Back to Journals » Infection and Drug Resistance » Volume 16
Comparative Genomic Analysis of Hypervirulence Carbapenem-Resistant Klebsiella pneumoniae from Inpatients with Infection and Gut Colonization, China
Authors He W ![]() , Wu C
, Wu C ![]() , Chen G, Zhang G, Zhao Z, Wen S, Zhou Y, Deng X, Feng Y, Zhong LL
, Chen G, Zhang G, Zhao Z, Wen S, Zhou Y, Deng X, Feng Y, Zhong LL ![]() , Tian GB, Dai M
, Tian GB, Dai M
Received 25 May 2023
Accepted for publication 12 July 2023
Published 14 August 2023 Volume 2023:16 Pages 5251—5261
DOI https://doi.org/10.2147/IDR.S416770
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Wan He,1,* Changbu Wu,2,3,* Guanping Chen,2– 4 Guili Zhang,2,3 Zihan Zhao,2,3 Shu’an Wen,2,3 Yuan Zhou,2,3 Xue Deng,2,3 Yu Feng,2,3 Lan-Lan Zhong,2,3 Guo-Bao Tian,2– 4 Min Dai1
1School of Laboratory Medicine, Chengdu Medical College, Chengdu, 610500, People’s Republic of China; 2Department of Immunology and Microbiology, Advanced Medical Technology Center, The First Affiliated Hospital, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, 510080, People’s Republic of China; 3Key Laboratory of Tropical Diseases Control (Sun Yat-sen University), Ministry of Education, Guangzhou, 510080, People’s Republic of China; 4Department of Immunology, School of Medicine, Sun Yat-Sen University, Shenzhen, 518107, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Min Dai, School of Laboratory Medicine, Chengdu Medical College, Chengdu, Sichuan Province, People’s Republic of China, Tel +86 28-62739128, Email [email protected] Guo-Bao Tian, Department of Immunology and Microbiology, Advanced Medical Technology Center, The First Affiliated Hospital, Zhongshan School of Medicine, Sun Yat-sen University, 74 Zhongshan 2nd Road, Guangzhou, 510080, People’s Republic of China, Tel/Fax +86 20 87335387, Email [email protected]
Background: The emergence and spread of hypervirulent carbapenem-resistant Klebsiella pneumoniae (hv-CRKP) is a potential epidemiological threat that needs to be monitored. However, the transmission and pathogenic characteristics of hv-CRKP in China remain unclear. We investigated the epidemiological characteristics of gut colonized hv-CRKP in a hospital in Guangdong Province, China.
Methods: A total of 46 gut colonized hv-CRKP isolates were collected from Sun Yat-Sen Memorial Hospital (Guangzhou, China) from August 31st to December 31st, 2021. Minimum inhibitory concentrations (MICs) were obtained for 15 antibiotics for 46 hv-CRKP isolates. BALB/C mice infection model and mucoviscosity assay was used to evaluate the virulence of the isolates. The characteristics of genome, phylogenetic relationship and the structure of the plasmid of 46 gut colonized hv-CRKP isolates were compared with pathogenic isolates from GeneBank based on whole-genome data.
Results: The hv-CRKP isolation rate of all gut colonized carbapenem-resistant Klebsiella pneumoniae was 17% (46/270), and the intestinal colonization rate of hv-CRKP was irrelevant to the sex, age, department of hospitalization, and history of antibiotic use of the host. The gut colonized hv-CRKP showed pandrug resistance and hypervirulence. The gut colonized hv-CRKP and pathogenic hv-CRKP prevalent in China were mainly ST11 hv-CRKP and had two major epidemic clades. The similarities in genomic characteristics between gut colonized hv-CRKP and pathogenic hv-CRKP were consistent. The gut colonized hv-CRKP carried an incomplete structure pK2044 virulence plasmid from hypervirulent K. pneumoniae NTUH-K2044 by analyzing the virulence plasmid structure.
Conclusion: Our results suggest that the gut colonized ST11 hv-CRKP may serve as a reservoir for the clinical pathogenic ST11 HV-CRKP. It is necessary to further strengthen the monitoring of gut colonized hv-CRKP and research the potential mechanism of infection caused by gut colonized hv-CRKP.
Keywords: K. pneumoniae, intestinal colonization, hypervirulent, genomic characterization, carbapenemase
Introduction
The worldwide spread of Klebsiella pneumoniae is a major global public health threat.1,2 K. pneumoniae is an opportunistic pathogen that colonizes the human gut and respiratory tract and possibly causes infection in immunocompromised hosts. Gut colonized K. pneumoniae is one of the important pathogens causing nosocomial infections, although the mechanism by which gut colonized K. pneumoniae breaks through the intestinal barrier is currently unknown;3 however, some studies have shown that gut colonized K. pneumoniae is a risk factor for secondary infection of inpatients.4,5 Therefore, surveillance of intestinal colonization by K. pneumoniae contributes to preventing infections, resulting in more serious clinical outcomes.
Hypervirulent carbapenem-resistant K. pneumoniae (hv-CRKP) is considered a superbug that exhibits hyperresistance and hypervirulence, posing a grave threat to human health. In 2018, a study first reported a hospital outbreak of hv-CRKP and the infection mortality rate was 100% in Zhejiang Province, China.6 Colonization of the intestinal tract of K. pneumoniae has become a critical population for the horizontal transmission of various genetic elements and genes. Particularly, colonization of hv-CRKP may develop into invasive infections resulting in disease.7 However, there is limited research on human colonization of hv-CRKP.8,9
In contrast to eukaryotes, the genome of prokaryotes is dynamically changing due to the mechanism of horizontal gene transfer evolution.10 Monitoring and analysis of the prokaryotic genome contributes to understanding the colonial structure and evolutionary characteristics.11,12
Herein, we conducted integrated genomic analysis to elucidate the epidemiological characteristics of gut colonized hv-CRKP. Further, we elucidated whether there is evolution at the genomic level between gut colonized hv-CRKP and pathogenic populations and provides the basis for the prevention of the spread and outbreak of hv-CRKP.
