Back to Journals » Cancer Management and Research » Volume 11
Clinical features and prognostic factors of primary bone marrow lymphoma
Authors Wang G, Chang Y, Wu X, Li X, Li L, Zhang L, Fu X, Sun Z, Zhang X, Zhang M
Received 14 September 2018
Accepted for publication 23 January 2019
Published 29 March 2019 Volume 2019:11 Pages 2553—2563
DOI https://doi.org/10.2147/CMAR.S187522
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Beicheng Sun
Gangjian Wang, Yu Chang, Xiaolong Wu, Xin Li, Ling Li, Lei Zhang, Xiaorui Fu, Zhenchang Sun, Xudong Zhang, Mingzh Zhang
Department of Oncology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China
Background: Primary bone marrow lymphoma (PBML) is a very uncommon neoplasm originally arising in the bone marrow system, and the most common pathological type is diffuse large B-cell lymphoma.
Patients and methods: To describe the clinical characteristics of PBML and evaluate the risk factors related to prognosis, we recruited and studied 66 patients from our center and the current published literature. Various symptoms are present at the onset of PBML, the most important of which is cytopenia, followed by fever. Forty-seven of these patients were included in our analysis.
Results: Univariate analysis suggested that B symptoms (P=0.024), a low serum platelet level (<75×109/L; P=0.032), an elevated serum LDH level (P=0.039), and not achieving a complete response (CR) following initial therapy (P=0.007) are associated with worse outcomes. Multivariate analysis showed that only a low serum platelet level (<75×109/L), B symptoms, and not achieving a CR following initial therapy are independent factors for prognosis. In addition, intensive regimens appear to be beneficial for prognosis.
Conclusion: PBML is a lymphoma with special clinical features, and its recognition is important for establishing a definitive prognosis model and searching for appropriate therapy.
Keywords: diffuse large B-cell lymphoma, primary bone marrow lymphoma, bone marrow, B symptoms, cytopenia
Background
Primary bone marrow lymphoma (PBML) is an extremely rare form of lymphoma with rapid disease progression and a poor prognosis.1,2 A previous case series study conducted by Martinez et al1 focused on the pathological features of PBML; however, only a few clinical features were found to be associated with the disease. Five different pathological types of lymphoma can originate in the bone marrow, including Hodgkin’s lymphoma (HL), diffuse large B-cell lymphoma (DLBCL), peripheral T-cell lymphoma, not otherwise specified lymphoma (PTCL, NOS), ALK-negative anaplastic large cell lymphoma (ALK-negative ALCL), and follicular lymphoma (FL).1 Among these types, DLBCL is the most common pathological subtype. However, due to the low incidence of the disease, large-scale and systematic case series studies are lacking; therefore, information regarding the clinical features of PBML is lacking. Additionally, the current treatments for PBML are not uniform and have not been standardized, and most treatments focus on only the pathological type and lack specificity and scientific evidence. Furthermore, no study has reported the risk factors affecting the outcomes of PBML. Thus, we reviewed some cases that had been diagnosed at our single center and analyzed previous studies. The present study aimed to investigate the specific clinical features and risk stratification effects on the outcomes of these patients and to discuss treatment strategies for PBML.
Patients and methods
Patient selection and data collection
The following criteria were used to define PBML: 1) pathologically confirmed bone marrow involvement, regardless of peripheral blood involvement; 2) absence of lymph node, spleen, liver, or other extra marrow involvement upon physical examination or imaging studies (including thoracic, abdominal, and pelvic enhanced computerized tomography [CT], systemic superficial lymph node B-scan ultrasonography, and systemic positron emission tomography/CT [PET/CT]; among these imaging studies, PET/CT is considered relatively authoritative); 3) no evidence of localized bone tumors; 4) bone marrow biopsy with no signs of bony trabecular destruction or PET/CT revealing diffuse enhanced bone marrow metabolism without a localized bone lesion; and 5) exclusion of leukemia/lymphoma cases, including chronic lymphocytic leukemia/small lymphocytic lymphoma, prolymphocytic leukemia, lymphoplasmacytic lymphoma, hairy cell leukemia, Burkitt lymphoma (Burkitt leukemia variant), and acute lymphoblastic leukemia.1
In addition to the above mentioned diagnostic criteria, we added the following exclusion criteria: 1) cases with a second tumor or a disease that could seriously influence survival and 2) B-cell lymphomas that could not be further diagnosed.
All patients’ clinical data, including sex, age, initial symptoms, peripheral blood indicators at first admission, LDH level, β-2 microglobulin level, international prognostic index, treatment modality, treatment response, and radiological findings, were collected. The bone marrow examination included a bone marrow smear cytologic examination, bone marrow biopsy, and bone marrow aspiration.
This study was approved by the ethics committee of the First Affiliated Hospital of Zhengzhou University and was conducted in accordance with the Declaration of Helsinki. Written informed consent for the collection of medical information was obtained from all patients. All procedures performed in the study were in accordance with the ethical standards of the institutional research committee.
Statistical analysis
Complete response (CR), partial response (PR), stable disease (SD), and progressive disease were used to define the classification of the treatment response according to the criteria for malignant lymphoma. The overall survival (OS) was defined from the date of pathological diagnosis to death or to the last date of follow-up. We divided the degree of cytopenia into the following four levels: Grade 0: leukocytes ≥4.0×109/L, hemoglobin ≥110 g/L, platelets ≥100×109/L; Grade 1: leukocytes (3.0–3.9)×109/L, hemoglobin (95–100) g/L, platelets (75–99)×109/L; Grade 2: leukocytes (2.0–2.9)×109/L, hemoglobin (80–94) g/L, platelets (50–74)×109/L; Grade 3: leukocytes (1.0–1.9)×109/L, hemoglobin (65–79) g/L, platelets (25–49)×109/L; and Grade 4: leukocytes (0–1.0)×109/L, hemoglobin <65 g/L, platelets <25×109/L. OS and survival curves were analyzed by the Kaplan–Meier method. The survival of patients with different prognostic variables was analyzed by the log-rank test, and multiple independent prognostic factors were analyzed using a Cox proportional hazards regression analysis. The correlations between two variables were analyzed by Pearson’s chi-square analysis. P-values <0.05 were considered statistically significant, and the statistical analyses were performed using SPSS 21.0.
Results
Patient characteristics in our center
Seven patients with PBML who were treated at the Lymphoma Diagnosis and Treatment Center of Henan Province were enrolled between July 2011 and June 2017 into this study; their clinical characteristics are listed in Table 1. The median follow-up time was 10.4 months (range: 1.0–25 months) and the median age at diagnosis was 56 years (range: 38–72 years). There were five deaths, accounting for 71.4%. Of these patients, three patients died from treatment failure and other two patients finally succumbed to recurrence of the disease. Notably, PET/CT was used in four cases, and all cases revealed disseminated bone marrow with diffuse hypermetabolism, and the median standard uptake value level was 5.9 (range: 4.8–7.5).
