")
Back to Journals » Journal of Inflammation Research » Volume 14
Comparison of the Characteristics of Cytokine Storm and Immune Response Induced by SARS-CoV, MERS-CoV, and SARS-CoV-2 Infections
Authors Liu T , Feng M, Wen Z, He Y, Lin W, Zhang M
Received 19 July 2021
Accepted for publication 30 September 2021
Published 23 October 2021 Volume 2021:14 Pages 5475—5487
DOI https://doi.org/10.2147/JIR.S329697
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Tong Liu,1,* Meng Feng,2,* Zexin Wen,3 Yijie He,1 Wei Lin,2 Min Zhang1
1Department of Medicine, Xizang Minzu University, Xianyang, Shaanxi, People’s Republic of China; 2School of Basic Medicine, Shandong First Medical University & Shandong Academy of Medical Sciences, Jinan, Shandong, People’s Republic of China; 3Department of Clinical Medicine, Fujian Medical University, Fuzhou, Fujian, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Min Zhang
Department of Medicine, Xizang Minzu University, Xianyang, People’s Republic of China
Tel +86 2918681997994
Email [email protected]
Wei Lin
School of Basic Medicine, Shandong First Medical University & Shandong Academy of Medical Sciences, Jinan, People’s Republic of China
Tel +86 53115098926353
Email [email protected]
Abstract: Cytokine storm (CS) is a significant cause of death in patients with severe coronavirus pneumonia. Excessive immune–inflammatory reaction, many inflammatory cell infiltration, and extreme increase of proinflammatory cytokines and chemokines lead to acute lung injury and acute respiratory distress syndrome (ARDS). This review compares the characters of cytokine storms and immune responses caused by three highly pathogenic and infectious coronaviruses (HCoVs), including severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome-coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and analyzes the possible mechanisms to guide clinical treatment in the future.
Keywords: cytokines, coronavirus pneumonia, cytokine storm, immune evasion, interferon
Introduction
Since December 2019, many new cases of human coronavirus infection have been reported globally. Then, the Coronaviridae Study Group (CSG) of the International Committee on Taxonomy of Viruses issued a statement naming novel coronavirus “severe acute respiratory syndrome coronavirus 2” (SARS-CoV-2).1 The SARS-CoV-2 infection causes an excessive immune response, including an increase in T helper 17 (Th17) cells and T follicular helper (Tfh) cells, a decrease in Th1 cells and high levels of cytokines (IFN, IL, chemokines, CSF, tumor necrosis factor (TNF), etc.) to be released into the circulation, with widespread and harmful effects on multiple organs; also, it can cause severe respiratory syndrome and even death.2,3 Severe inflammatory reactions caused by cytokine storms (CS) begin locally and spread throughout the body, leading to collateral damage in tissues.4
As of June 15, 2021, there had been 176,918,775 confirmed cases and 3,822,629 deaths worldwide. Considering its worldwide reach and severity, WHO declared the COVID-19 outbreak a global pandemic – the first pandemic sparked by a coronavirus.5 On January 24, 2020, the first clinical data report of novel coronavirus infection published in the Lancet showed that the severity of coronavirus disease 2019 (COVID-19) positively correlated with CS,3 resulting in most people dying of multiple organ failure. Hence, CS may be an important reason for acute respiratory distress syndrome (ARDS) caused by coronaviruses (HCoVs). Other previously reported coronaviruses, such as severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), can cause CSs.
The difference in the phenomenon and cause of CS between SARS-CoV-2 and other coronaviruses is still unclear. By collecting previous reports, we compared and reviewed the differences in CSs caused by SARS-CoV, MERS-CoV and SARS-CoV-2, as well as the different structural factors of the virus and the possible mechanism of immune activation.
Cytokines and CS
Cytokines are small molecular proteins with a wide range of biological activities synthesized and secreted by immune cells (monocytes, macrophages, T cells, B cells, and NK cells) and some nonimmune cells (endothelial cells, epidermal cells, and fibroblasts). Cytokine storm is life-threatening systemic inflammatory syndromes involving elevated levels of circulating cytokines and immune-cell hyperactivation that can be triggered by various therapies, pathogens, cancers, autoimmune conditions, and monogenic disorders.6 It may also occur after treatment with some types of immunotherapy. Symptoms include high fever, inflammation (redness and swelling), severe fatigue and nausea. Sometimes, cytokine storms can be serious or life-threatening and lead to multiple organ failure.
When the virus invades, the infected body initiates innate immune responses, and the pattern recognition receptors (PRRs) on the surface of the innate immune cells recognize the unique molecular structure of the invading virus, called pathogen-associated molecular patterns (PAMPs).7 PAMPs and PRRs trigger the body’s inflammatory response to the invading virus, which activates the signaling pathways to stimulate the production of IFN and other cytokines.8 The three most important inflammatory cytokines of the innate immune response are IL-1, IL-6, and TNF-α. They mainly come from macrophages, mast cells, epithelial cells, and endothelial cells in immune reactions. Like immune molecules, such as major histocompatibility complex, cluster of differentiation (CD), complement, adhesion molecules, and antibodies, cytokines are involved in disease occurrence and closely related to immune regulation.9 However, under specific conditions, many cytokines will increase rapidly in a short time to cause “CS,” seriously endangering life and safety.10
After coronavirus infection, human alveolar epithelial cells (AECs) proliferate and activate immune cells, such as CD4+ T cells. The activated CD4+ T cells further differentiate into T helper cell 1 (Th1) and T helper cell 2 (Th2). Th1 cells mainly produce IL-2, IFN-γ, TNF-β, and granulocyte-macrophage colony-stimulating factor (GM-CSF), which releases primary cytokines.11 After entering the blood, GM-CSF can recruit and activate more CD14+CD16+ inflammatory monocytes, eosinophils, neutrophils, and other immune cells. This will further secrete more cytokines, resulting in rising cytokine levels, thus releasing secondary cytokines. These inflammatory factors can activate more immune cells to release several cytokines, forming a positive feedback response.12 It leads to increased cytokines in a short time, resulting in ARDS and multiple organ dysfunction syndromes, leading to death.
CS Caused by Coronavirus Infection
Overview of Coronavirus
Coronaviruses belong to the family of Coronaviridae, the order Nidovirales, and the genus Coronavirus,13 Human coronaviruses belong to the subfamily Coronavirinae.14,15 According to their genomic structure and phylogenetic relationship, Coronavirinae subfamily can be divided into four genera: α-coronavirus, β-coronavirus, γ-coronavirus, and δ-coronavirus.16 α-coronavirus and β-coronavirus only infect mammals, whereas γ-coronavirus and δ-coronavirus can infect birds, and some of them also infect mammals.13 Presently, there are seven human coronaviruses: HCoV-229E, HCoV-OC43, SARS-CoV, HCoV-NL63, HCoV-HKU1, MERS-CoV, and SARS-CoV-2.17 SARS-CoV and MERS-CoV, two highly pathogenic viruses belonging to the β-coronavirus genus, can cause severe acute respiratory disease. However, the other four HCoVs (HCoV-NL63, HCoV-OC43, HCoV-229E, and HKU1) can cause mild upper respiratory diseases in immune hosts.18 According to the current gene sequence database, all HCoVs originate from animals. They include SARS-CoV, MERS-CoV, HCoV-NL63, and HCoV-229E, all thought to originate in bats; also, HCoV-OC43 and HKU1 may have originated from rodents.19 However, the origin of SARS-CoV-2 remains inconclusive. As human and animal activities increase, the chance of cross-species infection increases and livestock may also have a significant impact as an intermediate host. This allows the virus to spread from natural hosts to humans, so coronaviruses increasingly threaten humans.
