene cluster) were analyzed.
Diagnostic Signature Based on M2R-CRGs
To predict the incidence of osteomyelitis, a nomogram assessment model was constructed based on M2R-CRGs using the “rms” package. Calibration curves were plotted to evaluate the agreement between predicted and observed values. Clinical decision curves (DCA) were also utilized to assess the clinical utility of the nomogram model. Additionally, the diagnostic capability of M2R-CRGs for distinguishing between osteomyelitis and healthy samples was assessed by constructing receiver operating characteristic (ROC) curves and calculating the area under the curve (AUC). Furthermore, a logistic regression model for M2R-CRGs was developed using the “glmnet” package47 to predict sample types in the training set and calculate AUC values. The GSE30119 dataset served as a validation set to verify the diagnostic efficacy of the hub M2R-CRGs.
Validation of M2R-CRGs in Validation Set
To verify significant differences or trends in M2R-CRGs between osteomyelitis and control samples, gene expression data were extracted from the GSE30119 dataset. The expression levels of each M2R-CRG were validated between 39 osteomyelitis and 44 healthy samples.
Rat Models of S. Aureus-Induced Osteomyelitis
A total of twelve Sprague-Dawley (SD) rats weighing approximately 300–500g were procured from Kunming ChuShang Technology Co., Ltd. (Kunming, China), with six rats assigned to the experimental group and six rats to the control group. The rats were anesthetized via intravenous injection of pentobarbital sodium. After skin preparation and disinfection, the skin at the proximal third of the right tibia is fully exposed, and a small hole about 2 mm in size is drilled in the direction of the tibial medullary cavity. In the experimental group, about 1×106 colony-forming units of 6 μL S. aureus were injected into the right tibia of each experimental rat, while the control group received a sterile physiological saline injection at the same site on the right tibia. After the injection is complete, immediately seal the small hole with paraffin and suture the skin layer by layer. Detailed procedures for establishing the rat model of S. aureus-infected osteomyelitis can be found in previous publications by our research team. All experimental procedures were approved by the Nursing and Use of Experimental Animals Ethics Committee of the Joint Logistics Support Force 920 Hospital (2023-007-01) and conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, USA).
RT-qPCR Validation
Four weeks after establishing the animal model, TRIzol reagent (Invitrogen, USA) was employed to extract RNA from tibia tissues of both the experimental and control groups. The SweScript RT I First strand cDNA synthesis kit (Service Bio, Guangzhou, China) was utilized to reverse transcribe RNA into cDNA. Real-time fluorescence was employed for quantitative PCR to measure the amount of DNA present in each PCR cycle. PCR amplification was carried out in the CFX96 real-time quantitative fluorescence PCR instrument over 40 cycles, following the following conditions: pre-denaturation at 95 °C for 1 min; denaturation at 95 °C for 20s; annealing at 55 °C for 20s; and extension at 72 °C for 30s. GAPDH was used as the reference for expression normalization. Data analysis was performed using the 2-ΔΔCT method. The ethnographic primer sets used were as follows: SLC31A1: forward 5′-TATGACCTTCTACTTTGGC-3′ and reverse 5′-ACAGTTTTGTGTGTCTCCA-3′; DLD: forward 5′-GGTGCTGGAGAAATGGTGAA-3′ and reverse 5′-GCCTCTGATAAGGTCGGATG-3′; MTF1: forward 5′-CGGAAAGAAGTAAAGCGGTA-3′ and reverse 5′-GAGGCTGTAGGAGGTGAGGA-3′.
Immunohistochemistry Analysis
4% paraformaldehyde solution was employed to fix the right tibia tissues from both the experimental and control groups. The tissues were subsequently paraffin-embedded and deparaffinized for 15 minutes, followed by extensive rinsing in double-distilled water. Tibia tissues were then incubated overnight at 4°C with monoclonal antibodies against SLC31A1, DLD, and MTF1. These primary antibodies were diluted 1:500 in sterile phosphate-buffered saline (PBS; ServiceBio, Guangzhou, China) and visualized using the DAB substrate kit (Vector Laboratories, CA, USA) to observe the immune reaction. After sectioning the specimens, representative slices were chosen for microscopic examination of their staining patterns. Immunochemistry positive staining was defined as the presence of brown chromogen. The primary antibodies used were as follows: SLC31A1 (DF13356; Affinity Biosciences, Suzhou, China, 1:100), DLD (DF7322; Affinity Biosciences, Suzhou, China, 1:100), MTF1 (Bs-3601R; Bioss Antibodies, Beijing, China, 1:100)。
Statistics Analysis
Pearson’s test was used to reveal the relationship between the M2R-CRGs and immune infiltration. Student’s t-test was used for comparisons between the two groups. Cytoscape was conducted to visualize the PPI, TF-miRNA, and protein-drug network. P<0.05 was considered significant. All data analyses were performed in R software (version 4.1.3).
Results
Identification of DE-CRGs in Osteomyelitis
After merging and eliminating batch effects, 6 CRGs were identified based on 19 CRGs in 85 osteomyelitis and 35 healthy samples, and further differential expression analysis yielded 5 DE-CRGs (Figure 2A and B). The analysis results showed that SLC31A1, DLD, and MTF1 were highly expressed in the osteomyelitis group, while GLS and DBT were lowly expressed in the osteomyelitis group. Further correlation analysis suggested a significant positive correlation between SLC31A1 and MTF1, and negative correlations between SLC31A1 and DLD/GLS, as well as between MTF1 and DLD/DBT (Figure 2C). A circular plot was used to show the specific locations of the 5 DE-CRGs on chromosomes (Figure 2D). MTF1 and DBT are located on chromosome 1, and GLS, DLD, and SLC31A1 are located on chromosomes 2, 7, and 9, respectively.
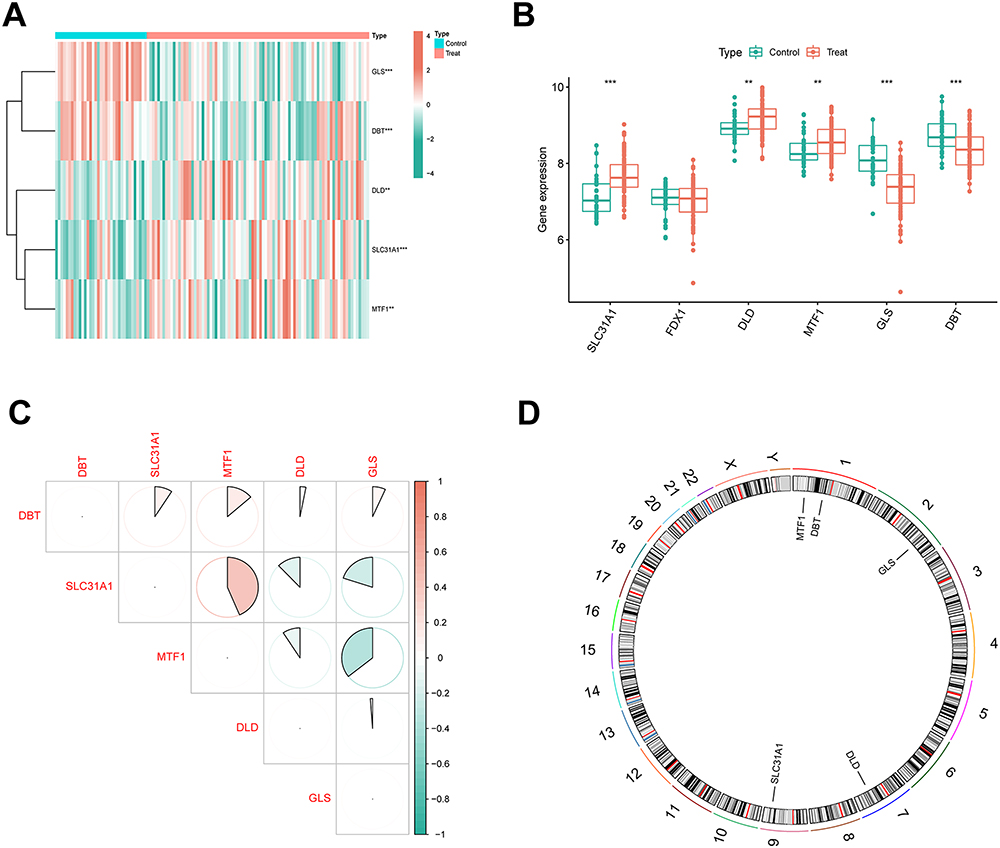 |
Figure 2 Identification and expression characteristics of differentially expressed-cuproptosis-related genes (DE-CRGs). (A) Heatmap displaying the differential expression of DE-CRGs, with red indicating upregulated genes and green indicating downregulated genes. (B) Differential expression of DE-CRGs between osteomyelitis patients and healthy controls. (C) Correlation analysis of DE-CRGs. (D) Chromosomal localization of DE-CRGs. Significance: **P < 0.01, ***P < 0.001. |
Function and KEGG Pathway Enrichment Analyses of DE-CRGs
We further explored the potential biological functions and pathways of 5 DE-CRGs in osteomyelitis through GO and KEGG enrichment analyses. The results of GO enrichment analysis indicated that cellular amino acid catabolic process, mitochondrial matrix, and oxidoreductase activity are significantly enriched modules of BP, CC, and BF, respectively (Figure 3A-C). In the KEGG pathway analysis, DE-CRGs were mainly involved in the enrichment of the propanoate metabolism, valine, leucine and isoleucine degradation, and arginine biosynthesis signaling pathways (Figure 3D). In conclusion, DE-CRGs may play a role in the pathogenesis of osteomyelitis through their involvement in the synthesis and metabolism of amino acids.
