")
Back to Journals » Patient Related Outcome Measures » Volume 13
Clinician and Patient Reporting of Symptomatic Adverse Events in Cancer Clinical Trials: Using CTCAE and PRO-CTCAE® to Provide Two Distinct and Complementary Perspectives
Authors Minasian LM , O'Mara A, Mitchell SA
Received 4 May 2022
Accepted for publication 12 November 2022
Published 8 December 2022 Volume 2022:13 Pages 249—258
DOI https://doi.org/10.2147/PROM.S256567
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Robert Howland
Lori M Minasian,1 Ann O’Mara,2,3 Sandra A Mitchell4
1Division of Cancer Prevention, National Cancer Institute, Bethesda, MD, USA; 2Consultant, ICF, Fairfax, VA, USA; 3Consultant to Division of Cancer Prevention, National Cancer Institute, Bethesda, MD, USA; 4Division of Cancer Control and Population Sciences, National Cancer Institute, Bethesda, MD, USA
Correspondence: Lori M Minasian, Division of Cancer Prevention, National Cancer Institute, 9609 Medical Center Drive, 5E-342, MSC-9784, Bethesda, MD, 20892-9784, USA, Tel +11 240 276 7053, Fax +11 240 276 7846, Email [email protected]
Abstract: Inclusion of the patient perspective in the reporting of symptomatic adverse events provides different and complementary information to clinician reporting using the Common Terminology Criteria for Adverse Events (CTCAE). The National Cancer Institute’s Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE®) is designed for patients to self-report their symptomatic adverse events in a manner that complements CTCAE reporting. Using CTCAE and PRO-CTCAE together offers the potential to refine our understanding of the prevalence and trajectory of lower grade AEs that can lead to elective discontinuation of therapy and diminished quality of life. This review addresses the development of PRO-CTCAE with an emphasis on the differences between PRO-CTCAE scores and CTCAE severity grades. This distinction is important when evaluating, grading and reporting toxicity and tolerability in cancer clinical trials.
Keywords: Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events, PRO-CTCAE, patient-reported outcomes, cancer therapy, toxicity, tolerability
Introduction
Significant advances in cancer treatment have been realized from investing in the basic and translational understanding of how cancer develops. Newer therapies, including molecularly targeted agents and immunotherapies have mechanisms of action sufficiently different from conventional chemotherapy such that the types and patterns of toxicity differ. Alongside these scientific developments, the approach to capturing, reporting, and analyzing adverse event (AE) data has also been evolving.1,2 While accurate capture of serious AEs remains essential, there is a growing need to recognize and address the chronic, cumulative lower grade AEs that may impair the patient’s ability to function, reduce quality of life, and potentially result in elective discontinuation and patient nonadherence. Many of these lower grade AEs are symptomatic, such as nausea, fatigue, pain, neuropathy, and anorexia. Characterizing these lower grade AEs is the first step in better understanding the risk-benefit profile of a given cancer treatment regimen and developing more tolerable treatment through strategies to mitigate adverse effects, thereby improving adherence to therapy, preserving dose density and ultimately the therapeutic efficacy of cancer treatment.
The Common Terminology Criteria for Adverse Events (CTCAE), a descriptive terminology and standardized criteria for AE reporting, is the widely accepted method used by clinicians to identify and grade AEs in cancer clinical trials.3 AE items in CTCAE can be characterized as: 1) laboratory-based events such as anemia or hypoglycemia; 2) observable or measurable events such as pleural effusion or hypotension; or 3) symptomatic AEs such as pain, nausea, and fatigue. It has long been recognized that the clinician reporting of symptomatic AEs through CTCAE may not align with the patient’s self-report of those same symptoms.2,4 While there are several reasons for this, capturing the patient’s perspective in the reporting of symptomatic AEs provides important information to complement clinician reporting. The National Cancer Institute (NCI) developed the Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE®). This PRO measurement system allows patients to self-report symptomatic AEs in a manner that complements clinician-reported CTCAE grading, thereby improving the precision and reliability in characterizing adverse treatment effects in cancer clinical trials.
This review will address the development of PRO-CTCAE as a companion to the CTCAE with an emphasis on the differences between PRO-CTCAE scores and CTCAE severity grades. This distinction is important when assessing toxicity and tolerability in cancer clinical trials. Using CTCAE and PRO-CTCAE together has the potential to refine the understanding of the prevalence and trajectory of the lower grade AEs that can lead to elective discontinuation of therapy and diminished quality of life. To clarify the differences between the clinician terminology and grading criteria and the companion self-report measurement system, this review will start with the development and use of CTCAE in cancer clinical trials.
CTCAE: Clinician Terminology for Reporting
In 1983, the National Cancer Institute (NCI) developed the Common Toxicity Criteria (CTC v1.0) as a tool for the systematic severity grading and reporting of AEs from chemotherapy agents in cancer clinical trials.3 The CTC has been revised over time to: 1) incorporate AE terms reflecting new acute adverse events and late effects, and grading criteria relevant to modalities such as radiation and surgery; 2) incorporate pediatric criteria, and a name change to the Common Terminology Criteria for Adverse Event Reporting (CTCAE); and 3) harmonize with the Medical Dictionary of Regulatory Activities (MedDRA) terminology.3,5–7 Through these revisions, new AE terms have been added to the library of AE items and infrequently used terms have been retired.
CTCAE includes a severity grading scale (Table 1)8 such that the report of any AE item always has a severity grade. This severity grading is used to report a safety signal requiring medical intervention to prevent a life-threatening event from happening. Not all AE items have all five severity grades associated with them. For example, the AE item “ear pain” has only grades 1–3 associated with it because “ear pain” does not result in a life-threatening event. The AE item “intra-abdominal hemorrhage” has only grades 2–5 associated with it because this event is never a mild condition and can result in death.
