Back to Journals » Infection and Drug Resistance » Volume 16
Clinical Efficiency of Metagenomic Next-Generation Sequencing in Sputum for Pathogen Detection of Patients with Pneumonia According to Disease Severity and Host Immune Status
Authors Chang C, Wang H, Zhang L, Hao J, Wang X ![]() , Wang Y, Qi F, Lou J, Zhao J
, Wang Y, Qi F, Lou J, Zhao J ![]() , Dong J
, Dong J
Received 26 May 2023
Accepted for publication 23 August 2023
Published 6 September 2023 Volume 2023:16 Pages 5869—5885
DOI https://doi.org/10.2147/IDR.S419892
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Can Chang,1 Huan Wang,1 Lianjun Zhang,1 Junling Hao,1 Xiaoning Wang,1 Yaoyao Wang,2 Fei Qi,2 Jingwei Lou,2 Jiangman Zhao,2 Junying Dong1
1Department of Respiratory and Critical Care Medicine, Shandong Provincial Hospital Heze Branch, Heze, Shandong, 274000, People’s Republic of China; 2Shanghai Zhangjiang Institute of Medical Innovation, Shanghai Biotecan Pharmaceuticals Co., Ltd, Shanghai, 201204, People’s Republic of China
Correspondence: Junying Dong, Department of Respiratory and Critical Care Medicine, Shandong Provincial Hospital Heze Branch, Caozhou Road 2888, Heze, Shandong, 274000, People’s Republic of China, Tel +86- 13723908695, Email [email protected] Jiangman Zhao, Shanghai Zhangjiang Institute of Medical Innovation, Shanghai Biotecan Pharmaceuticals Co., Ltd, Zhangheng Road 180, Shanghai, 201204, People’s Republic of China, Tel +86- 18817517482, Email [email protected]
Purpose: Severe pneumonia causes the highest mortality rate in immunocompromised patients. This study aimed to investigate the pathogen diagnostic efficacy of metagenomic next-generation sequencing (mNGS) using sputum sample in patients with pneumonia according to patients’ disease severity and immune conditions.
Patients and Methods: A total of 180 patients suffering from pneumonia were recruited, and sputum samples were collected in duplicate for pathogen detection by both conventional microbiological tests (CMT) and mNGS. Then, the performance of pathogen identification was examined between two methods, according to disease severity and patients’ immune status.
Results: In comparison to CMT, mNGS had higher positivity rates in all patients with pneumonia (85.0% vs 62.2%, P=9.445e-07). The most commonly detected microorganism in sputum of pneumonia patients was Acinetobacter baumannii (42/180, 23.3%) in bacterum level, Candida albicans in fungus level (44/180, 24.4%), and Human herpesvirus 1 (39/180, 27.5%) in virus level. However, for mNGS results, Candida albicans in 34.9% of positive patients, and Human herpesvirus 1 in 7.7% of positive cases were confirmed as pathogens causing pneumonia. Acinetobacter baumannii detected by mNGS in 75% of positive patients was diagnosed as pathogen of pneumonia. The microorganism profile of sputum mNGS differed according to disease severity and immune status of patients. Pneumocystis jirovecii was more likely to infect immunocompromised patients (P=0.002). Pseudomonas aeruginosa (14.8% vs 0.0%, P=0.008) and Human herpesvirus 1 (26.1% vs 5.3%, P=0.004) had higher infection rate in patients with severe pneumonia compared with non-severe cases. mNGS had overwhelming advantages over CMT in detecting a lot of microorganisms including Streptococcus pneumoniae, Enterococcus faecium, Pneumocystis jirovecii, and majority of viruses.
Conclusion: mNGS is a complementary tool of CMT for detecting suspected pathogens for patients with lower respiratory infections. The interpretation of opportunistic pathogens identified by mNGS is challenging, and needs comprehensive consideration of sequencing data and clinical factors.
Keywords: lower respiratory infections, severe pneumonia, immunocompromised patient, conventional microbiological test, metagenomic-next generation sequencing
Introduction
Lower respiratory infections (LRIs) are the world’s most deadly communicable disease and ranks fourth as the primary cause of death globally according to the World Health Organization (WHO) 2019 report.1,2 LRIs include hospital-acquired pneumonia (HAP), community-acquired pneumonia (CAP), bronchiolitis, bronchitis, and tracheitis.3,4 Immunocompromised patients have a higher risk of experiencing severe pneumonia, that is a major factor in mortality rate of these patients and a primary reason for admission to intensive care units (ICU).5,6 Fast and accurate verification of the causative pathogens is imperative as it helps avoid empirical misuse of wide-spectrum antibiotics and improves the clinical outcomes of immunocompromised patients with severe pneumonia.7
Pneumonia is heterogeneous and complex, caused by a broad spectrum of microbes, including bacteria, fungi, mycoplasma, chlamydia, and viruses.8 Conventional microbiological tests (CMT) are widely used for pathogen identification, which comprises culture-based methods and molecular detection procedures, for example polymerase chain reaction (PCR) and enzyme immunoassay.9 However, despite the extensive diagnostic workup, the etiology of 19% to 62% of pneumonia patients remains undiagnosed.10–12 Culture-based methods have disadvantages like being time-consuming, difficulty in culturing certain bacteria, and low sensitivity.13
Metagenomic next-generation sequencing (mNGS) is an unbiased technique of parallel sequencing that enables the simultaneous identification of all potentially infectious agents in samples, such as viruses, bacteria, fungi, and parasites, without presupposing a target.14,15 It is also used in the acquisition of novel microbial genome sequences. Increasing studies have proven mNGS have some advantages over CMT in pathogen detection in patients with LRIs.16–18 However, some problems of mNGS need to be solved, for example colonization differentiation from infection, contaminants of extraneous nucleic acid, method standardization, interpretation of results, detection efficiency of different sample types.
The purpose of this study was to assess and compare the clinical efficacy of pathogen detection between CMT and mNGS in a cohort of 180 pneumonia patients admitted to the Department of Respiratory and Critical Care Medicine. The microorganism distribution by mNGS detection in sputum of patients with pneumonia according to disease severity and immune status was analyzed. We also investigated the concordance between microorganisms detected by mNGS and pathogens confirmed by clinicians.
Materials and Methods
Patients and Sample Collection
A total of 180 patients diagnosed with pneumonia admitted to Department of Respiratory and Critical Care Medicine in Shandong Provincial Hospital Heze Branch (Shandong, China) were retrospectively included, from January 2021 to February 2022. The participants were eligible if they met the following inclusion criteria: (1) participants with clinically confirmed pneumonia diagnosis; (2) nucleic acid extracted from respiratory samples and constructed library satisfied quality control of mNGS; (3) patients receiving both CMT and mNGS for pathogen detection. The following criteria were used to exclude participants: (1) lack of patients’ clinical data; (2) patients who did not provide informed consent. All procedures were performed and approved according to the ethical standards of the Clinical Research Ethics Committee of Shandong Provincial Hospital Heze Branch (2022-KY011).
Sputum was collected from all 180 patients with pneumonia, which was used for pathogen identification by both CMT and mNGS. The samples for pathogen detection were mostly collected before medication or when patient’s condition was without improvement after empirical treatment. For some patients, multiple samples were collected for CMT, including plasma, bronchoalveolar lavage fluid (BALF), urine, peritoneal fluid, bile, and cerebrospinal fluid. The CMT mainly included microbiological culture, laboratory microscopy with staining, PCR and serologic immunoassay,9 Supplementary Figure 1 showed the positive rate of detected microorganisms by CMT using different sample types.