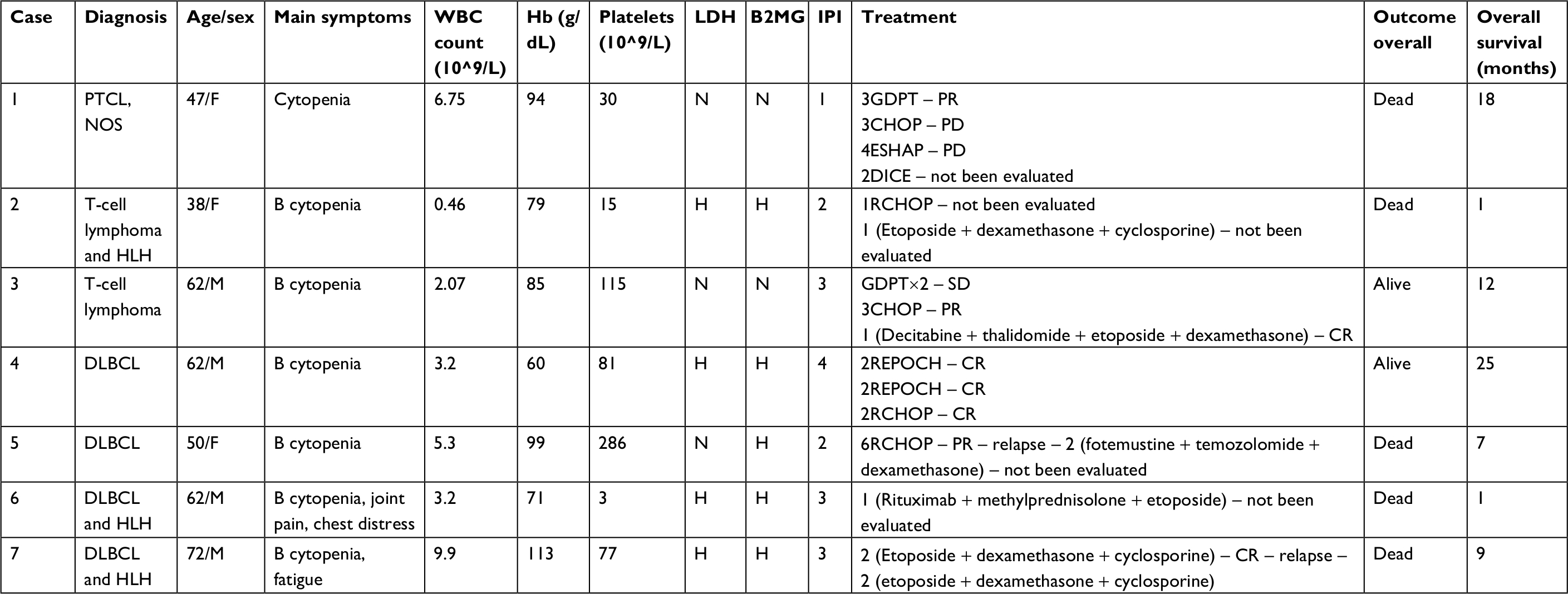 | Table 1 Clinical features of seven patients with PBML in our center Abbreviations: B, B symptoms; B2MG, β2-microglobulin; CHOP, cyclophosphamide, epirubicin, vincristine, prednisone; CR, complete response; DICE, dexamethasone, ifosfamide, mesna, etoposide; DLBCL, diffuse large B-cell lymphoma; ESHAP, etoposide, methylprednisolone, high-dose cytarabine, and cisplatin; GDPT, gemcitabine, cisplatin, prednisone, and thalidomide; H, high; Hb, hemoglobin; HLH, hemophagocytic lymphohistiocytosis; IPI, international prognostic index; N, normal; PBML, primary bone marrow lymphoma; PD, progressive disease; PR, partial response; PTCL, NOS, peripheral T-cell lymphoma, not otherwise specified; REPOCH, rituximab, etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin; SD, stable disease; WBC, white blood cells. |
Literature review and statistical analysis
Data of 66 cases of PBML were collected and analyzed as follows: 7 cases were among the cases at our center and 59 cases were identified through searching PubMed, China National Knowledge Infrastructure, and Wanfang Data from 1997 to 2018, and the specific information of these patients is presented in Table 2. Nineteen of them were excluded due to lack of specific follow-up time; finally, 47 cases were enrolled into this retrospective analysis. As shown in Table 2, the female/male ratio of the patients was 4:7 (24:42), and the age ranged between 29 and 81 years (median: 60.0 years; average: 57.6 years). PBML occurred in various pathological types of lymphoma. The most common PBMLs were DLBCLs with 40 cases (60.6%). The other types included HL (14/66, 21.2%), PTCL, NOS (3/66, 4.5%), ALK-negative ALCL (2/66, 3.0%), and FL (5/66, 7.6%). Six patients with PBML had hemophagocytic lymphohistiocytosis (HLH) at the same time, and the mean survival time was only 4 months. No examples of successful treatment were found in our study. Additionally, four patients had complicated cold agglutinin disease, and these patients usually present with cytopenia accompanied by elevated serum cold agglutinin levels. Finally, three patients were still alive and only one patient died from relapse 19 months after the initial chemotherapy. Of all the 40 cases of DLBCL, we obtained the immunohistochemical results of 39 patients, and these results are shown in Table 3. Thirty-three patients died during follow-up, including 20 patients who died from disease progression and 10 patients who died from chemotherapy-related complications. Additionally, 29/57 patients achieved CR, 9/57 patients achieved PR after initial therapy, and the overall response rate was 67.7% (50.9% CR +15.8% PR). PBML of different pathological types showed distinct prognoses. Most patients with HL were treated with doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) or ABVD-like regimens. The median survival period was 4 months, and HIV-negative HL had a poorer OS than HIV-positive HL (P=0.097). However, statistically significant indicators related to prognosis could not be obtained due to insufficient data. The treatment strategies and survival time of the T-cell lymphoma patients were diverse. The median survival period was 9.6 months. Among four out of five patients with FL, only one patient died. Most patients with DLBCL were treated with cyclophosphamide, epirubicin, vincristine, prednisone (CHOP) or a CHOP-like regimen. The median survival period was 19 months. The clinical features of 47 patients included in the retrospective analysis are summarized in Table 4. The median OS was 19 months. In the univariable analysis, the Kaplan–Meier method and log-rank test were used to analyze the influence of the following factors on survival: sex, age, degree of cytopenia, serum hemoglobin level, serum leukocyte level, serum platelet level, B symptoms, serum LDH level, serum β2-microglobulin level, and response to initial treatment; the degree of cytopenia, serum platelet level, B symptoms, serum LDH level, and response to initial treatment were found to be significantly correlated with OS (P<0.05; Table 5), and the survival curves are shown in Figure 1. Additionally, we attempted to identify the difference in prognosis between HL and non-Hodgkin’s lymphoma and between T-cell and B-cell lymphoma; both results were not statistically significant. Due to the limited number of cases, only those factors with P-values <0.05 were studied, including the serum LDH level, B symptoms, serum platelet level, degree of cytopenia, response to initial therapy, and degree of cytopenia* serum platelet level (which is defined as the interaction of two variables), and the results showed that there was an interaction between the two variables. Considering the P-value of the univariable analysis and clinical convenience, we excluded the variable of degree of cytopenia from the Cox regression model. Finally, the LDH level, B symptoms, serum platelet level, and response to initial therapy were included in the Cox regression analysis, and a low serum platelet level, B symptom, and not achieving CR following the initial therapy showed an independent association with an unfavorable OS (Table 5). In addition, because of the uniformity of the treatment options for DLBCL, we divided the patients into two groups: one group included 23 cases with RCHOP, RCHOP-like/CHOP, or CHOP-like regimens and the other group included 5 cases with intensive regimens, including HVPERCAVD, EPOCH, ALL, HD-CHOP, and VACOPB. The patients who had received intensive regimens showed a better OS (P=0.01; Figure 2).
