")
Back to Journals » Infection and Drug Resistance » Volume 16
Clinical Characteristics and Molecular Epidemiology of ST23 Klebsiella pneumoniae in China
Authors Liu Y , Jian Z , Wang Z, Yang A, Liu P, Tang B , Wang J, Yan Q, Liu W
Received 20 July 2023
Accepted for publication 6 December 2023
Published 11 December 2023 Volume 2023:16 Pages 7597—7611
DOI https://doi.org/10.2147/IDR.S428067
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Yanjun Liu,1 Zijuan Jian,1 Zhiqian Wang,1 Awen Yang,1 Peilin Liu,1 Bin Tang,1 Jiahui Wang,1,2 Qun Yan,1 Wenen Liu1,2
1Department of Clinical Laboratory, Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China; 2National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Changsha, Hunan, People’s Republic of China
Correspondence: Wenen Liu, Department of Clinical Laboratory, Xiangya Hospital, Central South University, Changsha, Hunan, 410000, People’s Republic of China, Tel +86 13973128689, Email [email protected]
Purpose: In clinical settings, CG23 Klebsiella pneumoniae (Kp) is the most virulent clonal group of Kp. Continuous fusions of hypervirulent (Hv) and highly resistant strains have been reported; however, few studies have analysed the molecular epidemiology and clinical characteristics of CG23 strains, especially MDR-sequence type ST23 strains. In this study, we investigated the molecular characteristics of ST23 Kp and analysed the clinical characteristics of ST23 Kp infections in a large teaching hospital of the third class in China.
Methods: ST23 Kp isolates were screened using whole-genome sequencing data from a large single centre. We compared the clinical characteristics of ST23 strains isolated from community-acquired infections (CAI) and hospital acquired infection (HAI). In addition, the infection characteristics of MDR and poor-prognosis isolates were investigated. We analysed genetic characteristics of ST23 Kp and further investigated the evolutionary relationship based on single-nucleotide polymorphism phylogenetic trees.
Results: We detected 184 ST23 strains between 2013 and July of 2018. There were no significant differences between the isolation rates of pulmonary, bloodstream, urinary tract, and cutaneous soft tissue infections in the community and hospitals, except for abscess infections. MDR strains primarily cause pulmonary infections and abscesses; infections with a poor prognosis are typically bloodstream and pulmonary infections. Fourteen MDR strains producing extended-spectrum or class C beta-lactamases, resulting in resistance to third-generation cephalosporins. In 3.8% of ST23 Kp strains, the clb locus was absent. The phylogenetic tree revealed that the isolates were primarily divided into three clades, and based on clinical data, it is inferred that three clonal transmission events have occurred, mainly in ICU causing lung infection.
Conclusion: This study demonstrates that virulence and drug-resistance fusion events of ST23 strains occur gradually, and that the hypervirulent clones facilitate the widespread dissemination of CAI and HAI, particularly pulmonary. Monitoring genomics and developing antivirulence strategies are essential.
Keywords: Klebsiella pneumoniae, ST23, multidrug-resistant, community-acquired infection, nosocomial infection, pneumonia
Introduction
Klebsiella pneumoniae (KP), a member of the Enterobacteriaceae family, inhabits aquatic environments and can colonise respiratory and gastrointestinal tracts in humans, making it a significant opportunistic pathogen. It may result in pneumonia, meningitis, urinary tract infections, and bloodstream infections. Hypervirulent Kp (HvKp) is believed to cause severe community-acquired invasive infections and is susceptible to most antimicrobials, whereas classic Kp is linked to HAI and is frequently multidrug-resistant (MDR).1
Recent research has uncovered a fusion event between hypervirulent and highly resistant strains. Several prevalent virulence clonal groups (CG), such as CG23, CG65, and CG86 were identified. Most contained prevalent Klebsiella spp capsule genotypes K1, K2, K5, K20, and K57.2 Two mechanisms were primarily responsible for the formation of MDR-Hv Klebsiella spp: mutation and horizontal gene transfer.3 Mutations in the quinolone resistance-determining region (QRDR mutations in gyrA and parC), membrane porin gene mutations (ompK36), and mutations that lead to overexpression of efflux pumps (acrAB, oqxxA, ramA, and rarA) were typically observed. As the most clinically virulent CG,4 CG23 fusion events continue to be reported, and their clinical impact demands immediate attention.
