Back to Journals » ImmunoTargets and Therapy » Volume 10
Complement Inhibition and COVID-19: The Story so Far
Received 24 May 2021
Accepted for publication 16 July 2021
Published 26 July 2021 Volume 2021:10 Pages 273—284
DOI https://doi.org/10.2147/ITT.S284830
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
Sofiane Fodil,1 Djillali Annane1– 3
1Department of Intensive Care, Hôpital Raymond Poincaré (APHP), Garches, 92380, France; 2Laboratory of Infection & Inflammation _ U1173, School of Medicine Simone Veil, University Versailles Saint Quentin _ University Paris Saclay, INSERM, Montigny-Le-Bretonneau, 78180, France; 3FHU SEPSIS (Saclay and Paris Seine Nord Endeavour to PerSonalize Interventions for SEPSIS), AP-HP, University Versailles Saint Quentin _ University Paris Saclay, INSERM, Garches, 92380, France
Correspondence: Djillali Annane
Department of Intensive Care, Hôpital Raymond Poincaré (APHP), 104 Boulevard Raymond Poincaré, Garches, 92380, France
Tel +33 147107787
Email [email protected]
Abstract: Acute respiratory distress syndrome (ARDS) is the most severe complication of COVID-19, a disease caused by severe acute respiratory syndrome coronavirus (SARS CoV) 2. The mechanisms underlying the progression from asymptomatic disease to pneumonia and ARDS are complex and by far unelucidated. As for bacterial sepsis, the release of damage associated molecular patterns and pathogen associated molecular patterns triggers activation of the complement cascade. Subsequently, overexpressed anaphylatoxins recruit inflammatory cells in the lung and other organs and contribute initiating and amplifying a vicious circle of thromboinflammation causing organs damage and eventually death. Preclinical and observational studies in patients with COVID-19 provided evidence that complement inhibition effectively may attenuate lung and systemic inflammation, restore the coagulation/fibrinolysis balance, improve organs function and eventually may save life. Ongoing Phase 2/3 trials should elucidate the benefit to risk profile of complement inhibitors and may clarify the optimal targets in the complement cascade.
Keywords: complement system, cytokines, anaphylatoxins, monoclonal antibodies
Introduction
Since December 2019, COVID-19, a disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) places a major burden on healthcare and economy systems in most if not all countries. As of May 15th 2021, COVID-19 has affected more than 160 million people and caused about 3.4 million deaths.1 It is now well established that following a phase of viral replication in airway and lung tissues, a deregulated immune response is the hallmark of COVID-19 alike bacterial sepsis.2 SARS-CoV-2 triggers immune responses that may vary quantitatively and qualitatively in-between individuals and overtime in the same individual. The very early – within 24 to 48 hours – response to SARS-CoV-2 involved mainly activation of plasmacytoid predendritic cells with subsequent upregulation of interferon type 1 (IFN-1) pathway3 which in turn may trigger TNF/IL-1β-driven inflammatory response.4 Distinct cytokine profiles may be associated with distinct clinical phenotypes with variable prognosis.5 Contrasting with the heterogeneity in clinical phenotypes, a core immune signature carried by peripheral blood cells is characterized by profound T-cells alteration and selective cytokines upregulation, specific antibodies and minimal alterations in B cells.6 The complement system plays a pivotal role in innate immunity.7–9 In sepsis, overactivation of the complement system may favour pro-coagulation over fibrinolysis and inflammation over immune suppression resulting in a vicious circle of self-amplification of thrombo-inflammation.10 Likewise, a dysregulated complement system may partly account for the progression from mild to severe form of COVID-19.11 This narrative review summarized the current knowledge about the activation and deregulation in the complement system during sepsis and COVID-19 and about modulating the complement cascade as a therapeutic approach.