Clinical Information Collection
We retrospectively collected each patient’s clinical data via hospital electronic medical record system. CAP was defined as patient with pneumonia-related clinical manifestations whose infection was acquired outside of the hospital setting. HAP was defined as an episode of pneumonia after at least 48 hours of hospitalization, which was not in the incubation period of community acquired infection. Severe pneumonia patients were diagnosed referencing the Infectious Diseases Society of America/American Thoracic Society criteria: met either one major criterion or at least three minor criteria.19 Major criteria: (1) septic shock with need for vasopressors; (2) respiratory failure requiring mechanical ventilation (due to pulmonary infection alone). Minor criteria: (1) respiratory rate ≥ 30 breaths/min; (2) PaO2/FiO2 ratio ≤ 250; (3) multilobar infiltrates; (4) confusion/disorientation; (5) uremia (blood urea nitrogen level ≥ 20 mg/dl); (6) leukopenia (white blood cell count < 4000 cells/μL, due to infection alone); (7) thrombocytopenia (platelet count < 100,000/μL); (8) hypothermia (core temperature < 36°C); (9) hypotension requiring aggressive fluid resuscitation. We defined patients with immunocompromised status according to the criteria established in a previous study.20 Patients were defined as immunocompromised when they had one or more of the following risk factors: (1) blood cancer; (2) solid tumor with either neutropenia or chemotherapy; (3) neutropenia; (4) chronic use of steroid or biologic drug for autoimmune diseases; (5) immunosuppressive therapy due to hematologic cancer or solid organ transplantation; (6) any immunocompromised state including congenital/genetic immunocompromise and asplenia. From 180 patients, 177 cases were administered empirical antibiotic treatment before sampling. Subsequently, treatment plans were modified according to the results of mNGS and CMT as well as imaging, clinical signs, and other indicators.
DNA Extraction
Sputum samples ranging from 1 to 3 mL were taken and conveyed in drilled dry sterile tubes for cryopreservation. Before extracting the nucleic acids, the sputum was rendered inactive in a 56°C water bath for 30 minutes. At room temperature, 0.1% DTT was used to liquefy sputum samples for 30 minutes. According to the instructions, DNA was extracted by a HostZEROTM Microbial DNA Kit (D4310, ZYMO RESEARCH).
Metagenomic Next-Generation Sequencing
The library was constructed using the commercial library preparation kit for the Illumina sequencing equipment. KAPA HyperPlus Kit was used to build the library in accordance with its instructions. The library’s fragment length was measured using Agilent 2100 Bioanalyzer, and the library’s concentration was managed by a Qubit™ dsDNA HS Assay Kit. It was sequenced using the NextSeq CN500 platform (Illumina). For each test, internal, positive, and negative controls were set. The use of no-template water as a negative control made it possible to identify contamination from the environment, reagents, and other samples. Real clinical samples that were confirmed to have well-known pathogens served as the positive control. It may be a sign that there were problems with the workflow of the laboratory experiment or bioinformatics process if the targeted pathogens were not found in positive controls.
Bioinformatics Analysis
To obtain high-quality sequencing data, reads with low-quality and short length (less than 35 bp) were removed. Afterwards, human host sequences mapping to the human reference genome (hg19) were eliminated using Burrows-Wheeler alignment. After reads containing repetitive sequences were removed, the data that were left-over were categorized by alignment to four microbial genome databases in the meantime, comprising of bacteria, viruses, fungi, and parasites. The National Center for Biotechnology Information’s genomes page can be accessed at ftp:/ftp.ncbi.nlm.nih.gov/genomes/ to get the classification reference datasets.
Criteria for a Positive Pathogen of mNGS
The in-house background database was consulted to eliminate the suspected background microbial organisms. For the residual microorganisms, we developed a set of requirements for a positive mNGS result based on prior research.21
- Bacteria (mycobacteria excluded): ≥50 reads mapped to pathogen species and with a reads number no less than 10 times of any other microorganism,17 or supported by CMT results.15
- Fungus/mycoplasma/chlamydia/virus: the reads mapped to pathogen species with a reads no less than five times of any other fungus,17 or supported by CMT results.22
- Mycobacterium tuberculosis (MTB): no less than one particular sequence was mapped to the reference genome of genus or species level, owing to the difficulty of nucleic acid extraction and the low likelihood23 of environmental contamination.13
- Nontuberculous Mycobacterium (NTM): a relative bacterial abundance ranking in the top 10 of bacterial list, in view of common environmental contamination.16
Antibiotic Resistance Gene Analysis
63 samples were selected for antibiotic resistance gene (ARG) analysis, which detects microorganism with ≥10,000 reads mapped to pathogen species. The sequences annotated to pathogenic microorganisms were aligned to the established drug resistance gene database by blastn (v 2.5.0+), based on the CARD (the Comprehensive Antimicrobial Resistance Database). The ARGs were retained, whose sequences met the following criteria: similarity identity ≥90% and length ≥70 bp. According to comparison of the starting position, the coverage of ARG was calculated. ARGs with coverage ≥90% and reads count ≥100 indicated that the drug-resistant gene was positive.
Statistical Analysis
The SPSS 26.0 (IBM, Armonk, NY, USA) software and R project with RStudio (R 4.0.2, R Core Team) were employed for data analyses and graphics plotting. The continuous variables between two groups were compared using the non-parametric Mann Whitney U-test, if they did not comply with the normal distribution. The distribution difference of categorical variables was tested by chi-square test or Fisher’s exact test. P<0.05 was considered significant. The Cohen’s kappa statistic was used to measure the agreement level between results of two methods. The interpretation of different values for Cohen’s Kappa refer to the following indications: a kappa value of 1 indicating perfect agreement, 0.81–0.99 indicating near perfect agreement, 0.61–0.80 indicating substantial agreement, 0.41–0.60 indicating moderate agreement, 0.21–0.40 indicating fair agreement, 0.01–0.20 slight agreement, <0 indicating less than chance agreement.
Results
Clinical Characteristics of Patients with Pneumonia
Clinical characteristics of 180 patients with pneumonia were shown in Table 1. There were 126 males and 54 females with a mean age of 69.7 years old. According to disease severity, 142 cases were diagnosed as severe pneumonia. According to immune status, 68 cases were immunocompromised. A total of 57 patients were diagnosed with immunocompromised combined severe pneumonia. Patients with severe pneumonia had a significantly lower BMI (P=0.047), and higher ratio of using breathing machine (90.8% vs 78.9%, P=0.042) than non-severe patients. No significant difference was found in age, gender, drinking and smoking history, underlying disease between severe and non-severe pneumonia (P >0.05). A higher ratio of immunocompromised patients had smoking history (45.6% vs 29.5%, P=0.028) than immunocompetent patients.