SARS-CoV and CS
SARS-CoV is a highly pathogenic respiratory virus with single-stranded RNA and is also a primary cause of SARS. It can infect tracheal epithelial cells and AECs, leading to acute lung injury and ARDS. The results of in vitro experiments show that When SARS-CoV infected AEC, many chemokines will be produced, such as C–C motif chemokine ligand 2 (CCL2), CCL3, CCL5, and C-X-C motif chemokine ligand 10 (CXCL10).11 These chemokines can recruit immune cells to infiltrate the inflammatory site, resulting in inflammatory injury. SARS-CoV binds with angiotensin-converting enzyme 2 (ACE2), expressed in alveolar epithelial type II cells (AT2), to invade cells. Also, various high-level viral replication-related genes were detected in AT2-expressing ACE2, including viral replication regulatory genes, viral life cycle regulatory genes, viral assembly genes, and viral genome replication genes.20 This shows that AT2-expressing ACE2 can promote the replication of HCoVs in lung tissue. However, when SARS-CoV infects dendritic cells (DCs), mononuclear macrophages, and other peripheral blood mononuclear cells, it cannot replicate in the cell to produce the virus, but it can induce infected cells to release a large number of cytokines (Figure 1).21,22 When SARS-CoV infects DCs, it induces the expression of the low levels of antiviral cytokine interleukin-12p40 (IL-12p40), IFN-α, IFN-β, and IFN-γ. This can appropriately upregulate the expression of proinflammatory cytokines TNF-α and IL-6 and significantly upregulate the expression of inflammatory chemokines CCL2, CCL3, CCL5, and CXCL10.21,22 Simultaneously, SARS-CoV induces macrophages to produce IFN.22 These proliferative cytokines and chemokines are the leading causes of severe lung injury in patients with SARS. Serum proinflammatory cytokines IFN-γ, IL-1, IL-6, and IL-12, transforming growth factor-β (TGF-β), and chemokine CCL2, CXCL10, CXCL9, and IL-8 in patients with severe infections were significantly higher than those in noncritical patients.23–25 The excessive release of these cytokines is the direct cause of CS. In summary, when AEC, DCs, and macrophages are infected with SARS-CoV, it leads to a surge in cytokines and chemokines, which plays a vital role in the pathogenesis of CS in patients with SARS (Figure 1). T cells recognize antigenic epitopes and are activated. SARS-CoV does not have the ability to attack T cells directly because T cells do not express the receptor (ACE2) they need. However, when monocyte–macrophages and DCs are infected by SARS-CoV, they release a large number of inflammatory cytokines to induce T cell apoptosis. The cells in the dotted frame can secrete various cytokines in the immune response, further forming a CS.
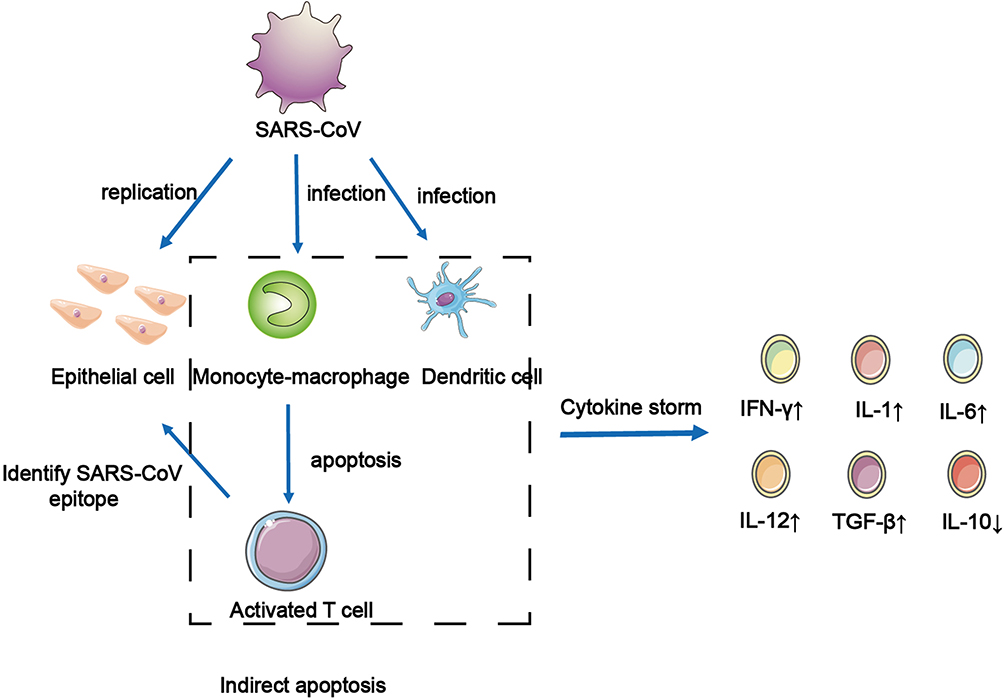 |
Figure 1 The infection and replication of SARS-CoV in epithelial cells and immune cells. SARS-CoV infect and replicate in epithelial cells. Monocyte-macrophage and DCs are infected by SARS-CoV to induced apoptosis of activated T cells. The cells in the dotted frame can secrete a variety of cytokines in the immune response, further forming a cytokine storm. |
MERS-CoV and CS
MERS is a respiratory disease caused by a single-stranded RNA virus, MERS-CoV. MERS-CoV specifically recognizes and binds to dipeptidyl peptidase 4 on the surface of AEC through S1 protein on the surface and then infects host cells.26 When MERS-CoV infects the body, besides common symptoms, such as cough and fever, it may also develop into acute respiratory failure and acute kidney injury (AKI).27,28 In vitro studies have shown that it has a similar infection mechanism to SARS-CoV. When MERS-CoV was infected with AEC, it could induce a strong response to IFN-γ and cytokines IL-1 β, IL-6, and IL-8, but the immune response was delayed.29 The only difference is that MERS-CoV can replicate in activated human T cells. However, whether it can replicate in human monocyte-derived macrophages and MDDCs is still controversial and needs to be further studied (Figure 2).30–32 When MERS-CoV infects monocyte line THP-1 cells, macrophages, and DCs derived from human peripheral blood monocytes, it can induce the production of proinflammatory cytokines and chemokines, such as CCL-2, CCL-3, CCL-5, IL-2, and IL-8, and form a positive feedback response, eventually leading to a CS.30,31 However, mononuclear macrophages and DCs do not induce the production of IFN-αβ; only plasmacytoid DCs can produce a large amount of IFN when they are infected by MERS-CoV.33 Recent studies have shown that the levels of cytokines and chemokines in the serum of patients with MERS positively correlate with the number of neutrophils and monocytes in their peripheral blood. The serum levels of cytokine IL-6, IL-8, and IFN-α, and chemokines CXCL10 and CCL5 in patients with severe infection were higher than those in patients with mild MERS.34,35 This is also the main factor causing CS in patients with MERS. Moreover, through the study of high mortality caused by CS in patients with MERS, it was shown that MERS-CoV could replicate in human macrophages and DCs, resulting in abnormal secretion of proinflammatory cytokines and chemokines. This induces delayed release of inflammatory cytokines, weakening the innate immune response and antiviral effect.29 MERS-CoV can infect and replicate the cells shown above, causing apoptosis in the dotted frame and releasing cytokines that cause CSs (Figure 2).