Evidence That COVID-19 is a Complement Mediated Disease
Physiological Basis
The complement system is composed by heat-labile plasma proteins that cross-talk in a catalytic cascade. After Bordet demonstration of the bacteriolytic activity of the serum alexin,7 Ehrlich introduced the term complement.12 A proportionate activation of the complement system is essential to immune homeostasis whereas overactivation may trigger thromboinflammation and end-organ damages and complement deficiency may cause susceptibility to life-threatening infections, and may precede autoimmune disorders. The complement system has about 40 proteins organized in seven functional components (Table 1). The complement system acts as a sentinel that rapidly and with high specificity detects, traces, targets and eradicates pathogens. Its activation involved the classical, mannose-binding lectin (MBL), and alternative pathways. The classical pathway is activated by direct association of C1q with pathogen surfaces or by binding of C1q to antigen-antibody complexes during an adaptive immune response. The MBL pathway is triggered by binding of MBL to mannose containing carbohydrate structures on bacteria or virus surface. The alternative pathway is activated by binding of spontaneously activated complement C3 protein (C3b fragment) to pathogen’s surface. These three pathways converge to the formation of C3 convertase that stimulates the formation of C3a, C3b, C5a, C5b, C6, C7, C8, and C9. The fragments C5b, C6, C7, C8, and C9 form the membrane attack complex (C5b-9, MAC) triggering bacteria lysis (Figure 1). Opsonins and anaphylatoxins boost pathogens phagocytosis and activate circulating white blood cells.9 The proinflammatory effects are finetuned by counter-balancing inhibitory factors such as inactive C3b.13 Each step of the catalytic cascade is tightly controlled by soluble and cell membrane bound regulatory factors (Table 1). Beyond immunity, the complement system has other targets including stem cells and progenitor cells, nervous system, and metabolic tissues modulating cellular stress, apoptosis and autophagy.9
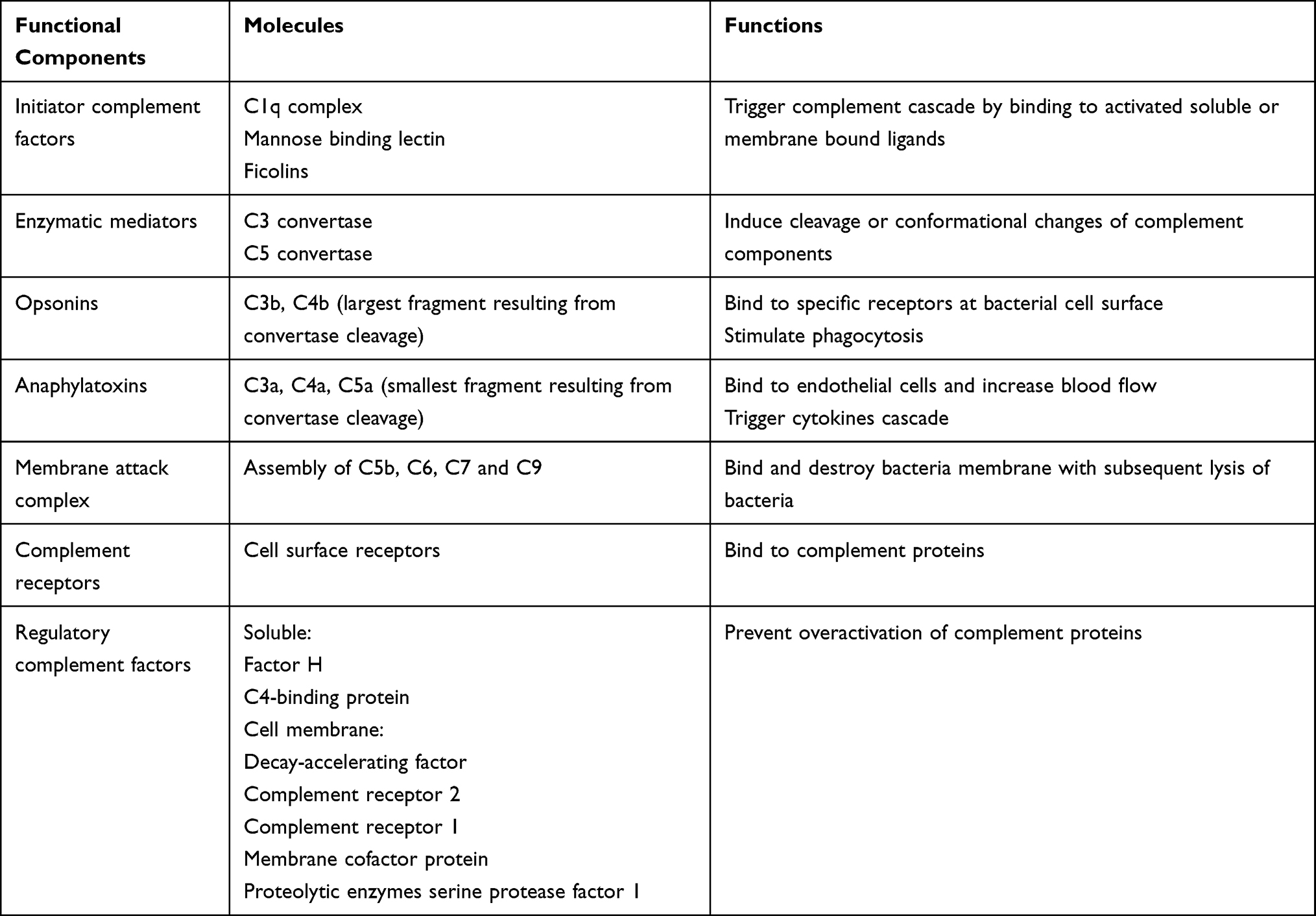 |
Table 1 Functional Components of the Complement System |
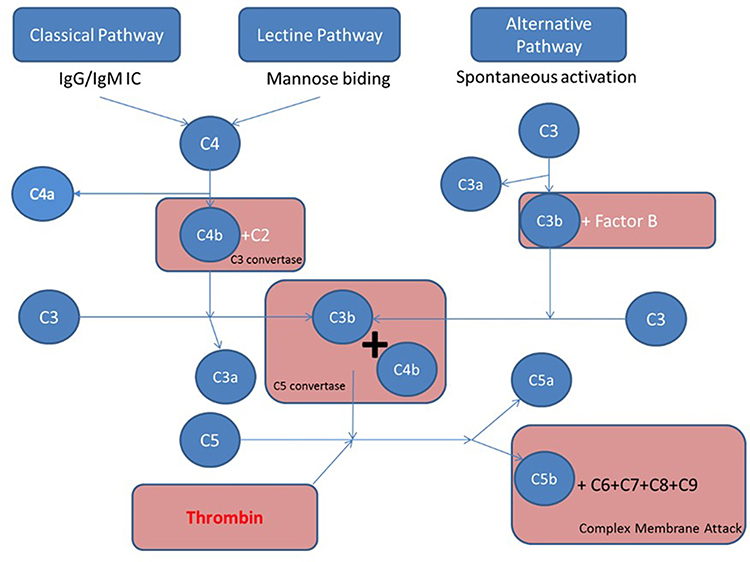 |
Figure 1 Four major pathways are involved in complement activation: classical, lectin, alternative, and coagulation pathways. The classical pathway is activated by direct association of C1q with pathogen surfaces or by binding of C1q to antigen-antibody complexes during an adaptive immune response. The mannose binding lectin pathway is triggered by binding of MBL to mannose containing carbohydrate structures on bacteria or virus surface. The alternative pathway is activated by binding of spontaneously activated complement C3 protein (C3b fragment) to pathogen’s surface. These three pathways converge to the formation of C3 convertase that stimulates the formation of C3a, C3b, C5a, C5b, C6, C7, C8, and C9. The fragments C5b, C6, C7, C8, and C9 form the membrane attack complex (C5b-9, MAC) triggering bacteria lysis, while C5a is a strong anaphylatoxin. The fourth activation pathway is through the action of thrombin, which catalysis C5 into C5a and C5b. |
Role of Anaphylatoxin C5 in the Physiopathology of Sepsis (Figure 2)
Human C5a is a 74 amino acid glycosylated peptide with anti-parallel α-helical structures that are crosslinked by disulphide bands, making the molecule quite stable, especially in the presence of oxidants. C5a is released from the N terminal region of the α chain of C5 by C5 convertases (C3b2•Bb or C4b•C2a•C3b) induced cleavage of C5 into C5a + C5b. C5b interacts with C6, C7, C8 and C9 to form the membrane attack complex C5b-9 (MAC). The release of C5a can also be triggered by neutral proteases derived from neutrophils or lung macrophages, and by thrombin (factor IIa).
