Back to Journals » International Journal of General Medicine » Volume 16
Clinical Manifestations, Genetic Variants and Therapeutic Evaluation in Sporadic Chinese Patients with Idiopathic Hypogonadotropic Hypogonadism
Authors He D ![]() , Sun H, Zhang M, Li Y, Liu F, Zhang Y, He M, Ban B
, Sun H, Zhang M, Li Y, Liu F, Zhang Y, He M, Ban B ![]()
Received 16 July 2023
Accepted for publication 18 September 2023
Published 29 September 2023 Volume 2023:16 Pages 4429—4439
DOI https://doi.org/10.2147/IJGM.S430904
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Dongye He,1,2,* Hailing Sun,1 Mei Zhang,1,3 Yanying Li,1,3 Fupeng Liu,1,2 Yanhong Zhang,1,3 Mingming He,1 Bo Ban1– 3,*
1Department of Endocrinology, Genetics and Metabolism, Affiliated Hospital of Jining Medical University, Jining, 272029, People’s Republic of China; 2Medical Research Center, Affiliated Hospital of Jining Medical University, Jining, 272029, People’s Republic of China; 3Chinese Research Center for Behavior Medicine in Growth and Development, Jining, 272029, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dongye He; Bo Ban, Department of Endocrinology, Genetics and Metabolism, Affiliated Hospital of Jining Medical University, Jining, 272029, People’s Republic of China, Tel/Fax +86 0537 2903579, Email [email protected]; [email protected]
Purpose: Genetic factors account for a large proportion of idiopathic hypogonadotropic hypogonadism (IHH) etiologies, although not necessarily a complete genetic basis. This study aimed to characterize the clinical presentations, genetic variants, and therapeutic outcomes of patients with sporadic IHH, which may be helpful for genetic counseling and treatment decisions.
Patients and Methods: Eleven Chinese patients with IHH were retrospectively analyzed. Rare genetic variants were evaluated using whole-exome sequencing and bioinformatics analysis and were further classified according to the ACMG-AMP guidelines. The therapeutic responses of patients were further evaluated.
Results: Six heterozygous variants of SOX10, WDR11, PROKR2, CHD7 and FGF17 were detected in five Kallmann syndrome (KS) patients, whereas two heterozygous variants of CHD7 and PROKR2 were detected in two normosmic IHH (nIHH) patients. Among these variants, a novel likely pathogenic variant in the SOX10 (c.429– 1G>C) was considered to cause the KS phenotype in patient 02, and two potential variants of uncertain significance (VUS) in CHD7 (c.3344G>A and c.7391A>G) possibly contributed to the KS phenotype in patient 05 and the nIHH phenotype in patient 07, which need to be confirmed by further evidence. Additionally, long-term testosterone or estradiol replacement treatment effectively improved the development of sexual characteristics in patients with IHH.
Conclusion: Next-generation sequencing is a powerful tool for identifying the molecular etiology and early diagnosis of IHH. Efficient therapeutic outcomes strongly indicate a need for timely treatment.
Keywords: Kallmann syndrome, normosmic idiopathic hypogonadotropic hypogonadism, luteinizing hormone, follicle-stimulating hormone, genetic variant, hormone treatment
Introduction
The roller coaster-like pulsatile secretory activity of gonadotropin-releasing hormone (GnRH) released by GnRH neurons in the mediobasal hypothalamus controls reproductive function during human lifespan. Notably, GnRH secretory activity lapses into silence throughout childhood until the onset of puberty. Its deficient synthesis, secretion, or action directly causes hypothalamic-pituitary-gonadal (HPG) axis hypofunctioning, defining hypogonadotropic hypogonadism (HH), which typically manifests as absent or impaired sexual development.1–3 A genetic cause that plays little role in acquired diseases affecting GnRH neuroendocrine function, accounts for almost half of idiopathic hypogonadotropic hypogonadism (IHH) cases, albeit not necessarily the complete genetic basis.4 Moreover, the reason why IHH is associated with olfactory impairment is that GnRH neurons located within the olfactory placode migrate alongside the olfactory tract to ultimately reach the mediobasal hypothalamus, where neuron numbers expand, form a functional network, and initiate pulsatile secretion during normal human fetal development. In terms of whether have a normal olfactory function, IHH patients were divided into normosmic IHH (nIHH) and Kallmann syndrome (KS) with anosmia or hyposmia.5 Hitherto, with the widespread application of next-generation sequencing (NGS) in clinical medicine, especially whole-exome sequencing (WES), genetic variants of nearly 60 genes have been found to be associated with IHH, either in respect of GnRH neuron migration and activation or GnRH secretion and action, wherein ANOS1, PROK2, PROKR2, KISS1R, FGFR1, GNRHR, FGF8, CHD7, SOX10, SEMA3A, TAC3 and TAC3R variants are the most frequent molecular etiologies.6–8
The incidence of IHH presents a very large gender difference, with an approximately 5-fold lower prevalence in female (1: 50,000) when compared to males (1: 10,000).9 Besides abnormal sexual development, non-reproductive non-olfactory abnormalities (hearing loss, cleft lip/palate, dental agenesis, cubitus valgus, low hairline) are also observed in some IHH patients, underpinning the peculiarity of genetic heterogeneity or gene pleiotropism.10 Crucially, sex hormone replacement therapy (testosterone or estradiol) can effectively improve delayed or absent sexual development in the majority of IHH patients, albeit only 10% cases achieved reversed reproductive axis function.11,12 Therefore, genetic counseling, early diagnosis and timely treatment of IHH is of great significance.