Materials and Methods
Sample Collection
We undertook a prospective cross-sectional and molecular epidemiological study to assess the prevalence of the gut colonized hv-CRKP in inpatients. The inpatient fecal samples were recovered from Sun Yat-sen Memorial Hospital in Guangzhou, Guangdong province, China from August 31st to December 31st, 2021. The inpatient submitting fecal samples during the study timeframe to the hospital microbiology laboratories for diagnostic purposes were randomly selected to participate in the study. Ultimately, a total of 631 non-duplicate fecal samples from patients were collected.
Isolation and Identification
The fresh fecal samples were cultured on Columbia Blood Agar (Oxoid, Hampshire, United Kingdom) with imipenem (4 mg/L) for 18–24 hours at 37°C. Subsequently, the imipenem-resistant colonies were selected, and species identification was conducted by MALDI-TOF MS (Bruker Daltonik GmbH, Bremen, Germany) and 16S rRNA sequencing. A total of 270 isolates of carbapenem-resistant K. pneumoniae (CRKP) were identified. The CRKP isolates were used to test for the presence of the virulence genes iucA, rmpA, and rmpA2 using PCR with specific primers (rmpA: Forward: TACATATGAAGGAGTAGTTAAT, Reverse: GAGCCATCTTTCATCAAC; rmpA2: Forward: TGTGCAATAAGGATGTTACATTAGT, reverse: TTTGATGTGCACCATTTTTCA; iucA: Forward: CCTGCGTGAAAAAGCGTTGA, reverse: CACGGTAGATAAGCCCGACC).6 Finally, 46 isolates of hv-CRKP were selected and subjected to whole-genome sequencing (WGS) as described previously.13
Antimicrobial Susceptibility Testing
Minimum inhibitory concentrations (MICs) were obtained for imipenem, meropenem, ertapenem, tigecycline, polymyxin, ciprofloxacin, gentamicin, amikacin, ceftazidime, cefepime, cefotaxime, aztreonam, piperacillin/tazobactam, cotrimoxazole, and fosfomycin for all hv-CRKP isolates using the methods in accordance with the Clinical and Laboratory Standards Institute guidelines, and we interpreted the results in accordance with the CLSI breakpoints (CLSI, 2020) except of tigecycline, which interpreted the results in accordance with the EUCAST breakpoints, version 9.0.
Mucoviscosity Assay
The mucovisosity of K. pneumoniae isolates was evaluated as described previously and with some modification.14 Briefly, the isolates were overnight cultured and sub-cultured to reach an OD600 reading of approximately 1.0 at 37 °C. After centrifuged for 5 min at 6000 rpm, the bacterial precipitation was resuspended with 1 mL of sterile PBS solution and then centrifuged for 5 min at 1000 rpm. Finally, the OD600 value of supernatant was measured. The experiment was conducted in triplicate.
Mice Infection Model
In vivo virulence of seven hv-CRKP isolates (KP04, KP06, KP08, KP22, KP26, KP27, and KP41) and K. pneumoniae ATCC700603 (as control group) were evaluated using mice intraperitoneal infection model. Female BALB/C mice weighing 16–18 g (6–8 weeks old) were used in this study. Fresh monoclonal strains grown on LB agar plates were picked and re-cultured in LB broth medium for 37°C overnight, respectively. Subsequently, the isolates were sub-cultured with 1:100 dilutions for 2.5 hours at 37 °C. The bacterial samples were collected by centrifugation at 6000 rpm for 5 min. Then, the bacterial pellets were resuspended with sterile saline to reach an OD600 reading of 1.0 and then diluted to an appropriate cell density. Each mouse was injected intraperitoneally with 100 μL of diluted bacteria fluid, and the number of bacteria injected was approximately 107 CFU. Followed by injection, the survival of mice was monitored for 72 hours. Eight mice were used for each stain. The results were analysed by Kaplan–Meier survival curves (GraphPad Prism 8.0 statistics software).
Genome Sequencing and Analysis
Short-read sequencing raw data is used for quality control with fastq v0.20 and de novo assembled with SPAdes v3.13.1.15,16 Long reads sequencing was used to sequence 3 KP strains by Pacbio Sequel II/Iie platform. De novo hybrid assembly of both short Illumina reads and long PacBio reads was performed using SPAdes v3.13.1. After assembly, contig-tools (https://gitlab.com/antunderwood/contig_tools) was used to remove contigs with length less than 500 bp. Kraken2 v2.1.2 was used to align the assembled contigs with a reference database with Kraken2.17
ABRicate v0.8.7 (https://github.com/tseemann/abricate) was used to identify the antibiotic resistance genes and virulence genes based on ResFinder database and VFDB (Virulence Factor Database).
For each de novo assembly, coding sequences were annotated using Prokka v1.12. The MLST were identified using mlst v2.23.0 (https://github.com/tseemann/mlst). Plasmid sequence alignment using Blastn and visualization using BLAST Ring Image Generator (BRIG) v0.95.18
Phylogenetic Analysis
Pan-genome analysis was performed by Roary v3.12.019 with the “- e -- mafft” parameter to use MAFFT software for alignment. SNP-sites v2.4.1 was used to extract the core-genome SNPs (cgSNPs).20 VCFtools v0.1.16 was used to filter cgSNPs.21 Hierarchical Bayesian Analysis of Population Structure (hierBAPS) is used to obtain the best sequence clusters (SCs) based on the cgSNPs. The phylogenetic tree based on the Maximum Likelihood (ML) method with GTR+G model and 1000 bootstrap was constructed by RAxML v8.2.1022 and was visualized by iTOL23 and ggtree.24
According to the pan-genome gene set constructed previously, those genes contained in 5–95% of the strains are retained as accessory genes. Scoary v1.6.16 was used to perform pangenome-wide association analysis (PGWAS),25 with the “-- collapse” parameter to merge the same operon to avoid false positives. The accessory genes meeting the following conditions are selected: (1) the Benjamini–Hochberg (BH) P-value < 0.05; (2) Best-pairwise-compared P-value < 0.05; Empirical P-value < 0.05. Furthermore, eggNOG v5.0 was used to annotate the cluster of homologous groups (COGs) of these accessory genes with significant differences.26
Heatmap was created by R package “ggplot2”, “pheatmap”.