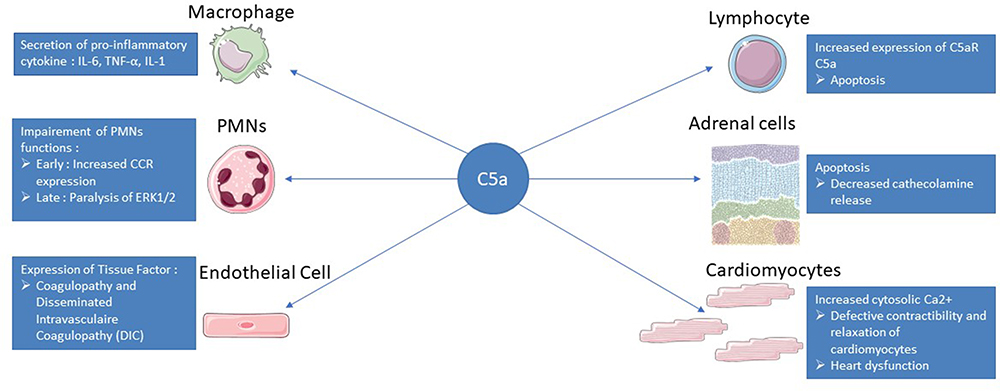 |
Figure 2 Summary of C5a action in sepsis. After binding to its C5aR and C5L2 receptors, C5a will have different effects on the immune system and different organs. The C5a pathway will lead to the secretion of pro-inflammatory cytokines by macrophages such as IL-6, IL-1 or TNF alpha allowing the recruitment of other immune cells. On PMNs, C5a will first increase the expression of CCR to facilitate their recruitment but will then induce a paralysis of ERK1/2 leading to a decrease in phagocytosis or respiratory burst. C5a-exposed endothelial cells obtain an activated phenotype with adhesion molecule and tissue factor expression that will initiate the coagulation cascade with a risk of DIC. Lymphopenia is a feature of sepsis and is related to increased lymphocyte apoptosis via the C5a pathway. C5a induces apoptosis of adrenomedullary cells, which are responsible for the bulk of endogenous catecholamines, leading to a disequilibrium that favors the development of septic shock. The binding of C5a to its receptors on the surface of the cardiomyocytes will lead to a defect in the repolarization of the cell and an accumulation of calcium in the cytoplasm, resulting in a defect in contractility and relaxation of the cardiomyocytes. |
The anaphylatoxin C5a contributes to the systemic overactivation of the proinflammatory cytokines that characterize sepsis. In cecal ligation and puncture (CLP) induced sepsis in mice, C5a induced the migration of interleukin-12+ dendritic cell (IL-12+DC) from the peritoneal cavity to blood stream and lymph nodes contributing to the systemic spreading of inflammation. IL-12+DC are key inducers of interferon (IFN)c+ Th1 and IL-17+ Th17 cells. The pleiotropic activities of IL-12 on natural killer (NK), T and B cells function require to be finetuned for an appropriate immune responses. C5a prevents toll-like receptor (TLR)4-induced synthesis of IL-12, IL-23, and IL-27 via extracellular signal-regulated kinase (ERK) and phosphoinositide 3-kinase (PI3K). In addition to its role in dampening TLR4-driven synthesis, C5a also regulates CD40-induced IL-12 production through ERK1/2. IFN regulatory factor 1 (IRF-1) and IFN consensus sequence binding protein (ICSBP; IRF-8) are also as crucial transcription factors down-streaming ERK and PI3K pathways, respectively. These data indicate that C5a modulates both innate (TLR4) and adaptive (CD40) immune responses that drive the production of IL-12 family cytokines. C5a also interferes with CD40/CD40 ligand axis involved in the communication between the antigen presenting cell (mostly IL-12+DC) and T cell preventing Th1 polarization.14,15 C5a/C5aR axis interacts synergistically with TLRs stimulating NK cells production of TNF-α and IFN-γ.16 C5a inhibits LPS-induced production of TNF-α in neutrophils and by contrast enhances TNF-α release by macrophages.17 There is also evidence for a synergistic relationship between C5a and LPS to produce neutrophil-derived antigenic IL-8.18 The release of IL-6 is closely linked to up-regulation of C5aR and activation of ERK1/2 and p38 as a final common step involving activation of mitogen and stress activated kinase 1/2 (MSK1/2).19,20 Interestingly, C5a exhibited anti-inflammatory effects by downregulating IL-17A and IL-23 via inhibition of CD11b(+)F4/80(+) macrophages and upregulating IL-10.21,22 Therefore, C5a down-modulates the more potent IL-17A while up-regulating the weaker IL-17F.
Activation of the C5a-C5aR pathway induced intrinsic (mitochondrial) and extrinsic (TNFα, Fas ligand) apoptosis of thymocytes, adrenal medullary and cortical cells.23 C5a inhibited spontaneous human neutrophil apoptosis via PI3-K/Akt signalling pathway.24,25 The local complement-dependent expansion of the effector repertoire results from C5a/C5aR-induced suppression of effector T-cell apoptosis, through PI-3Kγ–dependent alterations in Bcl-2 and Fas expression.26 Finally, upregulated C5a impairs neutrophils phagocytosis, reactive oxygen species (ROS) generation, and chemotactic migration.27 C5a and C5b collectively activate platelets and endothelial cells, with subsequent induction of expression of tissue factor, von Willebrand factor, prothrombinase assembly sites and P-selectin.28 E. coli-induced tissue factor upregulation is mainly dependent on C5a and much less on CD14.29,30 Finally, C5a plays a critical role in sepsis induced cardiomyopathy mainly by down-regulating sarcoplasmic reticulum Ca2+-ATPase and sarcolemma Na+-Ca2+ exchanger, key calcium regulatory proteins in left ventricle cardiomyocytes.31 Endotoxin upregulates C5a-C5aR system in a time-dependent manner subsequently within cerebral endothelium, microglial cells neighbouring the endothelium, and in deeper brain parenchyma.32 C5a triggers microglial cells release of proinflammatory cytokines and phagocytosis capacity, and activates astrocytes.
Role of Anaphylatoxins in the Physiopathology of Coronavirus Diseases
Acute respiratory distress syndrome (ARDS) is a severe complication of coronavirus infection.33–35 There are numerous cytokine-related molecules and pathways relevant to understanding the biological mechanisms underlying acute lung injury after viral infection. Complement activation and C5a play a key role in the development of virus-related ARDS.36 In C57BL/6J mice, mouse-adapted SARS-CoV infection resulted in high-titre virus replication within the lung, induction of inflammatory cytokines and chemokines, and immune cell infiltration within the lung.37 C3 activation products were detected in SARS-CoV-infected mice, but not in control mice, as early as 24 hours post-infection. Complement component 3 deposition was observed in the lungs two to four days following infection in wildtype mice. C3 knocked out mice were protected from SARSCoV-induced weight loss, had reduced lung damage, lung and systemic inflammation, and improved respiratory function. The unaltered kinetics of viral replication in the C3-deficient mice suggested that the protective effects related to the prevention of complement-mediated inflammatory processes and not to reduction of viral titre. In contrast, transgenic mice lacking alternative pathway proteins, Factor B or C4, were not protected from SARS CoV-mediated weight loss, suggesting that inhibition of the complement alternative pathway alone may not be sufficient. This implies that inhibition of C3 or C5 may be required. Likewise, Middle East respiratory syndrome (MERS)-CoV infection in mice caused severe acute respiratory failure and high mortality accompanied by elevated secretion of cytokines and chemokines.38 In this model, complement overactivation was characterized by increased concentrations in sera and lung tissue of C5a and C5b-9. The avian influenza virus H5N1 induced histopathological changes in the lungs are like those observed in SARS.39 In a mouse model of H5N1, complement activates immune effector cells and drives lung inflammation. C3a and C5a increased vascular permeability, activated leukocytes and endothelial cells, up-regulated adhesion molecule and cytokine expression, and induced goblet cell secretion of mucus. In these mice, deposition of C3, C5b-9, and mannose-binding lectin (MBL)-C was observed in lung tissue. Up-regulation of MBL-associated serine protease-2 (MASP-2) and complement receptors C3aR and C5aR was also detected. Specific inhibition of either C3aR or C5a in the infected mice reduced lung damage, inflammation and neutrophil infiltration, and improved survival.40 Upregulation of MASP-2 by extracellular release of the viral N-protein may be a common pathway of complement overaction by coronaviruses.41
Clinical evidence suggests that complement is activated during SARS infection and that the progression of severe pneumonia, acute lung injury, or ARDS in these patients is strongly associated with complement activation.42 Likewise, C5a is associated with the inflammatory response and severe lung damage that occurs in patients infected with the 2009 H1N1 influenza virus.43 It has also been shown that SARS-CoV can directly activate complement via the lectin pathway.44 Cytokine storm is thought to be a key step in the pathogenesis of ARDS following SARS-CoV-2 infection.45 Observational studies have shown evidence for over-activation of the complement pathway in patients with COVID-19. Lung and skin biopsies in patients with severe COVID-19 revealed microvascular damage and microthrombosis consistent with activation of the alternative and lectin pathways.46 Indeed, vascular injuries were associated with extensive of C5b-9, C4d and MASP-2. Clinical studies consistently reported increased levels of sC5b-9 correlating with severity of illness.47–50 Likewise, levels of C5a were gradually increased in serum and bronchoalveolar lavage from asymptomatic patients, patients with pneumonia and those with ARDS.51 Upregulation of the C5a-C5aR axis was sustained for at least 10 days after admission probably as a result of prolonged activation of the lectin and classical pathways. Overactivation of C5a-C5aR axis in the lung contributes directly and indirectly to recruitment of myeloid cells and subsequently to vasculitis and endothelialitis.