In the present study, we retrospectively collected and analyzed the physical, endocrinological, and radiological profiles of 11 patients with sporadic IHH and further uncovered their molecular defects and interlinked phenotyping. Subsequently, we evaluated the therapeutic outcomes of the patients (if available) through long-term follow-up. It is hoped that this study will help clinicians provide more prudent genetic counseling and valuable treatment decisions.
Materials and Methods
Study Subjects
Eleven unrelated IHH patients (nine males and two females) who visited the Department of Endocrinology, Affiliated Hospital of Jining Medical University (Shandong, China) between December 2013 and December 2020 were retrospectively included in the study. Eight patients were clinically diagnosed with a KS phenotype and three patients were diagnosed with an nIHH phenotype according to the classification criteria.13 All of whom showed low or inappropriately normal serum levels of luteinizing hormone (LH), follicle-stimulating hormone (FSH), and/or progesterone (P), testosterone (T), prolactin (PRL), and estradiol (E2), and failure to enter puberty spontaneously with partial or no development of secondary sex characteristics. Four male patients received testosterone undecanoate replacement treatment, one female patient received estradiol valerate hormonal treatment, and post-treatment follow-up data, including serum testosterone or estradiol levels and sexual development, were further collected and analyzed.
Clinical and Endocrinological Evaluations
The patients’ clinical parameters were obtained through a retrospective review of the medical records: age at diagnosis, sex, height, body weight, limb development, skin texture, baseline secondary sexual characteristics, cleft lip/palate, hearing impairment, karyotype, and family history. Olfactory function was evaluated either by olfaction testing or by patient self-reporting, depending on the clinical center. The olfactory system, including the olfactory bulb and tract structure, as well as the pituitary structure, was visualized using magnetic resonance imaging (MRI). Patients’ hearing status was evaluated using pure-tone audiometry and impedance audiometry. In male patients, testicular development was confirmed by physical and scrotal ultrasound examinations. A micropenis was defined as a stretched penile length (SPL) below −2.5 standard deviations (SD) of SPL-for-age.14 For female patients, uterine development was assessed using pelvic ultrasonography. Growth hormone (GH) concentration was measured using a chemiluminescence method (ACCESS2, Beckman Coulter) with an analytical sensitivity of 0.010 µg/L. Serum levels of insulin-like growth factor (IGF-1) were measured using a chemiluminescence immunometric method (DPC IMMULITE 1000 analyzer, SIEMENS). Gonadotropin, gonadal steroid hormones, free T3, free T4, thyroid-stimulating hormone (TSH), adrenocorticotropic hormone (ACTH), and cortisol rhythm were measured using a luminescence immunoassay system (Cobas e 602; Roche). In the triptorelin stimulation test, an intramuscular injection of triptorelin (100 mg) was used to determine the basal value, and at 15, 30, 45, 60, and 90 min after the injection; both LH and FSH levels in all patients were determined at the first visit (Supplementary Figure 1). Patient 01 did not undergo triptorelin stimulation test.
Whole-Exome Sequencing and Bioinformatic Analysis
Five milliliters of peripheral venous blood were collected from probands and their parents, and blood DNA was extracted using the Blood DNA Midi Kit (D3494-04, Omega Bio-Tek). Parental blood samples were available for only 4 of these 11 families. WES was performed using the BGI V4 capture kit on the MGISEQ-2000 platform (BGI, Shenzhen, China) with a mean 180 coverage of targeted region. Moreover, more than 95% of the targeted regions covered over 20 ×.
After quality control using fastQC, all clean reads were aligned to the human reference genome (GRCh37/hg19) using the Burrows-Wheeler Aligner software (BWA, version 0.7.15). Local realignment around insertions and deletions (InDels) and base quality score recalibration (BQSR) were performed using the Genome Analysis Toolkit (GATK) after removing duplicate reads. Genomic variations, including single nucleotide variants (SNVs) and indels, were detected using HaplotypeCaller of GATK and functionally annotated using SnpEff. Additionally, the R package ExomeDepth was used to detect the copy number variation (CNV). Genes and proteins were described using standard nomenclature, according to the Human Genome Variation Society (HGVS). Rare sequencing variants (RSVs) were nonsynonymous variants with minor allele frequency (MAF) of < 0.1% or absent in public databases (1000 Genomes, Exome Aggregation Consortium [ExAC], NHLBI Exome Sequencing Project [ESP6500], and Genome Aggregation Database [gnomAD]). The potential pathogenicity of the RSVs was predicted using several in silico prediction programs (SIFT, PolyPhen-2, PROVEAN, Condel, GERP++, and Splice AI). Protein sequence conservation across different species was analyzed using MEGA X version 10.1.8. Familial segregation analysis was performed using Sanger sequencing if parental blood samples were available. All IHH-associated RSVs with high-quality sequencing data (sequencing depth ≥ 80 and SNP mutation rate ≥ 40%) were screened and strictly classified as “pathogenic”, “likely pathogenic”, “uncertain significance”, “likely benign”, and “benign” according to the guidelines of the American College of Medical Genetics and Genomics (ACMG) Association for Molecular Pathology (AMP).15
Results
Clinical and Endocrinological Features
Detailed clinical data of the 11 patients with IHH are summarized in Table 1. The mean age at diagnosis was 18 years (range: 13–27 years) in males and 24 years (range: 17–31 years) in females. The height standard deviation score (SDS) was −0.63 ± 1.09. The body mass indices (BMI) of patients 02, 03, 05, 07, 09, and 11 were 24.13, 27.41, 24.13, 24.18, 30.11, and 28.73, respectively, indicating overweight (24 kg/m2 ≤ BMI < 28 kg/m2) or general obesity (28 kg/m2 ≤ BMI) in sex- and age-matched subjects.16 The karyotypes of all patients had normal karyotypes (46, XY [male] or 46, XX [female]). Patient 01, 02, 04, 06, 07, 08 and 09 showed low levels of growth hormone (GH) peak obtained from different provocative tests (all less than 10 ng/mL).17 Among them, compared with references ranges for healthy populations, patient 04 (211 ng/mL, reference median:315 ng/mL), 07 (178 ng/mL, reference median:280 ng/mL) and 09 (82.8 ng/mL, reference median:307 ng/mL) had lower level of IGF-1.18 However, no abnormal pituitary MRI finding was observed in patient 04, 07 and 09. All patients showed low or inappropriate basal serum levels of LH, FSH, P, T, PRL or E2, but had normal serum levels of FT3, FT4, TSH, ACTH, and cortisol. After triptorelin administration, serum LH and FSH levels were significantly increased in female patient 05, even within the normal range (Supplementary Figure 1). It should be noted that patient 04 was a 13-year-old boy at the first visit and still showed low basal serum levels of LH, FSH, P, T, PRL, and E2 when he was 16 years old. Therefore, patient 04 was clinically diagnosed with KS but not a constitutional delay of growth and puberty.