Dataset
Sequence data of hv-CRKP isolates in this study have been deposited in the NCBI database under BioProject no.PRJNA921948. The genomic sequences of 587 ST11 hv-CRKP isolated from infection patients and 20 ST11 hv-CRKP strains isolated from human intestinal tract between 2017 and 2022 in China were used for WGS analysis in this study (Figure S1 and Table S1). The two reference plasmids pK2044 (accession number: AP006726) and pKP20194a-p1 (accession number: CP054781) downloaded from NCBI database were used for virulence plasmid analysis.
Statistical Analysis
Data from the investigation were analyzed using SPSS version 23. Descriptive statistics and logistic regression were calculated at 95% of confidence interval to the different variables. In all cases, P-value less than 0.05 was considered as statistically significant.
Ethics Clearance
This study was conducted in accordance with the Declaration of Helsinki. Ethical approval for this study was sought and given by Zhongshan School of Medicine of Sun Yat-sen University (Approval No 068). Inpatients were individually contacted, either face-to-face or by phone and consent sought to use these samples/data for this study. The animal study was approved by the Institutional Animal Care and Use Committee of School of Zhongshan School of Medicine, Sun Yat-sen University (Approval No 2021044). The animal study was guided by the Russell-Burch tenet of Replacement, Reduction and Refinement (3Rs).
Result
Clinical Characteristics of Gut Colonized hv-CRKP
Among the 631 non-duplicate samples, 270 (42.8%) carbapenem-resistant K. pneumoniae (CRKP) were isolated. And the inpatients who carrying CRKP were free of primary K. pneumoniae infection in this study. Further virulence gene identification showed that 46 (17%) of the 270 CRKP isolates were hv-CRKP. Based on the clinical data, most patients (69.6%, 32/46) colonized by hv-CRKP were male (Table S2). In addition, those patients were distributed across 19 medical departments, and no significant department sources were found. Additionally, 67% of patients had a history of using antibiotics, but no significant differences were observed. The obtained results indicated that a high colonization rate of both CRKP and hv-CRKP was found among inpatients in Sun Yat-sen Memorial Hospital.
Molecular Epidemiological Characteristics of hv-CRKP
Then, 46 hv-CRKP underwent whole-genome sequencing. Among these isolates, 45 strains were ST11, and only 1 strain was ST23 (Figure 1). Of the ST11 strains, multiple antibiotic-resistance genes were detected, including carbapenem resistance genes, quinolone resistance genes, and aminoglycoside resistance genes. All ST11 strains harbored blaKPC-2, and one ST11 strain carried four carbapenem resistance genes (blaKPC-2, blaNDM-5, blaOXA-1, blaOXA-10) at the same time (Figure 1). The ST23 strain carried six drug resistance genes, including blaNDM-1, blaSHV-187, fosA6, mdf(A) and oqxAB (Figure 1). In addition, we performed antimicrobial susceptibility testing to confirm the drug resistance phenotype (Table S3). Low resistance rates were observed for colistin (4.3%, 2/46), tigecycline (23.9%, 11/46), and fosfomycin (2.2%, 1/46). In contrast, all strains were resistant to other antibiotics used in the test. The obtained results indicated that all strains exhibited pandrug resistance. Furthermore, we characterized the virulence level of 46 hv-CRKP. Most virulence factors, including iucABCD-iutA and rmpA, were identified among the 46 strains. Only the ST23 strain harbored iroBCDN (Figure 1). This result suggests that the virulence plasmid carried by the ST11 strain may be incomplete.
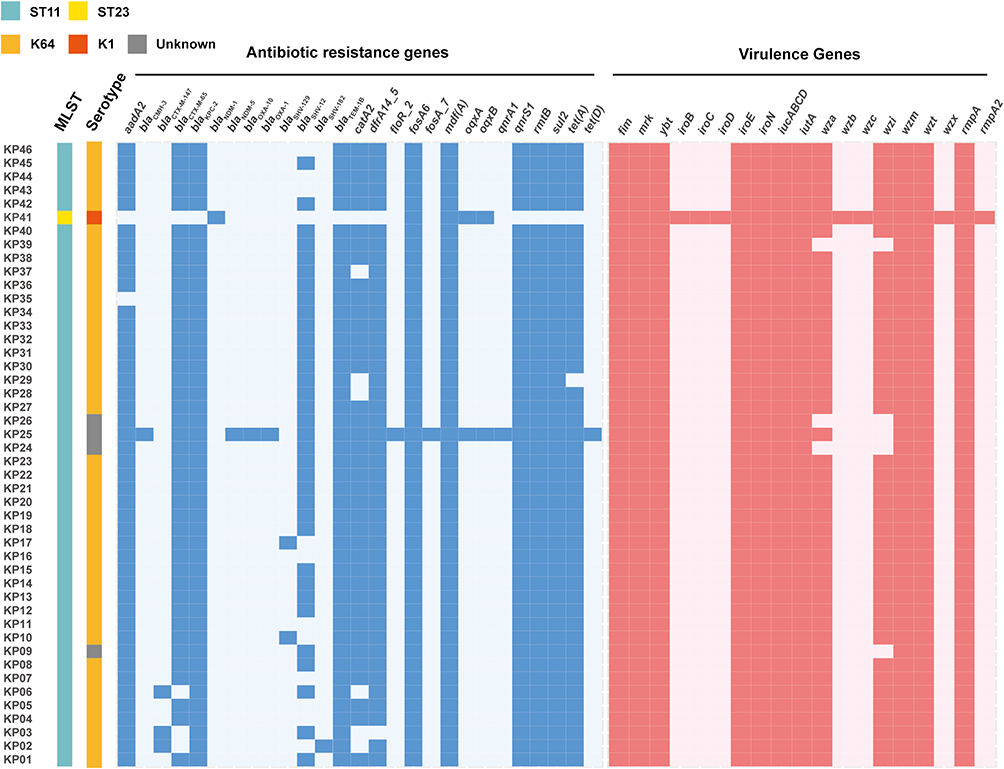 |
Figure 1 Genomic characteristics of 46 gut colonized hv-CRKP strains. Characteristics of virulence genes and drug resistance genes in 46 gut colonized hv-CRKP strains. |
Based on the SNPs in the core genome, 45 ST11 hv-CRKP strains were divided into 6 clones.27 To predict the toxicity of these strains, we used a mouse infection model and mucoviscosity assay. Six ST11 hv-CRKP strains, which were randomly selected from each clone, and one ST23 hv-CRKP strain were tested for virulence to mice and strain viscosity using ATCC700603 as the control strain with low virulence.28 The survival curve revealed that the ST23 hv-CRKP strain and most of the ST11 hv-CRKP strains (83.3%, 5/6) were highly virulent to mice, while the ST11 strains KP26 and ATCC700603 exhibited relatively low toxicity (Figure 2A). A semiquantitative viscosity test showed that all strains with high virulence to mice had high viscosity, except KP26 and ATCC700603 (Figure 2B). Our results suggested that gut colonized hv-CRKP could also exhibit a hypervirulent phenotype.