Collectively, preclinical and clinical observations suggest that COVID-19, alike other highly pathogenic viral diseases, is a complement-mediated disease, and that modulating the complement cascade may be an effective treatment option.
Complement System Inhibition
Theoretically, options for blocking the complement system may include targeting components of the lectin, classical or alternative pathways, or acting downstream at the level of C3-C3a or C5-C5a.52,53
Preclinical Studies
Main preclinical studies are summarized in Table 2. In E. coli challenged primates, selective inhibition of the C3 convertase prevented TLR-4 mediated inflammatory signals, tissues inflammation, preserved organs function and improved survival.54 These effects were preceded by inhibition of complement activation and deposition in tissues of MDL, C3b and C4b-9. In the same model, inhibition of C3 convertase protected against E. coli sepsis induced lung fibrosis by downregulating profibrogenic and matrix proteins genes.55 In rats with CLP induced sepsis, anti-C5a antibodies significantly reduced systemic and tissues inflammation, restored the coagulation/fibrinolytic balance, and improved survival.56,57 Likewise, in piglets with polymicrobial sepsis, the combination of the C5 inhibitor coversin and of an anti-CD14 significantly attenuated the inflammatory response and disseminated intravascular coagulopathy, improved hemodynamic, organ function and survival.58,59 In mice challenged with lethal load of H5N1, C3aR or C5aR inhibitors prevented neutrophils infiltration in the lung, downregulated local and systemic release of proinflammatory mediators with subsequent attenuation of lung injury and increased survival rate.40 The pharmacological approach of inhibiting C3a-C3aR or C5a-C5aR axes resulted in similar anti-inflammatory effects, organs protection and survival in H7N9 infected non-human primates60 and in MERS-CoV infected mice.38 Monoclonal antibodies that selectively blocked C5a-R1 prevented lung inflammation and injuries in a mice model of inhaled C5a induced acute lung injury mimicking COVID-19.51
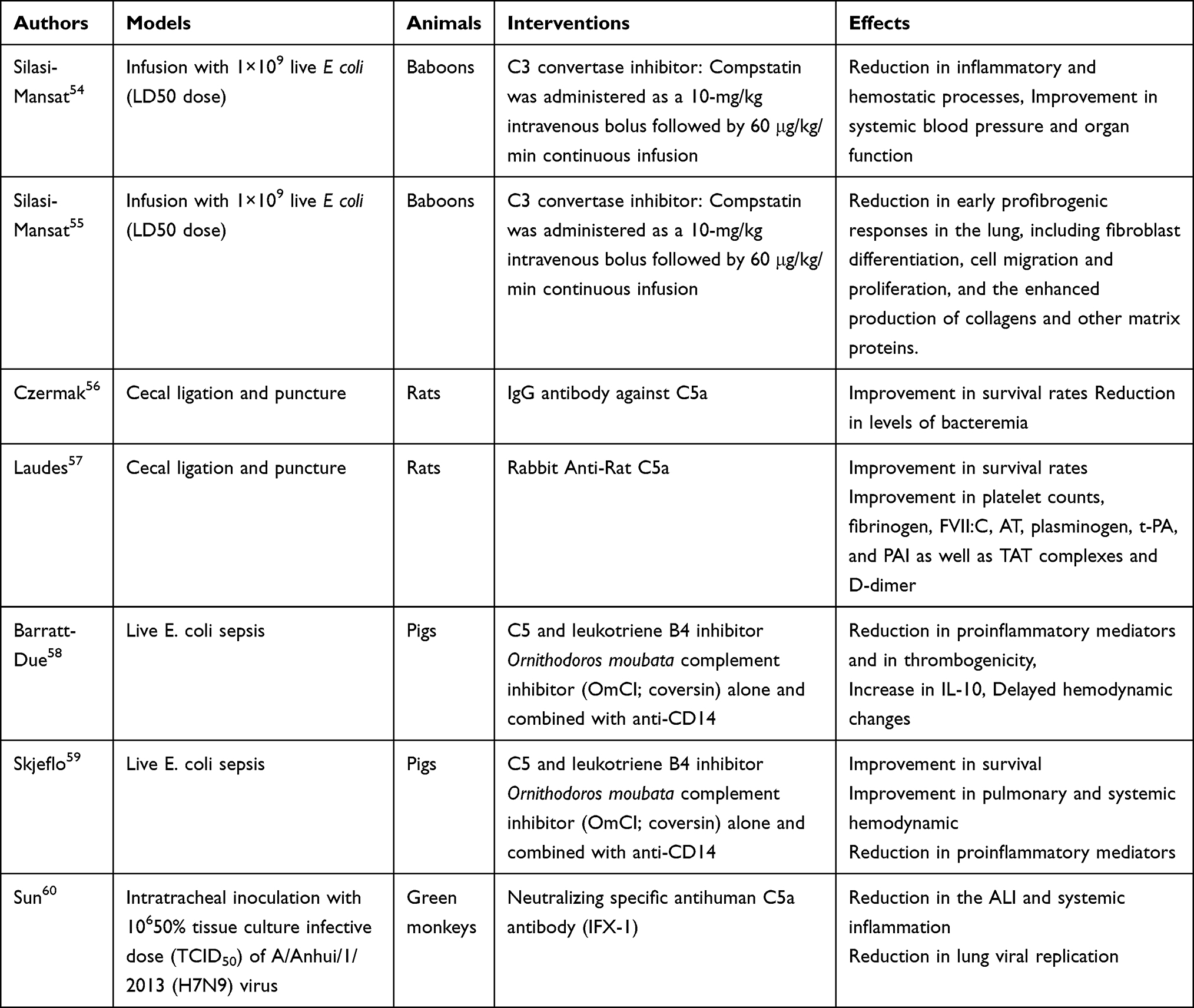 |
Table 2 Preclinical Studies on Complement Inhibition |
Clinical Studies
Main studies on the use of complement factor inhibitors in sepsis or COVID-19 are summarized in Table 3. Small sized observational studies suggested favorable outcomes in patients with sepsis treated with C1 esterase inhibitor.61,62 In a randomized trial, infusions of C1 esterase inhibitor reduced 28-day mortality by 33% from 45%.63 This trial has several limitations including a small sample size (61 patients), the lack of a placebo and of blinding of treatment administration and outcomes assessment. In a patient with sepsis and disseminated intravascular coagulopathy induced thrombotic microangiopathy, treatment with eculizumab, humanized monoclonal antibody that inhibits the cleavage of C5 to C5a and C5b, restored the coagulation/fibrinolytic balance and reduced sepsis associated terminal organs complications.64
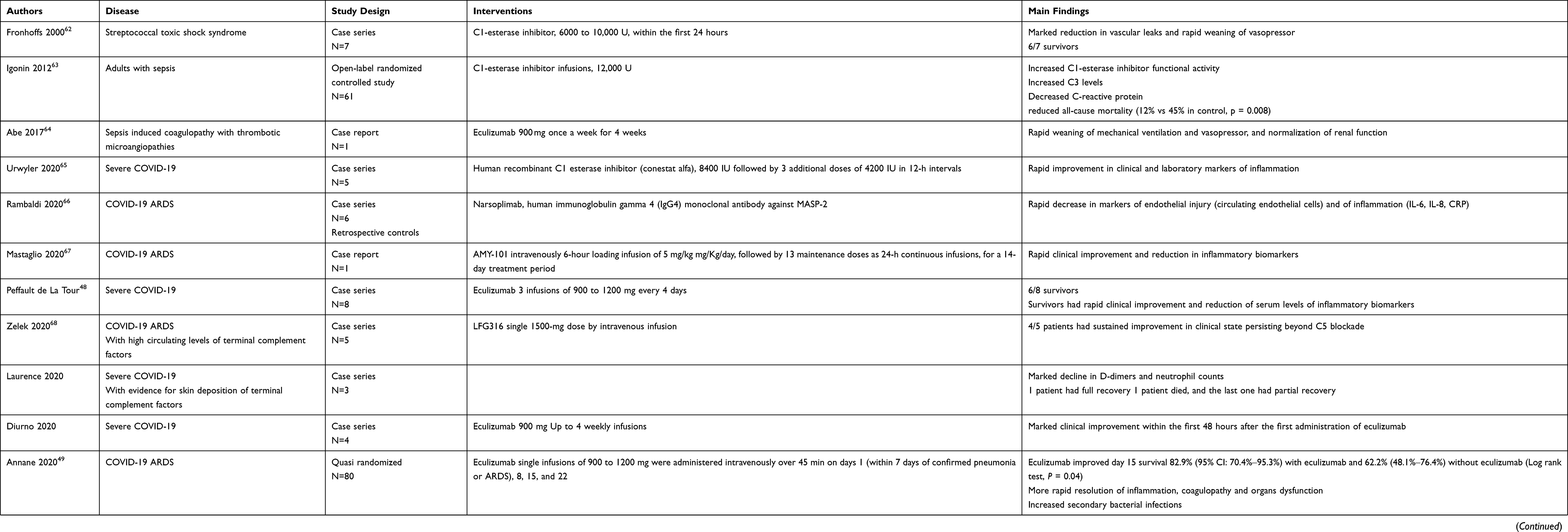 |
Table 3 Summary of Clinical Studies of Complement Inhibition |
Cases series have suggested promising effects on clinical outcomes in patients with severe COVID-19 treated with C1 esterase inhibitors,65 MASP-2 monoclonal antibodies,66 compstatin-based complement C3 inhibitor,67 anti-C5 drugs,48,68–70 or C5a-C5aR1 antagonists.41,51 A non-randomized study compared the complement C3 inhibitor AMY-101 and eculizumab -an anti-C5 monoclonal antibody- administered to 3 and 10 patients with severe COVID-19, respectively.71 Both drugs were associated with substantial reduction in circulating biomarkers of inflammation. The C3 inhibitor was