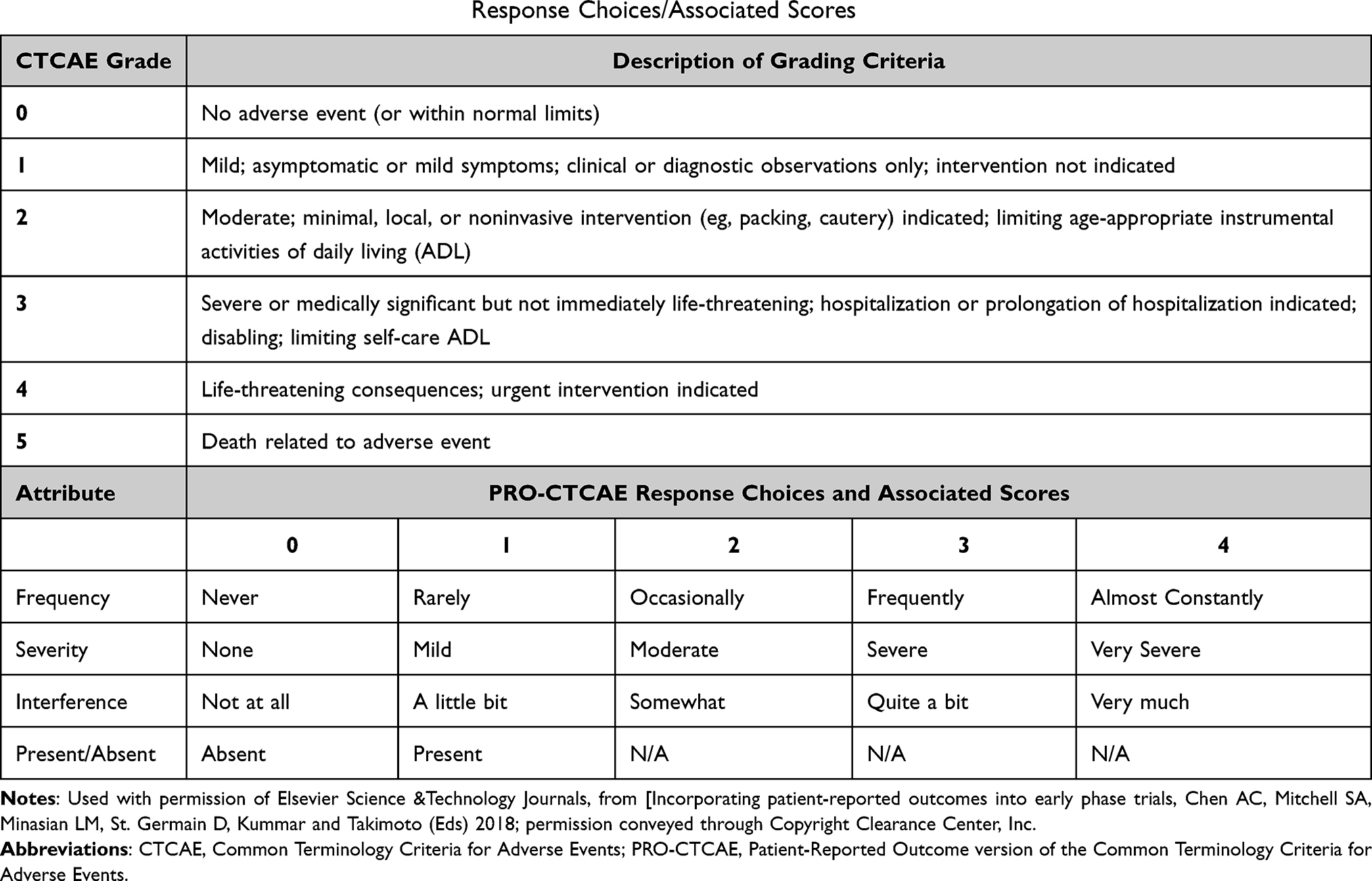 |
Table 1 CTCAE Grades and PRO-CTCAE® Response Choices/Associated Scores |
CTCAE grading of an AE item bundles the severity of the event, with the functional interference that event causes, and the extent to which medical intervention is needed to mitigate the specific event. For example, dysphagia, or trouble swallowing, can be experienced by the patient as severe, but the assignment of a grade 3 or 4 (severe or very severe) in CTCAE is dependent upon the level of functional interference and the extent to which a medical intervention is needed. If the patient is able to eat and drink, the dysphagia is graded as 1. If the patient is not able to eat but can drink fluids and is able to take liquid supplements, the dysphagia is graded as 2. However, if the patient is not able to swallow liquids and is hospitalized for intravenous hydration, the dysphagia is graded as 3 and considered a serious AE (SAE). This grade 3 dysphagia is considered a SAE because of the need for hospitalization. In each of these cases, however, the patient may experience the dysphagia as severe. There are many clinical scenarios where an AE can be severe without being a SAE. The CTCAE grade provides distinct information about safety. Nonetheless, the patient with the lower grade dysphagia may experience the trouble swallowing in a way that leads to diminished appetite, weight loss, and reduced quality of life or elective discontinuation of therapy. This information is not explicitly reflected in the reporting of the CTCAE grading criteria for dysphagia.