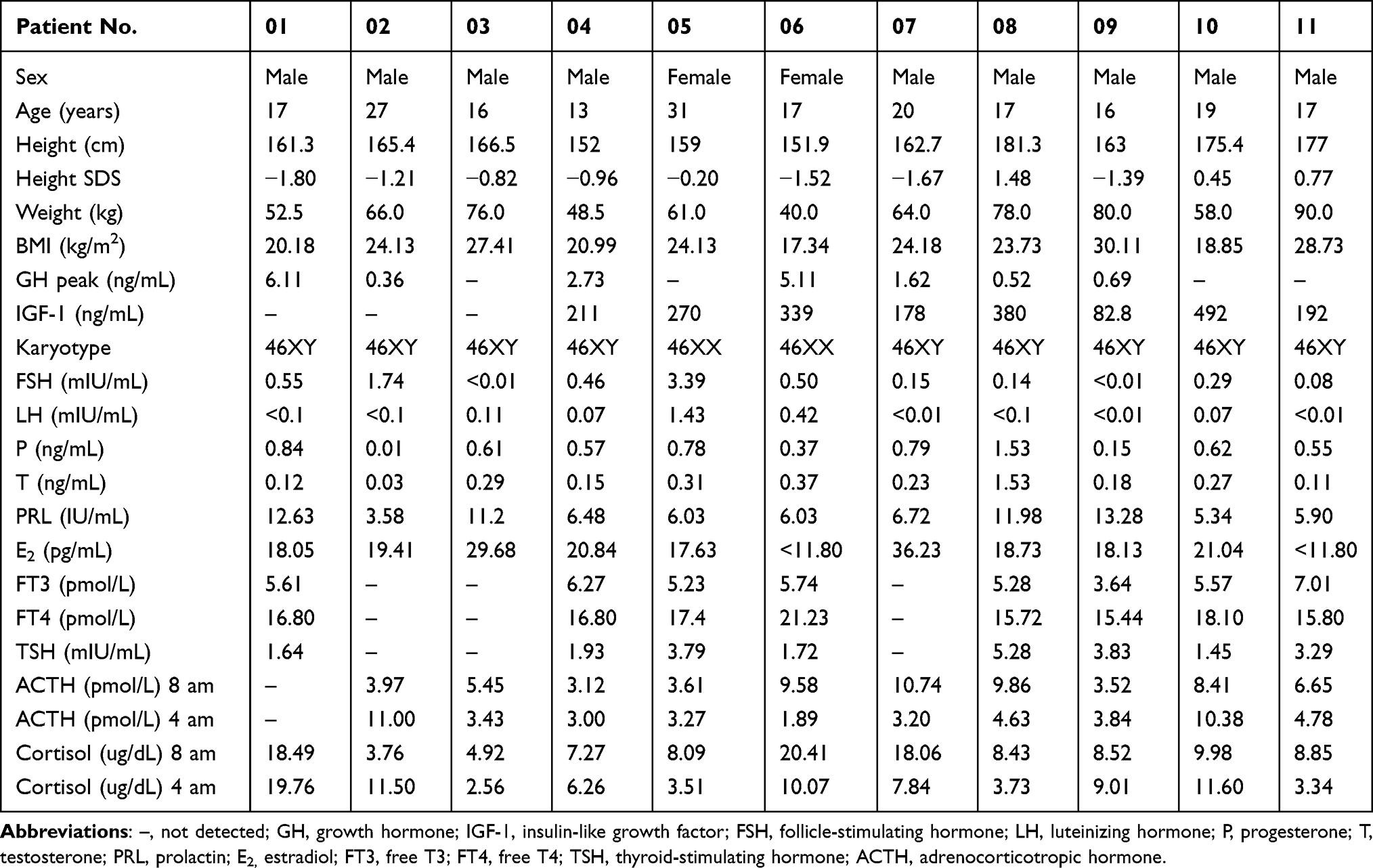 |
Table 1 Physical Characteristic and Laboratory Test Results of IHH Patients |
Genetic Variants in IHH Patients
The pedigree chart and identified genetic variants of the IHH patients are shown in Figure 1. Eight molecular variants, all in the heterozygous state, were identified in seven patients from a total of eleven sporadic IHH patients (Table 2). Among the two nIHH patients, patient 07 carried a CHD7 variant (c.7391A>G, p.K2464R) and patient 08 carried a PROKR2 variant (c.533G>C, p.W178S). Among the five KS cases, patient 02 carried the WDR11 variant (c.2931+6delC) and SOX10 variant (c.429–1G>C). Patient 04 and patient 05 had the PROKR2 (c.533G>C, p.W178S) and CHD7 (c.3344G>A, p.C1115Y) variants, respectively. Patient 06 carried two variants of PROKR2 (c. 337T>C, p.Y113H; c.1069C>T, p.R357W) inherited from her unaffected mother. Additionally, patient 11 carried an FGF17 variant (c.592G>C, p.V198L) inherited from his unaffected mother. These variations were categorized as likely pathogenic (LP), six variants of uncertain significance (VUS), and one as likely benign (LB).