associated with faster decrease in neutrophils count and NETosis and increase in lymphocytes and platelets count than eculizumab. Lung function and arterial haematosis improved similarly with both drugs. Unsurprisingly, AMY-101 and not eculizumab resulted in a rapid and sustained decrease in C3a serum levels. Of note, in contrast with AMY-101 effects, eculizumab was associated with transient decrease followed by a rebound in sC5b69 levels and residual AP-mediated complement haemolytic activity, suggesting transient and incomplete terminal pathway inhibition. These findings would support that in COVID-19, targeting C3 or its upstream activators may provide more benefits than the sole inhibition of C5-C5a-C5a-R axis via the regulation beyond the cytokine storm, of the Net generation and thrombotic microangiopathy. A quasi-randomized trial has investigated intravenous administration of 900mg of eculizumab at day 1, 8, 15 and 22 of ICU admission, in 80 critically ill COVID-19.49 Preliminary data found that these doses resulted in incomplete and transient inhibition of terminal pathway, and subsequent patients received higher doses at more frequent infusions. Eculizumab treated patients were more likely to survive at day-15 and day 28 and had prolonged survival time as compared to control. Eculizumab was associated with faster improvement in arterial oxygen tension and tissue oxygenation, more rapid increase in platelets count and prothrombin time, better renal function, and steeper decline in circulating levels of IL-6, IL-17 and IFN-α2. There were more patients with secondary bacterial infections in the eculizumab group versus controls. While levels of CH50 and sC5b-9 were dramatically decreased by eculizumab, levels of C5a did not differ from those of eculizumab-free patients.50 The incomplete terminal pathway inhibition by eculizumab in patients with severe COVID-19 is unclear. The strong activation of complement may have played a role in insufficient inhibition of the terminal complement pathway by eculizumab. Targeting C5a or C5a-R1 may offer the advantage over C5 antagonists, to preserve the formation of the membrane complex attack which is crucial to kill bacteria and to provide complete blockade of the anaphylatoxin C5a. The favorable benefit to risk ratio suggested in observational studies and small sized controlled studies should be tempered off by serious limitations inherent to the design of these studies, including intermediate outcomes and not patient-centered outcome and limited controls for selection and confounding biases. A proof-of-concept, phase 2 study has investigated IFX-1 - a selective antagonist of C5a- in 30 adults with severe COVID-19.72 There was no significant difference in changes in the ratio of arterial tension to the fraction of inspired oxygen (PaO2/FiO2) between IFX-1 treated and free patients (17% vs 41%; difference –24% [95% CI –58 to 9], p=0.15). Likewise, there were no difference in mortality by 28 days, 13% (95% CI 0–31) for IFX-1 and 27% (4–49) for controls (HR for death 0.65 [95% CI 0.10–4.14]). Patients in the experimental arm were more likely to have normalized lymphocytes count by day 15 (13 /15 [87%] versus 7/15 [47%], p=0.05). This adaptive design trial has now progressed to a Phase 3, placebo-controlled stage. Several ongoing randomized trials are investigating in severe COVID-19, the anti-C5 drugs zilucoplan,73,74 eculizumab,75 ravulizumab,76–78 the C3 inhibitors AMY-10179 and APL-9,80 the C1 esterase inhibitor conestat alpha,81–83 the novel peptide RLS-0071,84 the MASP-2 inhibitor narsoplimab.85 These trials are of variable size (ranging from 32 to 1500 participants) and most often compare active treatment to best supportive care.