Severity grading of 2 or greater is often used to define protocol parameters such as dose holds, dose modification, dose-limiting toxicity with consequent treatment discontinuation, and maximum tolerated dose. Some cancer treatment protocols may require a dose hold if a patient experiences a grade 2 or greater event with respect to a specific AE. Additionally, severity grades can serve as criteria for patient eligibility into a trial and help identify a tolerable dose when moving an investigational agent from Phase 1 into Phase 2 clinical trials. These grading criteria facilitate a consistent method for reporting safety data both in scientific publication and to regulatory agencies and permits the comparison of safety data across trials.
Typically, CTCAE data are displayed in a table that shows the worst grade of an AE experienced by the patient at any one point in time. These tables provide an efficient way to scan the data for serious and life-threatening AEs, but do not provide information on the trajectory of the AEs over time. Thus, information regarding the resolution or progression of an AE is absent. Low-grade AEs may be combined together as grade 1–2 in an AE table or omitted altogether. Such an approach makes it difficult to discern from a study report the extent to which these lower grade AEs occurred and accumulated over time. While these low-grade AEs may not result in hospitalization or serious consequences for organ function, they may over the course of sustained treatment persist or progress, resulting in elective discontinuation of therapy.9–11 Moreover, some AEs may be heralded by seemingly low-grade experiences that may not always be identified or reported. In addition, low-grade AEs may signal anticipated treatment response, particularly in the context of molecularly targeted agents and immunotherapies.12 Incorporating the patient’s perspective of symptomatic AEs may improve the characterization of treatment toxicity and improve the reporting of tolerability for that treatment regimen in clinical trials.
Incorporating the Patient’s Perspective: Health-Related Quality of Life (HRQOL) and Patient Reported Outcomes (PROs)
HRQOL assessments in cancer clinical trials have been a successful means of incorporating the patient’s perspective. While there is no uniformly agreed upon definition of HRQOL, it is generally comprised of self-reporting on four fundamental dimensions that include physical, mental/psychological, and social health, as well as global perceptions of function and well-being.13 Additional HRQOL domains considered important include pain, energy/vitality, sleep, appetite, and symptoms relevant to the intervention and to the natural history of the disease or condition.14 The term “PRO” is defined as a measurement based on a report that comes directly from the patient about the status of a patient’s health condition.15 Thus, PRO is a broad term encompassing any measurement reported by a patient, while HRQOL is a specific domain of PROs. Irrespective of the term, the overarching premise is that the patient provides a self-report that is independent of clinician interpretation.
Collecting HRQOL data in the setting of cancer clinical trials has been an ongoing endeavor for many decades. As far back as 1989, Moinpour et al noted that investigators saw the value in supplementing tumor response and survival data with information on patient perception of treatment impact.16 Since then, HRQOL and PRO fields have contributed significantly to a better understanding of how patients feel and function while undergoing cancer treatment and following its completion.17–22 Over time, methods for collecting and analyzing data as well as reducing missing data have improved.23–26
Collecting HRQOL data is not a substitute for toxicity reporting through CTCAE, but rather can complement AE reporting.2,27 Differences in scoring methodologies, timing of assessments, and sources of reporting make the comparison between HRQOL reports and clinician reports of symptomatic AEs difficult to interpret. Patient reports of symptom severity on HRQOL tools and clinician reports of the same events using CTCAE are not equivalent. Specifically, CTCAE grades 1–3 (Table 1) do not necessarily align with typical HRQOL scores of 1= “a little bit”, 2= “somewhat”, 3= “quite a bit”, and 4= “very much.” A CTCAE grade 2 reflects a moderate severity that impacts activities of daily living. This could mean moderate throat pain from mucositis that impairs the ability to eat and drink. A patient’s response of 2 (“somewhat”) on an HRQOL tool, may not have this same meaning. CTCAE Grade 3 reflects a severe AE that requires an intervention. Thus, a patient who reports severe throat pain with minimal impairment in eating or drinking would not be reported as grade 3 mucositis even though the patient’s experience is that of severe throat pain that would likely be scored as 3 on an HRQOL measure that ranges from 0 to 4.
Assessment time points are often not the same for CTCAE and HRQOL. Surveillance for AEs using CTCAE is initiated at baseline, and is conducted continuously through the trial, whereas HRQOL assessments are done less frequently to reduce patient burden. Clinicians prospectively monitor a limited number of protocol-identified AEs over the treatment course, but report any treatment emergent AEs when they occur. For example, a patient may develop throat pain after the first course of treatment that resolves prior to the next scheduled HRQOL assessment. The clinician would report the mucositis and grade the severity of the event at its occurrence. However, the patient would not necessarily report it at baseline or at the next HRQOL assessment time. As such, summary HRQOL assessments may miss the onset and offset of adverse events that occur during treatment.
One of the advantages in using PRO assessments in clinical trials is the systematic assessment of specific symptoms captured at baseline, followed prospectively through the course of treatment and provided directly from the patient. The pre-treatment assessment allows a comparison from baseline at specific time points that provides better attribution of a treatment-emergent AE from a pre-existing symptom.28 However, for most HRQOL assessments, the schedule of reporting (every 3–6 months) does not coincide with the need to report AEs as they occur in real time.
When investigators have compared patient reports of HRQOL and symptom-specific scales, such as pain, fatigue, or nausea, with clinician-reported AEs, the agreement between patient and clinician reporting is moderate at best. Patients frequently report greater symptom severity than what is seen for clinician-graded events.29–31 While the patient experiences the AE severity as high, if the event does not sufficiently impair function or require a clinical intervention, the event is assigned a lower CTCAE severity grade. In short, the patient’s experience through the HRQOL reporting of symptom severity is not well represented when looking only at CTCAE data.