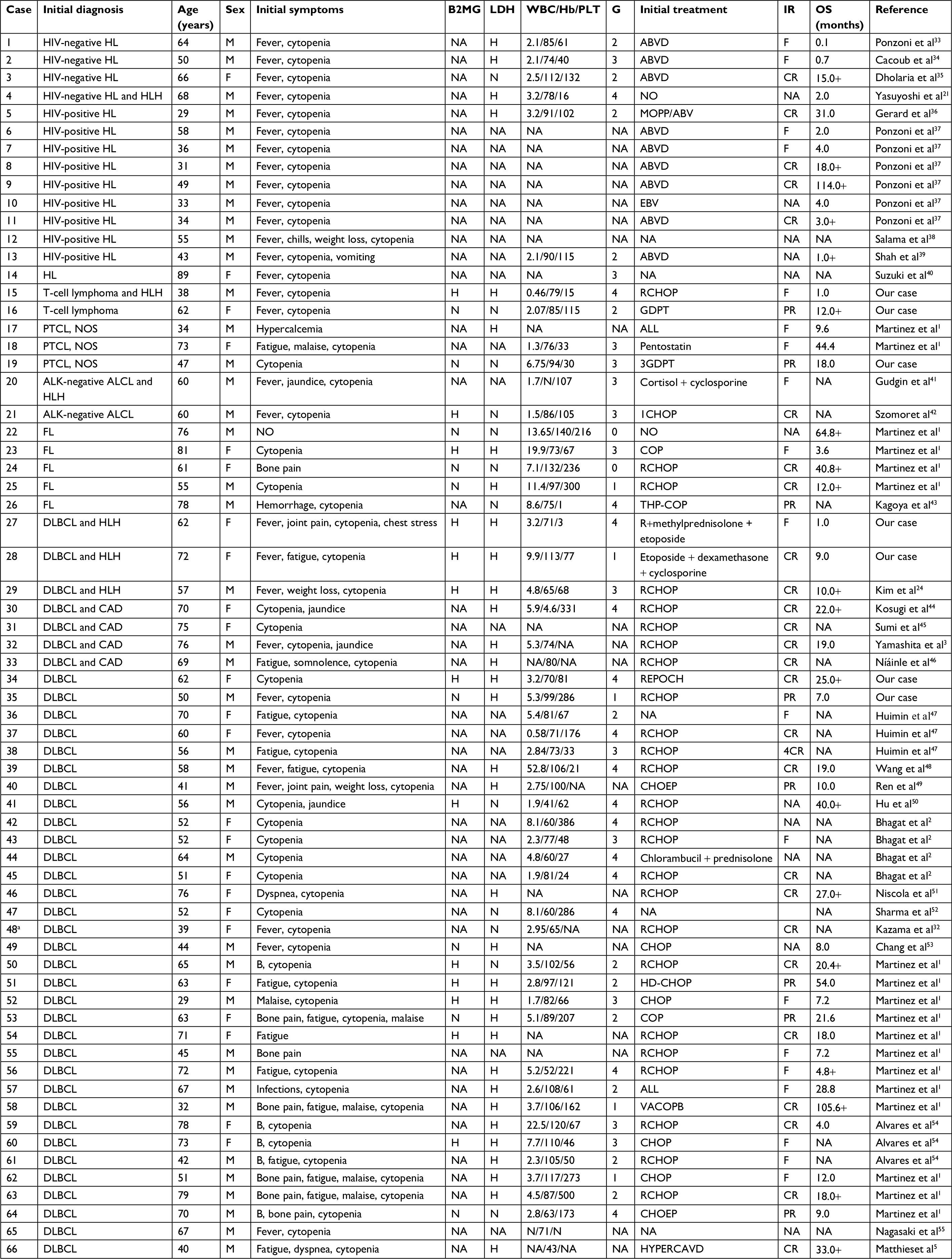 | Table 2 Clinical characteristics of 66 patients with PBML from literature (cases 1–66) Notes: aRecieved autologous stem cell transplantation. + Alive. Abbreviations: ALL, prednisone, vincristine, daunorubicin, L-asparaginase, cyclophosphamide, cytarabine, bleomycin, 6-mercaptopurine; ALK-negative ALCL, ALK-negative anaplastic large cell lymphoma; B, B symptoms; B2MG, β2-microglobulin; CAD, cold agglutinin disease; CHOEP, cyclophosphamide, doxorubicin, vincristine, prednisolone, etoposide; CHOP, cyclophosphamide, epirubicin, vincristine, prednisone; COP, cyclophosphamide, vincristine, prednisone; CR, complete response; DLBCL, diffuse large B-cell lymphoma; EBV, epiadriamicin, bleomicin, vinblastin; F, failure; FL, follicular lymphoma; G, hematopoietic function grade; GDPT, gemcitabine, cisplatin, prednisone, and thalidomide; H, high; Hb, hemoglobin; HD, high dose; HL, Hodgkin’s lymphoma; HLH, hemophagocytic lymphohistiocytosis; HYPERCAVD, cyclophosphamide, vincristine, dexamethasone, adriamycin/methotrexate, cytarabine; IR, initial response; MOPP/ABV, metchlorethamine, vincristine, cyclophosphamide, prednisone/adriamycin, bleomycin, vinblastin; N, normal; NA, not available; OS, overall survival; PLT, platelets; PR, partial response; PTCL, NOS, peripheral T-cell lymphoma, not otherwise specified; RCHOP, rituximab,cyclophosphamide, epirubicin, vincristine, prednisone; REPOCH, rituximab, etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin; THP-COP, pirarubicin, cyclophosphamide, vincristine, prednisolone; VACOPB, etoposide, doxorubicin, cyclophosphamide, vincristine, prednisone, bleomycin; WBC, white blood cells. |