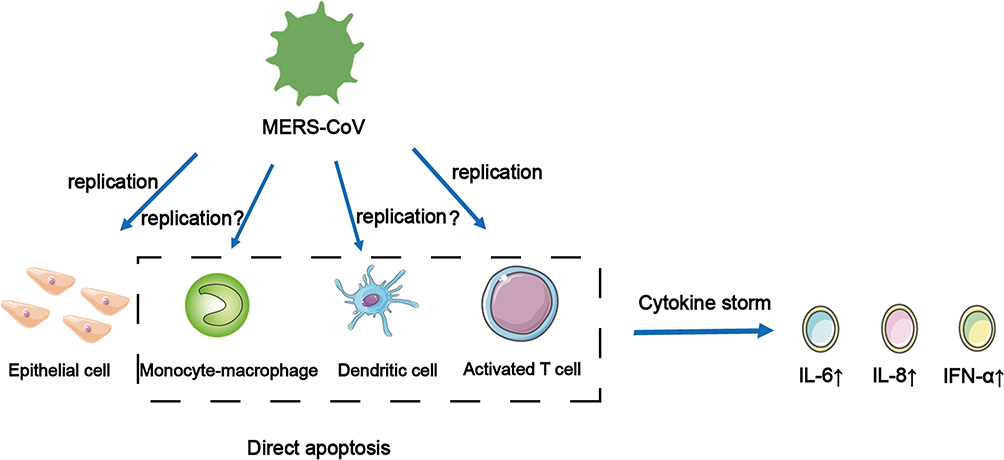 |
Figure 2 MERS-CoV can infect and replicate in the cells shown above, causing apoptosis in the dotted frame and releasing cytokines that cause cytokine storms. It is worth noting that whether it can replicate in human monocyte-derived macrophages and DCs is still not clear. |
SARS-CoV-2 and CS
The newly emerged SARS-CoV-2, which can infect humans, is a single-stranded RNA virus of the coronavirus family. The most common symptoms of COVID-19 are fever, fatigue, and respiratory symptoms; only a few people develop ARDS, acute cardiac injury, or secondary infection.3 Although SARS-CoV-2 can replicate in AT2, it has not been proven to replicate in alveolar macrophages and DCs.36 Most evidence suggests that some critical patients with COVID-19 had similar CSs in patients with SARS and MERS. Huang et al3 reported cytokine levels in 41 inpatients (including 13 patients in intensive care unit and 28 patients in nonintensive care unit). It was found that the levels of IL-7, IL-1RA, IL-1β, IL-10, IL-9, IL-8, fibroblast growth factor, GM-CSF, IFNγ, granulocyte colony-stimulating factor (G-CSF), interferon-γ inducible protein-10 (IP-10), platelet-derived growth factor, vascular endothelial growth factor, macrophage inflammatory protein-1α, monocyte chemotactic protein 1 (MCP1), and TNF-α were higher than those in healthy adults. Among them, IL-2, IL-7, IL-10, G-CSF, IP10, MCP1, MIP1α, and TNF-α were significantly increased in critical patients. This study points out that CS is related to the severity of disease caused by SARS-CoV-2 infection (Figure 3). It had been reported that IL-6 levels were also elevated in critical patients with COVID-19.37 Hence, it can be seen that IL-6 is an essential molecule in the occurrence of CS. However, SARS-CoV-2 infection, which is different from SARS-CoV infection, also leads to Th2 cytokine production that inhibits inflammation, such as increased secretion of IL-4 and IL-10; however, its mechanism is unclear. During SARS-CoV-2 infection, the production and mechanism of Th1 and Th2 cytokines must be further studied. SARS-CoV-2 infection of T cells can cause them to secrete several cytokines, which can cause CSs and damage to the heart, kidney, and lungs. However, it is unclear what other cells SARS can infect and replicate in them.
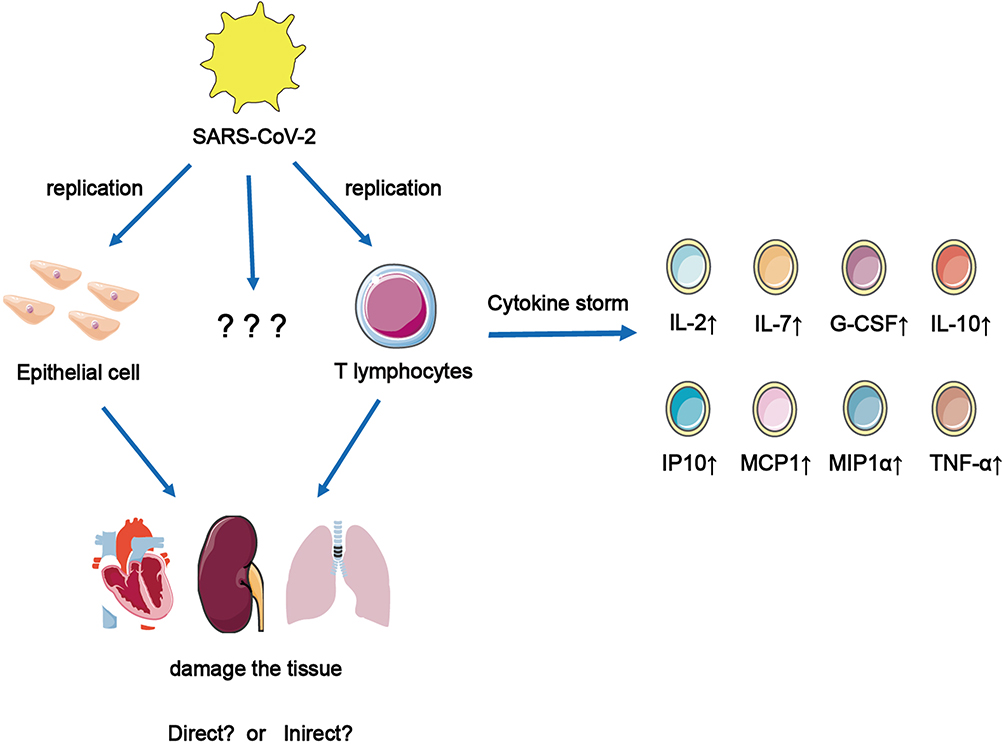 |
Figure 3 SARS-CoV-2 infect and replicate in epithelial cells and T cells and cause them to secrete a large number of cytokines, which can cause cytokine storms to cause the damage of heart, kidney and lungs. But it is not clear which other cells that SARS-CoV-2 can infect and replicate in them. |
There are differences in cytokines produced by the three coronaviruses after infecting humans (Table 1). For example, SARS-CoV can cause a decrease in antiinflammatory cytokines IL-10. In contrast, the antiinflammatory factors IL-4 and IL-10 of SARS-CoV-2 are increased. However, its mechanism is unclear, and other cytokines secreted can lead to immune cell proliferation and differentiation and promote inflammatory responses. Infection with SARS-CoV or MERS-CoV can stimulate mononuclear macrophages, DCs, and activated T cells to produce many cytokines. However, after SARS-CoV-2 infection, it can promote the secretion of related cytokines by epithelial and T cells.