Conclusion
Alike bacterial sepsis, severe COVID-19 is characterized by overactivation of the terminal complement pathway that contributes to airways and lung inflammation and injuries, disruption of endothelial function with subsequent spreading of the excessive inflammatory response to the periphery and imbalanced coagulation/fibrinolysis resulting in organs failure and eventually death. Collectively experimental data and preliminary results from observational studies shed light on the potential benefits from complement inhibition in severe COVID-19. Ongoing randomized trials should confirm a favorable benefit to risk ratio for complement inhibitors, and should precise the optimal targets in the complement cascade. Awaiting final results from these trials, complement inhibition should not be part of the routine management of patients with COVID-19.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Djillali Annane reports non-financial support from Alexion Pharmaceuticals Inc. to conduct a study on eculizumab for COVID-19. He is also an investigator for Alexion Pharmaceutical Inc sponsored trial on ravulizumab for COVID-19. The authors report no other potential conflicts of interest for this work.
References
1. John Hopkins University. Coronavirus resources center. Available from: https://coronavirus.jhu.edu/map.html.
2. Osuchowski MF, Winkler MS, Skirecki T, et al. The COVID-19 puzzle: deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir Med. 2021;S2213–2600(21):218–226. doi:10.1016/S2213-2600(21)00218-6
3. Onodi F, Bonnet-Madin L, Meertens L, et al. SARS-CoV-2 induces human plasmacytoid predendritic cell diversification via UNC93B and IRAK4. J Exp Med. 2021;218(4):e20201387. doi:10.1084/jem.20201387
4. Lee JS, Park S, Jeong HW, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol. 2020;5(49):eabd1554. doi:10.1126/sciimmunol.abd1554
5. Dorgham K, Quentric P, Gökkaya M, et al. Distinct cytokine profiles associated with COVID-19 severity and mortality. J Allergy Clin Immunol. 2021;S0091–S6749(21):651–6555. doi:10.1016/j.jaci.2021.03.047
6. Laing AG, Lorenc A, Del Molino Del Barrio I, et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat Med. 2020;26:623–1635. doi:10.1038/s41591-020-01186-5
7. Bordet J. Sur le mode d’action des sérums cytolytiques et sur l’unité de l’alexine dans un même sérum. Ann Inst Pasteur. 1901;15:303–318.
8. Cavaillon JM, Sansonetti P, Goldman M. 100th anniversary of Jules bordet’s Nobel prize: tribute to a founding father of immunology. Front Immunol. 2019;10:2114. doi:10.3389/fimmu.2019.02114
9. Bardhan M, Kaushik R. Physiology, Complement Cascade. StatPearls, Treasure Island (FL): StatPearls Publishing; 2021. PMID: 31855355.
10. Mollnes TE, Huber-Lang M. Complement in sepsis-when science meets clinics. FEBS Lett. 2020;594(16):2621–2632. doi:10.1002/1873-3468.13881
11. Ramlall V, Thangaraj PM, Meydan C, et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat Med. 2020;26(10):1609–1615. doi:10.1038/s41591-020-1021-2
12. Ehrlich P, Marshall HT. Ueber die complementophilen Gruppen der Amboceptoren. Berliner KlinWochenschr. 1902;39:585–587.
13. Kemper C, Köhl J. Novel roles for complement receptors in T cell regulation and beyond. Mol Immunol. 2013;56(3):181–190. doi:10.1016/j.molimm.2013.05.223
14. Hawlisch H, Belkaid Y, Baelder R, Hildeman D, Gerard C, Köhl J. C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity. 2005;22:415–426. doi:10.1016/j.immuni.2005.02.006
15. Ma N, Xing C, Xiao H, et al. C5a regulates IL-12+ DC migration to induce pathogenic Th1 and Th17 cells in sepsis. PLoS One. 2013;8(7):e69779. doi:10.1371/journal.pone.0069779
16. Fusakio ME, Mohammed JP, Laumonnier Y, Hoebe K, Köhl J, Mattner J. C5a regulates NKT and NK cell functions in sepsis. J Immunol. 2011;187(11):5805–5812. doi:10.4049/jimmunol.1100338
17. Riedemann NC, Guo RF, Bernacki KD, et al. Regulation by C5a of neutrophil activation during sepsis. Immunity. 2003;19(2):193–202. doi:10.1016/s1074-7613(03)00206-1
18. Strieter RM, Kasahara K, Allen RM, et al. Cytokine-induced neutrophil-derived interleukin-8. Am J Pathol. 1992;141(2):397–407.