 |
Table 2 Clinical Phenotypes and Genetic Variants of Patients with IHH Examinated by Whole-Exome Sequencing |
 |
Figure 1 Pedigree chart and genetic variants identified in patients affected with IHH. Notes: Squares and circles indicate males and females, respectively. Numbers (01–11) represent index cases, filled symbols represent affected individuals, open symbols represent unaffected individuals. +: wild type allele; -: mutated allele; n/a: not available for genetic test; n.d.: no variants were detected. Family 1 and Family 2 denote that parental DNA samples were available. Verification of PROKR2 variant in family 1 and FGF17 variant in family 2 were performed by Sanger sequencing. The variants found in patient 06 and patient 11 were inherited from their unaffected mother. |
The minor allele frequencies of all identified mutations were < 0.1% or absent in the 1000G, ExAC, ESP6500, and gnomAD databases (Supplementary Table 1). In addition, in silico prediction (SIFT, Polyphen-2, PROVEAN, Condel, and GERP++) indicated that PROKR2 (c.337T>C, c.553G>C) and CHD7 (c.3344G>A) were damaging, whereas PROKR2 (c.1069C>T), CHD7 (c.7391A>G), and FGF17 (c.592G>C) had little effect on gene function. The c.429–1G>C variant in SOX10 most likely affected the splicing pattern by altering the acceptor sites predicted by Splice AI (Supplementary Table 1). In contrast, the c. 2931+6delC variant in WDR11 probably has little impact on its splicing pattern. Amino acid alignment across different species using MEGA X revealed that PROKR2 Tyrosine 113, PROKR2 Tryptophan 178, CHD7 Lysine 2464, CHD7 Cysteine 1115, and FGF17 Valine 198 were located in a highly conserved region (Supplementary Figure 2). However, PROKR2 Arginine 357 was not conserved among the eight species.
Primary Within-Treatment and Follow-Up Outcomes
Male patients 01, 03, 04, 07, 08, and 10 received oral testosterone undecanoate replacement treatment at a dosage of 40 mg twice per day, whereas female patient 06 received oral estradiol valerate at a dosage of 1 mg once daily, from nearly half a year to several years. As shown in Figure 2a, during the follow-up period, all male patients showed increased serum testosterone levels, especially patient 08, from 0.5 ng/mL to 5.31 ng/mL. There were no changes in bilateral testicular volume for patient 01, 04 and 10 before and after treatment, but a increase in patient 03 (2.0 mL to 3.0 mL), patient 07 (0.5 mL to 2.0 mL) and 08 (1.0 mL to 3.0 mL) (Table 3). The stretched penile length of patient 01, 03, 04, 07, 08 and 10 improved by 4.0 cm, 5.0 cm, 4.5 cm, 1.0 cm, 3.5 cm and 3.0 cm, respectively. In addition, all the male patients showed obvious pubic hair development. In female, the serum estradiol level of patient 06 increased but showed great fluctuation throughout the follow-up period (Figure 2b). After more than three years of treatment with estradiol valerate, patient 06 displayed progressive sexual development, including breast Tanner stage (II to IV), pubic hair Tanner stage (I to II), and uterine volume (0 to 9.32 cm3). Nevertheless, all patients treated with hormones still showed low serum LH and FSH levels (Figure 2c and d) and did not meet the criterion for reversal of hypogonadism.19
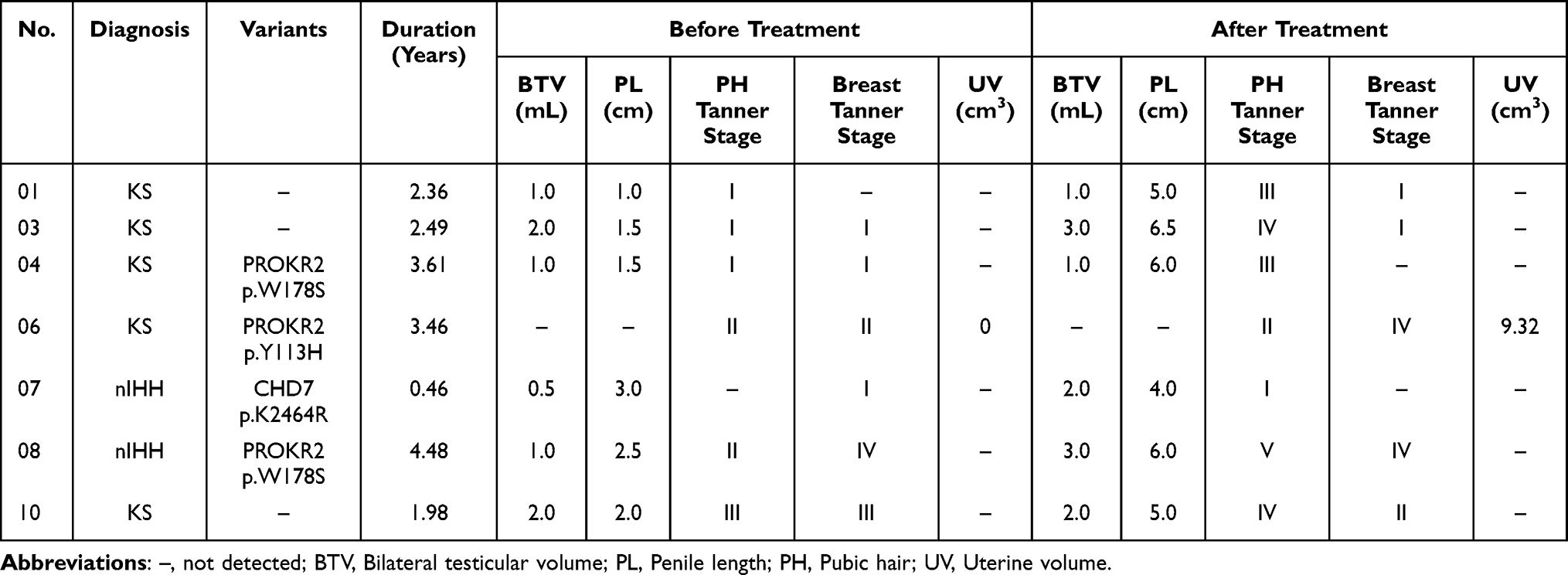 |
Table 3 Genetic Variants and Clinical Outcomes of Patients Affected with KS or nIHH After Hormonal Treatment |
 |