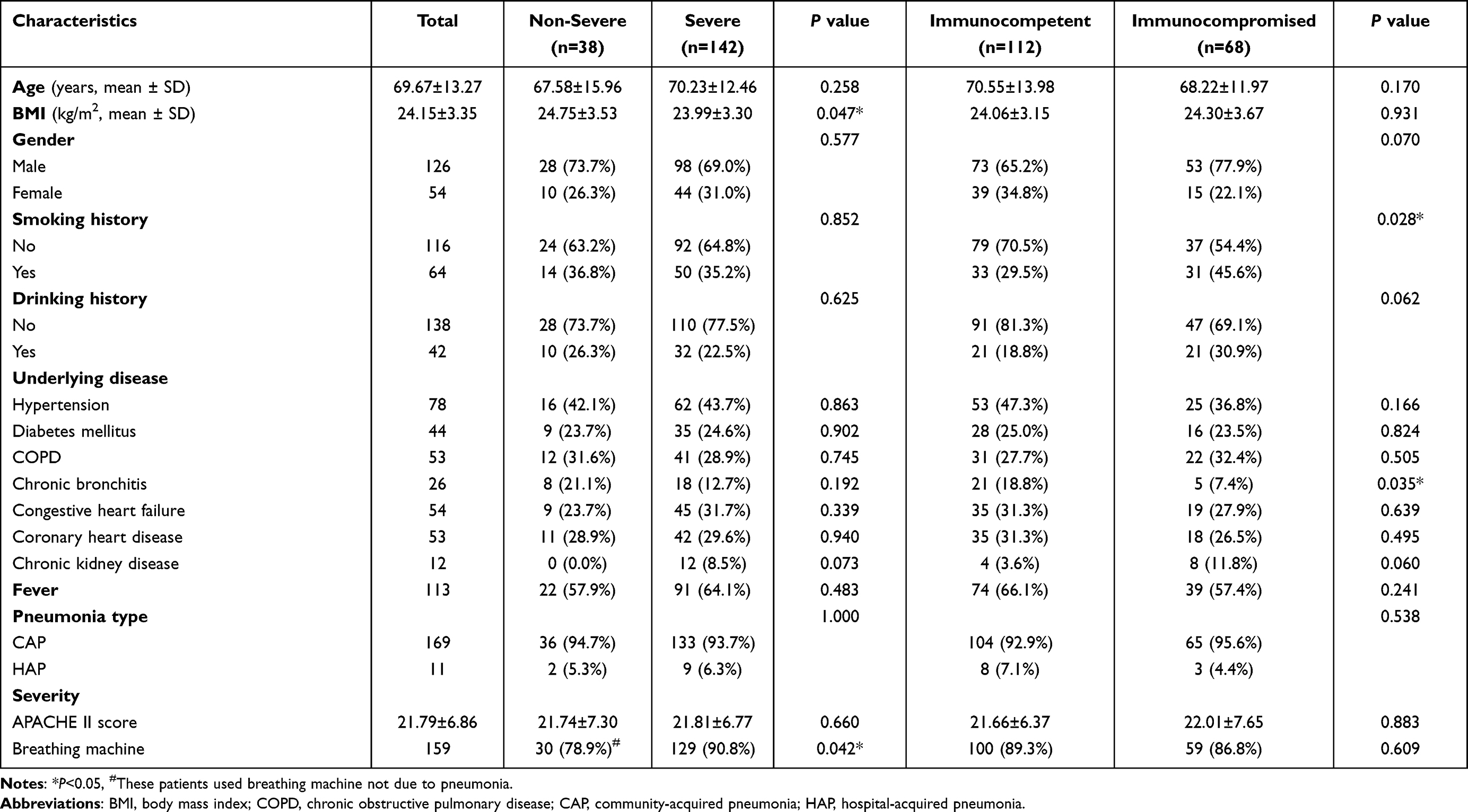 |
Table 1 Clinical Characteristics of 180 Patients with Pneumonia According to Immunocompromised Status and Pneumonia Severity |
Comparison of Overall Results Between CMT and mNGS
The positive rate of detected microorganism by mNGS was 85.0% (n=153), which was significantly higher than that of CTM results based on all samples (85.0% vs 62.2%, P=9.445e-07, Figure 1A) or only based on sputum (85.0% vs 50.6%, P=2.704e-12, Figure 1B). Out of 180 cases, our findings indicated that both mNGS and CMT were positive in 87 cases (48.33%) and negative in 23 cases (12.78%) (Figure 1C). For 66 patients’ results (36.67%), the CMT was negative, and mNGS was positive. Only 4 cases (2.22%) had positive CMT and negative mNGS results. For a total of 87 cases with both positive mNGS and CMT, we examined the results’ consistency of mNGS and CMT (Figure 1D). 12 cases showed complete microorganism overlap between the two methods, while 13 cases showed no overlap at all. The remaining 62 cases had partial overlap, meaning that at least one microorganism was detected by both mNGS and culture.
 |
Figure 1 The positive rate comparison of detected microorganisms by mNGS and CMT in patients with pneumonia. (A) Comparison of positive rate between pairwise mNGS and CMT in 180 patients with pneumonia, considering results of all sample types. (B) Comparison of positive rate between pairwise mNGS and CMT in 180 patients with pneumonia, only considering CMT results of sputum samples. (C) Pie chart demonstrates the positive and negative distribution of mNGS and CMT results. mNGS+: positive only by mNGS; CMT+: positive only by CMT; double+: both positive by CMT and mNGS; double-: both negative by mNGS and CMT. (D) For the double positive subgroup (grey in pattern c), 87 patients were divided into match (12/87), partial match (62/87) and mismatch (13/87). Match: both positive by CMT and mNGS and pathogens completely overlapped; mismatch: conflicts between mNGS and CMT; partial match: both positive by CMT and mNGS and partial overlapping of microorganisms. |
Comparison of Detected Microorganisms Between mNGS and CMT
Figure 2 showed the microorganism profile of sputum from 180 patients with pneumonia, according to mNGS and CMT methods. In total, 67 kinds of microorganisms were identified in 157 patients by mNGS or/and CMT method, including 41 bacteria, 14 fungi, and 12 viruses. Table 2 showed the concordance between microorganisms detected by mNGS and pathogens confirmed by clinicians.
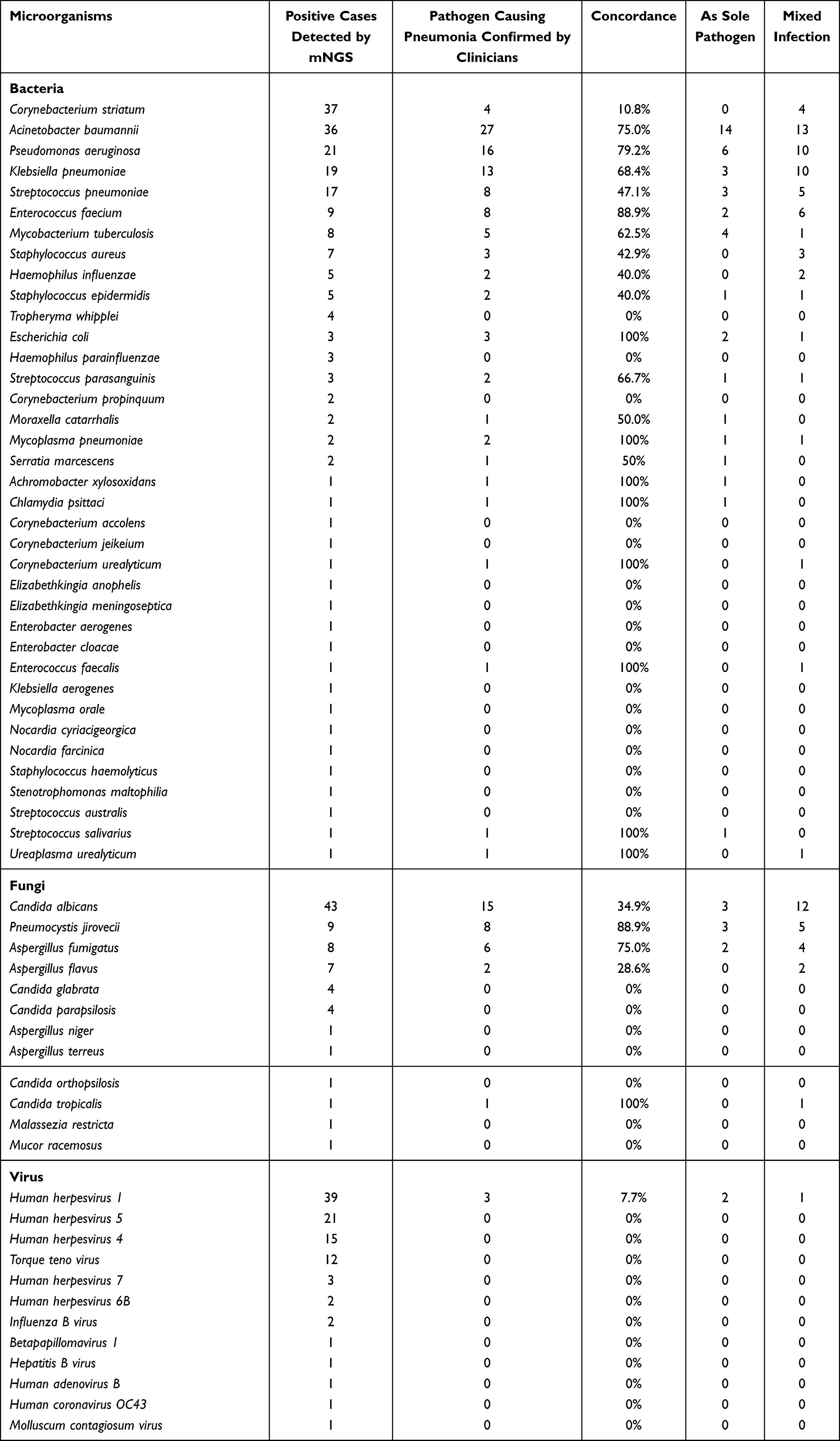 |
Table 2 The Concordance Between Microorganisms Detected by mNGS and the Finally Definitive Pathogens Confirmed by the Clinician |
 |
Figure 2 The overlap and comparison of positive microorganisms between mNGS and CMT in 180 patients with pneumonia. (A) Bacteria levels; (B) fungi level; and (C) virus level. |
In Bacterium Level
The most common bacteria (Figure 2A) were Acinetobacter baumannii (A. baumannii, n=42), followed by Corynebacterium striatum (C. striatum, n=38), Klebsiella pneumoniae (K. pneumoniae, n=22), Pseudomonas aeruginosa (P. aeruginosa, n=22). Eleven bacteria were positive in both CMT and mNGS results. We performed Cohen’s kappa statistics on top 10 bacterium to examine the results’ concordance between mNGS and CMT (Supplementary Table 1). The results showed there were substantial agreements of A. baumannii (Kappa value=0.731, P=8.87e-23), K. pneumoniae (Kappa value=0.715, P=5.343e-22), P. aeruginosa (Kappa value=0.790, P=8.652e-27), Escherichia coli (Kappa value=0.659, P=5.149E-21).