When HRQOL endpoints are captured in cancer clinical trials, they are often not fully reported alongside the toxicity data and thus, it is difficult for clinicians to evaluate both the toxicity data and the patient’s treatment experiences together. Most commonly, the HRQOL analysis is published separately, if it is published at all,32,33 such that clinicians are unaware of the trial results for all endpoints.
Development of PRO-CTCAE to Capture the Patient’s Experience
With the need to better understand how to optimize cancer treatment regimens, NCI developed a measurement system called PRO-CTCAE designed to capture symptomatic AEs by patient self-report in using terminologies that are consistent with clinician-reported CTCAE.34,35 CTCAE and PRO-CTCAE item structures are shown together in Table 1 and Figure 1. PRO-CTCAE was designed to be complementary to and used in conjunction with the CTCAE. As such, PRO-CTCAE serves to capture the patient’s experience of symptomatic AEs and its interference with day-to-day functioning, thus reflecting the patient perspective of treatment tolerability.
 |
Figure 1 CTCAE and PRO-CTCAE item structures: mucositis (CTCAE) and mouth/throat sores (PRO-CTCAE). Notes: Used with permission of Elsevier Science & Technology Journals, from Incorporating patient-reported outcomes into early phase trials, Chen AC, Mitchell SA, Minasian LM, St. Germain D, Kummar and Takimoto (Eds) 2018; permission conveyed through Copyright Clearance Center, Inc. |
In developing PRO-CTCAE, 78 AEs that are amenable to patient reporting were identified from version 4.0 of CTCAE.35,36 PRO-CTCAE has been demonstrated to be valid, reliable, and feasible for use in cancer clinical trials,36–39 has been translated and linguistically validated in more than 30 languages, and a pediatric module (Ped-PRO-CTCAE) is available.40
Each symptomatic AE has a different number of attributes (frequency, severity, interference, presence/absence) associated with it for a total of 124 unique questions.35 For example, the symptomatic AE “numbness and tingling” includes both severity and interference questions, generating a separate score for severity of “numbness and tingling” and a separate score for the interference from “numbness and tingling.” The patient scores each item on a scale from 0 to 4. The CTCAE term equivalent to “numbness and tingling” is peripheral sensory neuropathy, where the assignment of a grade is dependent upon the functional limitations resulting from that neuropathy. For example, a patient who reports moderate numbness and tingling, but repeatedly falls would have a higher CTCAE neuropathy grade than a patient who reports severe numbness and tingling but is able to perform his or her normal activities. In particular, the patient reporting of both severity and interference may help the clinician assign the most appropriate grade to that patient’s event of neuropathy.
It is important to note that PRO-CTCAE scores do not correspond directly to CTCAE grades. CTCAE and PRO-CTCAE were not developed for direct comparison. Differences between clinician and patient reporting of symptomatic AEs can arise due to different perceptions, and due to the fact that clinicians and patients may be providing their reports at slightly differing time points (in clinic versus between visits for example, or may be reporting on slightly different phenomena (eg, sadness vs depression). Patients may also under-report symptoms during clinic due to time constraints, a desire to avoid an appearance that they are complaining or bothering their clinician, as well as concerns about not qualifying for continued treatment, and/or being removed from a study protocol or receiving a lower dose of treatment, if they report toxicities. As such, differences between CTCAE and PRO-CTCAE reporting should not be over-interpreted as either under-reporting by clinicians, or over-reporting by patients. PRO-CTCAE was designed to be complementary to CTCAE, not to replace it, since each provides a unique and valuable source of information.
PRO-CTCAE is an item library of symptomatic AEs, and it is not expected that all items will be used in any given trial. Selecting which symptomatic AEs to evaluate and determining the time points for measurement are critical trial design decisions. Because PRO-CTCAE should be used and reported in conjunction with the CTCAE reports gathered by clinicians, PRO-CTCAE items are chosen for surveillance using an approach similar to that used to define the AE surveillance plan for the trial more broadly. The investigator chooses those symptomatic AE items that are anticipated from the agents involved in the trial to be prospectively monitored and captured over time.41
The choice of items should be informed by the anticipated adverse events based on previous preclinical data and regimen-specific information about the anticipated profile of symptomatic adverse events drawn from the Comprehensive Adverse Events and Potential Risks List (CAEPR).8 In a randomized trial with different agents and regimens, all participants should report on the same AE items across the different trial arms to reduce reporting bias. For example, if one arm of the trial includes agents for which the common AE is diarrhea and the other arm includes agents for which the common AE is a rash, all trial participants should report on both diarrhea and rash. Consideration should be given to inclusion of the PRO-CTCAE items that capture any other symptoms that the patient may wish to report, assessing the severity of that symptom at its worst. Inclusion of the free-text PRO-CTCAE item allows for the capture of unsolicited and unexpected symptomatic AEs, thereby supporting a conclusion that symptomatic AE event ascertainment is not biased by the specific subset of items selected for surveillance in the trial. With respect to the timing of assessments, weekly assessments should be captured during key periods in the trial (eg, first two or three cycles in an early phase trial), or at other crucial clinical assessment timeframes based upon knowledge of the anticipated toxicity profile of the regimen.
Patient-reported symptomatic AEs are not intended as safety signals that would be used by clinician investigators to take individual level protocol-specific clinical actions (such as dose holds, dose modifications, etc.).42 Rather, the trial may collect this information and analyze the data at the conclusion of the trial or include protocol-specific instructions for clinicians about how to use patient self-reported information in their grading and reporting of AEs for the trial. Protocol documents should clearly indicate that use of PRO-CTCAE is limited to describing in aggregate the safety and tolerability of a regimen, and should state explicitly that clinician CTCAE grades are to be used when making clinical decisions about trial eligibility, dose delays, dose reductions or treatment discontinuation.42
PRO-CTCAE Design Considerations in Cancer Clinical Trials
Since 20,155 when it became available for public use, PRO-CTCAE has been incorporated into randomized clinical trials43 with excellent patient compliance.39,44 Additional studies have helped to refine and clarify the use of the PRO-CTCAE measurement system in cancer clinical trials.