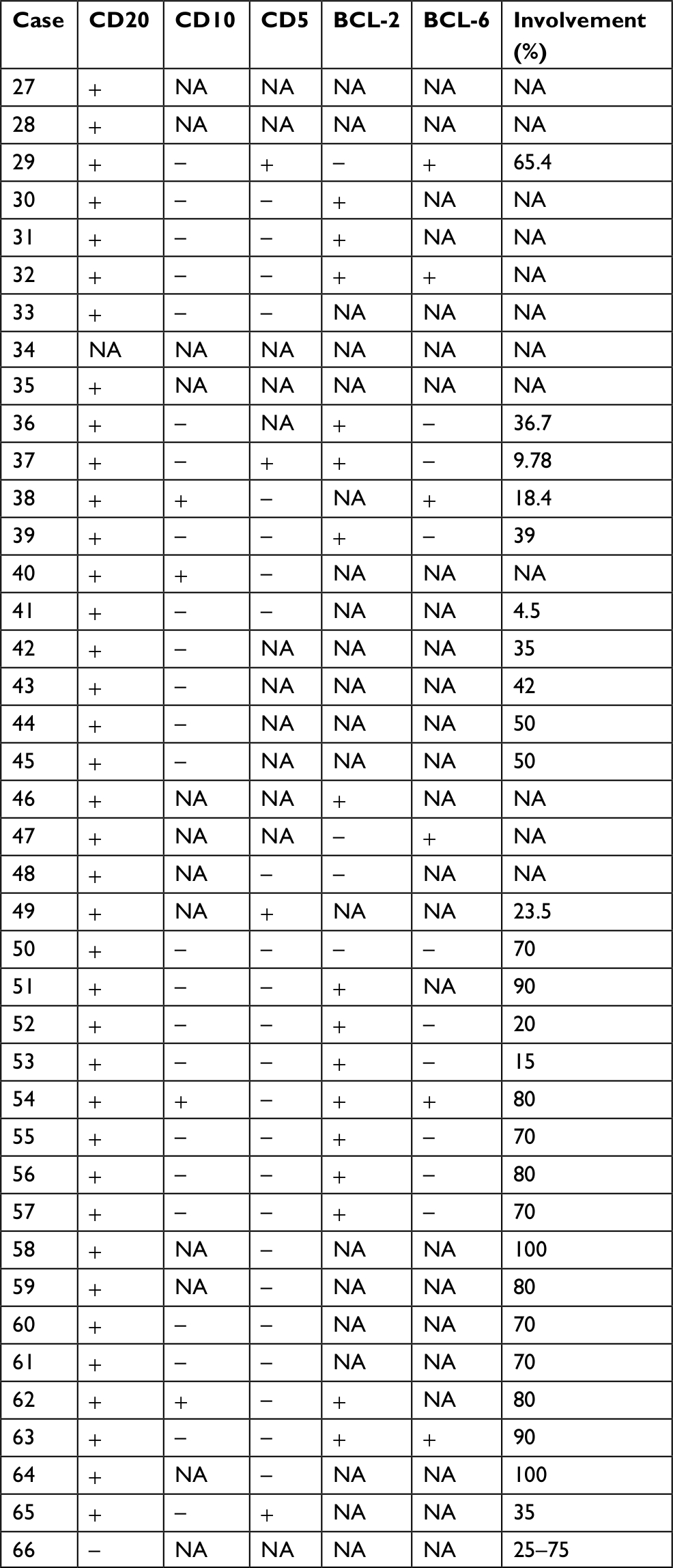 | Table 3 Pathological, phenotypic, and molecular features of DLBCL cases (n=34) Abbreviations: DLBCL, diffuse large B-cell lymphoma; NA, not applicable. |
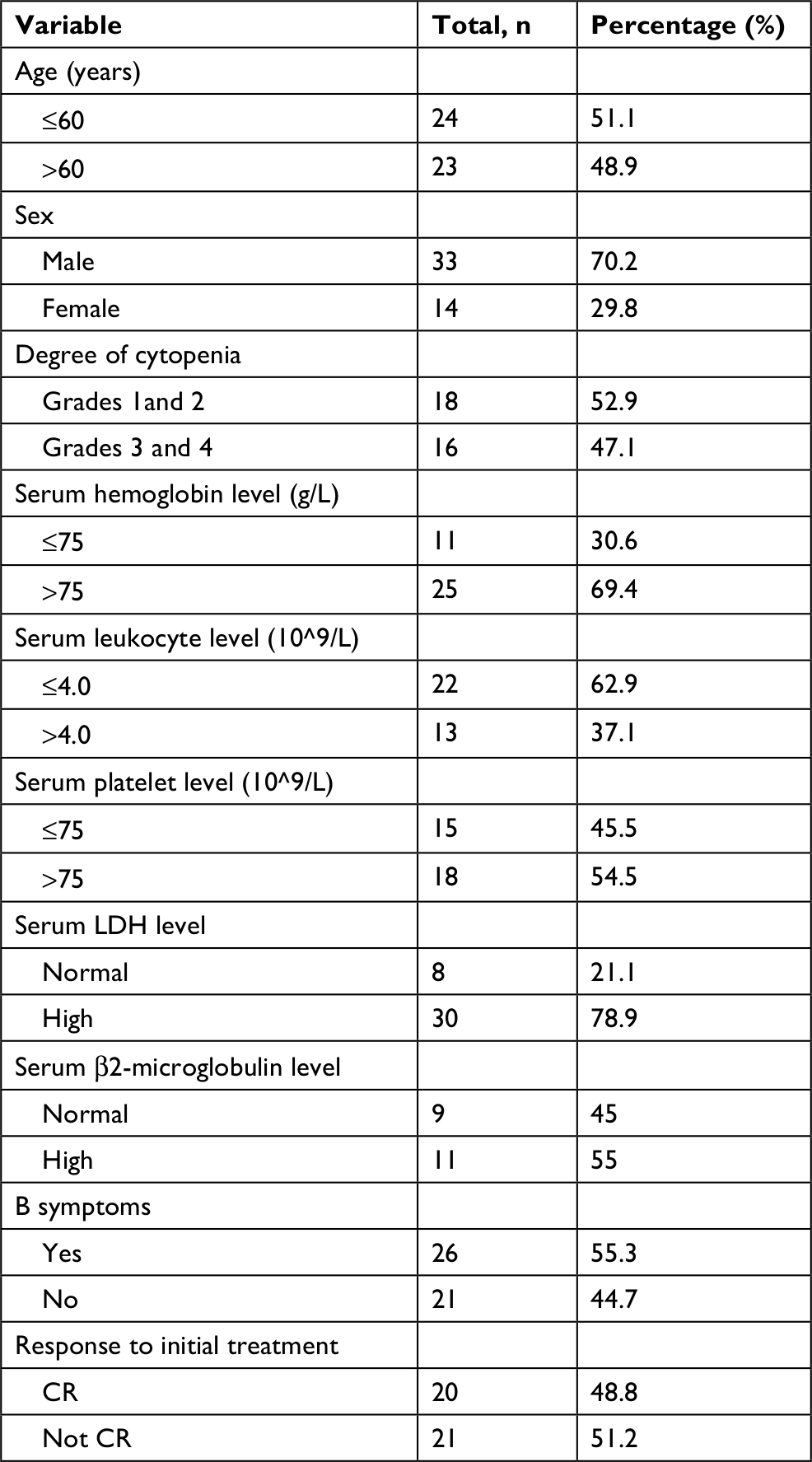 | Table 4 Clinical characteristics of 47 patients with PBML Abbreviations: CR, complete remission; PBML, primary bone marrow lymphoma. |
 | Table 5 Univariate and multivariate analyses for overall survival Abbreviation: CR, complete remission. |
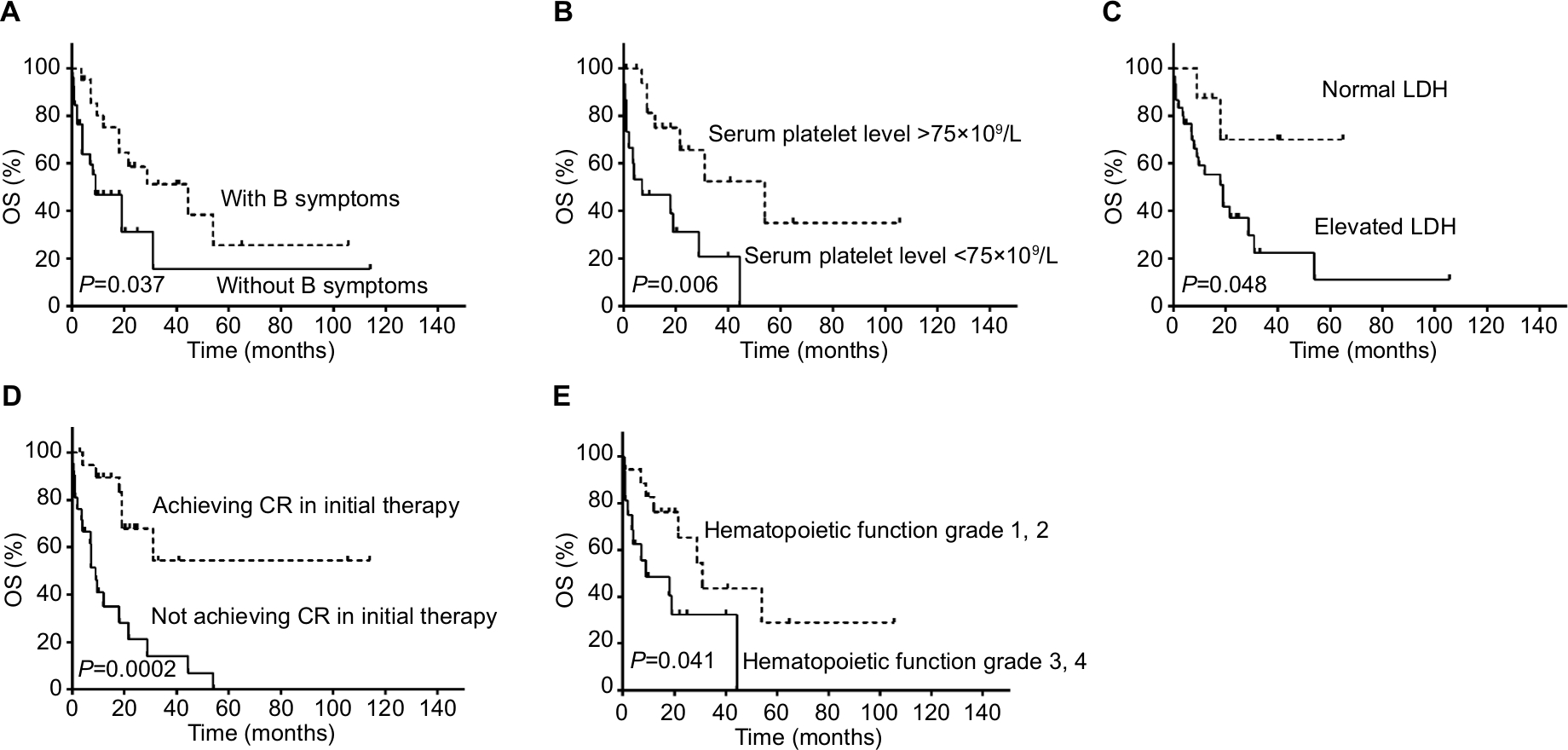 | Figure 1 Univariable analyses of prognostic factors for OS for our 66 patients with PBML. Notes: (A) Kaplan meier of OS in two groups with and without B symptoms. (B) Kaplan meier of OS in two groups with serum platelet level >75x109/L and with serum platelet level <75x109/L; (C) Kaplan meier of OS in two groups with normal LDH and with elevated LDH; (D) Kaplan meier of OS in two groups achieving CR and not achieving CR in the initial therapy; (E) Kaplan meier of OS in two groups with hematopoietic function grade 1,2 and hematopoietic function grade 3,4. Abbreviations: CR, complete response; OS, overall survival; PBML, primary bone marrow lymphoma. |
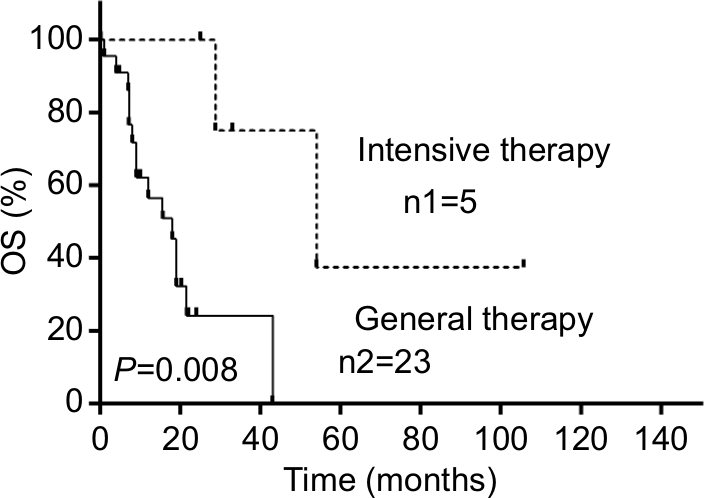 | Figure 2 Curve of cumulative survival of patients with PBML diagnosed as DLBCL with general therapy or with intensive therapy. Notes: n1 represents the initial number of recipients of the intensive therapy; n2 represents the initial number of recipients of the general therapy. Abbreviations: DLBCL, diffuse large B-cell lymphoma; OS, overall survival; PBML, primary bone marrow lymphoma. |
Discussion