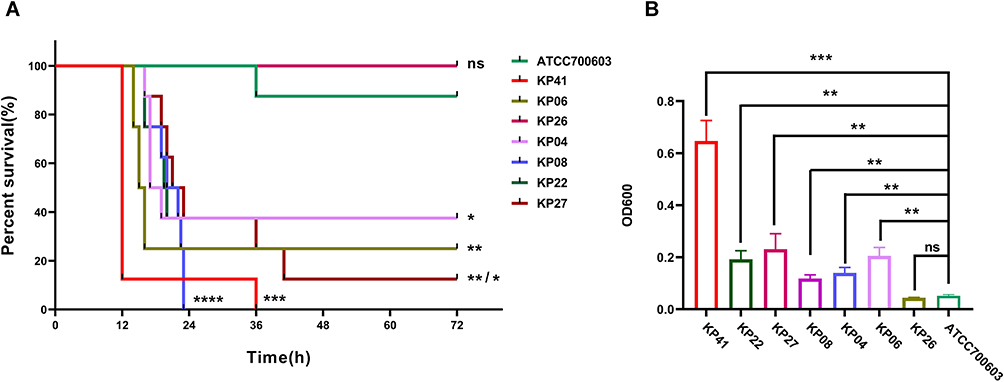 |
Figure 2 The virulence characteristics of 46 gut colonized hv-CRKP strains. (A) Virulence of 46 gut colonized hv-CRKP strains to BALB/C mice. (B) Viscosity characteristics of 46 gut colonized hv-CRKP strains. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. Abbreviation: ns, No statistical significance. |
The Epidemiological Characteristics of ST11 hv-CRKP in China Were Analyzed by Phylogenetic Tree
We performed ST-type analysis on the collected pathogenic bacteria data and found that the number of ST11 strains was dominant (90%, 587/652, Figure S1), which was consistent with the colonized hv-CRKP strains. To further compare the epidemiological characteristics of hv-CRKP between colonization and infection, we constructed pangenomic data of 652 strains ST11 hv-CRKP composed of 20,071 genes, in which the core genome was composed of 4376 genes. A phylogenetic tree was constructed based on the cgSNPs, and 6 sequence clusters (SCs) were identified by hierBAP analysis (Figure 3A). Six SCs allow further differentiation of the ST11 hv-CRKP strains into 2 different clades. Among them, SC2 contained the largest number of strains. We found that the colonized ST11 hv-CRKP strains in this study were mainly distributed in SC1, while the colonized strains collected from NCBI were scattered in other SCs, suggesting that the colonized strains and pathogenic strains had a close evolutionary relationship (Figure 3B). Notably, most of the ST11 hv-CRKP strains were isolated from blood samples, suggesting that ST11 hv-CRKP may mainly cause bloodstream infection (Figure 3B).
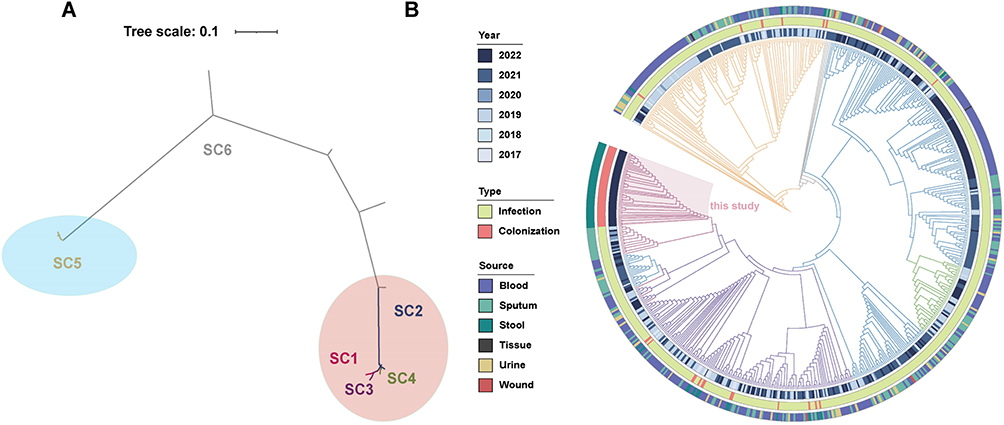 |
Figure 3 Phylogenetic tree of gut colonized hv-CRKP and pathogenic hv-CRKP. (A and B) Phylogenetic analysis of gut colonized ST11 hv-CRKP and pathogenic ST11 hv-CRKP based on core genome SNPs. SCs are distinguished by different colors. Clade1 (blue oval) was SC5, and Clade 2 (red oval) included SC1, SC2, SC3, and SC4. |
Comparison of Genomic Characteristics Between Colonized and Pathogenic Populations of ST11-hv-CRKP
We characterized the resistance genes and virulence genes of 652 ST11 strains of K. pneumoniae (Figure S2) and summarized the carrying rates of virulence genes and resistance genes of each SC (Figure 4A). All SCs possess the important virulence genes iucA and rmpA, but the salmonella synthesis gene cluster iroBCDN has a certain degree of diversity (Figure 4A). The diversity of virulence gene profiles of SC1 and SC3 contained in colonized bacteria has a high degree of similarity. This result reflects that the virulence gene of ST11 hv-CRKP is not significantly different between the colonized and infected population strains.