The mode through which patients report their data, whether using paper and pencil, interactive voice response, computer, tablet, or other electronic system, does not have an impact on the validity of patient responses.45,46 Thus, investigators may use any modality and patients may report using different modalities through the course of a trial (eg, tablet for in-clinic assessments, and home computer or other device for between-visit assessments).
Electronic data capture provides efficiency for survey scheduling and administration, reduces the need for manual data entry and permits centralized, real-time monitoring of missing data. PRO-CTCAE has been developed and tested with a conditional branching logic that helps to reduce respondent burden. Conditional branching should be employed for electronic administration of PRO-CTCAE symptom terms that have two or more attributes (eg, fatigue severity and interference, or pain frequency, severity, and interference). The logic branches from frequency, then to severity, and then to interference. Thus, if a patient reports no frequency, the survey does not present the severity or interference questions for a response.
During the development of PRO-CTCAE, investigators examined various recall periods (ie, past 7 days, past 2, 3, or 4 weeks) to explore the effects of recall period on measurement error.47 While patients were able to somewhat reliably recall their experiences over longer periods, there was some information loss associated with the longer recall periods. As such, the standard recall period for PRO-CTCAE is the past seven days. However, depending on the design of the clinical trial, longer or shorter recall periods may be logistically desirable. While use of longer recall periods may be an attractive approach to address burden on patient and study personnel, trialists must consider the fact that longer recall periods and less frequent assessments may introduce both measurement unreliability and under-reporting of important within-cycle treatment experiences. Notably, concerns about temporal breaks in reporting may be less pertinent in study contexts where symptomatic AEs are anticipated to be stable or to be changing only subtly, as, for example, with chronically administered oral therapies or during post-treatment follow-up.
Typically, PROs are included in randomized Phase 3 trials to compare the aggregate patient-reported symptomatic AEs between the different treatment regimens. In these late phase trials, the anticipated toxicity profile of a treatment regimen is usually well-described such that the symptomatic AEs to monitor are known. When HRQOL endpoints are included in trials, the aggregate summary scores for each domain (physical, psychosocial, social, and global perception of function) are also compared between treatment arms. Both summary scores as well as individual symptoms are displayed longitudinally to see the trajectory of those symptoms during and after completion of treatment.
In early phase trials, both the nature and the timing of AE occurrence is more difficult to anticipate. Thus, the ability to identify the optimal group of symptomatic AEs to monitor prospectively is more challenging. Investigators are exploring how best to incorporate patient-reported symptomatic AEs into early phase trials.48–50 In one study, patients in a phase 1 clinic completed the full library of questions at three time points.51 While there was reasonable compliance with the reporting, patients in phase 1 trials are often highly symptomatic and may not be able to sustain weekly reporting of 124 questions. Additionally, many patients do not stay on phase 1 trials for long periods of time. Thus, completing 124 questions weekly may not be practical.
Our recommended approach is to identify a limited number of symptomatic AEs that patients will report on weekly, and to solicit free-text responses to capture unanticipated symptomatic AEs. The selected symptomatic AEs should be consistent with the protocol-specified AEs which clinicians are monitoring, grading, and reporting using CTCAE. This approach provides distinct and complementary information to derived from both clinicians and patients on the same AE domains, thereby allowing conclusions about tolerability that incorporate both safety concerns and the patient’s treatment experiences. When provided with the opportunity to report free text, patients are able to meaningfully report additional symptoms and the majority of those additions can be mapped to existing PRO-CTCAE symptom terms or to CTCAE terminologies.52 While this approach is not consistent with how PRO data has been typically collected and interpreted, it is consistent with the overarching paradigm for the capture of AEs and could provide a systematic way to capture patient-reported treatment emergent symptomatic AEs in early phase trials.
Distinguishing Between Safety and Tolerability
While CTCAE has provided the standard for establishing the safety of cancer treatment drugs, no standard has been established for tolerability. In reporting the safety data of chemotherapeutic agents, the phrases “generally well tolerated” and “manageable toxicities” are often used but may mean very different things to clinicians and patients.53 Oncologists are not alone in using the term “tolerability” to indicate the absences of major safety issues. A review of 56 clinical trials (30 non-cancer) which reported on tolerability, found none of the trials defined specific events or parameters to evaluate and distinguish between safety and tolerability.54 These investigators acknowledge that the goal of most treatments is to improve patient quality of life, and that poor therapeutic tolerability negatively impacts patient function and adherence to treatment. They conclude that the term “tolerability” has been used too loosely and that the patient’s perspective on adverse drug effects is not given sufficient attention.
The inclusion of PRO-CTCAE along with other PRO measures (HRQOL and symptom-specific scales) provides a means to capture patients’ perceptions of how they feel and function and potentially can help clarify the term tolerability, although this may be challenging to achieve when self-report information is published separately from treatment efficacy outcomes. One example is the use of PRO-CTCAE to determine that a treatment regimen is tolerable. In one phase 3 trial evaluating a novel targeted agent for recurrent lung cancer, PRO-CTCAE data demonstrated a low to moderate level of symptomatic AEs,55 which confirmed the safety data derived from clinician reporting.56 Thus, a regimen that was reported as well-tolerated based on clinician-reporting was confirmed by patient self-report, although the impact of this observation would have been optimized if the patient self-report data were published in conjunction with the primary efficacy and tolerability data.