19. Riedemann NC, Guo RF, Hollmann TJ, et al. Regulatory role of C5a in LPS-induced IL-6 production by neutrophils during sepsis. FASEB J. 2004;18(2):370–372. doi:10.1096/fj.03-0708fje
20. Yu M, Blomstrand E, Chibalin AV, Krook A, Zierath JR. Marathon running increases ERK1/2 and p38 MAP kinase signalling to downstream targets in human skeletal muscle. J Physiol. 2001;536(Pt1):273–282. doi:10.1111/j.1469-7793.2001.00273.x
21. Bosmann M, Patel VR, Russkamp NF, et al. MyD88-dependent production of IL-17F is modulated by the anaphylatoxin C5a via the Akt signaling pathway. FASEB J. 2011;25(12):4222–4232. doi:10.1096/fj.11-191205
22. Bosmann M, Sarma JV, Atefi G, Zetoune FS, Ward PA. Evidence for anti-inflammatory effects of C5a on the innate IL-17A/IL-23 axis. FASEB J. 2012;26(4):1640–1651. doi:10.1096/fj.11-199216
23. Ward PA. Sepsis, apoptosis and complement. Biochem Pharmacol. 2008;76(11):1383–1388. doi:10.1016/j.bcp.2008.09.017
24. Perianayagam MC, Balakrishnan VS, King AJ, Pereira BJ, Jaber BL. C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3-kinase-signaling pathway. Kidney Int. 2002;61(2):456–463. doi:10.1046/j.1523-1755.2002.00139.x
25. Guo RF, Sun L, Gao H, et al. In vivo regulation of neutrophil apoptosis by C5a during sepsis. J Leukoc Biol. 2006;80(6):1575–1583. doi:10.1189/jlb.0106065
26. Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112(5):1759–1766. doi:10.1182/blood-2008-04-151068
27. Conway Morris A, Kefala K, Wilkinson TS, et al. C5a mediates peripheral blood neutrophil dysfunction in critically ill patients. Am J Respir Crit Care Med. 2009;180(1):19–28. doi:10.1164/rccm.200812-1928OC
28. Foley JH, Conway EM. Cross talk pathways between coagulation and inflammation. Circ Res. 2016;118(9):1392–1408. doi:10.1161/CIRCRESAHA.116.306853
29. Brekke OL, Waage C, Christiansen D, et al. The effects of selective complement and CD14 inhibition on the E. coli-induced tissue factor mRNA upregulation, monocyte tissue factor expression, and tissue factor functional activity in human whole blood. Adv Exp Med Biol. 2013;735:123–136. doi:10.1007/978-1-4614-4118-2_8
30. Landsem A, Fure H, Christiansen D, et al. The key roles of complement and tissue factor in Escherichia coli-induced coagulation in human whole blood. Clin Exp Immunol. 2015;182(1):81–89. doi:10.1111/cei.12663
31. Kalbitz M, Fattahi F, Herron TJ, et al. Complement destabilizes cardiomyocyte function in vivo after polymicrobial sepsis and in vitro. J Immunol. 2016;197(6):2353–2361. doi:10.4049/jimmunol.1600091
32. Annane D. Sepsis-associated delirium: the pro and con of C5a blockade. Crit Care. 2009;13(2):135. doi:10.1186/cc7754
33. Hammerschmidt DE, Weaver LJ, Hudson LD, Craddock PR, Jacob HS. Association of complement activation and elevated plasma-C5a with adult respiratory distress syndrome. Pathophysiological relevance and possible prognostic value. Lancet. 1980;1(8175):947–949. doi:10.1016/s0140-6736(80)91403-8
34. Rota PA, Oberste MS, Monroe SS, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300(5624):1394–1399. doi:10.1126/science.1085952
35. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–1820. doi:10.1056/NEJMoa1211721
36. Wang R, Xiao H, Guo R, Li Y, Shen B. The role of C5a in acute lung injury induced by highly pathogenic viral infections. Emerg Microbes Infect. 2015;4(5):e28. doi:10.1038/emi.2015.28
37. Gralinski LE, Sheahan TP, Morrison TE, et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio. 2018;9(5):e01753–e01818. doi:10.1128/mBio.01753-18
38. Jiang Y, Zhao G, Song N, et al. Blockade of the C5a-C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg Microbes Infect. 2018;7(1):77. doi:10.1038/s41426-018-0063-8
39. Ng WF, To KF, Lam WW, Ng TK, Lee KC. The comparative pathology of severe acute respiratory syndrome and avian influenza A subtype H5N1--a review. Hum Pathol. 2006;37(4):381–390. doi:10.1016/j.humpath.2006.01.015
40. Sun S, Zhao G, Liu C, et al. Inhibition of complement activation alleviates acute lung injury induced by highly pathogenic avian influenza H5N1 virus infection. Am J Respir Cell Mol Biol. 2013;49(2):221–230. doi:10.1165/rcmb.2012-0428OC
41. Gao T, Hu M, Zhang X, et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv. 2020. doi:10.1101/2020.03.29.20041962
42. Pang RT, Poon TC, Chan KC, et al. Serum proteomic fingerprints of adult patients with severe acute respiratory syndrome. Clin Chem. 2006;52(3):421–429. doi:10.1373/clinchem.2005.061689
43. Ohta R, Torii Y, Imai M, Kimura H, Okada N, Ito Y. Serum concentrations of complement anaphylatoxins and proinflammatory mediators in patients with 2009 H1N1 influenza. Microbiol Immunol. 2011;55(3):191–198. doi:10.1111/j.1348-0421.2011.00309.x
44. Ip WK, Chan KH, Law HK, et al. Mannose-binding lectin in severe acute respiratory syndrome coronavirus infection. J Infect Dis. 2005;191(10):1697–1704. doi:10.1086/429631
45. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the `Cytokine Storm’ in COVID-19. J Infect. 2020;80(6):607–613. doi:10.1016/j.jinf.2020.03.037
46. Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1–13. doi:10.1016/j.trsl.2020.04.007
47. Holter JC, Pischke SE, de Boer E, et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc Natl Acad Sci U S A. 2020;117(40):25018–25025. doi:10.1073/pnas.2010540117
48. Peffault de Latour R, Bergeron A, Lengline E, et al. Complement C5 inhibition in patients with COVID-19 - a promising target? Haematologica. 2020;105(12):2847–2850. doi:10.3324/haematol.2020.260117
49. Annane D, Heming N, Grimaldi-Bensouda L, et al. Eculizumab as an emergency treatment for adult patients with severe COVID-19 in the intensive care unit: a proof-of-concept study. EClinicalMedicine. 2020;28:100590. doi:10.1016/j.eclinm.2020.100590
50. Annane D, Grimaldi-Bensouda L, Fremeaux-Bacchic V. Complement inhibition in severe COVID-19 - blocking C5a seems to be key: author’s reply. EClinicalMedicine. 2021;35:100866. doi:10.1016/j.eclinm.2021.100866
51. Carvelli J, Demaria O, Vély F, et al. Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. Nature. 2020;588(7836):146–150. doi:10.1038/s41586-020-2600-6
52. Risitano AM, Mastellos DC, Huber-Lang M, et al. Complement as a target in COVID-19? [published correction appears. Nat Rev Immunol. 2020;20(6):343–344. doi:10.1038/s41577-020-0320-7
53. Garred P, Tenner AJ, Mollnes TE. Therapeutic targeting of the complement system: from rare diseases to pandemics. Pharmacol Rev. 2021;73(2):792–827. doi:10.1124/pharmrev.120.000072
54. Silasi-Mansat R, Zhu H, Popescu NI, et al. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood. 2010;116(6):1002–1010. doi:10.1182/blood-2010-02-269746
55. Silasi-Mansat R, Zhu H, Georgescu C, et al. Complement inhibition decreases early fibrogenic events in the lung of septic baboons. J Cell Mol Med. 2015;19(11):2549–2563. doi:10.1111/jcmm.12667