CG23 has been reported primarily in Asia, including Taiwan, Singapore, and Mainland China, where it is dominated by sequence type (ST) 23, along with ST26, ST57, and ST1633.5 The virulence plasmid (KPVP-1) is typically present in CG23 Kp, and the capsular serotype behaves as KL1. KPVP-1 is closely related to the capsule production regulatory genes rmpA/rmpA2 and the acquired siderophore genes iuc and iro.6,7
The majority lineage of the CG23Kp population is CG23-1, which was first detected in the 20th century and rapidly spread across five continents with population growth.8 ICEKp is an integrative conjugative element that aids in the propagation of the ybt locus encoding yersiniabactin. In contrast to the strain (Taiwan’s NTUH-K2044 reference strain) containing ICEKp1 encoding ybt-2, CG23-I contains ICEKp10 encoding ybt lineage 1 (ybt-1) and clb lineage 2.9 CG23-I contains the clb gene encoding the genotoxin colibactin, which increases bacterial pathogenicity.10
The current understanding of the drug resistance of ST23 Kp and infections caused by ST23 strains, particularly MDR strains, is insufficient. In this investigation, we sought to determine the molecular characteristics of ST23 Kp strains directly from second-generation-sequenced samples. We focused on the distribution of antibiotic resistance genes and virulence genes, as well as their evolution and dissemination, providing clinical characteristics of infection with the largest ST23 strain isolated from a single centre in China. Our findings may considerably expand our knowledge of Kp and provide insight for future HvKp research.
Materials and Methods
Klebsiella pneumoniae Strains Utilised in This Study
The results of whole-genome sequencing on 2193 Kp genomes were downloaded from China National GeneBank DataBase (accession number CNP0001198) (https://db.cngb.org/search/project/CNP0001198). The sequencing isolates were non-repetitive bacteria isolated between January 2013 and July 2018 from a teaching hospital in central China. We screened 202 ST23 Kp isolates out of 2193 Kp genomes, excluding lost or reactivated isolates, resulting in the inclusion of 184 ST23 isolates in the study. We utilised a software (https://cge.cbs.dtu.dk/services/MLST) to detect bacterial multilocus sequence typing (MLST).5
Antimicrobial Susceptibility Test, Clinical Collecting Information
The susceptibility to antimicrobials was determined using a VITEK-2 compact system (bioMérieux). The results were interpreted according to Clinical and Laboratory Standards Institute (CLSI) guidelines.11 According to the European Committee on Antimicrobial Susceptibility Testing (EUCAST) guidelines (accessible at http://www.eucast.org/clinical-breakpoints/), tigecycline and colistin drug susceptibility tests were detected using the microbroth dilution technique. MDR strains are resistant to three or more classes of antimicrobial drugs.12 These genotypes primarily confer resistance to third-generation cephalosporins. All sequenced isolates represented distinct patient infection episodes. According to the corresponding diagnostic criteria, an infection was diagnosed.13 HAI was defined as an infection occurring at least 48 hours after hospitalisation.14 CAI was defined as an infection that occurred within 48 hours of admission.15 The clinical outcomes of patients were divided into an improved group (patients, getting better and alive when being discharged) and a poor prognosis group (patients, death or terminal discharge).16
Detection of Drug Resistance Determinants, Virulence Determinants, and Plasmid Replicons
AMR genes, virulence genes, and plasmid replicon types were identified using ResFinder (https://cge.cbs.dtu.dk/services/ResFinder),17 Virulence Factor Database (VFDB; http://www.mgc.ac.cn/VFs), and the PlasmidFinder version 2.1 database, respectively.18 To identify AMR and virulence genes, the DNA sequence identity threshold and minimum length coverage were set to 90% and 80%, respectively.19 Through Kleborate (https://github.com/katholt/Kleborate), taxonomic assignment, capsule synthesis locus, lipopolysaccharide (O) locus, virulence locus genotypes, and ompK35 and ompK36 mutations were analysed.20
Phylogenetic Analysis
Core genes were identified in 184 isolates using Roary v3.13.0,21 and single-nucleotide polymorphisms (SNPs) were extracted from core genes for comparison. Snippy v4.6.0 was used to execute reference-based mapping and identify SNPs,14 with SGH10 Kp (CP025080) as the reference sequence.8 Fasttree 2.1.11 was used to construct a maximum likelihood tree,22 and iTOL version 3 was used to display and annotate the tree.23 KP isolates with fewer than 25 cgSNPs (core genome SNPs) were defined as belonging to the same clone.14
Statistical Analysis
Data analysis was performed using SPSS software (version 23.0). The χ2 test or Fisher’s exact test were used to analyse categorical variables, and the student’s t-test or Mann–Whitney U-test were used to compare continuous variables. All tests were two-tailed. Statistical significance was set at P < 0.05.
Ethical Approval
The Ethics Committee of Xiangya Hospital of Central South University authorised the collection of Klebsiella pneumoniae strains and clinical data from patients (anonymously). The inability to acquire the patient’s informed consent due to the retrospective nature of the study qualifies it for an informed consent exemption (ID 202212295, Changsha, Hunan Province, People’s Republic of China).