In a phase 3 radiation trial, the clinician safety data showed no differences in efficacy when comparing intensity-modulated radiation therapy (IMRT) with standard radiation therapy. However, the patient-reported symptomatic AEs showed a significant difference favoring IMRT in the occurrence of gastrointestinal symptomatic AEs, particularly diarrhea and fecal incontinence.57
Because of the lack of standard methods for determining tolerability, the NCI established a consortium of investigators to analyze clinician-reported AEs and patient-reported symptomatic AEs using the PRO-CTCAE.58 These investigators are developing approaches and methods for identifying tolerability of cancer treatment by defining the contributions that joint analysis of CTCAE and PRO-CTCAE data offer in determining tolerability, analyzing baseline factors that may predict the development of AEs, and exploring age-related and functional factors which affect treatment discontinuation and hospitalization.
Conclusion
The current standard for grading and reporting all AEs in cancer clinical trials is CTCAE. However, multiple studies have shown that patient self-report of symptom severity and its impact on function and overall quality of life contributes to a more complete understanding of the adverse effects of cancer treatment. Clinician-reported AEs and patient reporting of symptoms provide different and complementary information to better understand the therapeutic effectiveness and the toxicity profile of cancer treatment regimens. PRO-CTCAE scores provide additional information on the development and trajectory of symptomatic AEs during and following cancer treatment, which can be valuable to patients, their clinicians, trial sponsors, and regulators.
As newer anti-cancer agents become available for use, there is a need to understand the tolerability of those agents to improve patient adherence and ultimately patient outcomes. Oncology clinicians have used CTCAE to determine tolerability based upon an approach initially developed to capture the adverse effects of cytotoxic chemotherapy, which tends to minimize low-grade chronic and/or progressive AEs. Yet, those lower grade AEs may impact patient adherence to oral anti-cancer agents. By using both CTCAE and PRO-CTCAE together, patients and clinicians will have the data to better understand the tolerability of cancer treatment and improve the identification of symptomatic adverse events that should be monitored and managed. With continued use of PRO-CTCAE in cancer clinical trials, its role will evolve and the methods and approaches to analyzing the data along with other clinically relevant trial data will help patients and their clinicians understand the impact of cancer treatment on patient well-being and functioning.
Acknowledgments
The authors would like to thank Gwen Moulton for editorial assistance in preparing the manuscript.
Disclosure
The authors declare that they have no competing interests.
References
1. Thanarajasingam G, Minasian LM, Baron F, et al. Beyond maximum grade: modernising the assessment and reporting of adverse events in haematological malignancies. Lancet Haematol. 2018;5(11):e563–e598. doi:10.1016/S2352-3026(18)30051-6
2. Kluetz PG, Chingos DT, Basch EM, et al. Patient-reported outcomes in cancer clinical trials: measuring symptomatic adverse events with the national cancer institute’s patient-reported outcomes version of the common terminology criteria for adverse events (PRO-CTCAE). Am Soc Clin Oncol Educ Book. 2016;35(36):67–73. doi:10.1200/EDBK_159514
3. National Cancer Institute Division of Cancer Treatment and Diagnosis. Common Terminology Criteria for Adverse Events (CTCAE). Available from: https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_40.
4. Bruner DW. Should patient-reported outcomes be mandatory for toxicity reporting in cancer clinical trials? J Clin Oncol. 2007;25(34):5345–5347. doi:10.1200/JCO.2007.13.3330
5. Trotti A, Byhardt R, Stetz J, et al. Common toxicity criteria: version 2.0. An improved reference for grading the acute effects of cancer treatment: impact on radiotherapy. Int J Radiat Oncol Biol Phys. 2000;47(1):13–47. doi:10.1016/S0360-3016(99)00559-3
6. Trotti A, Colevas AD, Setser A, et al. CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol. 2003;13(3):176–181. doi:10.1016/S1053-4296(03)00031-6
7. Trotti A, Colevas AD, Setser A, et al. Patient-reported outcomes and the evolution of adverse event reporting in oncology. J Clin Oncol. 2007;25(32):5121–5127. doi:10.1200/JCO.2007.12.4784
8. Chen ACMS, Minasian LM, St. Germain D. Incorporating patient-reported outcomes into early phase trials. In: Takimoto K, editor. Novel Designs of Early Phase Trials for Cancer Therapeutics. London, UK: Elsevier; 2018:193–208.
9. Lee HS, Lee JY, Ah YM, et al. Low adherence to upfront and extended adjuvant letrozole therapy among early breast cancer patients in a clinical practice setting. Oncology. 2014;86(5–6):340–349. doi:10.1159/000360702
10. Creel PA. Optimizing patient adherence to targeted therapies in renal cell carcinoma. Clin J Oncol Nurs. 2014;18(6):694–700. doi:10.1188/14.CJON.694-700
11. Verbrugghe M, Verhaeghe S, Lauwaert K, et al. Determinants and associated factors influencing medication adherence and persistence to oral anticancer drugs: a systematic review. Cancer Treat Rev. 2013;39(6):610–621. doi:10.1016/j.ctrv.2012.12.014
12. Maher VE, Fernandes LL, Weinstock C, et al. Analysis of the association between adverse events and outcome in patients receiving a programmed death protein 1 or programmed death ligand 1 antibody. J Clin Oncol. 2019;37(30):2730–2737. doi:10.1200/JCO.19.00318
13. Wilson IB, Cleary PD. Linking clinical variables with health-related quality of life. A conceptual model of patient outcomes. JAMA. 1995;273(1):59–65. doi:10.1001/jama.1995.03520250075037
14. Berzon R, Hays RD, Shumaker SA. International use, application and performance of health-related quality of life instruments. Qual Life Res. 1993;2(6):367–368. doi:10.1007/BF00422214
15. U.S. Department of Health and Human Services. Food and drug administration guidance for industry: patient-reported outcome measures: use in medical product development to support labeling claims; 2009. Available from: https://www.Fda.Gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm193282.Pdf.