56. Czermak BJ, Sarma V, Pierson CL, et al. Protective effects of C5a blockade in sepsis. Nat Med. 1999;5:788–792. doi:10.1038/10512
57. Laudes IJ, Chu JC, Sikranth S, et al. Anti-c5a ameliorates coagulation/fibrinolytic protein changes in a rat model of sepsis. Am J Pathol. 2002;160(5):1867–1875. doi:10.1016/S0002-9440(10)61133-9
58. Barratt-Due A, Thorgersen EB, Egge K, et al. Combined inhibition of complement C5 and CD14 markedly attenuates inflammation, thrombogenicity, and hemodynamic changes in porcine sepsis. J Immunol. 2013;191(2):819–827. doi:10.4049/jimmunol.1201909
59. Skjeflo EW, Sagatun C, Dybwik K, et al. Combined inhibition of complement and CD14 improved outcome in porcine polymicrobial sepsis. Crit Care. 2015;19:415. doi:10.1186/s13054-015-1129-9
60. Sun S, Zhao G, Liu C, et al. Treatment with anti-C5a antibody improves the outcome of H7N9 virus infection in African green monkeys. Clin Infect Dis. 2015;60(4):586–595. doi:10.1093/cid/ciu887
61. Hack CE, Ogilvie AC, Eisele B, Eerenberg AJ, Wagstaff J, Thijs LG. C1-inhibitor substitution therapy in septic shock and in the vascular leak syndrome induced by high doses of interleukin-2. Intensive Care Med. 1993;19(Suppl 1):S19–s28. doi:10.1007/BF01738946
62. Fronhoffs S, Luyken J, Steuer K, Hansis M, Vetter H, Walger P. The effect of C1-esterase inhibitor in definite and suspected streptococcal toxic shock syndrome. Report of seven patients. Intensive Care Med. 2000;26(10):1566–1570. doi:10.1007/s001340000654
63. Igonin AA, Protsenko DN, Galstyan GM, et al. C1-esterase inhibitor infusion increases survival rates for patients with sepsis. Crit Care Med. 2012;40(3):770–777. doi:10.1097/CCM.0b013e318236edb8
64. Abe T, Sasaki A, Ueda T, Miyakawa Y, Ochiai H. Complement-mediated thrombotic microangiopathy secondary to sepsis-induced disseminated intravascular coagulation successfully treated with eculizumab: a case report. Medicine (Baltimore). 2017;96(6):e6056. doi:10.1097/MD.0000000000006056
65. Urwyler P, Moser S, Charitos P, et al. Treatment of COVID-19 with conestat alfa, a regulator of the complement, contact activation and kallikrein-kinin system. Front Immunol. 2020;11:2072. doi:10.3389/fimmu.2020.02072
66. Rambaldi A, Gritti G, Micò MC, et al. Endothelial injury and thrombotic microangiopathy in COVID-19: treatment with the lectin-pathway inhibitor narsoplimab. Immunobiology. 2020;225(6):152001. doi:10.1016/j.imbio.2020.15200
67. Mastaglio S, Ruggeri A, Risitano AM, et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol. 2020;215:108450. doi:10.1016/j.clim.2020.108450
68. Zelek WM, Cole J, Ponsford MJ, et al. Complement inhibition with the C5 blocker LFG316 in severe COVID-19. Am J Respir Crit Care Med. 2020;202(9):1304–1308. doi:10.1164/rccm.202007-2778LE
69. Laurence J, Mulvey JJ, Seshadri M, et al. Anti-complement C5 therapy with eculizumab in three cases of critical COVID-19. Clin Immunol. 2020;219:108555. doi:10.1016/j.clim.2020.108555
70. Diurno F, Numis FG, Porta G, et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharmacol Sci. 2020;24(7):4040–4047. doi:10.26355/eurrev_202004_20875
71. Mastellos DC, Pires da Silva BGP, Fonseca BAL, et al. Complement C3 vs C5 inhibition in severe COVID-19: early clinical findings reveal differential biological efficacy. Clin Immunol. 2020;220:108598. doi:10.1016/j.clim.2020.108598
72. Vlaar APJ, de Bruin S, Busch M, et al. Anti-C5a antibody IFX-1 (vilobelimab) treatment versus best supportive care for patients with severe COVID-19 (PANAMO): an exploratory, open-label, phase 2 randomised controlled trial. Lancet Rheumatol. 2020;2(12):e764–e773. doi:10.1016/S2665-9913(20)30341-6
73. Declercq J, Bosteels C, Van Damme K, et al. Zilucoplan in patients with acute hypoxic respiratory failure due to COVID-19 (ZILU-COV): a structured summary of a study protocol for a randomised controlled trial. Trials. 2020;21(1):934. doi:10.1186/s13063-020-04884-0
74. Wilkinson T, Dixon R, Page C, et al. A multicentre, seamless, phase 2 adaptive randomisation platform study to assess the efficacy and safety of multiple candidate agents for the treatment of COVID-19 in hospitalised patients: a structured summary of a study protocol for a randomised controlled trial. Trials. 2020;21(1):691. doi:10.1186/s13063-020-04584-9
75. CORIMUNO19-ECU: trial evaluating efficacy and safety of eculizumab (Soliris) in patients with COVID-19 infection, nested in the CORIMUNO-19 cohort full text view. Available from: ClinicalTrials.gov.
76. Kulkarni S, Fisk M, Kostapanos M, et al. Repurposed immunomodulatory drugs for Covid-19 in pre-ICu patients - mulTi-arm therapeutic study in pre-ICU patients admitted with Covid-19 - repurposed drugs (TACTIC-R): a structured summary of a study protocol for a randomised controlled trial. Trials. 2020;21(1):626. doi:10.1186/s13063-020-04535-4
77. Ravulizumab and COVID-19 full text view. Available from: ClinicalTrials.gov.
78. Smith K, Pace A, Ortiz S, Kazani S, Rottinghaus S. A phase 3 open-label, randomized, controlled study to evaluate the efficacy and safety of intravenously administered ravulizumab compared with best supportive care in patients with COVID-19 severe pneumonia, acute lung injury, or acute respiratory distress syndrome: a structured summary of a study protocol for a randomised controlled trial. Trials. 2020;21(1):639. doi:10.1186/s13063-020-04548-z
79. A study of the C3 Inhibitor AMY-101 in patients with ARDS due to COVID-19 (SAVE) - full text view. Available from: ClinicalTrials.gov.
80. A study of APL-9 in adults with mild to moderate ARDS due to COVID-19 - full text view. Available from: ClinicalTrials.gov.
81. Study to evaluate the benefit of RUCONEST in improving neurological symptoms in post COVID-19 infection full text view. Available from: ClinicalTrials.gov.
82. Urwyler P, Charitos P, Moser S, et al. Recombinant human C1 esterase inhibitor (conestat alfa) in the prevention of severe SARS-CoV-2 infection in hospitalized patients with COVID-19: a structured summary of a study protocol for a randomized, parallel-group, open-label, multi-center pilot trial (PROTECT-COVID-19). Trials. 2021;22(1):1. doi:10.1186/s13063-020-04976-x
83. Prevention of severe SARS-CoV-2 infection in hospitalized patients with COVID-19 full text view. Available from: ClinicalTrials.gov.
84. A study of RLS-0071 in patients with acute lung injury due to COVID-19 pneumonia in early respiratory failure full text view. Available from: ClinicalTrials.gov.
85. I-SPY COVID-19 TRIAL: an adaptive platform trial for critically ill patients full text view. Available from: ClinicalTrials.gov.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.