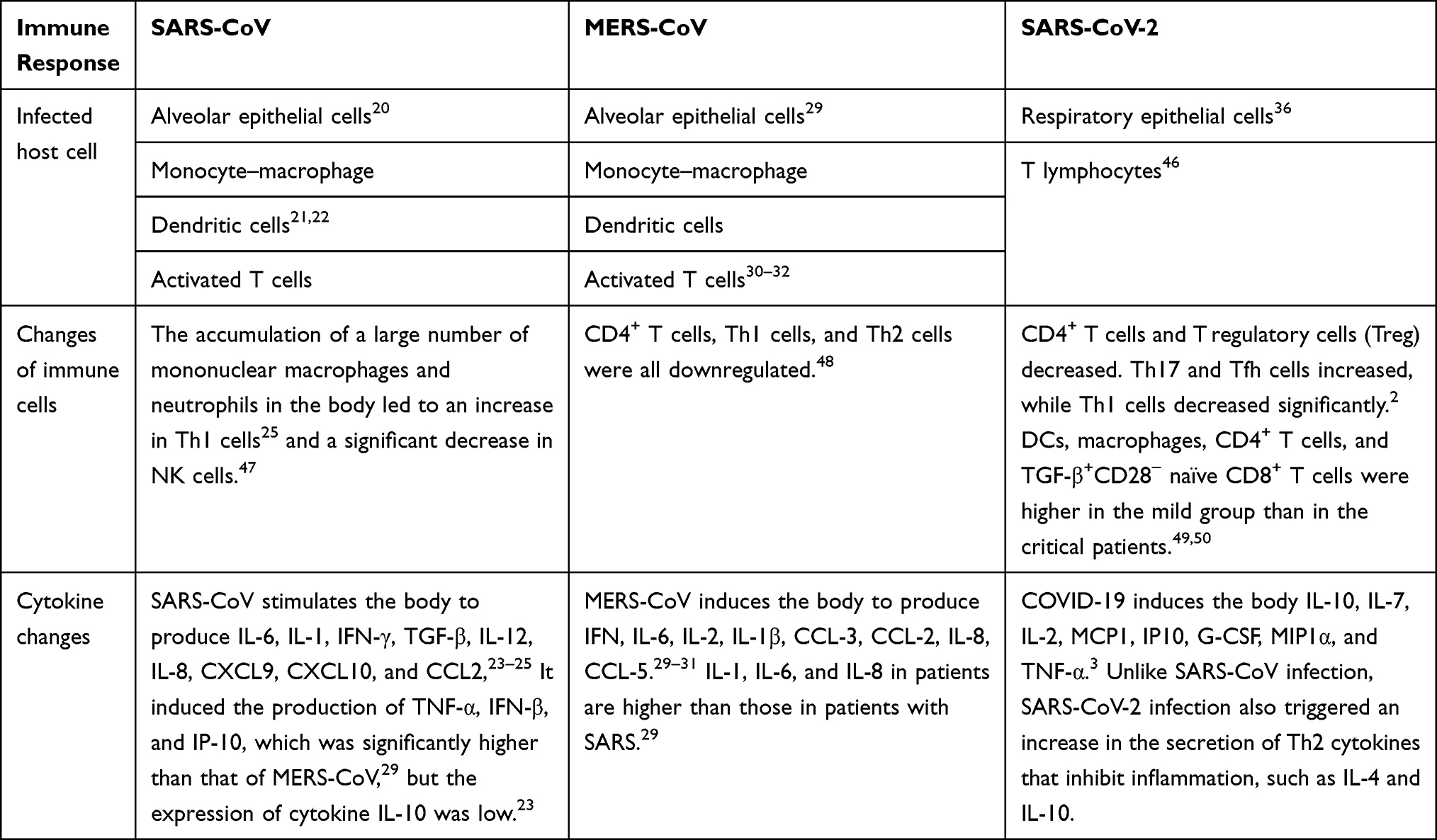 |
Table 1 Comparing Host Cells Infected with Different Types of Coronavirus and Changes in Immune Cells and Cytokines |
Comparing Clinical Immune Characteristics of CS Produced by Coronaviruses
The clinical symptoms and immune characteristics caused by different viruses are different. The main manifestations of patients with SARS are fever, dyspnea, and lymphocytopenia; some cases develop to respiratory failure, and the fatality rate is approximately 10%. The susceptible population was immunocompromised for older adults, and the fatality rate of elderly patients (>60 years old) increased significantly (from 6.8% to 43%).38 Although the infectious power of MERS-CoV in the population is not as high as that of SARS-CoV, the incidence of ARDS and AKI in patients with MERS is higher, and the fatality rate is as high as 34.4%.39 The lungs of patients with COVID-19 are mainly involved; also, there are more critically ill patients; approximately 20% and 25.2% of the patients have at least one underlying disease (such as hypertension and chronic obstructive pulmonary disease). The fatality rate was approved at 2%.3,40 In addition, most patients suffer from pneumonia, and the chest CT shows typical ground glass opacity of the lungs with reduced lymphatic vessels.3,41,42
Also, studies on the immune status of patients with COVID-19 showed that although lymphocytes in the peripheral blood of patients with severe COVID-19 decreased, they were still in the activation state. A study analyzed the changes in lymphocyte subsets and cytokines in the peripheral blood of 123 patients and found that all patients had reduced lymphocytes.43 The reduction rates of CD8+ T cells in the mild and severe groups were 28.43% and 61.9%, respectively. The reduction rates of NK cells were 34.31% and 47.62%. The level of serum NK cells in the severe group was significantly higher than that in the nonsevere group.43 Additionally, the expression of human leukocyte antigen-DR in CD4+ T and CD8+ T cells increased, and CD4+CCR4+CCR6+ T helper cell 17 also increased, and CD8+ T cells highly expressed cytotoxic particles, such as perforin and granzyme.44 Like other patients with COVID-19, most of the severe COVID-19 patients showed high C-reactive protein, erythrocyte sedimentation rate, and high levels of IL-6, tumor necrosis factor, IL-1, IL-2R, and IL-8, accompanied by ARDS, diffuse intravascular coagulation, and mainly thrombocytopenia, characterized by thrombosis and intractable gangrene. Th1 and Th2 decreased when the body was infected with MERS-CoV, whereas Th1 increased when the body was infected with SARS-CoV,25 and a significant decrease in NK cells was also observed (Table 1).
SARS-CoV stimulates the body to produce higher level of TNF-α, IFN-β, and IP-10 than MERS-CoV does.29 MERS-CoV also induces the body to produce IFN, IL-6, IL-2, IL-1β, CCL-3, CCL-2, IL-8, CCL-5, but it induces higher level of IL-1, IL-6, and IL-8 in patients than SARS.29–31 SARS-CoV-2 infection also triggered an increase in the secretion of Th2 cytokines that inhibit inflammation, such as IL-4 and IL-10.3
In COVID-19, IL-10 levels began to rise as early as the first week of the disease and decreased during the duration of the disease.45 However, the level of IL-10 only increased in convalescent patients with SARS. Thus, the early induction of IL-10 by COVID-19 may change the early infection process of the disease and play a certain role in the treatment of IL-10 in critically ill patients. Since this phenomenon may be unique to COVID-19, understanding how SARS-CoV-2 does this will benefit patient management, and there may be new treatments for COVID-19.
Analysis showed significant differences in immune cell distribution and cytokine expression induced by different coronaviruses, and the clinical symptoms and severity of patients with different coronavirus infections were also different (Table 1). This may be related to the infection mechanism of different coronaviruses and immune status change. The expression of different cytokines caused by different coronavirus infections provides an idea for accurate, personalized treatment of coronavirus.