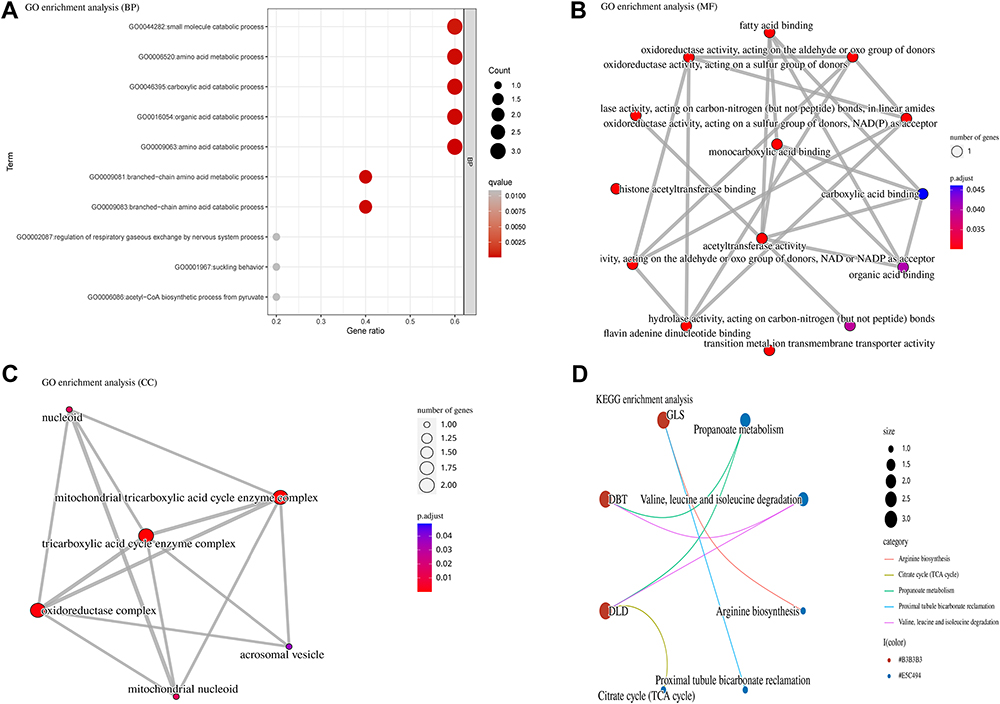 |
Figure 3 Functional enrichment analysis of DE-CRGs. (A) GO enrichment analysis of biological processes (BP). (B) Enriched GO terms for molecular functions (MF). (C) Enriched GO terms for cellular components (CC). (D) Top 5 enriched KEGG pathways. |
Immune Microenvironment Analysis and Identification of M2R-CRGs
In the microenvironment of S. aureus-infected osteomyelitis, there may be an induction of M0 macrophages towards a pro-inflammatory M1 phenotype, which could inhibit the anti-inflammatory effects of M2 macrophages. To explore changes in the immune microenvironment of osteomyelitis, we utilized the CIBERSORT algorithm to calculate immune cell scores for 22 types of immune cells in each osteomyelitis and healthy sample (Figure 4A and B). The results of the immune landscape analysis in osteomyelitis patients showed significant differences in 12 types of immune cells in the immune microenvironment of osteomyelitis patients, with 9 types of immune cells exhibiting increased immune infiltration in osteomyelitis patients, including M0 and M2 macrophages. Subsequently, further investigation was conducted into the relationship between 5 DE-CRGs and the infiltration proportions of 22 types of immune cells, and SLC31A1 (R=0.465, P<0.001), DLD (R=−0.304, P=0.005), and MTF1 (R=0.362, P<0.001) were screened as significantly correlated with M2 macrophages (Figure 4C). 3 DE-CRGs associated with M2 macrophages were presented in a Venn diagram (Figure 4D).
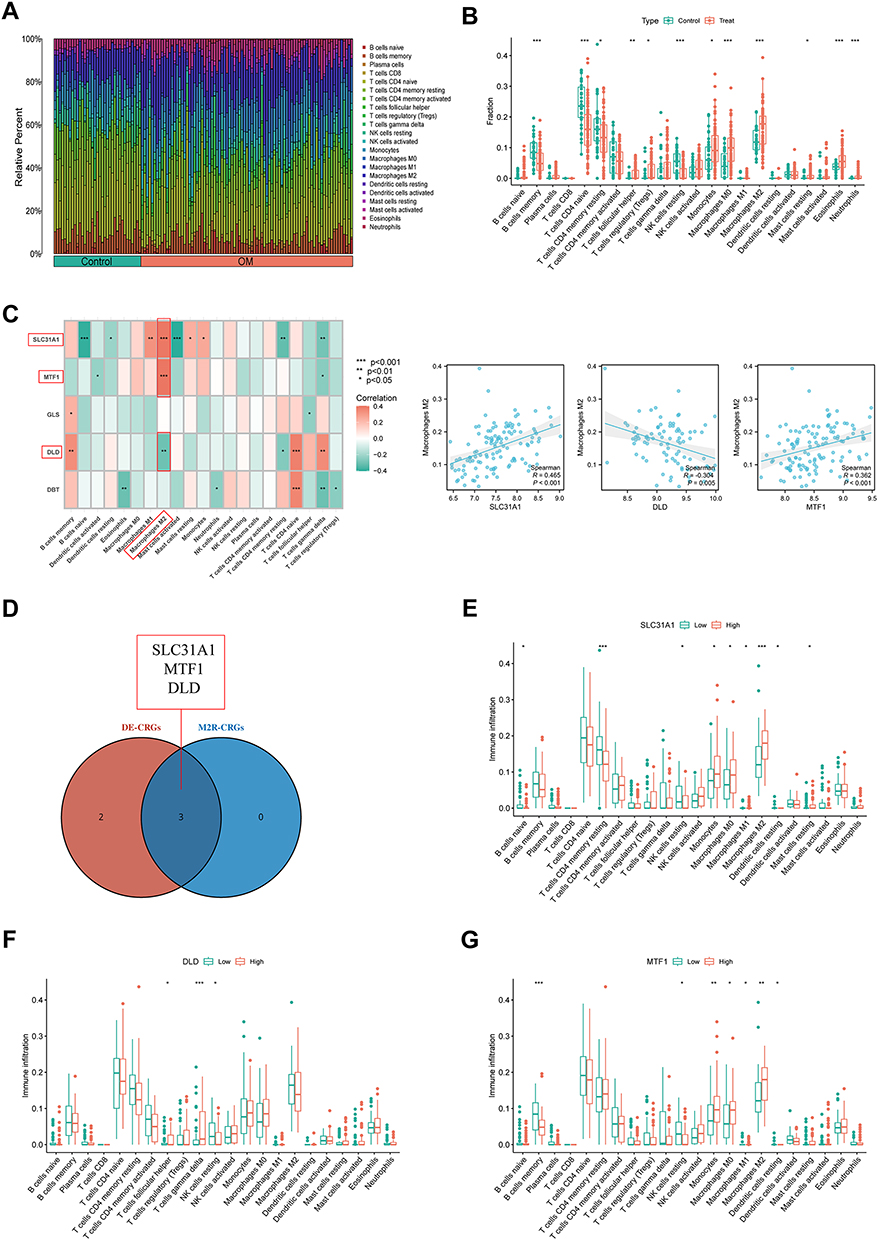 |
Figure 4 Immune microenvironment analysis and M2 macrophage-related CRGs (M2R-CRGs) selection. (A) Immune landscape of 22 immune cell types in osteomyelitis patients. (B) Significant differences in the infiltration levels of 12 immune cell types between osteomyelitis patients and healthy controls. (C) Correlation analysis of DE-CRGs with 22 immune cell types to select M2R-CRGs. (D) Venn diagram of M2R-CRGs. (E) Differences in the infiltration abundance of 22 immune cell types between the high expression group and low expression group of SLC31A1. (F) Differences in the infiltration abundance of 22 immune cell types between the high expression group and low expression group of DLD. (G) Differences in the infiltration abundance of 22 immune cell types between the high expression group and low expression group of MTF1. Significance: *P < 0.05, **P < 0.01, ***P < 0.001. |
Finally, to explore the specific relationship between the expression of key DE-CRGs and the proportions of 22 types of immune cells, the samples were divided into high- and low-expression groups based on the median expression level of key DE-CRGs (Figure 4E-G). The results indicated a significant correlation between SLC31A1 high-expression and high infiltration proportions of M0, M1, and M2 macrophages. T cells follicular helper and T cells gamma delta were significantly upregulated in the DLD high-expression group, while NK cells resting were significantly upregulated in the DLD low-expression group. M0 and M2 macrophages had a higher infiltration proportion in the MTF1 high-expression group, while M1 macrophages had a higher infiltration proportion in the MTF1 low-expression group.