Results
Clinical Distribution and Antimicrobial Susceptibility of 184 ST23 Kp Isolates
We analysed 184 ST23 isolates from a large teaching hospital between January 2013 and July 2018 (Supplemental Table 1). According to their clinical characteristics, all 184 ST23 Kp strains were diagnosed as infections, with 17, 24, 50, 34, 40, and 19 strains detected annually from 2013 to the first half of 2018 (Figure 1A). The majority of these 184 strains were isolated from internal medicine wards and intensive care units (ICUs), surgical wards, outpatient and emergency departments, with similar isolation rates. Figure 1B depicts that most of the samples were respiratory secretions, followed by drainage and puncture fluid, blood, abscess, urine, and wound secretions. Kp isolates were highly susceptible to a majority of antibiotics. Fourteen isolates were resistant to three or more antibiotic classes, including 7.6% (n = 14) that were resistant to ceftriaxone, 1.1% (n = 2) that were resistant to piperacillin/tazobactam, and 3.3% (n = 6) that were resistant to ciprofloxacin. One of the genotypes exhibited tigecycline resistance. Strains resistant to polymyxin B or carbapenems were not detected (Figure 1C). The rates of MDR sequentially from 2013 to 2018 were 5.8% (1/17), 8.3% (2/24), 6% (3/50), 5.8% (2/34), 10% (4/40), and 10.5% (2/19) (Figure 1A).
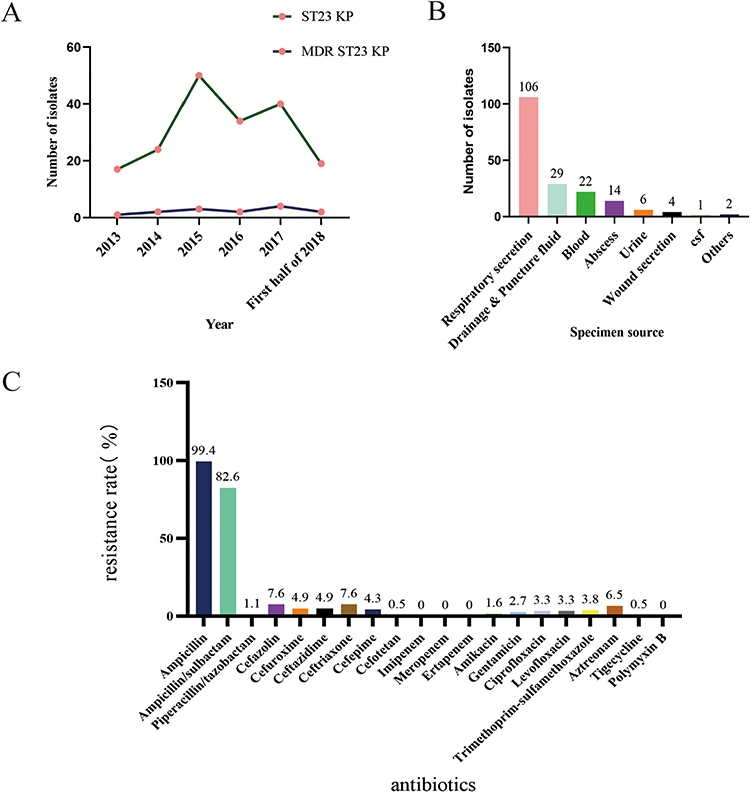 |
Figure 1 Clinical distribution and antimicrobial susceptibility of 184 ST23 Kp isolates. Notes: (A) Temporal distribution of ST23 Klebsiella pneumoniae and MDR ST23 Kp strains. (B) The number of ST 23 isolates from different specimen sources. (C) The resistance rate of 184 ST23 KP strains. |
Clinical Characteristics of Community-Acquired and Nosocomial Infections of ST23 Kp Isolates
We collected and analysed the clinical data of 184 ST23 Kp strains. Patients with ST23 Kp infection were predominantly of male sex (n = 135, 73.4%), with a median age of 55 years (interquartile range [IQR], 40–71 years). Of these infected patients, 27.2% (50/184) were elderly and 3.3% (6/184) were children. Infections were primarily found in internal medicine and ICU settings and were mainly CAI (65.2%). The most common manifestations were pneumonia (58.7%), followed by abscesses (19.57%), and bacteraemia (10.87%). In addition to common pyogenic liver abscesses, there were 27.8% (10/36) non-hepatic abscesses, including neck, brain, and skin abscesses (Supplemental Table 2). The most frequent comorbid conditions diagnosed were cerebral vascular disease (n = 35, 19.0%), malignant tumour (n = 29, 15.8%), pulmonary disease (n = 28, 15.2%), diabetes mellitus (n = 25, 13.5%), and trauma (n = 18, 9.8%). These results are shown in Table 1. Twelve patients had sepsis or septic shock, and seven patients had a poor prognosis (Supplemental Table 2). Our results showed a high incidence of abscesses in CAI (P<0.001), but primary bacteraemia and respiratory, urethral, skin, and soft tissue infections were not significantly associated with CAI (P = 0.98, 0.09, 0.72, and 0.72, respectively). Patients with comorbid diabetes had higher rates of CAI (P<0.001), patients with comorbid trauma had higher rates of HAI (P<0.001) (Table 1).