The present study is the largest study concerning PBML to date. To the best of our knowledge, regarding the clinical features and prognosis of primary marrow bone lymphoma, we are the first to perform a systematic and comprehensive review and establish a prognostic model of PBML. PBML has been sporadically reported in the literature since the 1970s. Compared with other lymphomas of the same pathological type, PBML is usually difficult to diagnose, progresses rapidly, and is easy to combine with multiple complications, such as severe infection and HLH, only part of people in the conventional treatment respond well, and OS is relatively short.1–3 In some cases, a clear diagnosis lacks complete evidence, and often, primary bone lymphoma (PBL) cases, which occur infrequently, are misdiagnosed as PBMLs. Most clinical manifestations of PBLs include bone pain and fractures; most of the common lesion features are destructive through localized radiological examination; and the disease often presents with a local single lesion.1,4 In contrast to PBL, the important symptoms of PBML include cytopenia and B symptoms, and the occurrence of bone pain is relatively rare based on our data. Additionally, in our study, the lesion was usually confined to the marrow cavity during the early stage, and the PET/CT results of 13 patients, not only from our center but also from the literature review, all presented diffuse abnormal fluorodeoxyglucose uptake in the bone marrow that appeared to be more intense in the axial bones; therefore, we believe that PET/CT has a certain importance in the diagnosis of PBML, and so, we added this item to the criteria proposed by Martinez et al1 in 2012. In addition, MRI usually reveals a diffusely abnormal marrow signal (low on T1-weighted images, high on T2-weighted images) in the bone marrow cavity.5 Thus, imaging data can provide important information for the identification of PBL and PBML.