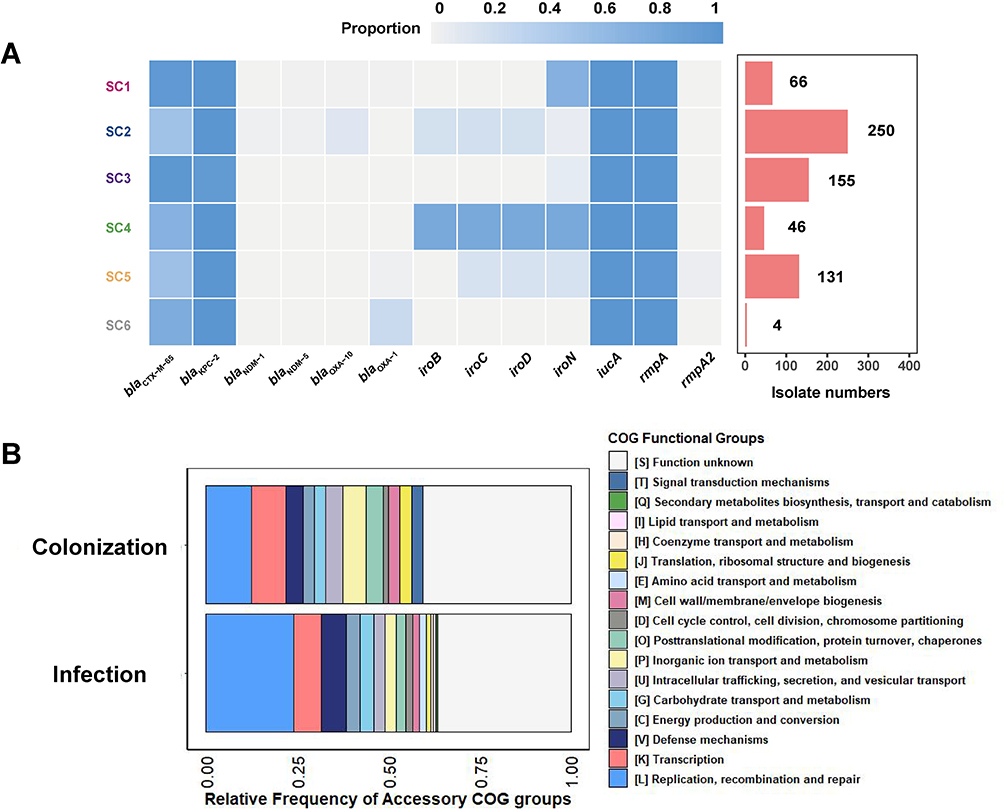 |
Figure 4 Genomic characteristics of gut colonized hv-CRKP and pathogenic hv-CRKP. (A) Virulence genes and carbapenem resistance genes carried by ST11 hv-CRKP among different SCs. (B) COG classification annotation results of accessory genome differential genes of gut colonized ST11 hv-CRKP and pathogenic ST11 hv-CRKP. |
The difference in accessory genomes may be a critical basis for colonized bacteria to break through the intestinal barrier and cause infection.29 We distinguished the differential genes between the pathogenic and colonized populations, of which 159 accessory genes were enriched in the pathogenic population and 64 genes were enriched in the colonized population (Table S4). After all genes enriched in the two populations were annotated using eggNOG-Mapper, it was found that there was no significant difference in specific functional types of genes between the two populations (Table S4, Figure 4B). The results of the comparison of functional COGs revealed that the enriched genes associated with infection or colonization were mainly distributed in these pathways (DNA replication, recombination and repair, transcription, defense mechanism, etc.) (Figure 4B). This result reflects that the accessory genome of ST11 hv-CRKP is not significantly different between the colonized and infected population strains.
Analysis of Plasmids Harbored by Gut Colonized hv-CRKP
The acquisition of virulence plasmids is the evolutionary mechanism responsible for the hypervirulent phenotype of most ST11 CRKP.3,6 Among the 652 strains ST11 hv-CRKP, we further compared the virulence plasmids of ST11 colonizers and ST11 pathogens and found that both colonizers and pathogens carried the same types of virulence plasmid replicons including IncFIIpHN7A8, IncFIBK, IncHI1B and IncR (Figure S3). In addition, the pathogen population also carries a fraction of other virulence replicons including IncFIBMar, IncFIIpKP91, IncFIA and IncQ (Figure S3).30,31 However, the carrying rates of the abovementioned four virulence plasmid replicons in the pathogenic bacterial population were quite low (≤5%). This result indicated that ST11 gut colonizers and ST11 pathogens carry the similar virulence plasmid replicon type.