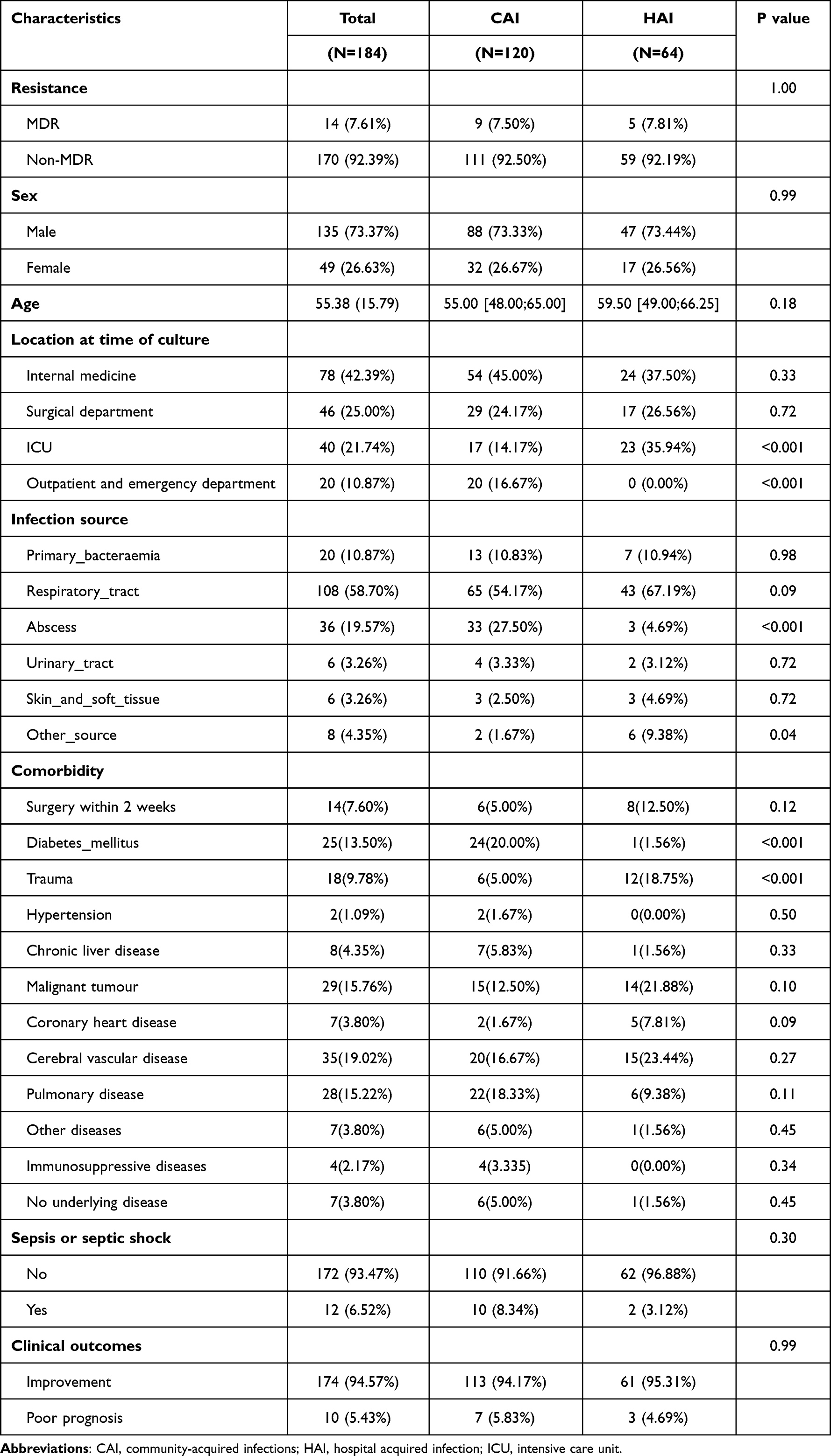 |
Table 1 Clinical Characteristics of 184 Strains in Community Acquired Infection and Nosocomial Infections |
Clinical Features of MDR and Non-MDR Strain Infections
This study compared the infection characteristics of MDR and non-MDR isolates and found no statistically significant differences (Supplementary Table 3-1). 50% (7/14) of the 14 MDR strain infections were respiratory tract infections, 28.6% (4/14) were abscess infections, 85.75% (12/14) were in males, and 64.3% (9/14) were CAI. Except for one strain, the preponderance of respiratory specimens were acquired in the community. It is noteworthy that in 2014, community-acquired MDR ST23 Kp caused severe pneumonia in an emergency department patient, which may have contributed to the patient’s mortality. The results are shown in Table 2.
 |
Table 2 Clinical Characteristics of 14 MDR ST23 Strains |
Clinical Outcomes of Infections Caused by ST23 Strains
We also analyzed the clinical outcomes of patients, and found that 174 patients with infection improved, and 10 patients with infection had poor prognosis. There was no significant difference in sex, age, acquired infection, location at time of culture, comorbidity, or resistance between two groups. The results showed primary bloodstream infection or sepsis has a stronger correlation with poor prognosis (P<0.001) (Table 3). Ten isolates resulted in infections with poor prognosis, one of which was MDR. Additionally, 50% percent of the specimens were from blood, 40% (4/10) from the respiratory tract, and 10% (1/10) from cerebrospinal fluid. All pulmonary infections and 70% of infections were CAI, 60% of bloodstream infections were community-based, 40% occurred in-hospital, and the only intracranial infection was nosocomial. A total of 40% of infections occurred in the ICU. Ninety percent of these patients had fever, and inflammatory marker testing revealed variably elevated white blood cell and procalcitonin levels in a large proportion of patients; all had elevated neutrophil percentage values. The results are presented in (Supplemental Table 3-2).