ssGSEA and ssGSVA Enrichment Analyses
We further focused on 3 M2R-CRGs (SLC31A1, MTF1 and DLD) and explored their potential molecular mechanisms and pathways (Figure 5A-C). Firstly, the results of the ssGSEA-pathway enrichment analysis indicated that SLC31A1 was mainly enriched in pathways related to basal cell carcinoma and ECM receptor interaction. The enrichment of MTF1 was significantly associated with pathways related to endocytosis, hypertrophic cardiomyopathy (HCM), melanogenesis, and neuroactive ligand receptor interaction. DLD was mainly enriched in pathways related to glycerophospholipid metabolism, melanogenesis, and cancer.
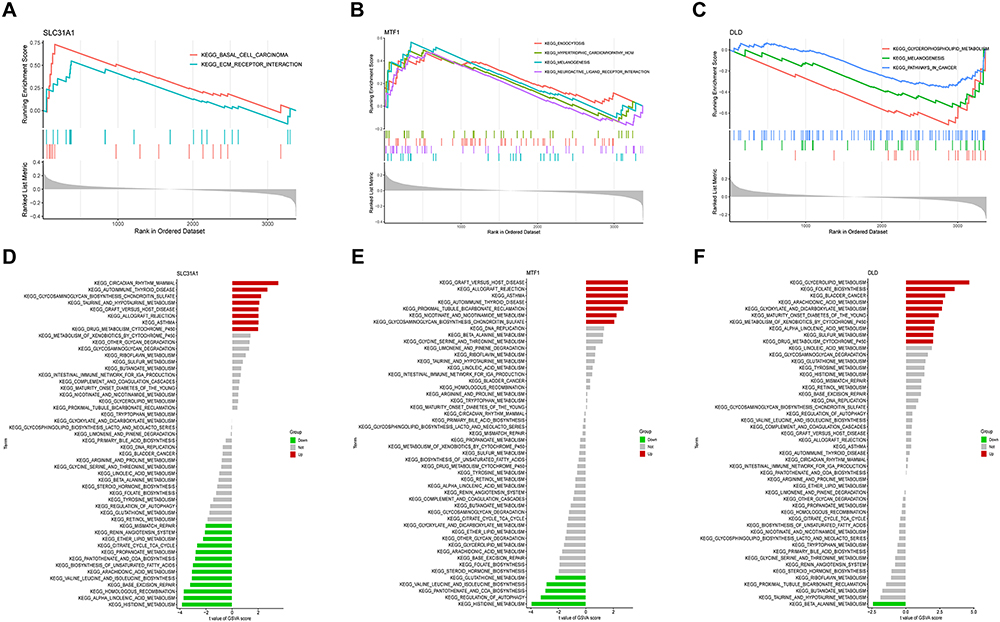 |
Figure 5 Single-gene GSEA-KEGG pathway analysis in SLC31A1 (A), DLD (B), MTF1 (C). Single-gene GSVA-KEGG pathway analysis in SLC31A1 (D), DLD (E), MTF1 (F). |
Next, based on the median expression values of each key DE-CRGs, osteomyelitis samples were divided into high- and low-expression groups, and ssGSVA enrichment analysis was used to further explore potential pathways that may be enriched (Figure 5D-F). The SLC31A1 high-expression group was mainly enriched in circadian rhythm mammal, autoimmune thyroid disease, and glycosaminoglycan biosynthesis chondroitin sulfate-related pathways, while the low-expression group was mainly enriched in histidine metabolism, alpha linolenic acid metabolism, and homologous recombination-related pathways. In the MTF1 high-expression group, pathways related to graft versus host disease, allograft rejection, and asthma were significantly enriched, while pathways related to histidine metabolism, regulation of autophagy, and pantothenate and CoA biosynthesis were significantly enriched in the low-expression group. The DLD high-expression group was mainly enriched in glycerolipid metabolism, folate biosynthesis, and bladder cancer-related pathways, while the low-expression group was only enriched in beta-alanine metabolism-related pathways. The aforementioned enrichment results suggested that SLC31A1, MTF1, and DLD may be involved in regulating the pathogenesis of amino acid metabolism and autoimmune diseases.
Construction of PPI, TF-miRNA, and Protein-Disease Networks
Using SLC31A1, MTF1, and DLD as seed genes, we constructed a protein-protein interaction (PPI) network consisting of 19 nodes and 148 edges, based on the String database, and visualized with the Cytoscape software (Figure 6A and B). To elucidate the molecular mechanisms of critical DE-CRGs, we constructed gene-TF and gene-miRNA regulatory networks, identifying a total of 63 TFs and 168 miRNAs that regulate these features (Figure 6C and D). Subsequently, a gene-gene interaction network was constructed consisting of 23 nodes and 183 edges, based on the GENMINIA database, using physical interaction, co-expression, and predicted interactions as reference criteria (Figure 6E). Finally, we predicted the pathological mechanisms in which SLC31A1 and DLD may be involved in 8 and 24 human diseases, respectively (Figure 6F).