ST11 hv-CRKP in our hospital had similar plasmid replicon types and virulence genes (Figure S4, Figure 1). We randomly selected KP01 as the representative ST11 hv-CRKP strain and the only colonized ST23-hv-CRKP strain KP41 for complete genome construction by hybrid assembly, and we obtained two virulence plasmids, pKP01_vir (193,050 bp) and pKP41_vir (231,501 bp). Referring to previous studies on virulence plasmids, we used the KPVR online tool to analyze the virulence plasmids pKP01_vir and pKP41_vir and found that KP01_vir and KP41_vir both contained two replicons, IncFIBK/IncHI1B, of which the IncFIBK subtype belonged to IncFIBK37,30 which was consistent with the virulence plasmid pK2044 (224,152 bp, AP006726) carried by hvkp NTUH-K2044. In addition, a BLASTn search in the NCBI database was performed, which revealed that the plasmid pKP20194a-p1 (195,031 bp, CP054781) from a sputum sample of a patient with pulmonary infection in China in 2020 was highly similar (99% identity, 98% coverage) to plasmid pKP01_vir in this study (Figure 5A). We found that pKP41_vir and pK2044 are almost identical, except for 1 kb of IS5 family transposase deletion (Figure 5A), while the deletion fragment of pKP01_vir up to 100 kb mainly includes part of the putative protein coding genes, silver, chromium, iron and other metal transporter-related genes (Figure 5A). These metal transport-related genes are not involved in the horizontal transfer or virulence level of virulence plasmids. These results further suggested that gut colonized hv-CRKP acted as a reservoir of pathogenic bacteria, sharing the same key virulence genes and virulence plasmids as the pathogenic strains. In addition, we characterized the structure of carbapenem-resistant plasmids isolating from the KP01 and KP41 strains (Figure S5). KP41 carries an IncX3-blaNDM-1-plasmid pNDM_KP41 of approximately 50 kb size, and the genetic structure of blaNDM-1 is demonstrated as blaNDM-1-IS5-IS3000-ΔTnpA_Tn2 (Figure 5B and Figure S5A). The carbapenem-resistant plasmid structure of KP01 is more complex and carries three resistance genes, rmtB, blaTEM-1B and blaKPC-2. Interestingly, the insertion sequence IS26 was present around all resistance genes (Figure 5B and Figure S5B, C), suggesting that IS26 may play an indispensable role in the integration of antibiotic resistance genes.
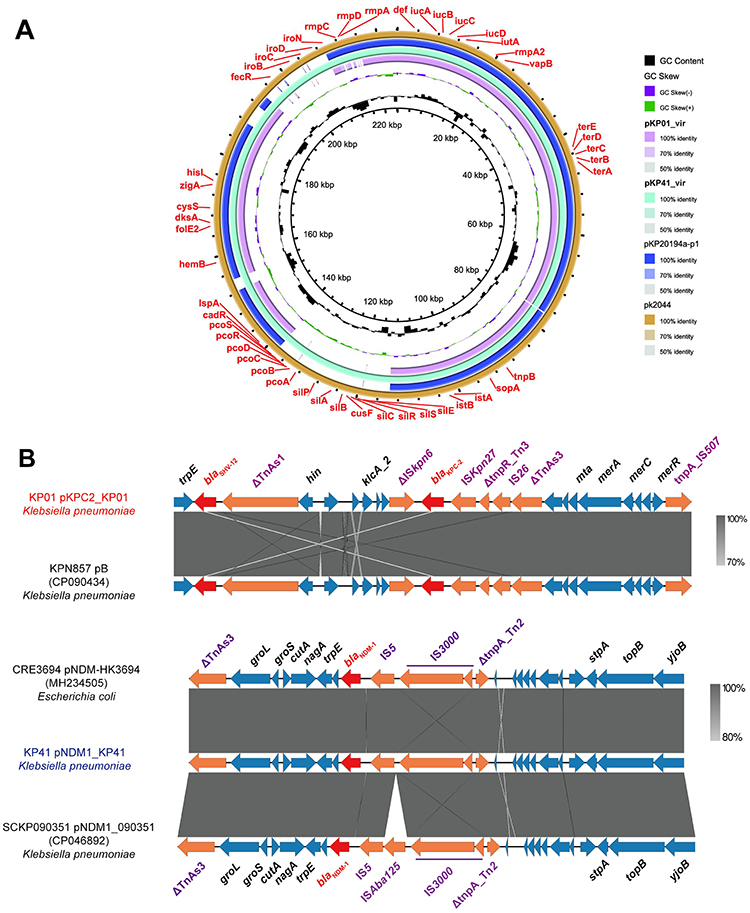 |
Figure 5 Gene map of virulence plasmids harbored by gut colonized hv-CRKP strains. (A) The structural comparison between virulence plasmid pKP01_vir, pKP41_vir, pKP20194a-p1 and pKP2044 (B) The genetic environment of the pKPC2_KP01 resistance gene blaKPC-2 and the pNDM_KP41 resistance gene blaNDM-1. |
Discussion
Intestinal colonization by K. pneumoniae is one of the risk factors for secondary infections in hospitalized patients.4,32 hv-CRKP has spread widely around the world and causes serious public health events,33,34 but there is a lack of characterization of the gut colonized population. ST11 hv-CRKP has been reported to cause nosocomial infections and fatal clinical outcomes in China. Experimental evidence from this study indicated a disturbing rate of carriage of ST11 hv-CRKP (17%) among hospitalized patients without primary K. pneumoniae infection. The colonization status of ST11 hv-CRKP in the intestinal tract of hospitalized patients is more critical than that in previous studies (8.6%).9 The results of this study did not investigate the directional colonization trend of ST11 hv-CRKP in different departments of hospitals, indicating that ST11 hv-CRKP may possess universal colonization characteristics.
Phylogenetic analysis based on whole-genome sequencing is a significant means to investigate the epidemiological characteristics of pathogens.35 Yang et al revealed that ST11 CRKP strains circulating in three hospitals in different areas of China could be divided into four epidemiological clusters with distant evolutionary relationships and contained a portion of ST11 hv-CRKP.27 Data from this study identified two epidemiological clades of ST11 hv-CRKP, including colonizers and pathogens. Further distinguishing the structure of each clade, we obtained 6 distinct ST11 hv-CRKP sequence clusters (SCs). One clade contained 4 SCs and was the dominant epidemic population of ST11 hv-CRKP. To avoid further widespread clinical dissemination of ST11 hv-CRKP, we suggest that close supervision of ST11 hv-CRKP in this clade is necessary. One noteworthy observation is that gut colonized ST11 hv-CRKP does not exist as a separate population in the phylogenetic tree. The gut colonized ST11 hv-CRKP in Sun Yat-sen Memorial Hospital was concentrated in SC1, and the strains that have been published were scattered in other SCs. The phylogenetic similarity between colonizers and pathogenic bacteria indicated that the colonizers have the potential to result in host infection. It may spread widely as a reservoir of the clinically infected population. This speculation was further supported by the fact that the key virulence genes on the plasmid and the accessory genome were not significantly different between colonizers and pathogens.