Comparing Coronavirus-Encoded Proteins
Releasing different cytokines may be related to different viral structures. Coronavirus particles contain four main structural proteins: spike protein (S), membrane protein (M), envelope protein (E), and nucleocapsid protein (N). The S protein of coronavirus is a trimer located on the surface of virions, which makes virions have a coronal appearance. The primary function is to establish a connection between the viral particles and the host cell membrane by interacting with various host cell receptors, thus mediating the entry of infectious viral particles into the cells.51 Additionally, it is an important factor in inducing host immune response.52 The M protein is the most abundant structural protein in coronavirus particles. Studies have shown that the M protein exists in the form of a dimer in coronaviruses and may use two conformations, promoting the bending of the host cell membrane and binding to the nucleocapsid.53 The E protein is the smallest structural protein in virions, which plays a multifunctional role in the pathogenesis, assembly, and release of viruses. The virulence of coronavirus is also related to the E protein.54 Furthermore, deleting the gene encoding E protein will slow down viral amplification, but the protein does not seem necessary for SARS-CoV replication (Table 2).55
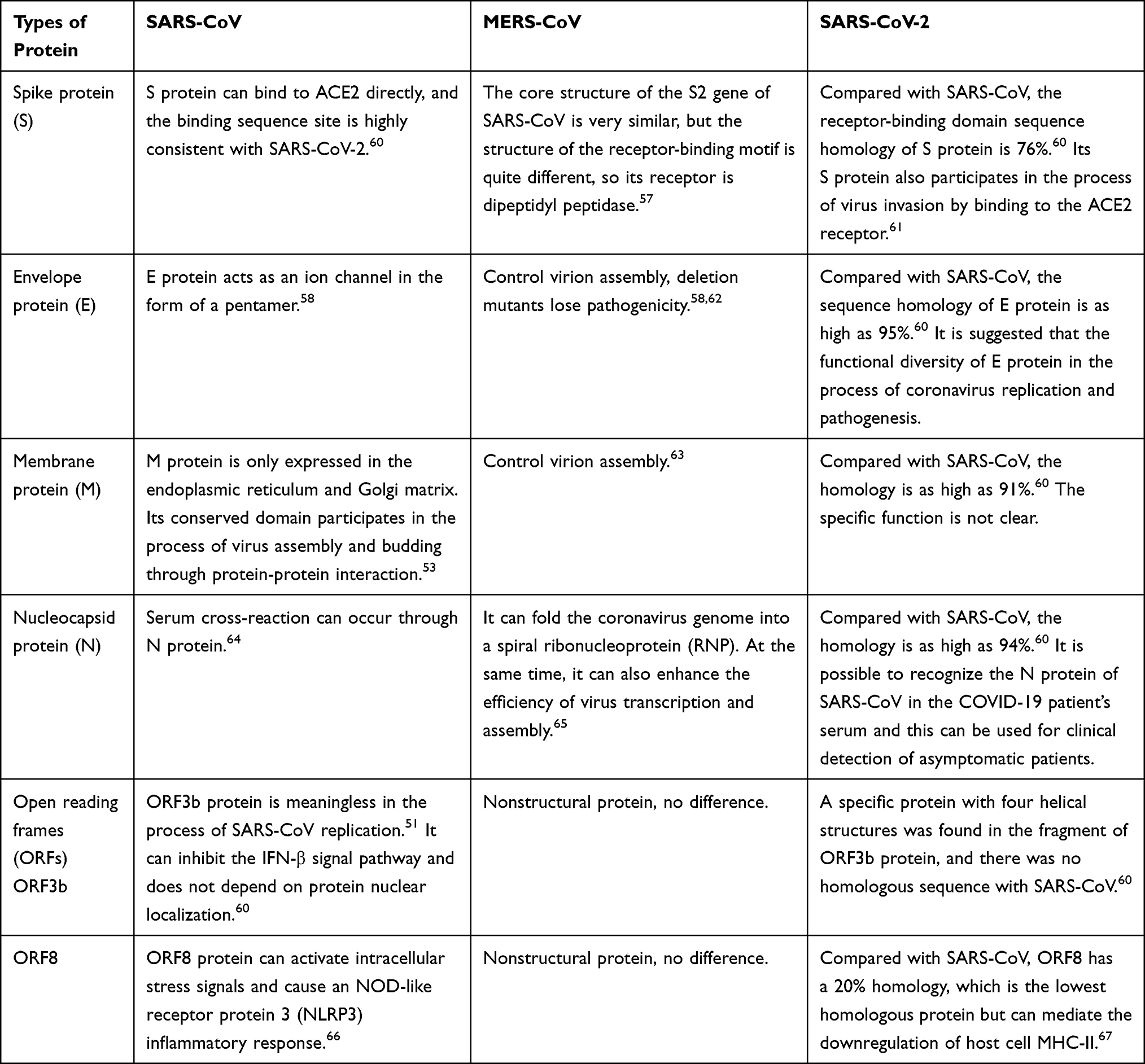 |
Table 2 Comparing Protein Differences in Coronavirus |
By comparing the structure of the three viruses with previous reports, it was found that the biggest difference among the structural proteins of the virus is the S protein. SARS-CoV, SARS-CoV-2, and MERS-CoV have different receptors that bind to the host cell because of the difference in S protein.56,57 This suggests that the ability of different viruses to recognize and invade cells and the types of host cells infected will be different. ORF3 and ORF8 are related to inflammation and escape after virus infection, and the differences in ORF3 and ORF8 genes in the three coronaviruses may be related to different inflammatory mechanisms. The difference in the E protein may be related to the mechanism of viral pathogenesis, assembly, and release. The E protein is less expressed in viruses and has ion channel activity.58 It is believed that the M protein is the most abundant protein and defines the shape of the viral envelope and the N protein is a multifunctional protein that can enhance viral transcription and assembly.59
Coronavirus Infection and Immune Response Mechanism
The excessive immune response caused by coronavirus invading the host is the primary pathogenic mechanism of its high clinical lethality. The viral genome and related viral proteins released by the coronavirus after infecting the host are recognized by PRRs in the host cells. Also, the CS caused by coronavirus infection is related to the activated cytokine signaling pathway.
During SARS-CoV-2 infection, Th1 inflammatory cytokines and Th2 inflammatory cytokines were activated, indicating that adaptive immunity was widely up-regulated in the process of anti-viral infection.3 It had been reported that there was an increased Th17 in the blood of severe COVID-19 patients, and Th17 type responses were speculated to be partially responsible for the local severe lung injuries and COVID-19 exacerbation.44 Strong Th17-mediated cytokine storms and activation of related pathways were also found in SARS and MERS infections, which play an important role in lung inflammatory injury. On this basis, the specific inhibitors of Th17 pathway signal may have certain therapeutic value.67,68
Apoptosis can be induced by multiple mechanisms in HCoV-infected cells. SARS-CoV was shown to induce caspase-dependent apoptosis, which is dependent on but not essential for viral replication, as treatment of pan-caspase inhibitor z-VAD-FMK or overexpression of Bcl2 did not significantly affect SARS-CoV replication.69 In contrast, although MERS-CoV infection of human primary T lymphocytes was abortive, apoptosis was induced via activation of both intrinsic and extrinsic pathways.32 Apoptosis in neuronal cells infected with HCoV-OC43 involved mitochondrial translocation of BAX but was independent of caspase activation.70
Toll-Like Receptors and Signal Pathways
Toll-like receptors (TLRs) play an essential role in the coronavirus, activating the immune system, and they are PRRs that participate in the innate immune response of extracellular pathogens. There are 13 members of the TLR family, among which TLR3, TLR7, TLR8, and TLR9 are located in intracellular vesicles and are transmembrane receptors that recognize viral nucleic acids.71 In the TLR3-mediated signaling pathway, TLR3, through the transporter TIR-containing adaptor molecule (TICAM-1/TRIF), mediates inflammation.72 TLR3 recruits and activates tumor necrosis factor receptor-associated factor 3 (TRAF3) through the TRIF pathway. The activated TRAF3 transmits signals to downstream TANK-binding kinase 1 (TBK1) and I-κ B kinase ε (IKK ε) to activate interferon regulatory factor 7 (IRF7) and IRF7 in the nucleus to induce IFN expression.73 The signal-binding proteins of TLR7, TLR8, and TLR9 are myeloid differentiation factor 88 (MyD88), in which TLR7 and TLR8 recognize single-stranded ribonucleic acid, whereas TLR9 preferentially recognizes DNA in viruses.72 After TLR7 or TLR8 binds to its specific ligand, MyD88 is recruited to the TIR domain and interacts with TIR; then, the TLR-MyD88 complex recruits IL-1R-associated kinase (IRAK) to phosphorylate it and is activated. Activated IRAK binds to TRAF6 and activates downstream effector molecules, resulting in the expression of several cytokines (interferon-stimulated genes).74 In summary, TLRs play a crucial role in restricting coronavirus replication and initiating immune responses.