mNGS had obvious advantage over CMT in detection efficacy of some bacterium such as C. striatum. C. striatum was detected in sputum of 37 patients by mNGS, but only 4 patients (10.8%, 4/37) were confirmed to have developed pneumonia caused by C. striatum combined with other pathogens. In addition, 26 bacteria were detected only by mNGS, such as Streptococcus pneumoniae (S. pneumoniae, n=17), Haemophilus influenzae (n=5), Tropheryma whipplei (T. whipplei, n=4), Haemophilus parainfluenzae (n=3) and so on (Figure 2A). S. pneumoniae was confirmed as pathogen causing pneumonia in 47.1% (8/17) infected patients (Table 2). However, Haemophilus influenzae, T. whipplei, Haemophilus parainfluenzae detected by mNGS were all not confirmed as pathogen of pneumonia.
In Fungi Level
Sputum of 44 patients tested positive of Candida albicans (C. albicans) as the most frequent fungi, and only mNGS was able to detect 36 of them (Figure 2B). From 43 patients with positive C. albicans of mNGS, pathogen of 15 cases (34.9%, 15/43) was confirmed as C. albicans (Table 2). Nine fungi were identified only by mNGS including Pneumocystis jirovecii (P. jirovecii, n=9), Aspergillus flavus (n=7), Candida parapsilosis (n=4), Candida glabrata (n=4), Aspergillus niger (n=1), Aspergillus terreus (n=1), Candida orthopsilosis (n=1), Malassezia restricta (n=1), Mucor racemosus (n=1). P. jirovecii is highly pathogenic for pneumonia, and the pathogens of 8 patients (88.9%, 8/9, Table 2) were confirmed as P. jirovecii. However, many fungi detected by mNGS such as Candida glabrata, Candida parapsilosis, Aspergillus niger, Aspergillus terreus, Candida orthopsilosis, Malassezia restricta, Mucor racemosus, were not supported as pathogens of pneumonia by clinicians.
In Virus Level
The most common virus was Human herpesvirus (HHV), (Figure 2C) including HHV-1 (n=39), HHV-5 (n=21), HHV-4 (n=15), HHV-7 (n=3), and HHV-6B (n=2). Except for Influenza B virus, 11 viruses were only detected by mNGS. It indicated mNGS had great advantage over CMT in virus detection. However, only 3 patients’ pulmonary infections were thought to be caused by HHV-1 (Table 2), other patients with positive viruses in sputum were not diagnosed with viral pneumonia.
Comparison of Detected Microorganisms Between Patients with Non-Severe and Severe Pneumonia
Figure 3 and Supplementary Table 2 displayed the microorganism positivity rates of non-severe and severe pneumonia groups between mNGS and CMT. The positive rate of mNGS was notably higher than that of CMT in both non-severe (84.2% vs 39.5%, P<0.05, Figure 3A) and severe patients (85.2% vs 53.5%, P<0.05, Figure 3A). No significant difference was found in positive rate (84.2% vs 85.2%, P=0.878) and mixed infection rate (60.5% vs 62.7%, P=0.808) among patients with non-severe and severe conditions, based on the results of mNGS (Figure 3B). Kappa test showed there was fair agreement between mNGS and CMT, both for patients with non-severe (kappa value=0.218, P=0.031, Table 3) and severe pneumonia (kappa value=0.215, P=0.001, Table 3). Figure 4 illustrated that the spectrum of identified microorganisms by mNGS varied between non-severe and severe pneumonia individuals. In total, 52 kinds of suspected pathogenic microorganisms were identified in the severe pneumonia patients using mNGS, including 31 kinds of bacterial, 11 kinds of fungi, and 10 kinds of viruses (Figure 4), while in non-severe pneumonia patients, including 18 kinds of bacterial, 6 kinds of fungi, and 7 kinds of viruses (Figure 4).