Figure 2 Changes in testosterone, estradiol, LH and FSH levels of patients affected with IHH during therapy. Abbreviations: LH, luteinizing hormone; FSH, follicle-stimulating hormone. Notes: (a) serum testosterone level of male patients treated with testosterone undecanoate through the entire follow-up period; (b) serum estradiol level of female patient treated with estradiol valerate through the entire follow-up period. (c and d) Serum LH and FSH level of all patients receiving hormone treatment during the entire follow-up period. |
Discussion
In this study, we identified eight rare heterozygous variants of SOX10, WDR11, PROKR2, CHD7 and FGF17 in 11 IHH patients with unrelated kindreds using WES. The results of the genetic analysis indicated that SOX10 c.429–1G>C was considered to cause the KS phenotype in patient 02, and two potential VUS variants in the CHD gene (c.3344G>A; c.7391A>G) possibly contributed to the KS phenotype in patient 05 and the nIHH phenotype in patient 07. However, the pathogenicity of these variants requires further investigation by more strong evidences. Long-term testosterone or estradiol replacement treatment effectively improved the development of sexual characteristics in patients with IHH regardless of whether they harbored potential variants.
PROKR2 plays an important role in the olfactory bulb interneurons development and GnRH neurons migration by binding to its ligand PROK2.20,21 PROKR2 variants, especially missense type, cause either KS or nIHH with variable expressivity and incomplete penetrance.22 Interestingly, only PROKR2−/− mice presented a phenotype reminiscent of IHH including hypoplasia of the olfactory bulb and atrophy of the reproductive system,23 albeit multiple heterozygous missense variants (Y113H, V115M, W178S, S188L, W251L) frequently identified in KS or nIHH patients could impair PROKR2 signaling in transfected HEK293 cells.24 This inconsistent finding brings new understanding of IHH, either in respect of digenic/oligogenic inheritance pattern or genetic basis of species differences. In the present study, three patients with IHH harboring PROKR2 variants presented similar phenotypic features in the reproductive and olfactory systems, including aplasia of the olfactory bulb and testicular or uterine dysplasia. However, they manifested variable non-reproductive non-olfactory abnormalities. Patients 04 and 08 carried the same PROKR2 variant (p.W178S), which is often reported in KS or nIHH patients and has a high frequency in East Asian populations (56/19,952).25 Even though the p.W178S variant has been proven to cause a loss-of-function of PROKR2 and impair its cell-surface targeting,26 regional predominance predisposes to a VUS classification. Patient 06 harbored non-compound double heterozygous PROKR2 variants (p.Y113H and p.R357W) that were inherited from her unaffected mother. As the same as the p.W178S variant, this variant has relatively high frequency in the East Asian population (28/19,954) and was also classified as VUS, albeit bioinformatic predictions and functional assessment mark it as pathogenic.27 Moreover, the p.R357W variant was considered as neutral in our study and previous report.28 Collectively, low pathogenicity of the three missense variants (p.W178S, p.Y113H, p.R357W) in PROKR2 are insufficient to explain the clinical phenotype of IHH patients, and there may be other undetected variations contributing to a digenic/oligogenic inheritance pattern, which needs to be further investigated.