Although coronavirus activates the TLR signaling pathway, the degree of activation is different. For example, the capability of coronaviruses to activate the TLR7/8 signaling pathway is SARS-CoV-2 > MERS-CoV > SARS-CoV. The low composition of human TLR7/8-stimulating RNA motifs in the MERS-CoV, SARS-CoV, and SARS-CoV-2 genomes suggests that they have a relatively low human TLR7/8-stimulating potential and a relatively high ability to evade human TLR7/8-mediated innate immune responses.75,76 TLR4 could also be involved in the pathogenesis of COVID-19.77,78 An in silico study revealed a close relationship between bat SARS-CoV and SARS-CoV-2 (based on the spike protein and ACE2) and found that SARS-CoV-2 spike protein binds with TLR1, TLR4, and TLR6, with a higher affinity for TLR4.79 TLR3 via the TIRF pathway leads to a protective response in SARS-CoV and MERS-CoV infections.80
Mechanism of Coronavirus Participating in Immune Escape
Host cells perceiving the invasion of pathogenic microorganisms can inhibit viral replication by regulating the immune signal pathway in vivo to induce IFN expression, proinflammatory factors, and various antiviral genes. To deal with the innate antiviral immune response of the host, most coronaviruses gradually form a complex immune escape mechanism during the long-term evolution of the infected host. Because of this, the following summarizes the pathogenic mechanism of coronavirus evading host innate immunity to provide theoretical reference for the research and development of human coronavirus prevention and treatment and novel coronavirus drugs.
For example, the SARS-CoV-induced p38MAPK pathway is involved in regulating host cell IL-8 secretion and its downstream STAT3 dephosphorylation to promote apoptosis. It can activate the PI3K pathway and phosphorylate intracellular Akt to resist apoptosis.81 Also, SARS-CoV can inhibit the innate immune response by inhibiting the I-κB of host cells and preventing NF-κB signal pathway activation through the deubiquitin enzyme activity of its nsp3 protein.82 The M protein of SARS-CoV can inhibit TRAF3-TANK-TBK1/IKKε formation, IRF-3 activation, and downregulate IFN expression.83 However, Nsp3 of MERES-CoV can inhibit IFN expression mediated by RIG-I and melanoma differentiation factors 5 of RIG-I-like receptors on the host cell membrane and inhibit NF-κB activation.84 The antagonistic effect of M protein encoded by MERS-CoV on IFN is similar to that of SARS-CoV; it can interact with TRAF3, inhibit the coupling of TRAF3-TBK1, reduce the activation and dimerization of IRF3, and decrease IFN expression.85 Nonstructural proteins Nsp1, Nsp6, Nsp13, ORF3a, ORF7a, and ORF7b encoded by SARS-CoV-2 can inhibit the phosphorylation of STAT1 and STAT2.86 Furthermore, Nsp13, Nsp14, and Nsp15 can effectively inhibit the entry of IRF3 into the nucleus. The expression of Nsp6 and Nsp13 can inhibit the phosphorylation of TBK1 and IRF3.87,88 Also, the N protein of SARS-CoV-2 can inhibit its phosphorylation and entry into the nucleus by binding STAT1 and STAT2, thus antagonizing the IFN-I signal pathway.89 The M protein of SARS-CoV-2 can interact with key proteins, such as RIG-I, MAVS, and TBK1, and inhibit the IFN-I pathway.90 Although immunity is suppressed by inhibiting IFN expression, the mechanism in several coronaviruses is different. Existing studies have shown that coronavirus-encoded proteins can antagonize the immune system in several ways, regulate virus replication, and play an essential role in viral pathogenicity and host pathological processes. However, a detailed mechanism for antagonizing the immune system needs to further study the interaction between coronavirus and the host to provide an effective scheme for antiviral treatment and disease control.
Conclusion and Prospects
Different coronaviruses produce different cytokines, and the clinical symptoms and immune responses of patients slightly vary. This may be related to the different structures of coronaviruses and different mechanisms of signal pathways acting on the body. This difference will provide new ideas for treating and diagnosing different coronaviruses.
Herein, we reviewed characteristics of cytokine storms caused by SARS-CoV, MERS-CoV and SARS-CoV-2 infection in humans. We then compared the clinical immune characteristics caused by three coronaviruses and clarified their immune response mechanism. In short, the three coronavirus infections have similar immune characteristics, which can be divided into three aspects: (1) virus infection in innate immune cells, (2) immune response disorder, (3) cytokine storm. Although SARS-CoV can infect DC, mononuclear macrophages and other peripheral blood mononuclear cells, it cannot replicate and produce virus in these cells. But, it can make the infected cells produce a large number of cytokines. SARS-CoV-2 infection also triggered an increase in the secretion of Th2 cytokines that inhibit inflammation, such as IL-4 and IL-10, as well as it can inhibit IFN-I signal pathway.
The purpose of this review is to compare the differences of cytokine storms caused by three coronaviruses and to provide research insights and supporting evidence for the treatment of cytokine storms. For example, the application of IL-10 therapy, blocking Th17 signal pathway and so on. Most of the immune mechanisms of coronavirus infection mentioned are preliminary. There are still many problems to be solved. For example, whether the MERS-CoV can replicate in human monocyte-derived macrophages and MDDCs is still controversial. The relationship between the replication of different coronaviruses and the apoptosis and cytokine release from host cells remains to be further studied. Comparing the immune response and cell signaling pathway caused by these three viruses will provide a strong molecular basis for animal research and can guide future experimental research. Some of these mechanisms have been proved to be closely related to clinical prognosis and disease outcome, thus highlighting their potential role in pathogenesis and clinical manifestations. In a review of most existing studies, great progress has been made in more and more research on coronavirus and other viral infections, which may be helpful for future clinical studies and large trials to verify laboratory-confirmed hypotheses.
Acknowledgments
The authors thank all the members of the Immunology Laboratory of Xizang Minzu University and Shandong First Medical University & Institute of Basic Medicine for encouraging the writing of this review. Tong Liu and Meng Feng are co-first authors.
Funding
This work was supported by the National Natural Science Foundation of China (81860719); Xizang Science and Technology Department (In 2021); the COVID-19 Epidemics Emergency Project of Xizang Minzu University (XZMDYJ04). The funding sources had no role in the study design, data collection, analysis, interpretation, or writing of the report.
Disclosure
No potential conflict of interest was reported by the authors.
References
1. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–544. doi:10.1038/s41564-020-0695-z
2. Schultheiss C, Paschold L, Simnica D, et al. Next-generation sequencing of T and B cell receptor repertoires from COVID-19 patients showed signatures associated with severity of disease. Immunity. 2020;53:442–455. doi:10.1016/j.immuni.2020.06.024
3. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi:10.1016/S0140-6736(20)30183-5
4. Gupta KK, Khan MA, Singh SK. Constitutive inflammatory cytokine storm: a major threat to human health. J Interferon Cytokine Res. 2020;40:19–23. doi:10.1089/jir.2019.0085
5. Cepeda-Valdes R, Carrion-Alvarez D, Trejo-Castro A, et al. Cutaneous manifestations in COVID-19: familial cluster of urticarial rash. Clin Exp Dermatol. 2020;45:895–896. doi:10.1111/ced.14290
6. Fajgenbaum DC, June CH, Longo DL. Cytokine Storm. N Engl J Med. 2020;383:2255–2273. doi:10.1056/NEJMra2026131
7. Moller AS, Ovstebo R, Haug KB, et al. Chemokine production and pattern recognition receptor (PRR) expression in whole blood stimulated with pathogen-associated molecular patterns (PAMPs). Cytokine. 2005;32:304–315. doi:10.1016/j.cyto.2005.11.008
8. Thompson MR, Kaminski JJ, Kurt-Jones EA, et al. Pattern recognition receptors and the innate immune response to viral infection. Viruses. 2011;3:920–940. doi:10.3390/v3060920
9. Dinarello CA. Historical insights into cytokines. Eur J Immunol. 2007;37(Suppl1):S34–S45. doi:10.1002/eji.200737772
10. Tavakolpour S, Mahmoudi H, Mirzazadeh A, et al. Pathogenic and protective roles of cytokines in pemphigus: a systematic review. Cytokine. 2020;129:155026. doi:10.1016/j.cyto.2020.155026
11. Yen YT, Liao F, Hsiao CH, et al. Modeling the early events of severe acute respiratory syndrome coronavirus infection in vitro. J Virol. 2006;80:2684–2693. doi:10.1128/JVI.80.6.2684-2693.2006
12. Wei YZBF. Aberrant Pathogenic GM-CSF+ T Cells and Inflammatory CD14+CD16+ Monocytes in Severe Pulmonary Syndrome Patients of a New Coronavirus (Published February 20, 2020). Atlanta: NewsRx; 2020.