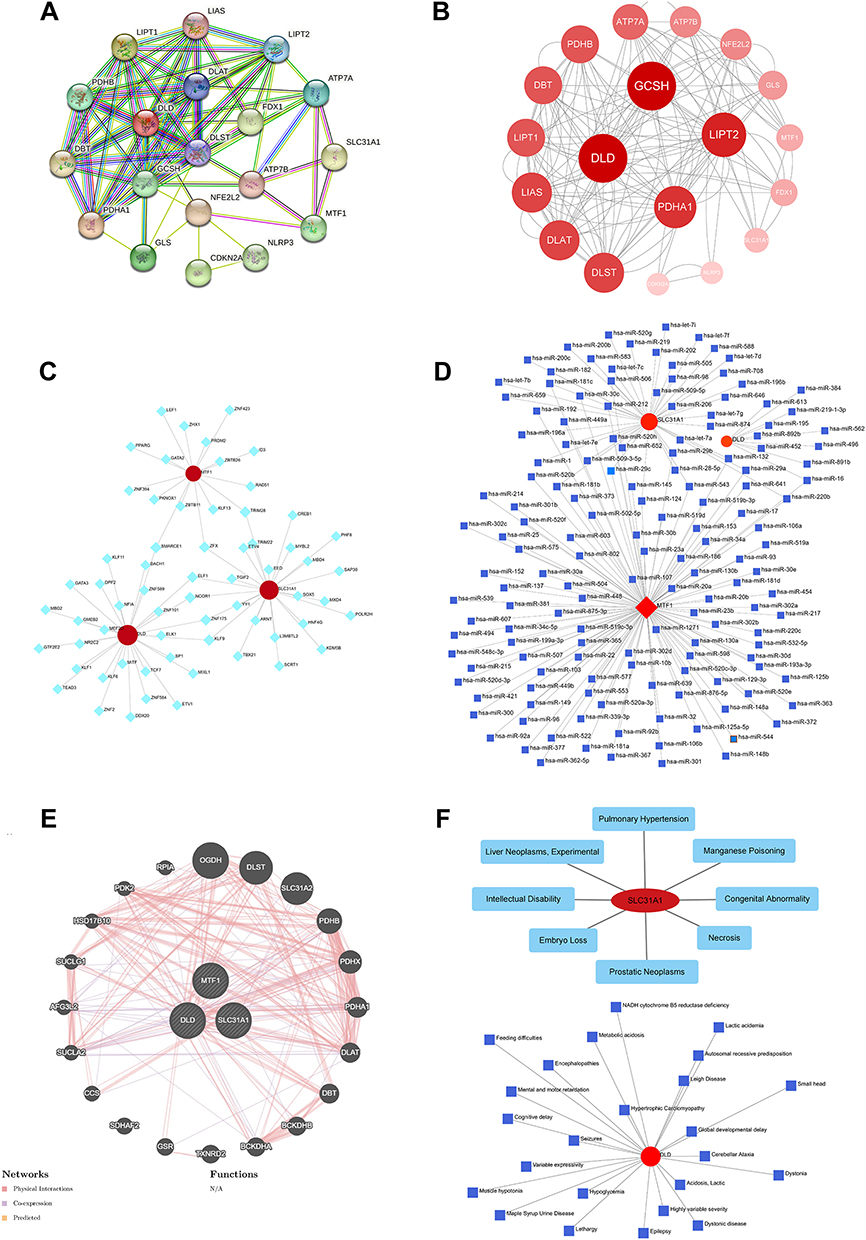 |
Figure 6 Protein-protein and gene-gene network analysis based on 3 M2R-CRGs. (A and B) Protein-protein interaction networks. (C) Gene-transcription factor interaction network. (D) Gene-miRNA interaction network. (E) Gene-gene interaction network. (F) Gene-disease and gene-drug interaction networks. |
Identification of Two Distinct CRG Patterns
Based on 3 M2R-CRGs, we identified CRG-related patterns in osteomyelitis patients using unsupervised clustering analysis. Consensus index, CDF curves, consistency scores, and PCA results suggested that K=2 was the optimal clustering subtype (cluster 1 and 2), with cluster 1 containing 55 samples and cluster 2 containing 30 samples (Figure 7A-D and Supplementary Figure S1). Except for DLD, SLC31A1 and MTF1 exhibited significant differences in expression levels between different clusters (Figure 7E and F). We further analyzed the differences in the proportions of 22 immune cell infiltrations among different CRG modification patterns. Compared with cluster 2, cluster 1 had a richer immune cell infiltration, including T cells gamma delta, macrophages M1 and mast cells activated, while the infiltration abundance of macrophages M2 was higher in cluster 2 (Figure 7G and H). These results suggest that cluster 1 may activate the pro-inflammatory response of osteomyelitis immune microenvironment, while cluster 2 plays an anti-inflammatory role in osteomyelitis. Furthermore, we explored the potential pathways in the two CRG patterns (Figure 7I), and the GSVA-pathway analysis results showed that ribosome, primary immunodeficiency, and taste transduction were significantly upregulated in cluster 2, while lysosome was significantly upregulated in cluster 1.
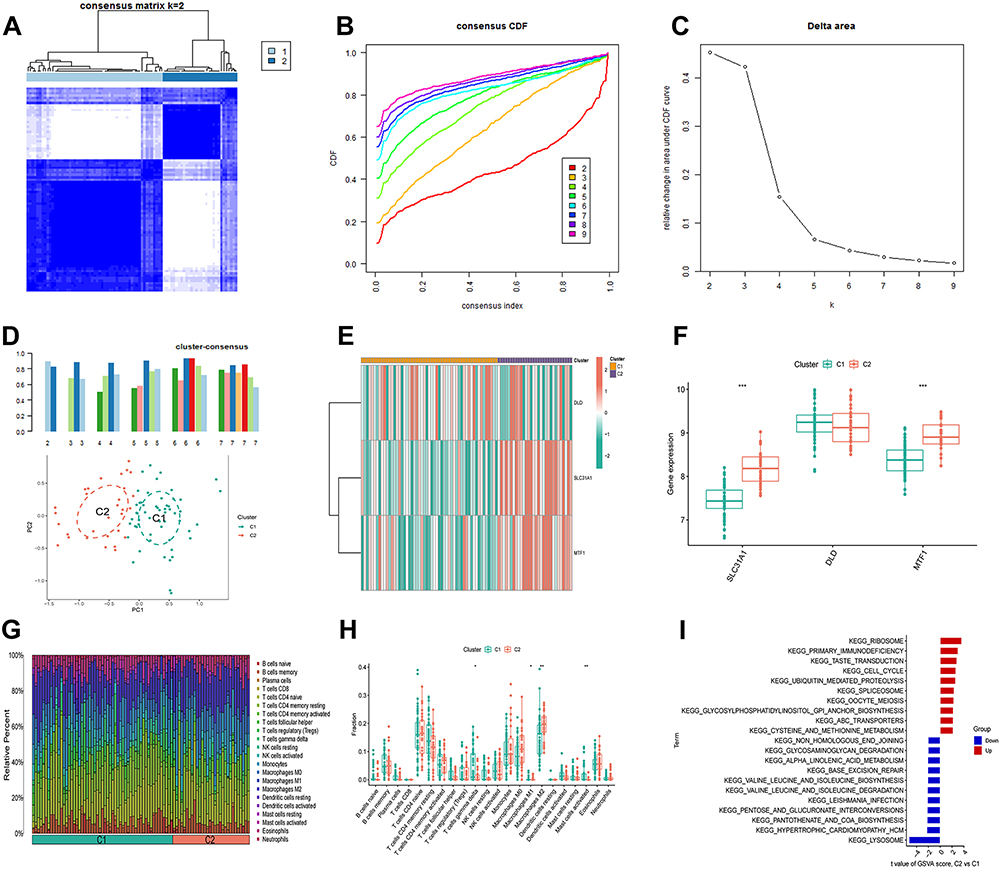 |
Figure 7 Identification of M2R-CRG patterns. (A) Clustering heatmap of 2 clusters (k = 2) based on M2R-CRGs. (B) Cumulative distribution function (CDF) of 2–9 clusters. (C) The area under CDF curve of 2–9 clusters. (D) Consistency scores of 2–7 clusters and PCA analysis of the 2 clusters. (E) Correlation heatmap of the 3 M2R-CRGs and 2 clusters. (F) Differential expression of the 3 M2R-CRGs in the 2 clusters. (G and H) Differential infiltration abundance of the 22 immune cells in the 2 clusters. (I) GSVA-KEGG pathway analysis between the 2 clusters: red indicates upregulated pathways in cluster 2 compared to cluster 1, while blue indicates downregulated pathways in cluster 2. Significance: *P < 0.05, **P < 0.01, ***P < 0.001. |
Generation of Gene Clusters and CRG Scores
To explore the underlying mechanisms of CRG-related subtypes, we identified a total of 134 DEGs between cluster 1 and cluster 2 based on unsupervised clustering analysis of osteomyelitis patients (Figure 8A). Based on the expression of these genes, we further clustered the samples into different gene clusters (Figure 8B-E). Consistent with the CRG patterns, clustering heatmap, clustering index, CDF curve, and PCA results indicated that K=2 was the optimal clustering subtype (cluster A and B), with 41 samples in cluster A and 44 samples in cluster B. The 134 DEGs could well distinguish gene clusters A and B. Except for DLD, SLC31A1 and MTF1 showed significant differences in expression levels between the different gene clusters (Figure 8F and G). In terms of immune infiltration analysis, there were significant differences in the proportions of immune cells between the two gene clusters (Figure 8H). Compared with gene cluster A, gene cluster B had a higher level of infiltration of activated CD8 T cells, eosinophils, immature dendritic cells, macrophages, mast cells, natural killer cells, neutrophils, and regulatory T cells, while gene cluster A had a higher proportion of activated B cells, CD56 dim natural killer cells, and immature B cells. Further GSVA-pathway analysis revealed that ribosome, small cell lung cancer, and oocyte meiosis were significantly upregulated in gene cluster B, while pentose and glucuronate interconversions, glycine serine and threonine metabolism, and regulation of actin cytoskeleton were significantly upregulated in gene cluster A (Figure 8I).