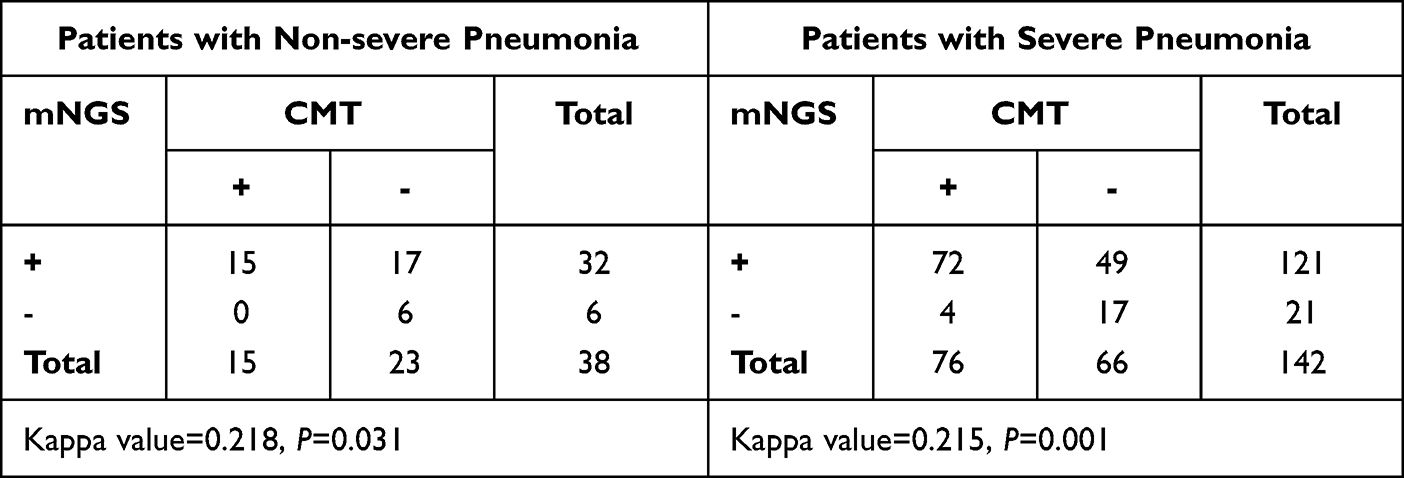 |
Table 3 The Concordance of Results Between mNGS and CMT Was Analyzed Using Cohen’s Kappa Statistics in Patients with Non-Severe and Severe Pneumonia |
 |
Figure 3 The positive rate comparison of detected microorganisms by mNGS and CMT between non-severe and severe pneumonia patients. (A) Comparisons of positive rates for pairwise mNGS and CMT test in non-severe and severe pneumonia patients. (B) Comparison of positive rate and mixed infection (at least two) rate by mNGS between non-severe and severe patients. |
 |
Figure 4 Comparison of microorganisms detected by mNGS between non-severe and severe pneumonia patients in bacteria (A), fungi (B) and virus (C) levels. |
C. striatum was the most common bacterium of severe patients’ sputum (n=31, 21.8%), and A. baumannii was the most common bacterium of non-severe patients’ sputum (n=8, 21.1%). 19 bacteria were only found in severe patients by mNGS, including P. aeruginosa, Staphylococcus aureus, Staphylococcus haemolyticus, T. whipplei, Streptococcus parasanguinis, and so on. P. aeruginosa had high infection rate (n=21, 14.8%) and very high bacterial virulence, infection of which all led to severe pneumonia (Figure 4A, Supplementary Table 2). From non-severe patients, 6 bacteria were exclusively detected by mNGS. In fungi level, C. albicans was the most frequent microorganism in both non-severe (13.2%) and severe patients (26.8%) with pneumonia. 6 fungi were detected only in severe group including Candida parapsilosis, Aspergillus niger, Candida orthopsilosis, Candida tropicalis, Malassezia restricta, and Mucor racemosus (Figure 4B). Aspergillus terreus was only found in non-severe patients. In severe patients, the virus that was most commonly identified was HHV-1, and it had significantly higher existence probability in severe pneumonia (P=0.004, Supplementary Table 2, Figure 4C).
Comparison of Detected Microorganisms Between Immunocompetent and Immunocompromised Patients with Pneumonia
mNGS had an obviously higher positive rate of detected microorganisms compared with CMT in either immunocompromised or immunocompetent patients (P<0.05, Figure 5A). The positive rate of mNGS was 80.9% and 87.5% in immunocompromised and immunocompetent patients respectively, with no significant difference (P=0.322). Surprisingly, immunocompetent patients had significantly higher possibility of being infected by mixed pathogens (P=0.013, Figure 5B). There were slight agreement and fair agreement between mNGS and CMT results in immunocompromised (Kappa value=0.178, P=0.036, Table 4) and immunocompetent patients (Kappa value=0.232, P=0.001, Table 4).
 |
Table 4 The Concordance of Results Between mNGS and CMT Was Analyzed Using Cohen’s Kappa Statistics in Immunocompetent and Immunocompromised Patients with Pneumonia |
 |
Figure 5 The positive rate comparison of detected microorganisms by mNGS and CMT in immunocompetent or immunocompromised patients with pneumonia. (A) Comparisons of positive rates for pairwise mNGS and CMT test in immunocompetent and immunocompromised patients with pneumonia. (B) Comparison of positivity rate of mixed infection based on mNGS between immunocompetent and immunocompromised patients with pneumonia. |
In immunocompetent patients with pneumonia, the most common microorganism in sputum was C. striatum (26.8%, Figure 6A), followed by C. albicans (25.0%, Figure 6B), A. baumannii (26.8%, Figure 6A), and HHV-1 (21.4%, Figure 6C). In immunocompromised pneumonia patients, the most common microorganisms were HHV-1 (22.1%, Figure 6C) and C. albicans (22.1%, Figure 6B), followed by A. baumannii (16.2%, Figure 6A) and C. striatum (10.3%, Figure 6A). Our findings showed the rate of P. jirovecii infection was significantly higher in immunocompromised patients compared to immunocompetent patients (11.8% vs 0.9%, P=0.002, Supplementary Table 3).
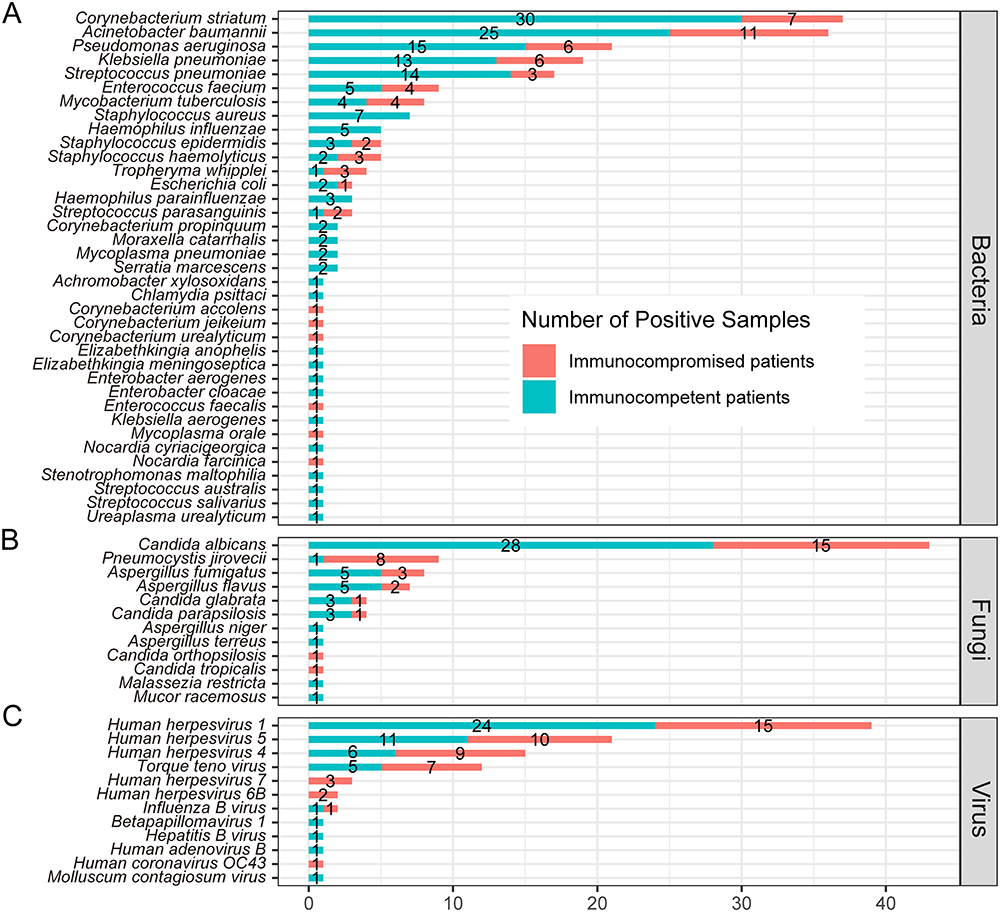 |
Figure 6 Comparison of microorganisms detected by mNGS between immunocompetent and immunocompromised patients with pneumonia in bacteria (A), fungi (B) and virus (C) levels. |
The Concordance Between mNGS Results and Clinician Diagnosis
Among 180 cases, the pathogens of 127 pneumonia patients (127/180, 70.6%) were confirmed by clinicians combining the mNGS/CMT results and comprehensive clinical manifestation. The pathogens causing pneumonia were unproven in 53 patients (53/180, 29.4%). The detected microorganisms by mNGS were finally proven by clinicians in 115 patients. For 59 patients, mNGS results were consistent with the initial diagnosis, and they continued to receive the original treatment regimens. For the other 56 patients, the results of mNGS gave new revelation about pathogens of pneumonia, and the patients’ treatment regimens were modified according to the mNGS results (Figure 7). In addition, the microorganisms detected by mNGS were not supported by clinicians in 12 cases.