13. Woo PCY, Lau SKP, Lam CSF, et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol. 2012;86:3995–4008.
14. Pal M, Berhanu G, Desalegn C, et al. Severe acute respiratory syndrome Coronavirus-2 (SARS-CoV-2): an update. Cureus. 2020;12:e7423.
15. Pellett PE, Mitra S, Holland TC. Basics of virology. Handb Clin Neurol. 2014;123:45–66.
16. Zhu Z, Lian X, Su X, et al. From SARS and MERS to COVID-19: a brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir Res. 2020;21:224. doi:10.1186/s12931-020-01479-w
17. Yin Y, Wunderink RG. MERS, SARS and other coronaviruses as causes of pneumonia. Respirology. 2018;23:130–137. doi:10.1111/resp.13196
18. Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of Coronaviruses. Trends Microbiol. 2016;24:490–502. doi:10.1016/j.tim.2016.03.003
19. Forni D, Cagliani R, Clerici M, et al. Molecular evolution of human coronavirus genomes. Trends Microbiol. 2017;25:35–48. doi:10.1016/j.tim.2016.09.001
20. Qian Z, Travanty EA, Oko L, et al. Innate immune response of human alveolar type II cells infected with severe acute respiratory syndrome-coronavirus. Am J Respir Cell Mol Biol. 2013;48:742–748. doi:10.1165/rcmb.2012-0339OC
21. Cheung CY, Poon LL, Ng IH, et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J Virol. 2005;79:7819–7826. doi:10.1128/JVI.79.12.7819-7826.2005
22. Law HKW, Cheung CY, Ng HY, et al. Chemokine up-regulation in SARS-coronavirus–infected, monocyte-derived human dendritic cells. Blood. 2005;106:2366–2374. doi:10.1182/blood-2004-10-4166
23. Chien JY, Hsueh PR, Cheng WC, et al. Temporal changes in cytokine/chemokine profiles and pulmonary involvement in severe acute respiratory syndrome. Respirology. 2006;11:715–722. doi:10.1111/j.1440-1843.2006.00942.x
24. Wang CH, Liu CY, Wan YL, et al. Persistence of lung inflammation and lung cytokines with high-resolution CT abnormalities during recovery from SARS. Respir Res. 2005;6:42. doi:10.1186/1465-9921-6-42
25. Wong CK, Lam CWK, Wu AKL, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. 2004;136:95–103. doi:10.1111/j.1365-2249.2004.02415.x
26. Raj VS, Mou H, Smits SL, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495:251–254. doi:10.1038/nature12005
27. Cha RH, Joh JS, Jeong I, et al. Renal complications and their prognosis in Korean patients with middle east respiratory syndrome-Coronavirus from the central MERS-CoV designated hospital. J Korean Med Sci. 2015;30:1807–1814. doi:10.3346/jkms.2015.30.12.1807
28. Khalid I, Alraddadi BM, Dairi Y, et al. Acute management and long-term survival among subjects with severe middle east respiratory syndrome Coronavirus pneumonia and ARDS. Respir Care. 2016;61:340–348. doi:10.4187/respcare.04325
29. Lau SKP, Lau CCY, Chan K, et al. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: implications for pathogenesis and treatment. J Gen Virol. 2013;94:2679–2690. doi:10.1099/vir.0.055533-0
30. Zhou J, Chu H, Li C, et al. Active replication of middle east respiratory syndrome Coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis. 2014;209:1331–1342. doi:10.1093/infdis/jit504
31. Tynell J, Westenius V, Ronkko E, et al. Middle East respiratory syndrome coronavirus shows poor replication but significant induction of antiviral responses in human monocyte-derived macrophages and dendritic cells. J Gen Virol. 2016;97:344–355. doi:10.1099/jgv.0.000351
32. Chu H, Zhou J, Wong BH, et al. Middle east respiratory syndrome coronavirus efficiently infects human primary T lymphocytes and activates the extrinsic and intrinsic apoptosis pathways. J Infect Dis. 2016;213:904–914. doi:10.1093/infdis/jiv380
33. Scheuplein VA, Seifried J, Malczyk AH, et al. High secretion of interferons by human plasmacytoid dendritic cells upon recognition of middle east respiratory syndrome Coronavirus. J Virol. 2015;89:3859–3869. doi:10.1128/JVI.03607-14
34. Kim ES, Choe PG, Park WB, et al. Clinical progression and cytokine profiles of middle east respiratory syndrome Coronavirus infection. J Korean Med Sci. 2016;31:1717. doi:10.3346/jkms.2016.31.11.1717
35. Min C, Cheon S, Ha N, et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci Rep. 2016;6:25359. doi:10.1038/srep25359
36. Abassi Z, Knaney Y, Karram T, et al. The lung macrophage in SARS-CoV-2 infection: a friend or a foe? Front Immunol. 2020;11. doi:10.3389/fimmu.2020.01312
37. Ruan Q, Yang K, Wang W, et al. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46:846–848. doi:10.1007/s00134-020-05991-x
38. Donnelly CA, Ghani AC, Leung GM, et al. Epidemiological determinants of spread of causal agent of severe acute respiratory syndrome in Hong Kong. Lancet. 2003;361:1761–1766. doi:10.1016/S0140-6736(03)13410-1
39. Baharoon S, Memish ZA. MERS-CoV as an emerging respiratory illness: a review of prevention methods. Travel Med Infect Dis. 2019;32:101520. doi:10.1016/j.tmaid.2019.101520
40. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi:10.1016/S0140-6736(20)30211-7
41. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi:10.1038/s41586-020-2008-3
42. Chan JF-W, Yuan S, Kok K-H. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395:514–523. doi:10.1016/S0140-6736(20)30154-9
43. Wan S, Yi Q, Fan S, et al. Characteristics of lymphocyte subsets and cytokines in peripheral blood of 123 hospitalized patients with 2019 novel coronavirus pneumonia (NCP). medRxiv. 2020;2020–2022. doi:10.1101/2020.02.10.20021832
44. Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8:420–422. doi:10.1016/S2213-2600(20)30076-X
45. Zhao Y, Qin L, Zhang P, et al. Longitudinal COVID-19 profiling associates IL-1RA and IL-10 with disease severity and RANTES with mild disease. JCI Insight. 2020;5:e139834. doi:10.1172/jci.insight.139834
46. Wang X, Xu W, Hu G, et al. RETRACTED ARTICLE: SARS-CoV-2 infects T lymphocytes through its spike protein-mediated membrane fusion. Cell Mol Immunol. 2020;17:894. doi:10.1038/s41423-020-0498-4
47. National Research Project for SARS. The involvement of natural killer cells in the pathogenesis of severe acute respiratory syndrome. Am J Clin Pathol. 2004;121:507–511. doi:10.1309/WPK7Y2XKNF4CBF3R
48. Alosaimi B, Hamed ME, Naeem A, et al. MERS-CoV infection is associated with downregulation of genes encoding Th1 and Th2 cytokines/chemokines and elevated inflammatory innate immune response in the lower respiratory tract. Cytokine. 2020;126:154895. doi:10.1016/j.cyto.2019.154895
49. Matic S, Popovic S, Djurdjevic P, et al. SARS-CoV-2 infection induces mixed M1/M2 phenotype in circulating monocytes and alterations in both dendritic cell and monocyte subsets. PLoS One. 2020;15:e241097. doi:10.1371/journal.pone.0241097
50. Wang W, Su B, Pang L, et al. High-dimensional immune profiling by mass cytometry revealed immunosuppression and dysfunction of immunity in COVID-19 patients. Cell Mol Immunol. 2020;17:650–652. doi:10.1038/s41423-020-0447-2
51. Beniac DR, Andonov A, Grudeski E, et al. Architecture of the SARS coronavirus prefusion spike. Nat Struct Mol Biol. 2006;13:751–752. doi:10.1038/nsmb1123
52. Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. 2016;3:237–261. doi:10.1146/annurev-virology-110615-042301
53. Neuman BW, Kiss G, Kunding AH, et al. A structural analysis of M protein in coronavirus assembly and morphology. J Struct Biol. 2011;174:11–22. doi:10.1016/j.jsb.2010.11.021
54. Nieto-Torres JL, Dediego ML, Verdia-Baguena C, et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014;10:e1004077. doi:10.1371/journal.ppat.1004077
55. Dediego ML, Alvarez E, Almazan F, et al. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J Virol. 2007;81:1701–1713. doi:10.1128/JVI.01467-06
56. Huang Y, Yang C, Xu XF, et al. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin. 2020;41:1141–1149. doi:10.1038/s41401-020-0485-4