 |
Table 3 Comparison of Clinical Infection Characteristics Caused by ST23 Kp Between the Improvement Group and the Poor Prognosis Group |
Antimicrobial Resistance Genes in ST23 Kp Isolates
All ST23 isolates were KpI (Kp, n = 184; Supplemental Table 4). We analysed the resistance genes of all ST23 strains and did not identify any genes encoding carbapenemases such as blaKPC-2 or blaNDM. The vast majority of strains were positive for blaSHV-190, fosA, oqxA, and oqxB. In addition, CTX-M beta-lactamases, which are among the most important extended-spectrum lactamases, were identified in 13 isolates. These were blaCTX-M-3 (n = 5, 2.7%), blaCTX-M-14 (n = 4, 2.2%), blaCTX-M-15 (n = 2, 1.1%), and blaCTX-M-55 (n = 3, 1.6%), with one strain containing both blaCTX-M-3 and blaCTX-M-14. Two (or 1.1%) of these strains contained the blaDHA-1 gene. Notably, we did not identify any isolates carrying the ompK35 or ompK36 mutations (Supplementary Table 4). Among 184 ST23 strains, resistance to aminoglycoside antibiotics was mediated by aminoglycoside transferases, including adenylyl-transferase aadA6 (n = 5, 2.7%), aadA1 (n = 2, 1.1%), and aadA5 (n = 1, 0.5%), phosphotransferases aph(3’)-Ib (n=5, 2.7%), and aph(6)-Id (n=5, 2.7%), and acetyltransferases aac(3)-IId (n = 4, 2.2%), aac(6’)-Ib-cr (n = 6, 3.3%), and aac(6’)-Ib3 (n = 1, 0.5%). In 2.7% (5/184) of isolates, we also identified mutations in the QRDR of the gyrA gene. In 3.8% (7/184), 1.1% (2/184), and 0.5% (1/184) of the isolates, the plasmid-mediated quinolone resistance-related qnrS1, qnrB4, and qnrB6 genes were detected, respectively. In addition, we identified genes associated with tetracycline resistance in 3.3% of isolates (tet(A)), trimethoprim resistance in 6.0% of isolates (dfrA), and sulphonamide resistance mediated by sul1 (4.9%) and sul2 (2%). We did not detect tet(X) and mcr genes linked to tigecycline and polymyxin resistance. Figure 2A demonstrates the results.
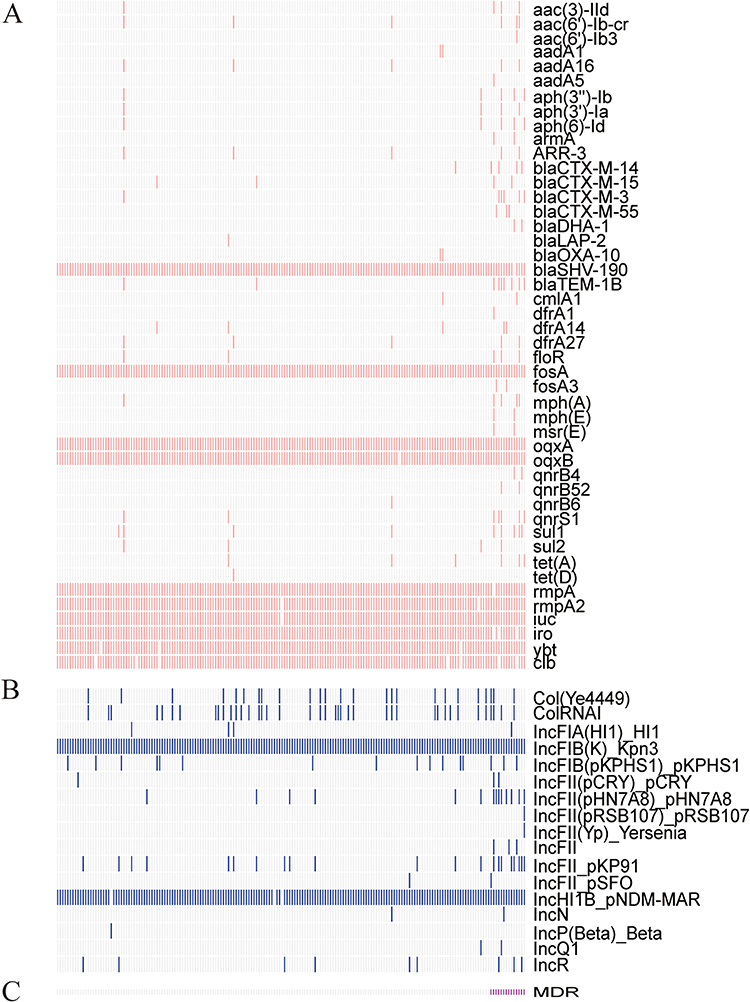 |
Figure 2 Molecular characteristics of 184 ST23 Klebsiella pneumoniae strains. Notes: The carriage of (A) resistance genes, virulence genes, and (B) plasmid replicon types is exhibited as follows: white colors represent the absence of genes or plasmid replicons, and vivid colors represent the presence of genes or plasmid replicons. The bottom rose color represents the MDR strain (C). |
Virulence Determinants in ST23 Kp Isolates
In addition to antimicrobial resistance, we investigated the genetic diversity of Kp isolates’ virulence determinants. All isolates carried the capsule synthesis locus KL1 and the O locus O1/O2v2 (Supplementary Table 4). We investigated the relationship between the capsular regulatory genes rmpA and rmpA2 and the hypermucoid phenotype in ST23 Kp isolates. The rmpA locus was present in 99.5% (n = 183) of isolates, whereas the rmpA2 locus was absent in two strains. Additionally, we analysed the siderophore genes encoding aerobactin, salmochelin, and yersiniabactin, which are acquired siderophore systems capable of enhancing bacterial