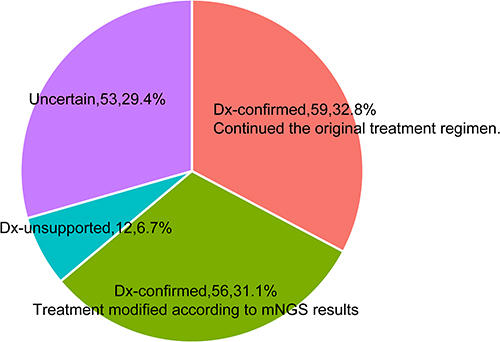 |
Figure 7 The concordance between microorganisms detected by mNGS and the finally definitive pathogens confirmed by the clinicians. Dx-confirmed: the microorganisms detected by mNGS were finally confirmed as pathogens of pneumonia by clinicians; Dx-unsupported: the microorganisms detected by mNGS were not confirmed as pathogens of pneumonia by clinician. Dx-uncertain: the pathogen of pneumonia was unproven. |
Antibiotic Resistance Gene Profile
The ARG was identified from sputum of 50 patients with pneumonia, including 134 ARGs of 37 AMR gene families. Supplementary Table 4 showed the positive patient number of every ARG, and its AMR gene family, resistance mechanism, and corresponding antibiotic. ErmX gene was the most frequent ARG which was positive in 25 cases, followed by APH(3”)-Ib (n=22), APH(6)-Id (n=22), APH(3’)-Ia (n=18), AbaF (n=15), abeM (n=15), adeA (n=15) and so on. ErmX gene, belonging to Erm 23S ribosomal RNA methyltransferase, conferred multi-drug resistance to Macrolide-Lincosamide-Streptogramin B (MLS B), its resistance mechanism is antibiotic target alteration. Among 50 patients, a total of 59 multi-drug resistance (MDR) genes were detected from 33 patients, the most frequent AMR gene family of MDR genes was RND efflux pump which plays a drug resistance role through antibiotic efflux.
Discussion
Pneumonia is responsible for the highest number of deaths among infectious diseases in the ICU, having considerable implications for healthcare systems worldwide.10,24 However, identification of the pathogenic microorganism is challenging because of various possible pathogens causing pneumonia, involving viral, bacterial or fungal pathogens.25 Therefore, a good tool is necessary to improve the accurate diagnosis and decision-making process in patients with infectious pneumonia, so as to avoid the worse results caused by over-diagnosis or underdiagnosis. Currently, the CMT methods for pathogen detection include bacterial, fungal and virus culture, serological testing, enzyme-linked immunosorbent assay (ELISA), and PCR. However, they are limited in their ability to identity pathogens with high sensitivity and specificity timely. And they need to make a prejudgment before test, it is hard to quickly identify rare or unknown microorganisms.26 High-throughput sequencing technology has made significant progress in the field of pathogen detection in recent years. In particular, mNGS has demonstrated notable advantages over CMT methods.
Bronchoalveolar lavage fluid (BALF) has been suggested as ideal sample by most researchers for pathogen detection by mNGS, which could provide a more accurate representation of the alveolar component and reduce the risk of contamination from the oropharyngeal flora.20,27,28 However, compared with BALF, sputum is the more accessible sample of patients with pulmonary infection for pathogen identification. An innovative advantage of this study was comparing the diagnostic efficacy of sputum mNGS with CMT in infectious pneumonia. In comparison to the CMT, sputum mNGS showed superior sensitivity in microorganism detection of pneumonia patients (85.0% vs 62.2%, P<0.05), which indicated that sputum mNGS detection can be a complementary method of CMT for pathogen detection in patients with pneumonia, especially those with negative CMT results. Previous studies have also verified it using small sample size.29,30
In the cohort of our study, A. baumannii was the most common bacteria causing infection (n=42), 36 of them were detected by mNGS. There was substantial agreement for A. baumannii detection between mNGS and CMT. A. baumannii is an opportunistic pathogen, that persists in nosocomial environment,31 which was mainly responsible for hospital-acquired nosocomial infections. In our study, although 169 cases (169/180) were diagnosed as CAP, majority of them received a period of hospitalized treatment in other departments such as Department of Respiratory Medicine, Emergency Ward, before ICU admission. From 36 patients in whom A. baumannii was detected by mNGS, 29 patients were identified as having mixed infections of A. baumannii combined with other suspected pathogens. This may be the primary reason why A. baumannii was the most commonly detected microorganism in our predominantly CAP cohort. Once considered benign, A. baumannii is currently viewed as an alarming threat to global healthcare as it has a high tendency to develop multidrug-resistance.17,32 In our study, among 36 patients with positive A. baumannii by mNGS, 75% (n=27) of them were finally confirmed by clinicians. The 9 unsupported patients with positive A. baumannii by mNGS had relatively low abundance of reads mapped to species. A previous study demonstrated that A. baumannii may colonize the sputum of patients with infectious diseases causing false-positive results.16 We suggest clinical interpretation should comprehensively consider a relative bacterial abundance rank in bacteria list.
HHV-1 was the most common virus, and we observed a significantly higher detection rate of this virus in severe patients’ sputum as compared to non-severe patients (26.1% vs 5.3%, P=0.004). HHV-1, also called Herpes simplex virus type 1 (HSV-1), is a frequently occurring virus in the respiratory secretions of critically-ill patients, that can cause a range of clinical manifestations, including oral and genital herpes, encephalitis, and pneumonia. Previous studies reported that HSV-1 pneumonia is relatively rare, it can occur in individuals who have a weakened immune system or those with underlying lung disease.33 In this study, among 39 patients with positive HSV-1, 34 cases were identified as having mixed infections by mNGS, 5 patients with HSV-1 as sole microorganism. Only 3 patients were diagnosed with HSV-1 pneumonia by clinician. Establishing a definite diagnosis of HSV-1 pneumonia can be challenging as radiological features, clinical criteria, and laboratory findings are not specific enough.34 Further research is necessary to accurately determine the pathogenicity of HSV-1 in the lower respiratory system of these individuals, enhance diagnostic methods, and evaluate the necessity of antiviral therapy for each patient.