16. Moinpour CM, Feigl P, Metch B, et al. Quality of life end points in cancer clinical trials: review and recommendations. J Natl Cancer Inst. 1989;81(7):485–496. doi:10.1093/jnci/81.7.485
17. Ganz PA, Gotay CC. Use of patient-reported outcomes in Phase III cancer treatment trials: lessons learned and future directions. J Clin Oncol. 2007;25(32):5063–5069. doi:10.1200/JCO.2007.11.0197
18. O’Mara AM, Denicoff AM. Health related quality of life in NCI-sponsored cancer treatment trials. Semin Oncol Nurs. 2010;26(1):68–78. doi:10.1016/j.soncn.2009.11.009
19. Secord AA, Coleman RL, Havrilesky LJ, et al. Patient-reported outcomes as end points and outcome indicators in solid tumours. Nat Rev Clin Oncol. 2015;12(6):358–370. doi:10.1038/nrclinonc.2015.29
20. Efficace F, Collins GS, Cottone F, et al. Patient-reported outcomes as independent prognostic factors for survival in oncology: systematic review and meta-analysis. Value Health. 2021;24(2):250–267. doi:10.1016/j.jval.2020.10.017
21. Trask PC, Hsu MA, McQuellon R. Other paradigms: health-related quality of life as a measure in cancer treatment: its importance and relevance. Cancer J. 2009;15(5):435–440. doi:10.1097/PPO.0b013e3181b9c5b9
22. Rouette J, Blazeby J, King M, et al. Integrating health-related quality of life findings from randomized clinical trials into practice: an international study of oncologists’ perspectives. Qual Life Res. 2015;24(6):1317–1325. doi:10.1007/s11136-014-0871-9
23. Osoba D. Lessons learned from measuring health-related quality of life in oncology. J Clin Oncol. 1994;12(3):608–616. doi:10.1200/JCO.1994.12.3.608
24. Bottomley A, Aaronson NK. International perspective on health-related quality-of-life research in cancer clinical trials: the European organisation for research and treatment of cancer experience. J Clin Oncol. 2007;25(32):5082–5086. doi:10.1200/JCO.2007.11.3183
25. Bell ML, Fairclough DL. Practical and statistical issues in missing data for longitudinal patient-reported outcomes. Stat Methods Med Res. 2014;23(5):440–459. doi:10.1177/0962280213476378
26. Calvert M, King M, Mercieca-Bebber R, et al. Spirit-PRO extension explanation and elaboration: guidelines for inclusion of patient-reported outcomes in protocols of clinical trials. BMJ Open. 2021;11(6):e045105. doi:10.1136/bmjopen-2020-045105
27. Basch E, Jia X, Heller G, et al. Adverse symptom event reporting by patients vs clinicians: relationships with clinical outcomes. J Natl Cancer Inst. 2009;101(23):1624–1632. doi:10.1093/jnci/djp386
28. George GC, Barata PC, Campbell A, et al. Improving attribution of adverse events in oncology clinical trials. Cancer Treat Rev. 2019;76:33–40. doi:10.1016/j.ctrv.2019.04.004
29. Falchook AD, Green R, Knowles ME, et al. Comparison of patient- and practitioner-reported toxic effects associated with chemoradiotherapy for head and neck cancer. JAMA Otolaryngol Head Neck Surg. 2016;142(6):517–523. doi:10.1001/jamaoto.2016.0656
30. Quinten C, Maringwa J, Gotay CC, et al. Patient self-reports of symptoms and clinician ratings as predictors of overall cancer survival. J Natl Cancer Inst. 2011;103(24):1851–1858. doi:10.1093/jnci/djr485
31. Xiao C, Polomano R, Bruner DW. Comparison between patient-reported and clinician-observed symptoms in oncology. Cancer Nurs. 2013;36(6):E1–E16. doi:10.1097/NCC.0b013e318269040f
32. St Germain D, Denicoff A, Torres A, et al. Reporting of health-related quality of life endpoints in national cancer institute-supported cancer treatment trials. Cancer. 2020;126(11):2687–2693. doi:10.1002/cncr.32765
33. Saleh RR, Meti N, Ribnikar D, et al. Associations between safety, tolerability, and toxicity and the reporting of health-related quality of life in phase III randomized trials in common solid tumors. Cancer Med. 2020;9(21):7888–7895. doi:10.1002/cam4.3390
34. Minasian L, Rosen O, Auclair D, et al. Optimizing dosing of oncology drugs. Clin Pharmacol Ther. 2014;96(5):572–579. doi:10.1038/clpt.2014.153
35. National Cancer Institute Patient-Reported outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE); 2022. Available from: https://healthcaredelivery.cancer.gov/pro-ctcae/.