iron uptake. The genotoxicity-related clb gene was also identified. Figure 2A depicts the detection rates of the iuc gene, iro gene, ybt, and clb genes as 99.5%, 98.4%, 98.4%, and 96.2%, respectively. Further examination of the virulence locus genotypes revealed that ICEKp10 with ybt1 was the most prevalent subtype of ICEKp (Figure 3A). ICEKp1 with ybt-2 was detected in three digestive system and respiratory secretion isolates. One abscess isolate contained ICEKp3, encoding ybt-9 (Supplementary Table 4). Intriguingly, the two isolates that carried ICEKp1 and ybt-2 were MDR strains. The absence of the iuc1 gene was most prevalent in MDR strains (Figure 3B).
 |
Figure 3 Virulence genes and subtypes among the 184 ST23 strains. Notes: The sequence gene clusters encoding aerobactin, colibactin, salmochelin, and yersiniabactin, and the corresponding subtypes of the genes, including iuc, clb, iro, and ybt locus. (A) 184 ST23 isolates. (B) 14 MDR ST23 isolates. |
Plasmid Replicons in ST23 Kp Isolates
We analysed 17 plasmid replicon types, including ColRNAI, IncFII, IncFIB, IncFIA, IncP, and IncR, to determine the significance of plasmid-borne elements in ST23 Kp (Figure 2B). We detected 16 plasmid replicon types in MDR strains and 14 replicon types in non-MDR strains. Nearly every strain contained two plasmid replicons, IncFIBK (n=184) and IncHI1B (n=182). The majority of MDR strains had an IncFII replicon (Figure 2C).
Phylogenetic Analysis of ST23 Kp Isolates
The phylogenetic tree analysis revealed that 184 ST23 Kp strains were primarily divided into three clades, with clade A containing 19 isolates, clade B containing 33 isolates, and clade C containing the maximum number of isolates (124). The remaining eight isolates did not form a cluster with them. CG23-1 (178/184) was the most prevalent sublineage (Supplementary Table 4), while the remaining six non-CG23-1 strains formed a minor branch within clade C. In terms of hospital-acquired infections, clade A had an isolation rate of 42.1% (8/19), clade B had an isolation rate of 42.4% (14/33), and clade C had an isolation rate of 29.8% (37/124). In the three evolutionary branches, the isolation rates of MDR strains were 5.3% (1/19) for clade A, 6% (2/33) for clade B, and 8.9% (11/124) for clade C. The paired SNP matrix heatmap and hospitalisation data indicated the occurrence of three clonal dissemination events, including Kp788 (HAI, August 29, 2015) to Kp939 (HAI, November 14, 2015), Kp832 (HAI, September 16, 2015) to Kp836 (HAI, September 17, 2015), and Kp975 (HAI, December 11, 2015) to Kp977 (HAI, December 13, 2015) (Supplementary Figure 1). The range of SNP differences was between 0 and 4. The first clone diffusion occurred in clade B, and the subsequent two occurred in clade C. These six strains originated in the respiratory system and caused pulmonary infections in their hosts. One strain was detected in the department of internal medicine, and five strains were isolated in the intensive care unit (Supplemental Table 2). The results are presented in Figure 4.