The microorganism most frequently detected in severe patients or those who were immunocompromised was C. striatum. Immunocompromised patients had a considerably higher infection rate than immunocompetent patients (26.8% vs 10.3%, P=0.008). However, only 34.9% of infected patients’ pneumonia was thought to be caused by C. striatum. C. striatum frequently colonizes both the skin and mucous membranes. Although previously considered a contaminant when isolated from clinical specimens, it is now increasingly being recognized as a pathogen that causes infectious disease.35 Factors that increase the risk of C. striatum infection include the patient’s immune status, a prolonged stay in hospital, continuous exposure to antibiotics, and the use of invasive medical equipment.36 Similarly, P. jirovecii was found to be more frequently associated with pneumonia in immunocompromised patients than in immunocompetent ones (11.8% vs 0.9%, P=0.002). P. jirovecii is an opportunistic fungal pathogen capable of being transmitted from asymptomatic carriers to immunocompromised individuals, potentially leading to the development of pneumonia, such as those with HIV/AIDS, organ transplant recipients, and patients receiving immunosuppressive therapy.37 CMT for detecting P. jirovecii involves staining and microscopy of respiratory specimens, but these methods have limited sensitivity and specificity. From Figure 2A, we can see mNGS has overwhelming advantages over CMT for detection of C. striatum and P. jirovecii (Figure 2A). And P. jirovecii infection was highly pathogenic (Table 2). These results suggest mNGS as a key tool for C. striatum and P. jirovecii of immunocompromised patients with negative CMT result.
Compared to BALF, sputum affected by open oral environment has complex microbiota. In Tianlai Lin’s study,21 based on 83 BALF, 89 blood and 33 sputum samples of 205 CAP patients from ICU, the top 3 common bacteria detected by mNGS were Escherichia coli, Mycobacterium tuberculosis and K. pneumoniae, followed by C. striatum and A. baumannii. However, in our study, based on sputum of 180 pneumonia patients from ICU, the top 3 common bacteria detected by mNGS were C. striatum, A. baumannii and Pseudomonas aeruginosa, followed by K. pneumoniae and S. pneumoniae. These results indicated that opportunistic and colonized microorganisms had higher positive rate in sputum than BALF. High sensitivity of mNGS for microorganism detection was two-sided for pathogen identification, especially in sputum samples. On the one hand, mNGS improved the pathogen detection rate of patients. On the other hand, mNGS discovered lots of opportunistic and colonized microorganisms in sputum which challenged clinicians’ judgment.
Apart from pathogen identification, mNGS also has great potential in antibiotic resistance prediction. In our study, after strict filtering, 134 ARGs of 37 AMR gene families were detected from sputum samples from 50 ICU admission patients with pneumonia, and 33 patients had MDR genes. ErmX gene was the most frequent ARG which was positive in 25 cases, which is a common ARG detected in C. striatum. The ErmX gene could be found in plasmid, chromosomes or transposon, encoding the rRNA methylase enzyme, which brings about MLSB resistance.38,39 The cmx gene encoding efflux protein is another ARG in C. striatum, which confers resistance to phenicol through antibiotic efflux. In A. baumannii, a group of ARGs were detected, such as adeH, adeL, adeG, adeF, adeI, adeJ, adeK, adeN, which are MDR genes. MDR strains of A. baumannii are attributed to the extensive use of wide-spectrum antimicrobial drugs in hospitals, cross infection between inpatients, and invasive ICU procedures.40
Our research also has some limitations. First, mNGS was performed only based on DNA-seq, which may have lost the information of RNA virus. In patients with negative CMT and DNA mNGS results, complementary mNGS based on RNA-seq may improve pathogen detection sensitivity. In addition, the participants included in this study included majority of CAP patients and few HAP patients, but we did not carry out analysis according to type of pneumonia because of small sample size of HAP patients.
Conclusion
In conclusion, this study accumulated evidence that sputum mNGS is complementary to CMT in detecting pathogens for patients diagnosed with pneumonia, especially in situations when the CMT yields negative results or when the patients’ condition worsens despite treatment. The microorganism profile of sputum mNGS differed according to disease severity and immune status. This study suggested mNGS as a potential tool for pathogen detection especially for immunocompromised patients. There are a lot of colonized, opportunistic pathogenic and contaminative microorganisms in sputum of pneumonia patients. The interpretation of mNGS results should comprehensively consider microorganism’s relative abundance, the type of microorganism and its virulence, host immune status and disease history, clinical symptoms. mNGS is also a powerful tool in antibiotic resistance prediction. All these factors can help to guide appropriate treatment decisions.
Data Sharing Statement
The datasets generated for this study can be found in the NCBI BioProject database (BioProject ID: PRJNA955402).
Ethics Approval and Informed Consent
The study was conducted in accordance with the Declaration of Helsinki. All procedures were performed in accordance with the ethical standards of the ethics committee of Shandong Provincial Hospital Heze Branch, and informed consent was obtained from all individuals included in the study.
Funding
This work was supported by National Key Research & Development Plan (2018YFE0102400), Shanghai Special Project for Artificial Intelligence Innovation and Development (2020-RGZN-02039) and Science and Technology Development Project of Shandong Provincial Hospital Heze Branch (2021YN02).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Feldman C, Shaddock E. Epidemiology of lower respiratory tract infections in adults. Expert Rev Respir Med. 2019;13(1):63–77. doi:10.1080/17476348.2019.1555040
2. Bonell A, Azarrafiy R, Huong VTL, et al. A systematic review and meta-analysis of ventilator-associated pneumonia in adults in Asia: an analysis of national income level on incidence and etiology. Clin Infect Dis. 2019;68(3):511–518. doi:10.1093/cid/ciy543
3. Collaborators GL. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory tract infections in 195 countries: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect Dis. 2017;17(11):1133–1161. doi:10.1016/S1473-3099(17)30396-1
4. Collaborators GL. Age-sex differences in the global burden of lower respiratory infections and risk factors, 1990–2019: results from the Global Burden of Disease Study 2019. Lancet Infect Dis. 2022;22(11):1626–1647. doi:10.1016/S1473-3099(22)00510-2
5. Azoulay E, Russell L, Van de Louw A, et al. Diagnosis of severe respiratory infections in immunocompromised patients. Intensive Care Med. 2020;46(2):298–314. doi:10.1007/s00134-019-05906-5
6. Azoulay E, Lemiale V, Mokart D, et al. Effect of high-flow nasal oxygen vs standard oxygen on 28-day mortality in immunocompromised patients with acute respiratory failure: the high randomized clinical trial. JAMA. 2018;320(20):2099–2107. doi:10.1001/jama.2018.14282
7. Hill AT. Management of community-acquired pneumonia in immunocompromised adults: a consensus statement regarding initial strategies. Chest. 2020;158(5):1802–1803. doi:10.1016/j.chest.2020.08.003
8. Diao Z, Han D, Zhang R, Li J. Metagenomics next-generation sequencing tests take the stage in the diagnosis of lower respiratory tract infections. J Adv Res. 2022;38:201–212. doi:10.1016/j.jare.2021.09.012
9. Miller JM, Binnicker MJ, Campbell S, et al. A guide to utilization of the microbiology laboratory for diagnosis of infectious diseases: 2018 update by the Infectious Diseases Society of America and the American Society for Microbiology. Clin Infect Dis. 2018;67(6):813–816. doi:10.1093/cid/ciy584
10. Cilloniz C, Martin-Loeches I, Garcia-Vidal C, San Jose A, Torres A. Microbial etiology of pneumonia: epidemiology, diagnosis and resistance patterns. Int J Mol Sci. 2016;17(12):2120.
11. Katz SE, Williams DJ. Pediatric community-acquired pneumonia in the United States: changing epidemiology, diagnostic and therapeutic challenges, and areas for future research. Infect Dis Clin North Am. 2018;32(1):47–63. doi:10.1016/j.idc.2017.11.002
12. Jin X, Li J, Shao M, et al. Improving suspected pulmonary infection diagnosis by bronchoalveolar lavage fluid metagenomic next-generation sequencing: a multicenter retrospective study. Microbiol Spectr. 2022;10(4):e0247321. doi:10.1128/spectrum.02473-21
13. Buchan BW, Windham S, Balada-Llasat J-M, et al. Practical comparison of the BioFire FilmArray Pneumonia Panel to routine diagnostic methods and potential impact on antimicrobial stewardship in adult hospitalized patients with lower respiratory tract infections. J Clin Microbiol. 2020;58(7):10–128.