CHD7 is widely expressed in fetal and adult tissues and its harmful variants are implicated in multisystem disorders including iris coloboma, coloboma, heart malformation/defects, atresia choanae, retardation of growth and/or development, genito-urinary defects and/or hypogonadism, and ear anomalies/deafness as the hallmarks of CHARGE syndrome.29 A large majority of CHD7 allelic variants occurs de novo, sporadic and nonsense or frameshift,30 and approximately 6% of KS or nIHH patients carried predominantly CHD7 missense variants, whereas 60 ~ 70% CHARGE syndrome patients had mainly nonsense or frameshift variants capable of causing complete loss-of-function.31 At present, some IHH-associated CHD7 missense variants including p.S834F, p.K907T and p.T917M, had been functionally validated via attenuating both ATPase and nucleosome remodeling activities by CHD7.32 Accordingly, in our study, both patient 05 with KS and patient 07 with nIHH carried missense CHD7 variant. The p.C1115Y variant is located in the highly conserved DEXHc region within the SNF2-like ATPase/helicase domain, and a larger side chain residue (hydroxyphenyl group) may alter the conformation of the α-helix in the ATP-binding region. This variant was previously recorded in the ClinVar database as a VUS classification (VCV000588616.4). Although the p.K2464R variant was not located in a catalytically active domain, it was absent in public databases and the site was conserved across different species, suggesting its potential as a cause of IHH onset. This novel variant was submitted to the Leiden Open Variation Database (#00430229). The p.C1115Y and p.K2464R variants in CHD7 might be involved in the KS phenotype in patient 05 and the nIHH phenotype in patient 07, respectively. Their pathogenicity undoubtedly needs to be further verified by stronger evidence, including functional assessment or co-segregation analysis within family members. In addition to the typical clinical features of nIHH, patient 07 carrying the p.K2464R variant also presented partial phenotypic overlap with CHARG syndrome, such as cleft lip/palate and hearing loss, but no ocular coloboma, heart malformations, or atresia of the choanae, which are the decisive diagnostic criteria for CHARGE syndrome.33 Therefore, detailed clinical evaluation and systematic screening of genetic variants of CHARGE syndrome-like features are indispensable for a more precise diagnosis of IHH (KS/nIHH) and CHARGE syndrome.
SOX10 heterozygous variants cause Waardenburg syndrome (WS) type 2, WS type 4, PCWH syndrome and KS.34 Another similar pathogenic variant in SOX10 gene (c.429–1G>A) associated with WS type 4A was recorded in the ClinVar database (VCV000590850.3). In practice, WS-like features, such as hearing loss and pigmentation abnormalities, frequently occur in IHH individuals with pathogenic SOX10 RSVs.35,36 Additionally, the role of SOX10 in GnRH neuron migration and organization of the olfactory bulb nerve layer has been experimentally demonstrated using SOX10−/− mice.37 SOX10 loss-of-function variants showed a good genotype-phenotype correlation in KS patients with hearing loss. However, SOX10 heterozygous variants displayed obvious phenotypic differences between humans and mice.6,38 Overall, the phenotypic spectrum resulting from SOX10 variants might be more variable than previously understood. In our study, two heterozygous splice variants, SOX10 (c.429–1G>C) and WDR11 (c.2931+6delC), were identified in a patient with KS and hearing loss. They were classified as LP and VUS and submitted to the Leiden Open Variation Database (#00430227; #00434898). Although WDR11 interacts with EMX1, a homeodomain transcription factor involved in the development of olfactory neurons, and heterozygous missense variants were found in patients with KS,39 the low pathogenicity of WDR11 splicing variant (c.2931+6delC) indicated that SOX10 (c.429–1G>C) was more likely to be considered the molecular etiology of the KS phenotype in patient 02.
Much less common, but equally strong in terms of causation, is FGF17 that has highly homologous to FGF8 and plays a critical role in GnRH neuron ontogeny though interaction with FGFR1c.40 Previous report showed that the prevalence of FGF17 variants in a 145 IHH probands was approximately 2.07%.41 Heterozygous FGF17 missense variant (p.V198L) found in patient 11 was inherited from unaffected mother, and has been recorded in ClinVar database as a VUS classification (VCV002287919.1). Therefore, the current evidence does not support the hypothesis that the p.V198L variant causes KS in patient 11.
Seven patients with available follow-up information showed improved sexual characteristics, including bilateral testicular volume, penile length, and pubic hair Tanner stage in male or breast Tanner stage, and uterine volume in female with variable degrees after testosterone undecanoate or estradiol valerate replacement treatment. They did not achieve reversal of the IHH. Six male showed low basal LH level (< 0.11 mIU/mL), low testosterone level (< 1.53 ng/mL) and small testicular volume (< 2.0 mL), which seems to consistent with previous reports that higher basal LH and stimulated LH levels as well as larger testicle size are favorite parameters for reversal.11,19 Currently, pulsatile GnRH pump treatment, testosterone/estradiol treatment and human chorionic gonadotropin (hCG)/human menopausal gonadotropin (hMG) combination treatment are three main treatment options for KS or nIHH, wherein testosterone/estradiol only improve sexual characteristics and GnRH pump is restricted to hospital grade and high cost, by contrast, hCG/hMG combination was recommended as the optimal therapy for IHH patients, either in respect of sexual development or reproductive demands.42 Practically, the choice of treatment strategies will depend on multi-factors including patient’ age, sex, individual desire, etc. Unfortunately, the lack of follow-up and fertility potential data in some patients has hindered systematic evaluation. In terms of treatment efficacy in this and previous studies, testosterone or estradiol effectively improved the development of sexual characteristics in patients with IHH, regardless of whether they harbored potential variants.
This study has a few limitations. First, the number of IHH cases was relatively small and did not represent the full genetic etiology of local IHH. Second, DNA samples from family members were often unavailable and functional studies of candidate variants were not performed. Finally, non-coding regions (introns, promoters, and enhancers), large-fragment deletions, and mitochondrial genomes could not be captured using the WES method, thus probably missing certain types of pathogenic variation.