The widespread of two common virulence plasmids, pK2044 and pLVPK, led to the evolution of the hypervirulence phenotype in ST11 K. pneumoniae.36,37 We compared the characteristics of virulence plasmids of the colonized bacteria in our hospital and pathogenic bacteria in China and found that the replicon types of virulence plasmids (IncFIBK/IncHI1B) and virulence genes carried by the colonized bacteria in our hospital were highly similar to those of pathogenic bacteria. The virulence plasmid of gut colonized ST11 hv-CRKP was highly homologous to the virulence plasmid pK2044 carried by ST23 K1 serotype hypervirulent K. pneumoniae. Previous research has revealed that pK2044 exists stably in ST23 K. pneumoniae as a large nontransfer plasmid, but recently, the horizontal transfer mechanism of pK2044 was explored.38,39 Interestingly, pK2044 transferred to the ST11 population basically no longer had a complete virulence plasmid structure with different degrees of fragment deletion. These results suggest that pK2044 can lead to the evolution of a hypervirulent phenotype as long as it has some key structures. The IncX3-type blaNDM-1 plasmid found in KP41 in this study has now been widely reported in various clinical carbapenem-resistant pathogens. The insertion sequence IS26 around the plasmid resistance gene of KP01 is an essential factor for mediating horizontal transfer and integration of drug resistance genes. Yang et al found that IS26 mediates the integration of virulence genes into K. pneumoniae chromosomes.40 The presence of IS26 in gut colonized hv-CRKP may complicate the evolution and transmission mechanism of hv-CRKP. We propose the need to take the necessary measures to closely monitor the structural changes of plasmids to prevent wider horizontal transmission, which is more important in colonized groups with a wider spread than nosocomial pathogens.
A limitation of this study was the small number of hospitals. Our research provided insights into the epidemiological study of colonized hv-CRKP in inpatients. The study demonstrates the need for further studies on the relationship between gut colonized hv-CRKP and pathogenic populations.
Conclusion
In conclusion, we described the distribution and molecular epidemiological characteristics of hv-CRKP colonized the intestinal tract of inpatients. Our data indicated that the hv-CRKP population in China was dominated by ST11, which had two primary epidemic clades. It is worrisome that ST11 hv-CRKP has a high colonization rate in the intestine of hospitalized patients and the gut colonized ST11 hv-CRKP may serve as a clinical pathogen reservoir. Therefore, it is urgent to strengthen the prevention of secondary infection and horizontal transmission of gut colonized ST11 hv-CRKP.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (grant number 82061128001, 81830103 to G.-BT), Project of High-level Health Teams of Zhu Hai in 2018 (The Innovation Team for Antimicrobial Resistance and Clinical Infection to G.-BT).
Disclosure
The authors report no competing interests in this work.
References
1. Munoz-Price LS, Poirel L, Bonomo RA, et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis. 2013;13(9):785–796. doi:10.1016/S1473-3099(13)70190-7
2. Shrivastava SR, Shrivastava PS, Ramasamy J . World health organization releases global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. J Med Soc. 2017;32(1):76–77. doi:10.4103/jms.jms_25_17
3. Russo TA, Marr CM. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev. 2019;32(3):e00001–e00019. doi:10.1128/CMR.00001-19
4. Gorrie CL, Mirceta M, Wick RR, et al. Gastrointestinal carriage is a major reservoir of Klebsiella pneumoniae infection in intensive care patients. Clin Infect Dis. 2017;65(2):208–215. doi:10.1093/cid/cix270
5. Ferreira AM, Moreira F, Guimaraes T, et al. Epidemiology, risk factors and outcomes of multi-drug-resistant bloodstream infections in haematopoietic stem cell transplant recipients: importance of previous gut colonization. J Hosp Infect. 2018;100(1):83–91. doi:10.1016/j.jhin.2018.03.004
6. Gu D, Dong N, Zheng Z, et al. A fatal outbreak of ST11 carbapenem-resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: a molecular epidemiological study. Lancet Infect Dis. 2018;18(1):37–46. doi:10.1016/S1473-3099(17)30489-9
7. Kang JTL, Teo JJY, Bertrand D, et al. Long-term ecological and evolutionary dynamics in the gut microbiomes of carbapenemase-producing Enterobacteriaceae colonized subjects. Nat Microbiol. 2022;7(10):1516–1524. doi:10.1038/s41564-022-01221-w
8. Juan CH, Chou SH, Chen IR, et al. Intestinal colonisation with hypervirulent or third-generation cephalosporin-resistant Klebsiella pneumoniae strains upon hospital admission in a general ward in Taiwan. Int J Antimicrob Agents. 2022;60(2):106624 . doi:10.1016/j.ijantimicag.2022.106624
9. Zheng B, Xu H, Lv T, et al. Stool samples of acute diarrhea inpatients as a reservoir of ST11 hypervirulent KPC-2-producing Klebsiella pneumoniae. mSystems. 2020;5(3):e00498–e00520. doi:10.1128/mSystems.00498-20
10. Koonin EV, Makarova KS, Wolf YI. Evolution of microbial genomics: conceptual shifts over a quarter century. Trends Microbiol. 2021;29(7):582–592. doi:10.1016/j.tim.2021.01.005