In our studies, three clinical variables were proved to be independent prognostic factors, including B symptoms, serum platelet levels, and response to initial treatment. In several studies about lymphoma, B symptoms have been found to be an important prognostic variable for OS.6–8 The degree of cytopenia is clearly closely related to disease severity. Therefore, cytopenia likely affects prognosis. However, our results indicate that platelet count was also an independent factor affecting prognosis. In fact, based on our data, thrombocytopenia does not affect treatment or increase the number of bleeding events. Some early studies9–11 have revealed that thrombocytopenia affects the survival results in lymphomas with bone marrow involvement, including PBML. In addition, a similar prognostic result was reported in early-stage B-cell gastric lymphoma12 and DLBCL.13,14 We believe that autoimmune thrombocytopenia-associated PBML may be a potential cause because autoimmune-induced thrombocytopenia predicted relapse in one-third of lymphoma patients in a study.15 Age, which is a classic risk factor, was no longer associated with the OS in our study. In some conventional studies, older age-associated poor prognosis may be caused by the following three potential factors: multiple comorbidity, lower tolerance to therapy, and multiple organ dysfunction, including bone marrow function.16,17 However, our study indicated that the incidence of complications was nearly equal between the older age group and the younger age group. Additionally, PBML is a disease that severely influences and impairs various organ functions, especially bone marrow function, thereby reducing chemotherapy tolerance. Thus, age seems to be less important than disease malignancy.
Compared with the data from literature review, the incidence of T-cell lymphoma in our center is relatively higher, and the data showed that OS seems to be shorter, which may be associated with a higher rate of B symptoms and HLH. Lymphoma-associated HLH is a relatively vicious disease associated with the uncontrolled activation of the normal immune system,18,19 and another important reason is that only two people achieved CR after initial treatment.
Patients with leukopenia or thrombocytopenia were all administered granulocyte-colony stimulating factor (G-CSF) or recombinant human thrombopoietin, but without success. Hammerstrøm reported that three patients with neutropenia secondary to lymphoid bone marrow involvement responded well to G-CSF before chemotherapy.20 However, we found no other literature confirming this problem. Based on our single-center experience, G-CSF or recombinant human thrombopoietin treatment for cytopenia involving or originating in the bone marrow is usually ineffective.
Most patients with HL were treated with ABVD or an ABVD-like regimen. Unfortunately, more than half of these patients died in the short term, and disease progression was the dominant cause of death. Morita et al21 reported that HIV-negative cases tended to progress rapidly and resulted in worse outcomes, which is consistent with our analysis. Regarding T-cell lymphoma, we first used the gemcitabine, cisplatin, prednisone, and thalidomide regimen in our patients because this regimen was proven to be more efficient than CHOP for the treatment of PTCL in a prospective, randomized, controlled, and open-label clinical trial;22 however, the results were not satisfactory and the results of the CHOP regimen were also not satisfactory. Some reports had mentioned that lymphomas, especially T-cell lymphomas, were the main cause of secondary HLH and were associated with a poorer prognosis.23,24 This condition also appeared in PBML. Among the patients with primary bone marrow T-cell lymphomas, two out of seven (28.6%) patients had HLH complications and died within a month. The clinical use of decitabine in T-cell PBML is a ground-breaking initiative, and the result that patients had increased long-term survival is also promising. Among all PBML cases, the FL cases seemed to show the mildest symptoms and best prognosis, likely due to the slow development of the disease itself. Martinez et al reported a patient who had leukocytosis as the only abnormal indicator at the time of onset and survived for >5 years until the end of follow-up. In the past two decades, most reported PBMLs have been DLBCLs. Regarding treatment, CHOP or the CHOP-like regimen was usually used as the first-line regimen, and only a small subset of patients (two cases) died from chemotherapy-related side effects during the initial therapy; our results showed that intensive regimens seem to be more effective. We suggest that this effectiveness might be because continuous low concentrations of drugs increased the effectiveness of killing aggressive cancer cells and decreased MDR-1–mediated resistance.25–28 Additionally, in some studies investigating aggressive non-Hodgkin’s lymphoma with high-risk factors, the patients who received intensive chemotherapies actually showed a better OS than those treated with CHOP.29,30 In addition, some studies have published the following results in non-Hodgkin’s lymphoma with bone marrow infiltration: the response rate, OS, and progression-free survival of patients treated with intensive regimens were significantly higher than those of patients treated with the standard CHOP regimen.11,31 In conclusion, we believe that intensive therapy may indeed be conducive to survival in PBML. However, another problem that cannot be ignored is that intensive treatment may lead to greater risks in PBML than other high-risk non-Hodgkin lymphomas. Thus, safety and chemotherapy tolerance require extensive data, and the CHOP or CHOP-like regimen remains a relatively safe and moderate regimen. Kazama et al32 reported that a patient who underwent autologous stem cell transplantation (Auto-SCT) after eight cycles of RCHOP survived for >7 years, representing another clinically significant therapeutic initiative illustrating the possibility that Auto-SCT can improve prognosis. Five cases (case 35, case 39, case 55, case 57, and case 59) developed central nervous system involvement during follow-up, and case 59 developed neurological lesions despite intrathecal prophylaxis, indicating not only that intrathecal treatment is necessary, but also that more valuable therapeutic measures need to be investigated to prevent intracranial progression.