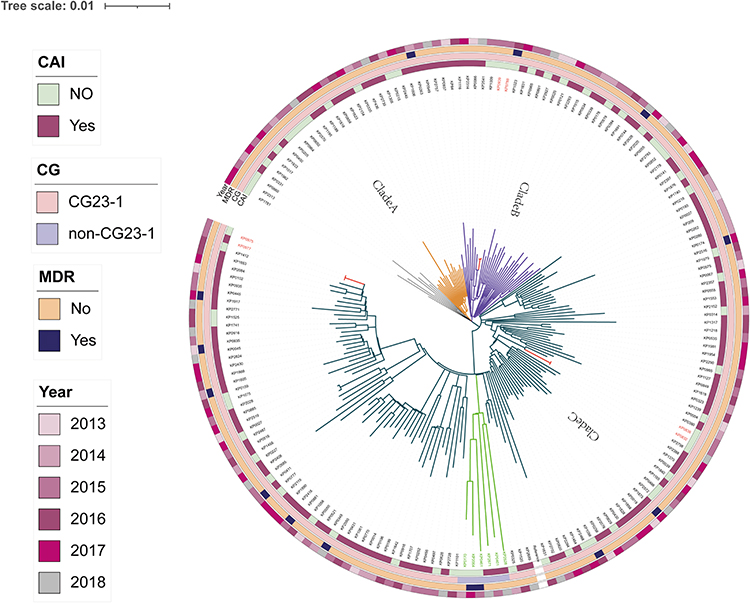 |
Figure 4 Phylogenetic analysis of 184 ST23 Klebsiella pneumoniae strains. Notes: The maximum likelihood phylogenetic tree constructed by core genome single-nucleotide polymorphisms. Circles outside the tree, from inside to outside, indicate CAI (yes or no), CG sublineages, MDR (yes or no), year. The specific information represented by different colors in each circle of the heatmap is presented in the left note. Within circles orange yellow represents clade A, purple represents clade B, dark blue represents clade C, red represents clonally disseminated strains, and green represents the non-CG23-1 sublineage. |
Trends of ST23 Infections from 2013–2018
This study examined the fluctuating trend of ST23 Kp infections over the past five years. Figure 5 is a Sankey diagram illustrating the gradual increase in the detection rate of MDR strains from 5.8% in 2013 to 10.0% in 2018. Despite the prevalence of CAI (65.2%), nosocomial infections continue to occur. Although the predominant lineage CG23-1 was responsible for a wide variety of infections, it was found that other lineages also caused pulmonary, systemic, and abscess infections in multiple hospital departments. Figure 5 demonstrates the results.
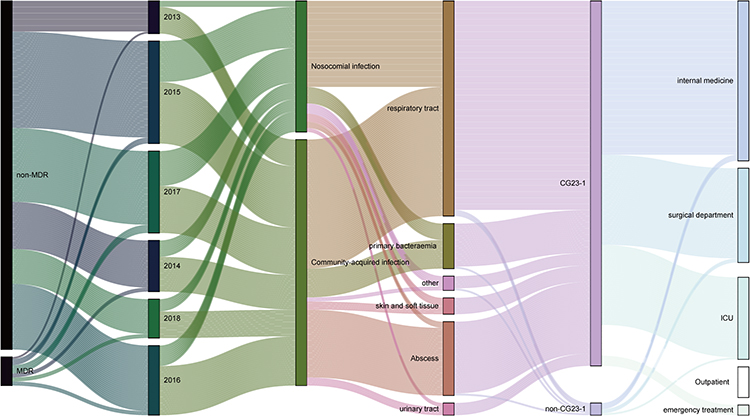 |
Figure 5 Trend diagram of infection characteristics of ST23 KP strains between 2013–2018. Notes: Strains were divided into MDR and non-MDR, CG23-1 lineages and non-CG23-1 lineages. From the clinical perspective, community-acquired and nosocomial infections, as well as infection and department sources were included. |
Discussion
HvKp strains frequently cause life-threatening CAIs in young, healthy hosts, but are typically susceptible to antibiotics. CG23 Kp is one of the most prevalent Kp varieties in clinical settings and is typically associated with hypervirulence. We analysed the clinical characteristics and molecular genetic characteristics of 184 ST23 isolates from the last five years, which may provide a solid foundation for clinicians to diagnose and treat infectious diseases.
We compared CAI and HAI infections caused by ST23 strains. Our investigation revealed that ST23 Kp genotypes were widely responsible for clinical infections. MDR and non-MDR strains of CAI and HAI did not exhibit significant differences. Notably, the rate of abscess was consistent with previous findings from Taiwan,24 and there was no significant nosocomial dissemination (P<0.001). Pulmonary, bloodstream, urinary tract, and cutaneous soft tissue infections (CAI vs HAI, P=0.98, 0.09, 0.72, and 0.72, respectively) exhibited obvious community-to-hospital transmission. This implies that strains that were formerly believed to be primarily responsible for CAI have been linked to other types of infections in hospitals.
We further analysed the origin of the abscesses: 72.2% (26/36) were from pyogenic liver abscesses, and 27.6% were from areas such as the abdominal cavity, neck, and pelvic cavity. We speculated that abscesses are more likely to be diagnosed early by clinicians and that early use of antibiotics and effective drainage make abscesses less prone to nosocomial dissemination. Several studies24,25 have reported that HvKp strains can colonise the gut and cause liver abscesses, but HvKp can also colonise the nasopharynx or skin surface.2 We speculated that the damage to the skin barrier caused by an invasive operation (use of ventilator, catheter, or intravenous catheter) may contribute to hospital infection by HvKp. Therefore, any reduction in invasive operations may reduce the occurrence of HvKp in hospitals.