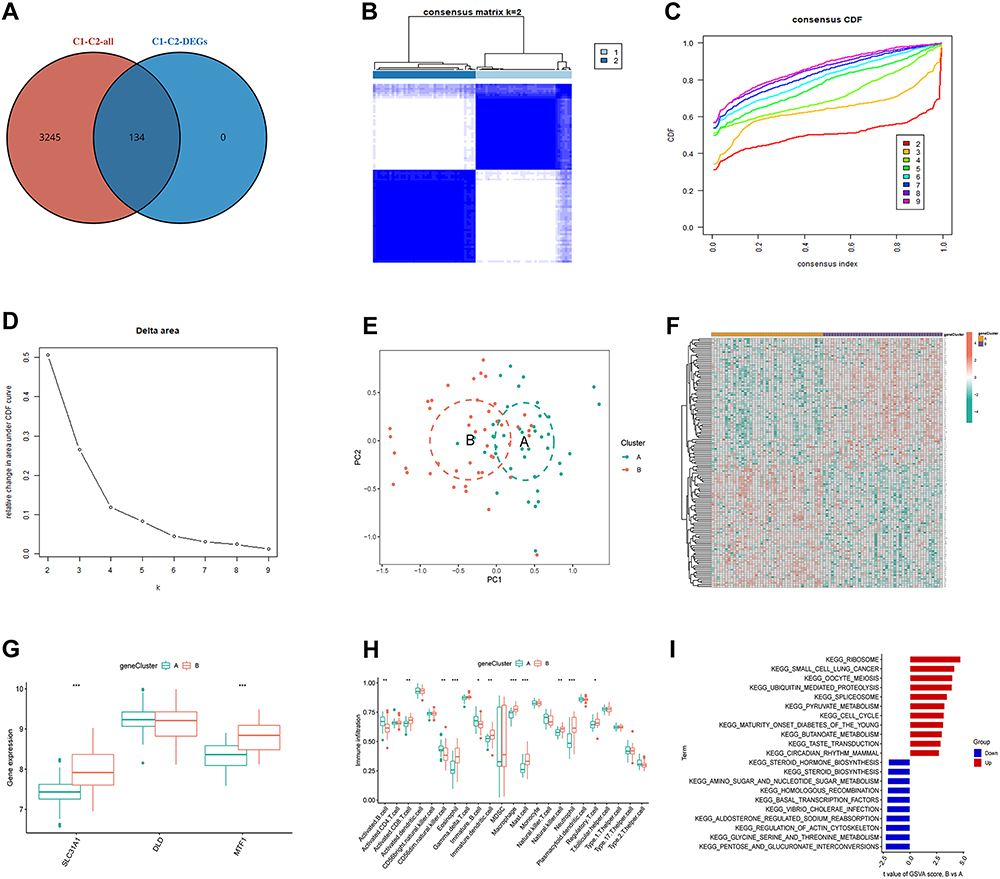 |
Figure 8 Identification of gene patterns. (A) Intersection of DEGs in cluster 1 and cluster 2. (B) Heatmap based on the intersection genes for classification (cluster k=2). (C) Cumulative distribution function (CDF) of 2–9 clusters. (D) Area under the CDF curve of 2–9 clusters. (E) PCA analysis of the 2 clusters. (F) Correlation heatmap of the intersection DEGs and the 2 clusters. (G) Differential expression of the 3 M2R-CRGs in the 2 clusters. (H) Expression differences of 22 immune cell types between the two different gene clusters. (I) GSVA-KEGG pathway analysis between the 2 clusters: red indicates upregulated pathways in cluster 2 compared to cluster 1, while blue indicates downregulated pathways in cluster 2. Significance: *P < 0.05, **P < 0.01, ***P < 0.001. |
Based on the expression of DEGs between the two different clusters, PCA was used to score the osteomyelitis samples (CRG score), and the differences in the scores within the two different clusters were compared (Figure 9A and B). We observed significant differences in CRG scores in the internal comparisons of different CRG clusters and gene clusters, with higher CRG scores in CRG cluster 2 and gene cluster B. Sankey diagrams showed that there was consistency in trends between CRG clusters, gene clusters, and CRG scores in the training set (Figure 9C). Additionally, boxplots showed that IL7R and IL21R were highly expressed in CRG cluster 1 and gene cluster A, while IL6R and IL17RC were upregulated in CRG cluster 2 and gene cluster B (Figure 9D and E). The COL4A4 gene was only highly expressed in CRG cluster 1, with no significant difference in expression in the gene cluster (Figure 9F and G). BMP2K was significantly highly expressed in both CRG cluster 2 and gene cluster B (Figure 9H and I). Overall, different CRG clusters and gene clusters were related to osteomyelitis inflammation and bone-related phenotypes.
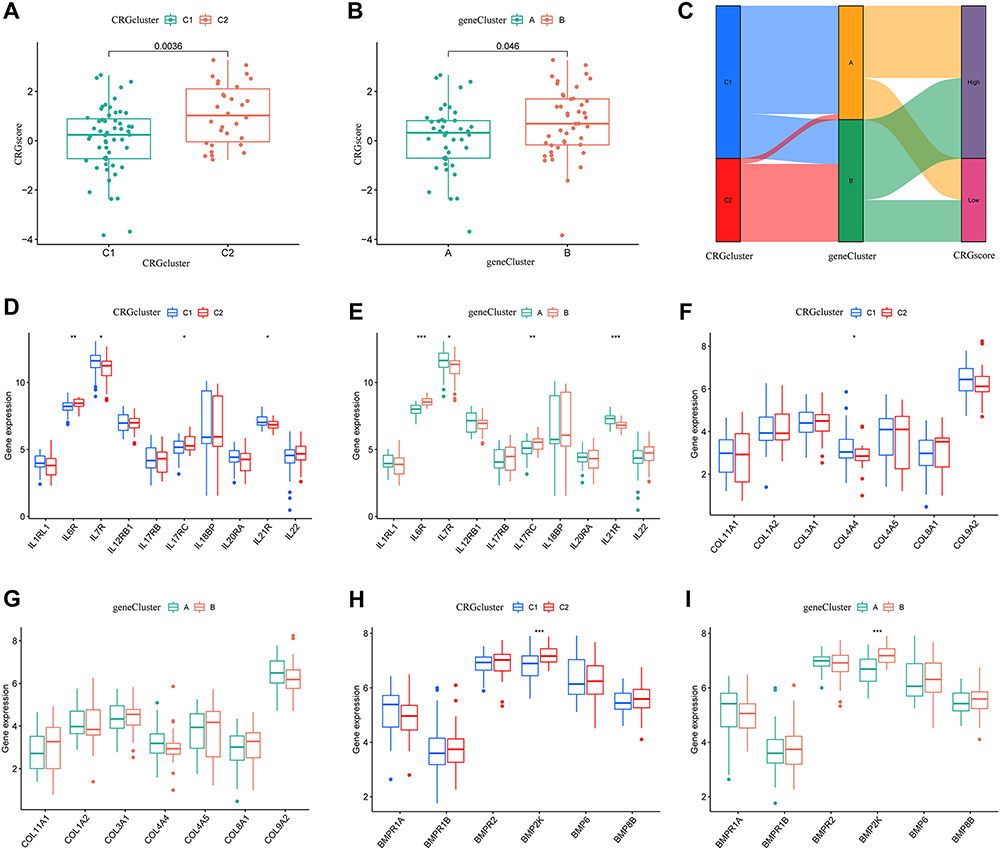 |
Figure 9 CRG scores of CRG clusters and gene clusters, differences in interleukin family, collagen fiber family, and bone morphogenetic protein family-related genes. (A) Differences in CRG scores between cluster 1 and cluster 2. (B) Differences in CRG scores between cluster A and cluster B. (C) Sankey diagram depicting CRG scores, CRG clusters, and gene clusters. Differential expression of 10 interleukin-related genes in different CRG clusters (D) and gene clusters (E). Differential expression of 7 collagen fiber-related genes in different CRG clusters (F) and gene clusters (G). Differential expression of 6 bone morphogenetic protein-related genes in different CRG clusters (H) and gene clusters (I). Significance: *P < 0.05, **P < 0.01, ***P < 0.001. |
Construction of Osteomyelitis Prediction Model Based on M2R-CRGs
To predict the incidence of osteomyelitis, we constructed a nomogram model based on 3 M2R-CRGs and evaluated its predictive ability (Figure 10A-C). The calibration curve and DCA curve demonstrated that the nomogram model was an ideal predictor for osteomyelitis, and clinical decision-making may be beneficial for patients. Next, we further assessed the diagnostic value of the 3 M2R-CRGs for osteomyelitis and plotted ROC curves for each gene, with AUC values all greater than 0.65, indicating good diagnostic performance. A logistics regression model for the three genes was established using the “glmnet” package (Figure 10D and E), and ROC curves confirmed that the model could distinguish well between osteomyelitis patients and healthy individuals. Additionally, we extracted expression data for M2R-CRGs from an independent GSE30119 dataset and established ROC curves to validate the ability of the 3 genes to distinguish between osteomyelitis patients and healthy controls (Figure 10F and G. AUC values were all greater than 0.6, indicating relatively good diagnostic value of the M2R-CRGs for osteomyelitis in the validation set.