This study had some limitations. First, although we expanded the sample size by reviewing the literature, this study was still a relatively small-scale study, which might have influenced the accuracy of our results. Second, some data were missing in this retrospective study. Third, this study involved less discussion and analysis of target molecules, pathology, and biology.
Conclusion
PBML is a type of lymphoma with a relatively poor prognosis compared to other lymphomas. Patients usually have poor general condition at the time of onset and a poor tolerance to initial chemotherapy during the acute phase. The survival period is generally short. B symptoms, a low serum platelet level (<75×109/L), and not achieving CR following initial therapy are unfavorable prognosis factors. Additionally, some intensive regimens that differ from traditional regimens or Auto-SCT are worth considering and further exploring. Furthermore, additional and larger prospective multicenter studies are required in the future.
Acknowledgments
This study was supported by The National Natural Science Foundation of China (No. 81570203). Gangjian Wang and Yu Chang share first authorship.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Martinez A, Ponzoni M, Agostinelli C, et al. Primary bone marrow lymphoma: an uncommon extranodal presentation of aggressive non-Hodgkin lymphomas. Am J Surg Pathol. 2012;36(2):296–304. | ||
Bhagat P, Sachdeva MU, Sharma P, et al. Primary bone marrow lymphoma is a rare neoplasm with poor outcome: case series from single tertiary care centre and review of literature. Hematol Oncol. 2016;34(1):42–48. | ||
Yamashita T, Ishida M, Moro H, et al. Primary bone marrow diffuse large B-cell lymphoma accompanying cold agglutinin disease: a case report with review of the literature. Oncol Lett. 2014;7(1):79–81. | ||
Liu Y. The role of 18F-FDG PET/CT in staging and restaging primary bone lymphoma. Nucl Med Commun. 2017;38(4):319–324. | ||
Matthies A, Schuster SJ, Alavi A. Staging and monitoring response to treatment in primary non-Hodgkin’s lymphoma of bone marrow using (18)F-fluorodeoxyglucose positron emission tomography. Clin Lymphoma. 2001;1(4):303–306. | ||
Mu S, Ai L, Fan F, et al. Prognostic role of neutrophil-to-lymphocyte ratio in diffuse large B cell lymphoma patients: an updated dose–response meta-analysis. Cancer Cell Int. 2018;18:119. | ||
Akhtar S, El Weshi A, Rahal M, et al. High-dose chemotherapy and autologous stem cell transplant in adolescent patients with relapsed or refractory Hodgkin’s lymphoma. Bone Marrow Transplant. 2010;45(3):476–482. | ||
Park S, Lee J, Ko YH, et al. The impact of Epstein–Barr virus status on clinical outcome in diffuse large B-cell lymphoma. Blood. 2007;110(3):972–978. | ||
Bloomfield CD, Mckenna RW, Brunning RD. Significance of haematological parameters in the non-Hodgkin’s malignant lymphomas. Br J Haematol. 1976;32(1):41–46. | ||
Conlan MG, Armitage JO, Bast M, Weisenburger DD. Clinical significance of hematologic parameters in non-Hodgkin’s lymphoma at diagnosis. Cancer. 1991;67(5):1389–1395. | ||
Yi SH, Xu Y, Zou DH, et al. [Prognostic impact of bone marrow involvement (BMI) and therapies in diffuse large B cell lymphoma]. Zhonghua Xue Ye Xue Za Zhi. 2009;30(5):307–312. Chinese. | ||
Ferreri AJ, Freschi M, Dell’oro S, et al. Prognostic significance of the histopathologic recognition of low- and high-grade components in stage I-II B-cell gastric lymphomas. Am J Surg Pathol. 2001;25(1):95–102. | ||
Ochi Y, Kazuma Y, Hiramoto N, et al. Utility of a simple prognostic stratification based on platelet counts and serum albumin levels in elderly patients with diffuse large B cell lymphoma. Ann Hematol. 2017;96(1):1–8. | ||
Chen LP, Lin SJ, Yu MS. Prognostic value of platelet count in diffuse large B-cell lymphoma. Clin Lymphoma Myeloma Leuk. 2012;12(1):32–37. | ||
Liebman H. Other immune thrombocytopenias. Semin Hematol. 2007;44(4 Suppl 5):S24–S34. | ||
Kolb GF. [Myelopoiesis and bone marrow function in elderly tumor patients]. Z Gerontol Geriatr. 2012;45(3):197–200. German. | ||
Bertini M, Boccomini C, Calvi R. The influence of advanced age on the treatment and prognosis of diffuse large-cell lymphoma (DLCL). Clin Lymphoma. 2001;1(4):278–284. | ||
Chang Y, Cui M, Fu X, et al. Lymphoma associated hemophagocytic syndrome: a single-center retrospective study. Oncol Lett. 2018;16(1):1275–1284. | ||
Lee DE, Martinez-Escala ME, Serrano LM, et al. Hemophagocytic lymphohistiocytosis in cutaneous T-cell lymphoma. JAMA Dermatol. 2018;154(7):828–831. | ||
Hammerstrøm J. [Granulocyte colony-stimulating factor in neutropenia secondary to lymphoid bone marrow infiltration]. Tidsskr Nor Laegeforen. 1996;116(4):484–486. Norwegian. | ||
Morita Y, Emoto M, Serizawa K, et al. HIV-negative primary bone marrow Hodgkin lymphoma manifesting with a high fever associated with hemophagocytosis as the initial symptom: a case report and review of the previous literature. Intern Med. 2015;54(11):1393–1396. | ||
Li L, Duan W, Zhang L, et al. The efficacy and safety of gemcitabine, cisplatin, prednisone, thalidomide versus CHOP in patients with newly diagnosed peripheral T-cell lymphoma with analysis of biomarkers. Br J Haematol. 2017;178(5):772–780. | ||
Rosado FG, Kim AS. Hemophagocytic lymphohistiocytosis: an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013;139(6):713–727. | ||
Kim MS, Cho YU, Jang S, et al. A case of primary bone marrow diffuse large B-cell lymphoma presenting with fibrillar projections and hemophagocytic lymphohistiocytosis. Ann Lab Med. 2017;37(6):544–546. | ||
Wilson WH, Teruya-Feldstein J, Fest T, et al. Relationship of p53, Bcl-2, and tumor proliferation to clinical drug resistance in non-Hodgkin’s lymphomas. Blood. 1997;89(2):601–609. | ||