14. Simner PJ, Miller S, Carroll KC. Understanding the promises and hurdles of metagenomic next-generation sequencing as a diagnostic tool for infectious diseases. Clin Infect Dis. 2018;66(5):778–788. doi:10.1093/cid/cix881
15. Wang J, Han Y, Feng J. Metagenomic next-generation sequencing for mixed pulmonary infection diagnosis. BMC Pulm Med. 2019;19(1):1–8.
16. Miao Q, Ma Y, Wang Q, et al. Microbiological diagnostic performance of metagenomic next-generation sequencing when applied to clinical practice. Clin Infect Dis. 2018;67(suppl_2):S231–S240. doi:10.1093/cid/ciy693
17. Lu H, Ma L, Zhang H, et al. The comparison of metagenomic next-generation sequencing with conventional microbiological tests for identification of pathogens and antibiotic resistance genes in infectious diseases. Infect Drug Resist. 2022;15:6115–6128. doi:10.2147/IDR.S370964
18. Sun T, Wu X, Cai Y, et al. Metagenomic next-generation sequencing for pathogenic diagnosis and antibiotic management of severe community-acquired pneumonia in immunocompromised adults. Front Cell Infect Microbiol. 2021;11:661589. doi:10.3389/fcimb.2021.661589
19. Metlay JP, Waterer GW, Long AC, et al. Diagnosis and treatment of adults with community-acquired pneumonia. An Official Clinical Practice Guideline of the American Thoracic Society and Infectious Diseases Society of America. Am J Respir Crit Care Med. 2019;200(7):e45–e67. doi:10.1164/rccm.201908-1581ST
20. Wu X, Li Y, Zhang M, et al. Etiology of severe community-acquired pneumonia in adults based on metagenomic next-generation sequencing: a prospective multicenter study. Infect Dis Ther. 2020;9(4):1003–1015. doi:10.1007/s40121-020-00353-y
21. Lin T, Tu X, Zhao J, et al. Microbiological diagnostic performance of metagenomic next-generation sequencing compared with conventional culture for patients with community-acquired pneumonia. Front Cell Infect Microbiol. 2023;13:1136588. doi:10.3389/fcimb.2023.1136588
22. Chen X, Ding S, Lei C, et al. Blood and bronchoalveolar lavage fluid metagenomic next-generation sequencing in pneumonia. Can J Infect Dis Med Microbiol. 2020;2020:6839103. doi:10.1155/2020/6839103
23. van Ingen J, Kohl TA, Kranzer K, et al. Global outbreak of severe Mycobacterium chimaera disease after cardiac surgery: a molecular epidemiological study. Lancet Infect Dis. 2017;17(10):1033–1041. doi:10.1016/S1473-3099(17)30324-9
24. Edwardson S, Cairns C. Nosocomial infections in the ICU. Anaesth Intens Care Med. 2019;20(1):14–18. doi:10.1016/j.mpaic.2018.11.004
25. Li H, Gao H, Meng H, et al. Detection of pulmonary infectious pathogens from lung biopsy tissues by metagenomic next-generation sequencing. Front Cell Infect Microbiol. 2018;8:205. doi:10.3389/fcimb.2018.00205
26. Liang M, Fan Y, Zhang D, et al. 14Metagenomic next-generation sequencing for accurate diagnosis and management of lower respiratory tract infections. Int J Infect Dis. 2022;122:921–929. doi:10.1016/j.ijid.2022.07.060
27. Chen S, Kang Y, Li D, Li Z. Diagnostic performance of metagenomic next-generation sequencing for the detection of pathogens in bronchoalveolar lavage fluid in patients with pulmonary infections: systematic review and meta-analysis. Int J Infect Dis. 2022;122:867–873. doi:10.1016/j.ijid.2022.07.054
28. Yang A, Chen C, Hu Y, et al. Application of Metagenomic Next-Generation Sequencing (mNGS) Using Bronchoalveolar Lavage Fluid (BALF) in diagnosing pneumonia of children. Microbiol Spectr. 2022;10(5):e0148822. doi:10.1128/spectrum.01488-22
29. He Y, Fang K, Shi X, et al. 7 Enhanced DNA and RNA pathogen detection via metagenomic sequencing in patients with pneumonia. J Transl Med. 2022;20(1):195. doi:10.1186/s12967-022-03397-5
30. Qu J, Zhang J, Chen Y, et al. Aetiology of severe community acquired pneumonia in adults identified by combined detection methods: a multi-centre prospective study in China. Emerg Microbes Infect. 2022;11(1):556–566. doi:10.1080/22221751.2022.2035194
31. Harding CM, Hennon SW, Feldman MF. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat Rev Microbiol. 2018;16(2):91–102. doi:10.1038/nrmicro.2017.148
32. Giammanco A, Cala C, Fasciana T, Dowzicky MJ. Global assessment of the activity of tigecycline against multidrug-resistant gram-negative pathogens between 2004 and 2014 as Part of the tigecycline evaluation and surveillance trial. mSphere. 2017;2(1):e00310–e00316.
33. Jellinge ME, Hansen F, Coia JE, Song Z. Herpes simplex virus type 1 pneumonia-a review. J Intensive Care Med. 2021;36(12):1398–1402. doi:10.1177/0885066620965941
34. Simoons-Smit AM, Kraan EM, Beishuizen A, Strack van Schijndel RJ, Vandenbroucke-Grauls CM. Herpes simplex virus type 1 and respiratory disease in critically-ill patients: real pathogen or innocent bystander? Clin Microbiol Infect. 2006;12(11):1050–1059. doi:10.1111/j.1469-0691.2006.01475.x
35. Lee YW, Huh JW, Hong SB, et al. Severe Pneumonia Caused by Corynebacterium striatum in Adults, Seoul, South Korea, 2014–2019. Emerg Infect Dis. 2022;28(11):2147–2154. doi:10.3201/eid2811.220273
36. Nudel K, Zhao X, Basu S, et al. Genomics of Corynebacterium striatum, an emerging multidrug-resistant pathogen of immunocompromised patients. Clin Microbiol Infect. 2018;24(9):1016 e7–1016 e13. doi:10.1016/j.cmi.2017.12.024
37. Ponce CA, Gallo M, Bustamante R, Vargas SL. Pneumocystis colonization is highly prevalent in the autopsied lungs of the general population. Clin Infect Dis. 2010;50(3):347–353. doi:10.1086/649868
38. Chen H, Bai X, Gao Y, Liu W, Yao X, Wang J. Profile of bacteria with ARGs among real-world samples from ICU admission patients with pulmonary infection revealed by metagenomic NGS. Infect Drug Resist. 2021;14:4993–5004. doi:10.2147/idr.S335864
39. Ramos JN, Souza C, Faria YV, et al. Bloodstream and catheter-related infections due to different clones of multidrug-resistant and biofilm producer Corynebacterium striatum. BMC Infect Dis. 2019;19(1):1.
40. Ibrahim ME. Prevalence of Acinetobacter baumannii in Saudi Arabia: risk factors, antimicrobial resistance patterns and mechanisms of carbapenem resistance. Ann Clin Microbiol Antimicrob. 2019;18(1):1.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Outcomes and Predictors of Severe Community-acquired Pneumonia Among Adults Admitted to the University of Gondar Comprehensive Specialized Hospital: A Prospective Follow-up Study
Kassaw G, Mohammed R, Tessema GM, Yesuf T, Lakew AM, Tarekegn GE
Infection and Drug Resistance 2023, 16:619-635
Published Date: 28 January 2023