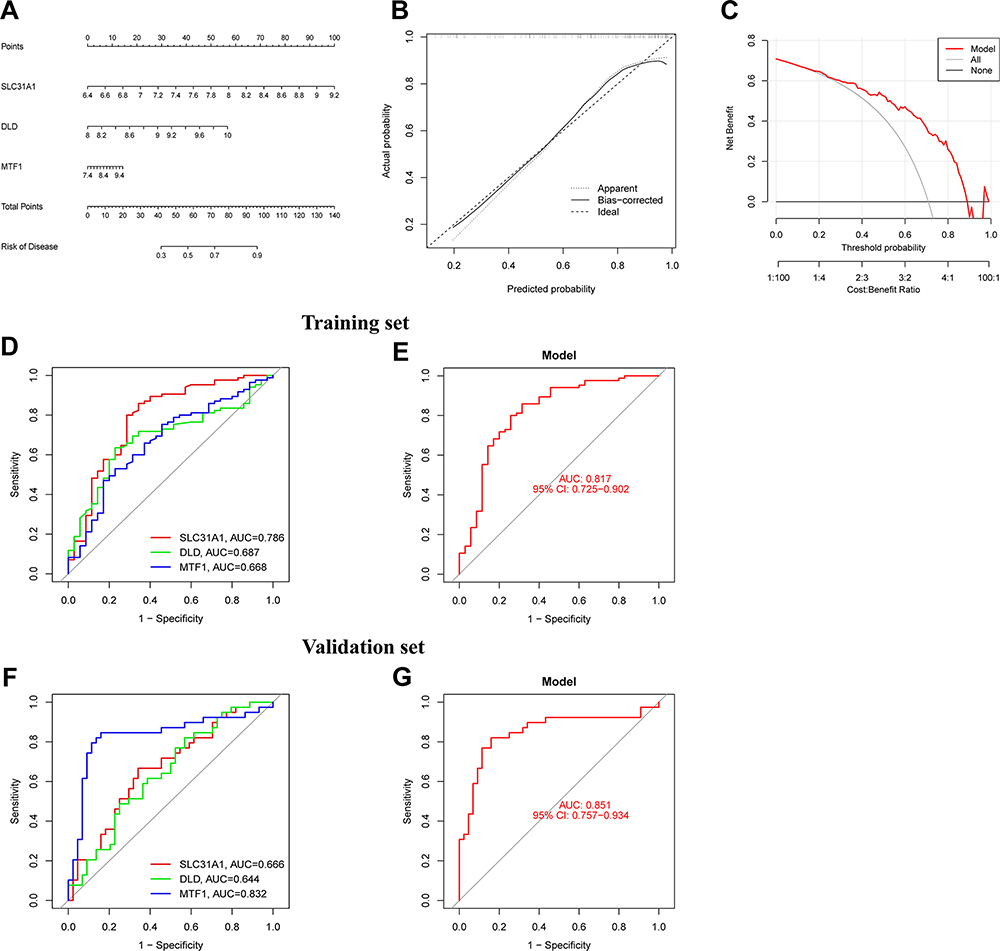 |
Figure 10 Nomogram model and ROC curve based on 3 M2R-CRGs. (A) Nomogram model based on 3 M2R-CRGs. (B) Calibration curve. (C) Decision curve analysis. (D) ROC curves of 3 M2R-CRGs for the diagnosis of osteomyelitis patients in training set. (E) A Logistic regression model was constructed to distinguish the osteomyelitis samples in training set. (F) ROC curves of 3 M2R-CRGs for the diagnosis of osteomyelitis patients in validation set. (G) A Logistic regression model was constructed to distinguish the osteomyelitis samples in validation set. |
Validation of M2R-CRGs Expression Levels
Given the excellent performance of the 3 M2R-CRGs in osteomyelitis clustering and diagnosis, we validated their expression levels using the GSE3019 dataset. Consistent with the training set results, the expression levels of SLC31A1, MTF1, and DLD were significantly upregulated in osteomyelitis samples (Figure 11A-C). RT-qPCR results indicated that mRNA levels of SLC31A1, MTF1, and DLD were significantly upregulated in an osteomyelitis rat model (Figure 11D-F). Further evaluation of the protein levels of the 3 M2R-CRGs using immunohistochemical staining revealed that the positivity rates of SLC31A1, MTF1, and DLD were significantly higher in the osteomyelitis group than in the healthy control group (Figure 11G-I), consistent with the bioinformatics results.
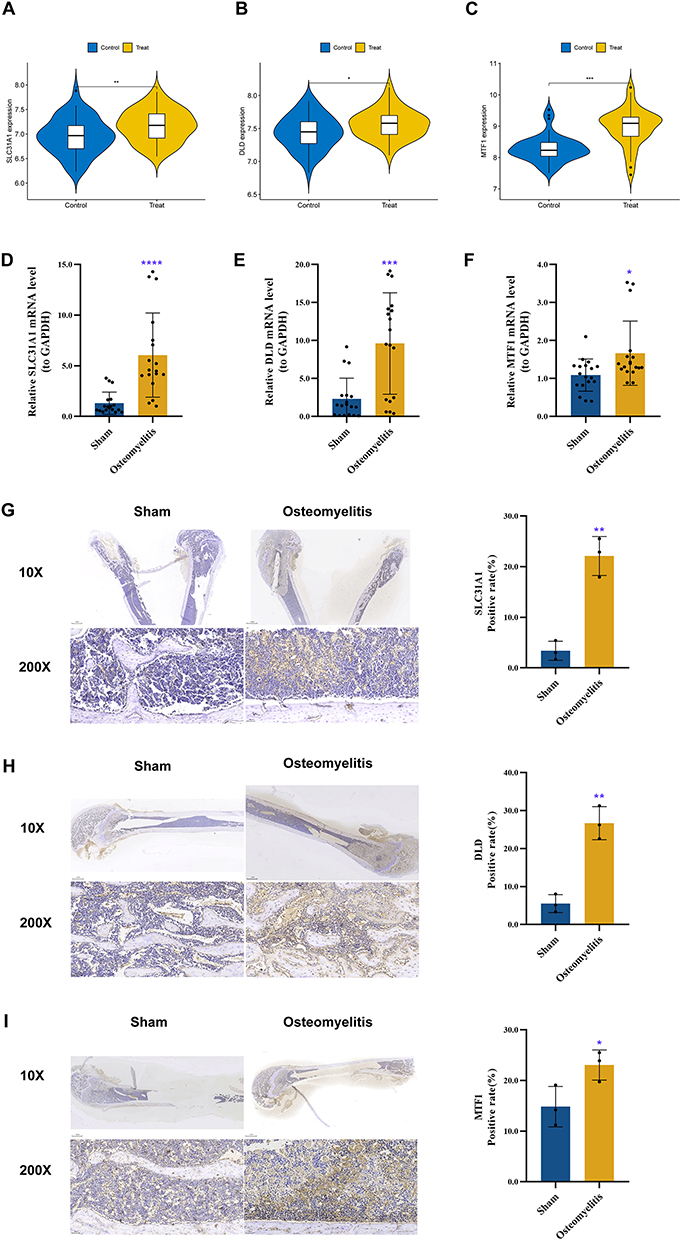 |
Figure 11 Validation for 3 M2R-CRGs through independent dataset (GSE30119), RT-qPCR, and immunohistochemistry. In the independent dataset, SLC31A1 (A), DLD (B), and MTF1 (C) were all significantly upregulated in the osteomyelitis rat group. Total RNA and protein were extracted from both the osteomyelitis rat group and the sham operation group. The mRNA expression levels of SLC31A1 (D), DLD (E), and MTF1 (F) were significantly increased in the osteomyelitis rat group. Furthermore, the protein levels of SLC31A1 (G), DLD (H), and MTF1 (I) showed a significant upregulation in the osteomyelitis rat group. Significance: *: P<0.05, **: P<0.01, ***: P<0.001, ****: P<0.0001. |
Discussion
Osteomyelitis is a common infectious disease in orthopedics, characterized by a prolonged and recurrent course that poses significant health burdens on both patients and society.48 The nonspecific clinical manifestations and radiological examinations, along with the invasive nature of the current “gold standard” diagnostic methods, contribute to the absence of recognized biomarkers for predicting S. aureus-infected osteomyelitis.10 Cuproptosis primarily relies on mitochondrial stress to regulate cell death, with a focus on lipidation of mitochondrial enzymes and loss of Fe-S cluster proteins.32,49 As a novel type of cell death, cuproptosis has been implicated in the development of various diseases.50,51 However, its regulatory role in infectious bone diseases remains unclear. Previous studies have indicated a slight upregulation of genes related to mitochondrial fission in animal models of S. aureus-infected osteomyelitis, suggesting mitochondrial dysfunction. Consequently, this study aimed to elucidate the regulatory role of CRGs in S. aureus-infected osteomyelitis and its relationship with the immune microenvironment.