Moskowitz CH, Schöder H, Teruya-Feldstein J, et al. Risk-adapted dose-dense immunochemotherapy determined by interim FDG-PET in advanced-stage diffuse large B-cell lymphoma. J Clin Oncol. 2010;28(11):1896–1903. | ||
Lai GM, Chen YN, Mickley LA, Fojo AT, Bates SE. P-glycoprotein expression and schedule dependence of adriamycin cytotoxicity in human colon carcinoma cell lines. Int J Cancer. 1991;49(5):696–703. | ||
Jermann M, Jost LM, Taverna C, et al. Rituximab-EPOCH, an effective salvage therapy for relapsed, refractory or transformed B-cell lymphomas: results of a Phase II study. Ann Oncol. 2004;15(3):511–516. | ||
Intragumtornchai T, Bunworasate U, Nakorn TN, Rojnuckarin P. Rituximab-CHOP-ESHAP vs CHOP-ESHAP-high-dose therapy vs conventional CHOP chemotherapy in high-intermediate and high-risk aggressive non-Hodgkin’s lymphoma. Leuk Lymphoma. 2006;47(7):1306–1314. | ||
Tilly H, Lepage E, Coiffier B, et al. Intensive conventional chemotherapy (ACVBP regimen) compared with standard CHOP for poor-prognosis aggressive non-Hodgkin lymphoma. Blood. 2003;102(13):4284–4289. | ||
Li QC, Yuan XL, Wang YF, et al. [Clinical outcomes of different regimens for non-Hodgkin’s lymphoma with bone marrow involvement: analysis of 148 cases]. Zhonghua Yi Xue Za Zhi. 2008;88(4):254–257. Chinese. | ||
Kazama H, Teramura M, Yoshinaga K, Masuda A, Motoji T. Long-term remission of primary bone marrow diffuse large B-cell lymphoma treated with high-dose chemotherapy rescued by in vivo rituximab-purged autologous stem cells. Case Rep Med. 2012;2012:957063:1–3. | ||
Ponzoni M, Ciceri F, Crocchiolo R, Famoso G, Doglioni C. Isolated bone marrow occurrence of classic Hodgkin’s lymphoma in an HIV-negative patient. Haematologica. 2006;91(3):Ecr04. | ||
Cacoub L, Touati S, Yver M, et al. Isolated bone marrow Hodgkin lymphoma in a human immunodeficiency virus-negative patient: a second case. Leuk Lymphoma. 2014;55(7):1675–1677. | ||
Dholaria B, Alapat D, Arnaoutakis K. Primary bone marrow Hodgkin lymphoma in an HIV-negative patient. Int J Hematol. 2014;99(4):503–507. | ||
Gérard L, Oksenhendler E. Hodgkin’s lymphoma as a cause of fever of unknown origin in HIV infection. AIDS Patient Care STDS. 2003;17(10):495–499. | ||
Ponzoni M, Fumagalli L, Rossi G, et al. Isolated bone marrow manifestation of HIV-associated Hodgkin lymphoma. Mod Pathol. 2002;15(12):1273–1278. | ||
Salama ME, Perkins SL, Mariappan MR. Images in HIV/AIDS. primary bone marrow presentation of Epstein–Barr virus-driven HIV-associated Hodgkin lymphoma. AIDS Read. 2007;17(12):604–605. | ||
Shah BK, Subramaniam S, Peace D, Garcia C. HIV-associated primary bone marrow Hodgkin’s lymphoma: a distinct entity? J Clin Oncol. 2010;28(27):e459–e460. | ||
Suzuki T, Kusumoto S, Masaki A, et al. CD30-positive primary bone marrow lymphoma mimicking Hodgkin lymphoma. Int J Hematol. 2015;101(2):109–111. | ||
Gudgin E, Rashbass J, Pulford KJ, Erber WN. Primary and isolated anaplastic large cell lymphoma of the bone marrow. Leuk Lymphoma. 2005;46(3):461–463. | ||
Szomor A, Al Saati T, Delsol G, et al. Primary bone marrow T-cell anaplastic large cell lymphoma with triple M gradient. Pathol Oncol Res. 2007;13(3):260–262. | ||
Kagoya Y, Sahara N, Matsunaga T, et al. A case of primary bone marrow B-cell non Hodgkin’s lymphoma with severe thrombocytopenia: case report and a review of the literature. Indian J Hematol Blood Transfus. 2010;26(3):106–108. | ||
Kosugi S, Watanabe M, Hoshikawa M. Primary bone marrow lymphoma presenting with cold-type autoimmune hemolytic anemia. Indian J Hematol Blood Transfus. 2014;30(Suppl 1):271–274. | ||
Sumi M, Ichikawa N, Shimizu I, et al. [Primary diffuse large B-cell lymphoma of the bone marrow complicated with autoimmune hemolytic anemia and erythroid hypoplasia]. Rinsho Ketsueki. 2007;48(7):571–575. Japanese. | ||
Níáinle F, Hamnvik OP, Gulmann C, et al. Diffuse large B-cell lymphoma with isolated bone marrow involvement presenting with secondary cold agglutinin disease. Int J Lab Hematol. 2008;30(5):444–445. | ||
Liu H, Yi S, Liu E, et al. [Primary bone marrow diffuse large B cell lymphoma: three case reports and literature review]. Zhonghua Xue Ye Xue Za Zhi. 2014;35(10):914–917. Chinese. | ||
Wang W, Zhou GY, Zhang W. Early relapse in a case of primary bone marrow diffuse large B-cell lymphoma treated with rituximab-CHOP. Immunotherapy. 2017;9(5):379–385. | ||
Ren S, Tao Y, Jia LU, et al. Fever and arthralgia as the initial symptoms of primary bone marrow diffuse large B-cell lymphoma: a case report. Oncol Lett. 2016;11(5):3428–3432. | ||
Hu Y, Chen SL, Huang ZX, Gao W, An N. Case report diffuse large B-cell lymphoma in the primary bone marrow. Genet Mol Res. 2015;14(2):6247–6250. | ||
Niscola P, Palombi M, Fratoni S, Perrotti A, de Fabritiis P. Primary diffuse large B-cell lymphoma of the bone marrow in a frail and elderly patient successfully treated with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone. Blood Res. 2013;48(4):296–297. | ||
Sharma P, Ahluwalia J, Sachdeva MU, et al. Primary bone marrow T-cell/histiocyte-rich large B-cell lymphoma: a diagnostic challenge. Hematology. 2013;18(2):85–88. | ||
Chang H, Hung YS, Lin TL, et al. Primary bone marrow diffuse large B cell lymphoma: a case series and review. Ann Hematol. 2011;90(7):791–796. | ||
Alvares CL, Matutes E, Scully MA, et al. Isolated bone marrow involvement in diffuse large B cell lymphoma: a report of three cases with review of morphological, immunophenotypic and cytogenetic findings. Leuk Lymphoma. 2004;45(4):769–775. | ||
Nagasaki A, Hiroo H, Taira N, Takasu N, Masuda M. Primary malignant lymphoma of bone marrow. Intern Med. 2004;43(6):524–525. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.