Conclusion
Genetics variants in the SOX10 (c.429-1G>C) and CHD7 (c.3344G>A; c.7391A>G) possibly contributed to the IHH phenotype found in this study. NGS-based identification of genetic variants in patients with sporadic IHH has extended our understanding of the biological implications of IHH pathogenesis, continuously promoting the clinical application of NGS for the early diagnosis of IHH. Efficient therapeutic outcomes strongly indicate the need for a precise diagnosis and timely treatment.
Acknowledgments
We thank the patients with IHH and their parents, who donated blood samples for this study.
Ethics Approval and Consent to Participate
This study was approved by the Ethics Committee of the Affiliated Hospital of Jining Medical University (2021C123, Jining, Shandong, China) in accordance with the principles outlined in the Declaration of Helsinki. All participants and/or their legal guardians provided written informed consent for participation in clinical and genetic studies.
Funding
This work was supported by the Research Fund for Academician Lin He New Medicine (JYHL2019FZD01) and Shandong Traditional Chinese Medicine Science and Technology Development Plans Project (2019-0486). This study was partially supported by the PhD Research Foundation of the Affiliated Hospital of Jining Medical University (2018-BS-007).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Boehm U, Bouloux PM, Dattani MT, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015;11(9):547–564. doi:10.1038/nrendo.2015.112
2. Bonomi M, Vezzoli V, Krausz C, et al. Characteristics of a nationwide cohort of patients presenting with isolated hypogonadotropic hypogonadism (IHH). Eur J Endocrinol. 2018;178(1):23–32. doi:10.1530/EJE-17-0065
3. Good DJ. New gene targets in the study of hypogonadotropic hypogonadism. Mol Cell Endocrinol. 2021;520:111077. doi:10.1016/j.mce.2020.111077
4. Mitchell AL, Dwyer A, Pitteloud N, Quinton R. Genetic basis and variable phenotypic expression of Kallmann syndrome: towards a unifying theory. Trends Endocrinol Metab. 2011;22(7):249–258. doi:10.1016/j.tem.2011.03.002
5. Bassi I, Luzzani F, Marelli F, et al. Knocking-down of the Prokineticin receptor 2 affects reveals its complex role in the regulation of the hypothalamus-pituitary-gonadal axis in the zebrafish model. Sci Rep. 2020;10(1):7632. doi:10.1038/s41598-020-64077-2
6. Maione L, Dwyer AA, Francou B, et al. Genetics in endocrinology: genetic counseling for congenital hypogonadotropic hypogonadism and Kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing. Eur J Endocrinol. 2018;178(3):R55–R80. doi:10.1530/EJE-17-0749
7. Amato LGL, Montenegro LR, Lerario AM, et al. New genetic findings in a large cohort of congenital hypogonadotropic hypogonadism. Eur J Endocrinol. 2019;181(2):103–119. doi:10.1530/EJE-18-0764
8. Cangiano B, Swee DS, Quinton R, Bonomi M. Genetics of congenital hypogonadotropic hypogonadism: peculiarities and phenotype of an oligogenic disease. Hum Genet. 2021;140(1):77–111. doi:10.1007/s00439-020-02147-1
9. Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. 2009;5(10):569–576. doi:10.1038/nrendo.2009.177
10. Butz H, Nyiro G, Kurucz PA, Liko I, Patocs A. Molecular genetic diagnostics of hypogonadotropic hypogonadism: from panel design towards result interpretation in clinical practice. Hum Genet. 2021;140(1):113–134. doi:10.1007/s00439-020-02148-0
11. Mao JF, Xu HL, Duan J, et al. Reversal of idiopathic hypogonadotropic hypogonadism: a cohort study in Chinese patients. Asian J Androl. 2015;17(3):497–502. doi:10.4103/1008-682X.145072
12. Turan I, Hutchins BI, Hacihamdioglu B, et al. CCDC141 mutations in idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2017;102(6):1816–1825. doi:10.1210/jc.2016-3391
13. Pitteloud N, Hayes FJ, Dwyer A, Boepple PA, Lee H, Crowley WF. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002;87(9):4128–4136. doi:10.1210/jc.2002-020518
14. Xu D, Lu L, Xi L, et al. Efficacy and safety of percutaneous administration of dihydrotestosterone in children of different genetic backgrounds with micropenis. J Pediatr Endocrinol Metab. 2017;30(12):1285–1291. doi:10.1515/jpem-2016-0400
15. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
16. Zhang LL, Bao L, Li YQ, et al. Age at menopause, body mass index, and risk of type 2 diabetes mellitus in postmenopausal Chinese women: the Henan Rural Cohort study. Nutr Metab Cardiovas. 2020;30(8):1347–1354. doi:10.1016/j.numecd.2020.04.003
17. Israel E, Attie KM, Bengtsson BA, et al. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. J Clin Endocr Metab. 2000;85(11):3990–3993.