In this study, the expression profiles of CRGs and the immune microenvironment were comprehensively analyzed between S. aureus-infected osteomyelitis patients and healthy controls, leading to the identification of three CRGs strongly associated with M2 macrophages. M2 macrophages were found to exhibit a positive correlation with SLC31A1 and MTF1, while a significant negative correlation was shown with DLD. Notably, high expression of MTF1 correlated with a high infiltration abundance of M2 macrophages, whereas low expression of MTF1 was associated with a high infiltration ratio of M1 macrophages. These findings suggest that MTF1 may modulate the immune microenvironment of S. aureus-infected osteomyelitis by regulating the states of macrophages (M1 and M2).
To construct a diagnostic model for predicting the incidence of S. aureus-infected osteomyelitis, ROC curves were built based on three M2R-CRGs. The logistic model yielded an AUC of 0.817, indicating good diagnostic performance for these biomarkers. In a previous study, a diagnostic model based on ferroptosis-related genes was constructed for osteomyelitis patients, achieving an AUC of 0.993.52 Additionally, comprehensive clustering analysis was performed on S. aureus-infected osteomyelitis patients, resulting in the identification of two distinct subclassification patterns (CRG pattern and gene pattern). Analysis of the immune microenvironment revealed that cluster 1 might activate pro-inflammatory responses, while cluster 2 exhibited anti-inflammatory effects within the osteomyelitis immune microenvironment. In the gene pattern, cluster B was associated with macrophage activation. Based on these findings, it is hypothesized that different CRG and gene patterns may regulate the pathogenesis of S. aureus-infected osteomyelitis by modulating macrophage states.
Copper ions, essential trace elements in the human body, have been shown to be relevant to bone homeostasis, mediated by osteoblasts and osteoclasts. The hypoxic bone environment and excessive cellular glycolysis may inhibit cuproptosis, subsequently promoting proliferation in immune cells (macrophages and effector T cells) as well as in osteoblasts. Consequently, CRGs are recognized as potential therapeutic targets for treating bone loss associated with osteomyelitis. The three M2R-CRGs (SLC31A1, MTF1, and DLD) have been identified as characteristic biomarkers in S. aureus-infected osteomyelitis. SLC31A1, as a member of the copper transporter family, regulates cellular copper homeostasis. It has been linked to cisplatin chemoresistance in cancer treatments and may serve as a potential molecular target for tumor therapy.53–55 Feng et al 2023,56 discovered that CRGs promote immune activation and evasion, and these genes can be used to predict the prognosis and response to immune therapy in glioblastoma patients. Moreover, they found that SLC31A1 was positively correlated with most immunosuppressive cells, including M2 macrophages. In alignment with these findings, our study also demonstrated a strong positive correlation between SLC31A1 and M2 macrophages in osteomyelitis patients. This suggests that SLC31A1 may play a role in regulating the immune microenvironment in S. aureus-infected osteomyelitis by modulating M2 macrophages.
MTF-1 is essential for regulating metallothionein (MT) related genes in human monocyte-derived macrophages (MDMs). Lahiri et al 2014,57 identified MTF-1 as a key transcription factor that regulates the expression of MT-related genes in macrophages. Targeting MTF-1 can increase intracellular zinc and induce autophagy, thus enhancing bacterial clearance. In line with the positive correlation observed between SLC31A1 and M2 macrophages, high expression of MTF1 is significantly associated with an increased infiltration of M2 macrophages, suggesting that MTF1 may exert anti-inflammatory effects in S. aureus-infected osteomyelitis by activating M2 macrophages. DLD, an essential gene involved in cellular copper death and the decarboxylation of pyruvate,58 is linked to cell apoptosis and the generation of reactive oxygen species, making it relevant in the production of anticancer agents. As a positive regulator of cuproptosis, DLD promotes copper-dependent cell death. Interestingly, in this study, high expression of DLD correlated significantly with low expression of M2 macrophages, indicating that DLD may promote inflammatory reactions in the osteomyelitis immune microenvironment and accelerate osteoblast cell death by inhibiting M2 macrophage activation. Additionally, the mRNA and protein levels of SLC31A1, MTF1, and DLD were further validated and found to be significantly upregulated in the osteomyelitis rat model, suggesting a potential role for CRGs in S. aureus-infected osteomyelitis.
However, this study is subject to several limitations that warrant emphasis. Firstly, the data were sourced from public databases, which may introduce batch effects from variations in sample collection times and conditions, potentially impacting the analysis results. Secondly, the dataset lacks detailed clinical data; future studies should include comprehensive clinical information, such as osteomyelitis staging, to enhance the diagnostic performance of the model. Thirdly, the small sample sizes in the osteomyelitis datasets and rat models limit the generalizability of our findings; larger sample sizes are needed to validate the applicability of these conclusions. Lastly, the expression of the three M2R-CRGs was identified using RT-qPCR and immunohistochemistry. Further experiments are needed to explore the specific mechanisms by which these characteristic genes regulate macrophages in the microenvironment of S. aureus-infected osteomyelitis.
Conclusion
This study reveals the expression profile of CRGs and the immune infiltration landscape in patients with S. aureus-infected osteomyelitis. SLC31A1, MTF1, and DLD have been identified as characteristic biomarkers, Furthermore, a diagnostic model based on the three M2R-CRGs accurately predicts the risk of S. aureus-infected osteomyelitis in patients. The clustering patterns suggest significant associations between different CRG clusters and gene clusters with inflammation and bone-related phenotypes in osteomyelitis. This study is the first to explore the role of cuproptosis from an immunological perspective in S. aureus-infected osteomyelitis, potentially offering molecular targets for early diagnosis and personalized immune therapy for patients with osteomyelitis in the future.
Data Sharing Statement
The microarray data used to support the findings of this study can be downloaded from the GSE6269, GSE16129 and GSE30119 datasets (https://www.ncbi.nlm.nih.gov/geo).
Ethics Approval
All experimental procedures were approved by the Nursing and Use of Experimental Animals Ethics Committee of the Joint Logistics Support Force 920 Hospital (2023-007-01) and conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, USA).
Acknowledgments
Xiangwen Shi, Haonan Ni, and Linmeng Tang are co-first authors for this study. We appreciate the constructive suggestions provided by Dr. Xianjun Chen for this article and are grateful to the authors who contributed to public GEO databases.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by National Natural Science Foundation of China (Grant No. 82072392); the Yunnan Traumatology and Orthopedics Clinical Medical Center (Grant No. ZX20191001); the Yunnan Orthopedics and Sports Rehabilitation Clinical Medicine Research Center (Grant No. 202102AA310068); the Scientific Research Fund Project Department of Education of Yunnan Province (Grant No. 2024Y251).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Walter G, Kemmerer M, Kappler C, Hoffmann R. Treatment algorithms for chronic osteomyelitis. Dtsch Arztebl Int. 2012;109(14):257–264.
2. Wang X, Zhang M, Zhu T, Wei Q, Liu G, Ding J. Flourishing Antibacterial Strategies for Osteomyelitis Therapy. Adv Sci (Weinh). 2023;10(11):e2206154.
3. Hofstee MI, Muthukrishnan G, Atkins GJ, et al. Current Concepts of Osteomyelitis: from Pathologic Mechanisms to Advanced Research Methods. Am J Pathol. 2020;190(6):1151–1163.
4. Lew DP, Waldvogel FA. Osteomyelitis. Lancet. 2004;364(9431):369–379.
5. Campoccia D, Montanaro L, Speziale P, Arciola CR. Antibiotic-loaded biomaterials and the risks for the spread of antibiotic resistance following their prophylactic and therapeutic clinical use. Biomaterials. 2010;31(25):6363–6377.