11. Moradigaravand D, Martin V, Peacock SJ, et al. Evolution and epidemiology of multidrug-resistant Klebsiella pneumoniae in the United Kingdom and Ireland. mBio. 2017;8(1):e01976–16. doi:10.1128/mBio.01976-16
12. David S, Reuter S, Harris SR, et al. Epidemic of carbapenem-resistant Klebsiella pneumoniae in Europe is driven by nosocomial spread. Nat Microbiol. 2019;4(11):1919–1929. doi:10.1038/s41564-019-0492-8
13. Yang Y, Yang Y, Chen G, et al. Molecular characterization of carbapenem-resistant and virulent plasmids in Klebsiella pneumoniae from patients with bloodstream infections in China. Emerg Microbes Infect. 2021;10(1):700–709. doi:10.1080/22221751.2021.1906163
14. Walker KA, Miner TA, Palacios M, et al. A Klebsiella pneumoniae regulatory mutant has reduced capsule expression but retains hypermucoviscosity. mBio. 2019;10(2):e00089–19. doi:10.1128/mBio.00089-19
15. Chen S, Zhou Y, Chen Y, et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–i890. doi:10.1093/bioinformatics/bty560
16. Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477. doi:10.1089/cmb.2012.0021
17. Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20(1):257. doi:10.1186/s13059-019-1891-0
18. Alikhan NF, Petty NK, Ben Zakour NL, et al. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genom. 2011;12:402. doi:10.1186/1471-2164-12-402
19. Page AJ, Cummins CA, Hunt M, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31(22):3691–3693. doi:10.1093/bioinformatics/btv421
20. Page AJ, Taylor B, Delaney AJ, et al. SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb Genom. 2016;2(4):e000056. doi:10.1099/mgen.0.000056
21. Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–2158. doi:10.1093/bioinformatics/btr330
22. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi:10.1093/bioinformatics/btu033
23. Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–W296. doi:10.1093/nar/gkab301
24. Yu G, Lam TT, Zhu H, Guan Y. Two methods for mapping and visualizing associated data on phylogeny using Ggtree. Mol Biol Evol. 2018;35(12):3041–3043. doi:10.1093/molbev/msy194
25. Brynildsrud O, Bohlin J, Scheffer L, et al. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016;17(1):238. doi:10.1186/s13059-016-1108-8
26. Huerta-Cepas J, Szklarczyk D, Heller D, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47(D1):D309–D314. doi:10.1093/nar/gky1085
27. Yang X, Sun Q, Li J, et al. Molecular epidemiology of carbapenem-resistant hypervirulent Klebsiella pneumoniae in China. Emerg Microbes Infect. 2022;11(1):841–849. doi:10.1080/22221751.2022.2049458
28. D’Apolito D, Arena F, Conte V, et al. Phenotypical and molecular assessment of the virulence potential of KPC-3-producing Klebsiella pneumoniae ST392 clinical isolates. Microbiol Res. 2020;240:126551. doi:10.1016/j.micres.2020.126551
29. Mageiros L, Méric G, Bayliss SC, et al. Genome evolution and the emergence of pathogenicity in avian Escherichia coli. Nat Commun. 2021;12(1):765. doi:10.1038/s41467-021-20988-w
30. Tian D, Wang M, Zhou Y, et al. Genetic diversity and evolution of the virulence plasmids encoding aerobactin and salmochelin in Klebsiella pneumoniae. Virulence. 2021;12(1):1323–1333. doi:10.1080/21505594.2021.1924019
31. Du P, Liu C, Fan S, et al. The role of plasmid and resistance gene acquisition in the emergence of ST23 multi-drug resistant, hypervirulent Klebsiella pneumoniae. Microbiol Spectr. 2022;10(2):e0192921. doi:10.1128/spectrum.01929-21
32. Holmes CL, Anderson MT, Mobley HLT, et al. Pathogenesis of gram-negative bacteremia. Clin Microbiol Rev. 2021;34(2):e00234–20. doi:10.1128/CMR.00234-20
33. Ernst CM, Braxton JR, Rodriguez-Osorio CA, et al. Adaptive evolution of virulence and persistence in carbapenem-resistant Klebsiella pneumoniae. Nat Med. 2020;26(5):705–711. doi:10.1038/s41591-020-0825-4
34. Arato V, Raso MM, Gasperini G, et al. Prophylaxis and treatment against Klebsiella pneumoniae: current insights on this emerging anti-microbial resistant global threat. Int J Mol Sci. 2021;22(8):4042. doi:10.3390/ijms22084042
35. Raskin DM, Seshadri R, Pukatzki SU, et al. Bacterial genomics and pathogen evolution. Cell. 2006;124(4):703–714. doi:10.1016/j.cell.2006.02.002
36. Yang X, Wai-Chi Chan E, Zhang R, et al. A conjugative plasmid that augments virulence in Klebsiella pneumoniae. Nat Microbiol. 2019;4(12):2039–2043. doi:10.1038/s41564-019-0566-7
37. Du FL, Huang QS, Wei DD, et al. Prevalence of carbapenem-resistant Klebsiella pneumoniae co-harboring blaKPC-carrying plasmid and pLVPK-like virulence plasmid in bloodstream infections. Front Cell Infect Microbiol. 2021;10:556654. doi:10.3389/fcimb.2020.556654
38. Tian D, Liu X, Chen W, et al. Prevalence of hypervirulent and carbapenem-resistant Klebsiella pneumoniae under divergent evolutionary patterns. Emerg Microbes Infect. 2022;11(1):1936–1949. doi:10.1080/22221751.2022.2103454
39. Xu Y, Zhang J, Wang M, et al. Mobilization of the nonconjugative virulence plasmid from hypervirulent Klebsiella pneumoniae. Genome Med. 2021;13(1):119. doi:10.1186/s13073-021-00936-5
40. Yang X, Ye L, Li Y, et al. Identification of a chromosomal integrated DNA fragment containing the rmpA2 and iucABCDiutA virulence genes in Klebsiella pneumoniae. mSphere. 2020;5(6):e01179–20. doi:10.1128/mSphere.01179-20
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.