18. Isojima T, Shimatsu A, Yokoya S, et al. Standardized centile curves and reference intervals of serum insulin-like growth factor-I (IGF-I) levels in a normal Japanese population using the LMS method. Endocr J. 2012;59(9):771–780. doi:10.1507/endocrj.EJ12-0110
19. Raivio T, Falardeau J, Dwyer A, et al. Reversal of idiopathic hypogonadotropic hypogonadism. New Engl J Med. 2007;357(9):863–873. doi:10.1056/NEJMoa066494
20. Stamou MI, Georgopoulos NA. Kallmann syndrome: phenotype and genotype of hypogonadotropic hypogonadism. Metabolism. 2018;86:124–134. doi:10.1016/j.metabol.2017.10.012
21. Wen Y, Zhang ZZ, Li Z, et al. The PROK2/PROKR2 signaling pathway is required for the migration of most olfactory bulb interneurons. J Comp Neurol. 2019;527(18):2931–2947. doi:10.1002/cne.24719
22. Dode C, Rondard P. PROK2/PROKR2 signaling and Kallmann syndrome. Front Endocrinol. 2013;4:19. doi:10.3389/fendo.2013.00019
23. Matsumoto S, Yamazaki C, Masumoto KH, et al. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. P Natl Acad Sci USA. 2006;103(11):4140–4145. doi:10.1073/pnas.0508881103
24. Sbai O, Monnier C, Dode C, Pin JP, Hardelin JP, Rondard P. Biased signaling through G-protein-coupled PROKR2 receptors harboring missense mutations. FASEB J. 2014;28(8):3734–3744. doi:10.1096/fj.13-243402
25. Wang Y, Qin M, Fan LJ, Gong CX. Correlation analysis of genotypes and phenotypes in Chinese male pediatric patients with congenital hypogonadotropic hypogonadism. Front Endocrinol. 2022;13:846801. doi:10.3389/fendo.2022.846801
26. Monnier C, Dode C, Fabre L, et al. PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling activity. Hum Mol Genet. 2009;18(1):75–81. doi:10.1093/hmg/ddn318
27. Martinez-Mayer J, Perez-Millan MI. Phenotypic and genotypic landscape of PROKR2 in neuroendocrine disorders. Front Endocrinol. 2023;14:1132787. doi:10.3389/fendo.2023.1132787
28. Wang XY, Chen DN, Zhao YG, et al. A functional spectrum of PROKR2 mutations identified in isolated hypogonadotropic hypogonadism. Hum Mol Genet. 2023;32(10):1722–1729. doi:10.1093/hmg/ddad014
29. Cho HJ, Shan YF, Whittington NC, Wray S. Nasal placode development, GNRH neuronal migration and Kallmann syndrome. Front Cell Dev Biol. 2019;7:121.
30. Brajadenta GS, Bilan F, Gilbert-Dussardier B, Kitzis A, Thoreau V. A functional assay to study the pathogenicity of CHD7 protein variants encountered in CHARGE syndrome patients. Eur J Hum Genet. 2019;27(11):1683–1691. doi:10.1038/s41431-019-0465-7
31. Li JD, Wu JY, Zhao YG, et al. Phenotypic spectrum of idiopathic hypogonadotropic hypogonadism patients with CHD7 variants from a large Chinese cohort. J Clin Endocr Metab. 2020;105(5):1515–1526.
32. Balasubramanian R, Crowley WF. Reproductive endocrine phenotypes relating to CHD7 mutations in humans. Am J Med Genet C. 2017;175(4):507–515. doi:10.1002/ajmg.c.31585
33. Zentner GE, Layman WS, Martin DM, Scacheri PC. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A. 2010;152A(3):674–686. doi:10.1002/ajmg.a.33323
34. Pingault V, Zerad L, Bertani-Torres W, Bondurand N. SOX10: 20 years of phenotypic plurality and current understanding of its developmental function. J Med Genet. 2022;59(2):105–114. doi:10.1136/jmedgenet-2021-108105
35. Dai WT, Wu JY, Zhao YG, et al. Functional analysis of SOX10 mutations identified in Chinese patients with Kallmann syndrome. Gene. 2019;702:99–106. doi:10.1016/j.gene.2019.03.039
36. Rojas RA, Kutateladze AA, Plummer L, et al. Phenotypic continuum between Waardenburg syndrome and idiopathic hypogonadotropic hypogonadism in humans with SOX10 variants. Genet Med. 2021;23(4):629–636. doi:10.1038/s41436-020-01051-3
37. Barraud P, St John JA, Stolt CC, Wegner M, Baker CVH. Olfactory ensheathing glia are required for embryonic olfactory axon targeting and the migration of gonadotropin-releasing hormone neurons. Biol Open. 2013;2(7):750–759. doi:10.1242/bio.20135249
38. Pingault V, Bodereau V, Baral V, et al. Loss-of-Function mutations in SOX10 cause Kallmann syndrome with deafness. Am J Hum Genet. 2013;92(5):707–724. doi:10.1016/j.ajhg.2013.03.024
39. Kim HG, Ahn JW, Kurth I, et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2010;87(4):465–479. doi:10.1016/j.ajhg.2010.08.018
40. Valdes-Socin H, Almanza MR, Fernandez-Ladreda MT, Debray FG, Bours V, Beckers A. Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes. Front Endocrinol. 2014;5:109. doi:10.3389/fendo.2014.00109
41. Men MC, Wu JY, Zhao YG, et al. Genotypic and phenotypic spectra of FGFR1, FGF8, and FGF17 mutations in a Chinese cohort with idiopathic hypogonadotropic hypogonadism. Fertil Steril. 2020;113(1):158–166. doi:10.1016/j.fertnstert.2019.08.069
42. Yang L, Zhang SX, Dong Q, Xiong ZB, Li X. Application of hormonal treatment in hypogonadotropic hypogonadism: more than ten years experience. Int Urol Nephrol. 2012;44(2):393–399. doi:10.1007/s11255-011-0065-0
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.