6. Fantoni M, Taccari F, Giovannenze F. Systemic antibiotic treatment of chronic osteomyelitis in adults. Eur Rev Med Pharmacol Sci. 2019;23(2 Suppl):258.
7. Stewart PS, Costerton JW. Antibiotic resistance of bacteria in biofilms. Lancet. 2001;358(9276):135–138.
8. Conterno LO, Turchi MD. Antibiotics for treating chronic osteomyelitis in adults. Cochrane Database Syst Rev. 2013;2013(9):Cd004439.
9. Bury DC, Rogers TS, Dickman MM. Osteomyelitis: diagnosis and Treatment. Am Fam Physician. 2021;104(4):395–402.
10. Jha Y, Chaudhary K. Diagnosis and Treatment Modalities for Osteomyelitis. Cureus. 2022;14(10):e30713.
11. Urish KL, Cassat JE. Staphylococcus aureus Osteomyelitis: bone, Bugs, and Surgery. Infect Immun. 2020;88(7).
12. Pääkkönen M, Peltola H. Acute osteomyelitis in children. N Engl J Med. 2014;370(14):1365–1366.
13. Connolly LP, Connolly SA, Drubach LA, Jaramillo D, Treves ST. Acute hematogenous osteomyelitis of children: assessment of skeletal scintigraphy-based diagnosis in the era of MRI. J Nucl Med. 2002;43(10):1310–1316.
14. Hudson MC, Ramp WK, Nicholson NC, Williams AS, Nousiainen MT. Internalization of Staphylococcus aureus by cultured osteoblasts. Microb Pathog. 1995;19(6):409–419.
15. Garzoni C, Kelley WL. Return of the Trojan horse: intracellular phenotype switching and immune evasion by Staphylococcus aureus. EMBO Mol Med. 2011;3(3):115–117.
16. Bai J, Feng Y, Li W, et al. Alternative Copper-Based Single-Atom Nanozyme with Superior Multienzyme Activities and NIR-II Responsiveness to Fight against Deep Tissue Infections. Research. 2023;6:0031.
17. Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339(8):520–532.
18. Garzoni C, Kelley WL. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol. 2009;17(2):59–65.
19. von Eiff C. Staphylococcus aureus small colony variants: a challenge to microbiologists and clinicians. Int J Antimicrob Agents. 2008;31(6):507–510.
20. Thurlow LR, Hanke ML, Fritz T, et al. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J Immunol. 2011;186(11):6585–6596.
21. Scherr TD, Roux CM, Hanke ML, Angle A, Dunman PM, Kielian T. Global transcriptome analysis of Staphylococcus aureus biofilms in response to innate immune cells. Infect Immun. 2013;81(12):4363–4376.
22. Jiang J, Wang F, Huang W, et al. Mobile mechanical signal generator for macrophage polarization. Exploration. 2023;3(2):20220147.
23. Claro T, Widaa A, O’Seaghdha M, et al. Staphylococcus aureus protein A binds to osteoblasts and triggers signals that weaken bone in osteomyelitis. PLoS One. 2011;6(4):e18748.
24. Claro T, Widaa A, McDonnell C, Foster TJ, O’Brien FJ, Kerrigan SW. Staphylococcus aureus protein A binding to osteoblast tumour necrosis factor receptor 1 results in activation of nuclear factor kappa B and release of interleukin-6 in bone infection. Microbiology. 2013;159(Pt 1):147–154.
25. Mendoza Bertelli A, Delpino MV, Lattar S, et al. Staphylococcus aureus protein A enhances osteoclastogenesis via TNFR1 and EGFR signaling. Biochim Biophys Acta. 2016;1862(10):1975–1983.
26. Josse J, Velard F, Gangloff SC. Staphylococcus aureus vs. Osteoblast: relationship and Consequences in Osteomyelitis. Front Cell Infect Microbiol. 2015;5:85.
27. Das T, Sa G, Chattopadhyay S, Ray PK. Protein A-induced apoptosis of cancer cells is effected by soluble immune mediators. Cancer Immunol Immunother. 2002;51(7):376–380.
28. Marriott I. Apoptosis-associated uncoupling of bone formation and resorption in osteomyelitis. Front Cell Infect Microbiol. 2013;3:101.
29. Varoga D, Wruck CJ, Tohidnezhad M, et al. Osteoblasts participate in the innate immunity of the bone by producing human beta defensin-3. Histochem Cell Biol. 2009;131(2):207–218.
30. Chen Q, Hou T, Luo F, Wu X, Xie Z, Xu J. Involvement of toll-like receptor 2 and pro-apoptotic signaling pathways in bone remodeling in osteomyelitis. Cell Physiol Biochem. 2014;34(6):1890–1900.
31. Widaa A, Claro T, Foster TJ, O’Brien FJ, Kerrigan SW. Staphylococcus aureus protein A plays a critical role in mediating bone destruction and bone loss in osteomyelitis. PLoS One. 2012;7(7):e40586.
32. Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–1261.
33. Mendelsohn DH, Niedermair T, Walter N, Alt V, Rupp M, Brochhausen C. Ultrastructural Evidence of Mitochondrial Dysfunction in Osteomyelitis Patients. Int J Mol Sci. 2023;24(6):5709.
34. Li K, Chen Y, Lin Y, et al. PD-1/PD-L1 blockade is a potent adjuvant in treatment of Staphylococcus aureus osteomyelitis in mice. Mol Ther. 2023;31(1):174–192.
35. Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–883.
36. Zhang Y, Parmigiani G, Johnson WE. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom Bioinform. 2020;2(3):lqaa078.
37. Zhao J, Guo S, Schrodi SJ, He D. Cuproptosis and cuproptosis-related genes in rheumatoid arthritis: implication, prospects, and perspectives. Front Immunol. 2022;13:930278.
38. Li D, Gao Z, Li Q, Liu X, Liu H. Cuproptosis-a potential target for the treatment of osteoporosis. Front Endocrinol. 2023;14:1135181.
39. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
40. Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol Biol. 2018;1711:243–259.
41. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550.
42. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7.
43. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–d13.
44. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504.
45. Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572–1573.
46. David CC, Jacobs DJ. Principal component analysis: a method for determining the essential dynamics of proteins. Methods Mol Biol. 2014;1084:193–226.
47. Li Y, Lu F, Yin Y. Applying logistic LASSO regression for the diagnosis of atypical Crohn’s disease. Sci Rep. 2022;12(1):11340.
48. Momodu II, Savaliya V. Osteomyelitis. Treasure Island (FL): StatPearls Publishing; 2022.
49. Tang D, Chen X, Kroemer G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. 2022;32(5):417–418.
50. Chen L, Min J, Wang F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. 2022;7(1):378.
51. Tong X, Tang R, Xiao M, et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J Hematol Oncol. 2022;15(1):174.
52. Shi X, Tang L, Ni H, Li M, Wu Y, Xu Y. Identification of Ferroptosis-Related Biomarkers for Diagnosis and Molecular Classification of Staphylococcus aureus-Induced Osteomyelitis. J Inflamm Res. 2023;16:1805–1823.
53. Wee NK, Weinstein DC, Fraser ST, Assinder SJ. The mammalian copper transporters CTR1 and CTR2 and their roles in development and disease. Int J Biochem Cell Biol. 2013;45(5):960–963.
54. Ishida S, McCormick F, Smith-McCune K, Hanahan D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell. 2010;17(6):574–583.
55. Wu G, Peng H, Tang M, et al. ZNF711 down-regulation promotes CISPLATIN resistance in epithelial ovarian cancer via interacting with JHDM2A and suppressing SLC31A1 expression. EBioMedicine. 2021;71:103558.
56. Feng S, Zhang Y, Zhu H, et al. Cuproptosis facilitates immune activation but promotes immune escape, and a machine learning-based cuproptosis-related signature is identified for predicting prognosis and immunotherapy response of gliomas. CNS Neurosci Ther. 2023.
57. Lahiri A, Abraham C. Activation of pattern recognition receptors up-regulates metallothioneins, thereby increasing intracellular accumulation of zinc, autophagy, and bacterial clearance by macrophages. Gastroenterology. 2014;147(4):835–846.
58. Duarte IF, Caio J, Moedas MF, et al. Dihydrolipoamide dehydrogenase, pyruvate oxidation, and acetylation-dependent mechanisms intersecting drug iatrogenesis. Cell Mol Life Sci. 2021;78(23):7451–7468.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.