57. Hulswit RJ, de Haan CA, Bosch BJ. Coronavirus spike protein and tropism changes. Adv Virus Res. 2016;96:29–57.
58. Verdia-Baguena C, Nieto-Torres JL, Alcaraz A, et al. Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids. Virology. 2012;432:485–494. doi:10.1016/j.virol.2012.07.005
59. Malik YA. Properties of Coronavirus and SARS-CoV-2. Malays J Pathol. 2020;42:3–11.
60. Chan JF, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9:221–236. doi:10.1080/22221751.2020.1719902
61. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi:10.1038/s41586-020-2012-7
62. Surya W, Li Y, Verdia-Baguena C, et al. MERS coronavirus envelope protein has a single transmembrane domain that forms pentameric ion channels. Virus Res. 2015;201:61–66. doi:10.1016/j.virusres.2015.02.023
63. Ujike M, Taguchi F. Incorporation of spike and membrane glycoproteins into coronavirus virions. Viruses. 2015;7:1700–1725. doi:10.3390/v7041700
64. Wang N, Li SY, Yang XL, et al. Serological evidence of bat SARS-related Coronavirus infection in Humans, China. Virol Sin. 2018;33:104–107. doi:10.1007/s12250-018-0012-7
65. Wu A, Peng Y, Huang B, et al. Genome composition and divergence of the Novel Coronavirus (2019-nCoV) originating in China. Cell Host Microbe. 2020;27:325–328. doi:10.1016/j.chom.2020.02.001
66. Shi CS, Nabar NR, Huang NN, et al. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019;5:101. doi:10.1038/s41420-019-0181-7
67. Zhang Y, Chen Y, Li Y, et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-Ι. Proc Natl Acad Sci U S A. 2021;118:e2024202118. doi:10.1073/pnas.2024202118
68. Wu D, Yang XO. TH17 responses in cytokine storm of COVID-19: an emerging target of JAK2 inhibitor Fedratinib. J Microbiol Immunol Infect. 2020;53:368–370. doi:10.1016/j.jmii.2020.03.005
69. Fung TS, Huang M, Liu DX. Coronavirus-induced ER stress response and its involvement in regulation of coronavirus-host interactions. Virus Res. 2014;194:110–123. doi:10.1016/j.virusres.2014.09.016
70. Favreau DJ, Meessen-Pinard M, Desforges M, et al. Human coronavirus-induced neuronal programmed cell death is cyclophilin d dependent and potentially caspase dispensable. J Virol. 2012;86:81–93. doi:10.1128/JVI.06062-11
71. Yamamoto M, Takeda K. Current views of toll-like receptor signaling pathways. Gastroenterol Res Pract. 2010;2010:1–8. doi:10.1155/2010/240365
72. Mahla RS, Reddy MC, Prasad DVR, et al. Sweeten PAMPs: role of sugar complexed PAMPs in innate immunity and vaccine biology. Front Immunol. 2013;4:248. doi:10.3389/fimmu.2013.00248
73. Matsumoto M, Oshiumi H, Seya T. Antiviral responses induced by the TLR3 pathway. Rev Med Virol. 2011;21:67–77. doi:10.1002/rmv.680
74. Gay NJ, Symmons MF, Gangloff M, et al. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol. 2014;14:546–558. doi:10.1038/nri3713
75. Yang C, Chen M. Low compositions of human toll-like receptor 7/8-stimulating RNA motifs in the MERS-CoV, SARS-CoV and SARS-CoV-2 genomes imply a substantial ability to evade human innate immunity. PeerJ. 2021;9:e11008. doi:10.7717/peerj.11008
76. Moreno-Eutimio MA, López-Macías C, Pastelin-Palacios R. Bioinformatic analysis and identification of single-stranded RNA sequences recognized by TLR7/8 in the SARS-CoV-2, SARS-CoV, and MERS-CoV genomes. Microbes Infect. 2020;22:226–229. doi:10.1016/j.micinf.2020.04.009
77. Cicco S, Cicco G, Racanelli V, et al. Neutrophil Extracellular Traps (NETs) and Damage-Associated Molecular Patterns (DAMPs): two potential targets for COVID-19 treatment. Mediators Inflamm. 2020;2020:1–25. doi:10.1155/2020/7527953
78. Onofrio L, Caraglia M, Facchini G, et al. Toll-like receptors and COVID-19: a two-faced story with an exciting ending. Future Sci OA. 2020;6:FSO605. doi:10.2144/fsoa-2020-0091
79. Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS‐CoV‐2 spike glycoprotein with ACE‐2 receptor homologs and human TLRs. J Med Virol. 2020;92:2105–2113. doi:10.1002/jmv.25987
80. Birra D, Benucci M, Landolfi L, et al. COVID 19: a clue from innate immunity. Immunol Res. 2020;68:161–168. doi:10.1007/s12026-020-09137-5
81. Mizutani T. Signal transduction in SARS-CoV-infected cells. Ann N Y Acad Sci. 2007;1102:86–95. doi:10.1196/annals.1408.006
82. Frieman M, Ratia K, Johnston RE, et al. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J Virol. 2009;83:6689–6705. doi:10.1128/JVI.02220-08
83. Siu KL, Kok KH, Ng MJ, et al. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J Biol Chem. 2009;284:16202–16209. doi:10.1074/jbc.M109.008227
84. Alfuwaires M, Altaher A, Kandeel M. Molecular dynamic studies of interferon and innate immunity resistance in MERS CoV non-structural protein 3. Biol Pharm Bull. 2017;40:345–351. doi:10.1248/bpb.b16-00870
85. Yang Y, Zhang L, Geng H, et al. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell. 2013;4:951–961. doi:10.1007/s13238-013-3096-8
86. Xia H, Cao Z, Xie X, et al. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020;33:108234. doi:10.1016/j.celrep.2020.108234
87. Lei X, Dong X, Ma R, et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun. 2020;11:3810. doi:10.1038/s41467-020-17665-9
88. Konno Y, Kimura I, Uriu K, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 2020;32:108185. doi:10.1016/j.celrep.2020.108185
89. Mu J, Fang Y, Yang Q, et al. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov. 2020;6:65. doi:10.1038/s41421-020-00208-3
90. Zheng Y, Zhuang MW, Han L, et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct Target Ther. 2020;5:299. doi:10.1038/s41392-020-00438-7
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.