36. Basch E, Reeve BB, Mitchell SA, et al. Development of the National Cancer Institute’s patient-reported outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). J Natl Cancer Inst. 2014;106:9. doi:10.1093/jnci/dju244
37. Dueck AC, Mendoza TR, Mitchell SA, et al. Validity and reliability of the Us National Cancer Institute’s Patient-Reported Outcomes version of the Common Terminology Criteria For Adverse Events (PRO-CTCAE). JAMA Oncol. 2015;1(8):1051–1059. doi:10.1001/jamaoncol.2015.2639
38. Basch E, Dueck AC, Rogak LJ, et al. Feasibility assessment of patient reporting of symptomatic adverse events in multicenter cancer clinical trials. JAMA Oncol. 2017;3(8):1043–1050. doi:10.1001/jamaoncol.2016.6749
39. Basch E, Dueck AC, Rogak LJ, et al. Feasibility of implementing the patient-reported outcomes version of the common terminology criteria for adverse events in a multicenter trial: ncctg n1048. J Clin Oncol. 2018;36(31):Jco2018788620. doi:10.1200/JCO.2018.78.8620
40. PRO-CTCAE certified translations and countries in which they have been tested. Available from: https://healthcaredelivery.cancer.gov/pro-ctcae/countries-pro.html.
41. Basch E, Rogak LJ, Dueck AC. Methods for implementing and reporting Patient-Reported Outcome (PRO) measures of symptomatic adverse events in cancer clinical trials. Clin Ther. 2016;38(4):821–830. doi:10.1016/j.clinthera.2016.03.011
42. Kim J, Singh H, Ayalew K, et al. Use of PRO measures to inform tolerability in oncology trials: implications for clinical review, IND safety reporting, and clinical site inspections. Clin Cancer Res. 2018;24(8):1780–1784. doi:10.1158/1078-0432.CCR-17-2555
43. Giesinger JM, Efficace F, Aaronson N, et al. Past and current practice of patient-reported outcome measurement in randomized cancer clinical trials: a systematic review. Value Health. 2021;24(4):585–591. doi:10.1016/j.jval.2020.11.004
44. Thorpe CS, DeWees TA, Golafshar MA, et al. Patient-reported outcomes version of the common terminology criteria for adverse events and quality-of-life linear analogue self-assessment in breast cancer patients receiving radiation therapy: single-institution prospective registry. J Patient Rep Outcomes. 2022;6(1):3. doi:10.1186/s41687-021-00408-9
45. Knoerl R, Gray E, Stricker C, et al. Electronic versus paper-pencil methods for assessing chemotherapy-induced peripheral neuropathy. Support Care Cancer. 2017;25(11):3437–3446. doi:10.1007/s00520-017-3764-y
46. Bennett AV, Dueck AC, Mitchell SA, et al. Mode equivalence and acceptability of tablet computer- interactive voice response system-, and paper-based administration of the U.S. National Cancer Institute’s Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). Health Qual Life Outcomes. 2016;14:24. doi:10.1186/s12955-016-0426-6
47. Mendoza TR, Dueck AC, Bennett AV, et al. Evaluation of different recall periods for the US National Cancer Institute’s PRO-CTCAE. Clin Trials. 2017;14(3):255–263. doi:10.1177/1740774517698645
48. Kennedy F, Shearsmith L, Ayres M, et al. Online monitoring of patient self-reported adverse events in early phase clinical trials: views from patients, clinicians, and trial staff. Clin Trials. 2021;18(2):168–179. doi:10.1177/1740774520972125
49. Sedhom R, Ferrell B, Ruel N, et al. Using patient-reported outcomes to describe the patient experience on Phase I clinical trials. JNCI Cancer Spectr. 2020;4(6):pkaa067. doi:10.1093/jncics/pkaa067
50. Retzer A, Aiyegbusi OL, Rowe A, et al. The value of patient-reported outcomes in early-phase clinical trials. Nat Med. 2022;28(1):18–20. doi:10.1038/s41591-021-01648-4
51. Shepshelovich D, McDonald K, Spreafico A, et al. Feasibility assessment of using the complete Patient-Reported Outcomes Version of the Common Terminology Criteria For Adverse Events (PRO-CTCAE) item library. Oncologist. 2019;24(4):e146–e148. doi:10.1634/theoncologist.2018-0332
52. Chung AE, Shoenbill K, Mitchell SA, et al. Patient free text reporting of symptomatic adverse events in cancer clinical research using the National Cancer Institute’s Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). J Am Med Inform Assoc. 2019;26(4):276–285. doi:10.1093/jamia/ocy169
53. Sacks CA, Miller PW, Longo DL. Talking about toxicity - “what we’ve got here is a failure to communicate”. N Engl J Med. 2019;381(15):1406–1408. doi:10.1056/NEJMp1908310
54. Stanulović V, Hodolic M, Mitsikostas DD, et al. Drug tolerability: how much ambiguity can be tolerated? A systematic review of the assessment of tolerability in clinical studies. Br J Clin Pharmacol. 2022;88(2):551–565. doi:10.1111/bcp.15016
55. Sebastian M, Rydén A, Walding A, et al. Patient-reported symptoms possibly related to treatment with osimertinib or chemotherapy for advanced non-small cell lung cancer. Lung Cancer. 2018;122:100–106. doi:10.1016/j.lungcan.2018.05.003
56. Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR t790m-positive lung cancer. N Engl J Med. 2017;376(7):629–640. doi:10.1056/NEJMoa1612674
57. Yeung AR, Pugh SL, Klopp AH, et al. Improvement in patient-reported outcomes with intensity-modulated radiotherapy (RT) compared with standard RT: a report from the NRG oncology RTOG 1203 study. J Clin Oncol. 2020;38(15):1685–1692. doi:10.1200/JCO.19.02381
58. Analyzing and interpreting clinician and patient adverse event data to better understand tolerability RFA-CA-17-052. Available from: https://grants.nih.gov/grants/guide/rfa-files/rfa-ca-17-052.html.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.