Compared to the 17–25% of ST23 Kp-caused infections occurring at other sites than abscesses reported in a previous study,8 in which pulmonary infections accounted for 58.7%; our study revealed that more than 80% of ST23 Kp-caused infections were found at sites in addition to abscesses. Kp is a significant cause of community-acquired pneumonia (CAP) in Asia, is associated with high mortality in this region, and is also a primary cause of nosocomial pneumonia in the United States.26 Our previous study27 demonstrated that 28.6% of ventilator-associated pneumonia was caused by HvKp; however, this proportion increased to 44.6% in the study of Liu et al,28 with ST23 strains predominating. Our results confirmed that ST23 Kp has caused a substantial number of pneumonia cases in clinical practice, and in nosocomial acquired infections ST23 Kp occurrence reached 39.8% (43/108).
In addition, the characteristics of MDR and poor-prognosis strains were investigated. The 14 MDR strains were primarily isolated from the respiratory tract and abscesses, and only the KP159 strain was associated with a poor prognosis in pulmonary infections. Only one of the ten genotypes associated with a poor prognosis was resistant to third-generation cephalosporins. Ninety percent of infections were pulmonary and bloodstream, with all CAI presenting as pulmonary infections. This result prompted us to consider not only infections caused by influenza A virus, adenovirus, and Chlamydia psittaci, but also infections caused by ST23 Kp in clinical settings involving patients with community-derived refractory fever. Our study also suggests that CAP and bloodstream infections can be fatal even when there is an abundance of sensitive antibiotics; therefore, the development of new antivirulence therapies (eg, siderophore inhibitors, quorum-sensing system inhibitors, and phage therapy) or other novel anti-infection strategies may already be warranted in the face of HvKp infections.
In terms of antibiotic resistance and mobile components related to drug resistance, this study revealed that 7.6% of the 184 ST23 Kp isolates belonged to MDR strains, which is slightly less than the 10.4% found in a previous study.29 All fourteen of these isolates were resistant to third-generation cephalosporins. Previous research29 demonstrates that IncFII plasmids and/or IS26 contribute to the acquisition of AMR genes in ST23 Kp. In our study, MDR strains had a higher incidence of IncFII plasmids than non-MDR strains. We did not identify any ST23 carbapenem-resistant HvKp (CR-HvKp) strains. Currently, the majority of CR-HvKp infections reported worldwide originate in China, primarily as a result of ST11-KPC HvKp,3,30 whereas ST23 only accounts for a minor proportion. In a multicentre investigation in China, only 11 strains of ST23 CR-HvKp were collected.31 This strain has also been reported sporadically in other countries, such as Poland32 and Switzerland.33 Previous studies8 have speculated that the physical barrier or CRISPR/CAS system may have affected the acquisition of drug-resistant plasmids by bacteria. Dong et al34 revealed that the acquisition of resistance plasmids by ST23 may generate a limited fitness cost in virulence and drug resistance; however, the acquisition of virulence plasmids by ST11-CRKP without the development of adaptive changes may be one cause of the clinical dissemination of ST11-HvKp. Overall, during the five years of the study, ST23 Kp was relatively restrained in its acquisition of drug resistance, but it still requires vigilance and attention.
The virulence factors of ST23 Kp, which are prevalent in almost all ST23 isolates, were examined in greater detail. In addition to fimbriae and lipopolysaccharide, it is known that the enhancement of bacterial virulence depends on siderophores or capsular regulatory genes.2 Among them, the iro and iuc genes are highly specific siderophore genes that can assist microbes in acquiring iron from the environment, thereby enhancing their ability to survive.7 In this study, the rates of carrying the iuc gene, the iro gene, the ybt gene, and the clb gene in ST23 Kp strains were all greater than 96%. MDR strains lacked the iro1 gene, consistent with Peng et al’s study.29 However, further investigation into the cause of this absence is required. Previous research has demonstrated a correlation between the classification of CG23 sublineages and the acquisition of ICEKp, with CG23-1 being the most prevalent and rapidly disseminating sublineage in the human population. Six additional sublines devoid of yersiniabactin and/or colibactin were found to cause clinical infection rather than asymptomatic transmission.8 Moreover, it was demonstrated that no specific virulence factor is required for CG23 Kp to cause its invasive infection.
Finally, we analysed the evolutionary relationships and clonal dissemination events among the 184 ST23 strains. We discovered that the SNP phylogenetic tree separated 184 into three main clades, with clade C having the largest branch. Clade A (42.1%, 8/19) and clade B (42.4%, 14/33) exhibited a higher prevalence of nosocomial infections compared to clade C (29.8%, 37/124), which displayed greater genetic divergence. In addition, this study identified three transmission events that led to pulmonary infections in 2015, all of which were nosocomial infections. All six strains primarily caused pulmonary infections in the ICU, where they were found in abundance. We hypothesised that the use of ventilators or tracheal catheters facilitated the spread of clones, necessitating vigilance on the part of the clinician. Notably, sample collection and hospitalisation data indicate that clone transmissions peaked in the latter half of 2015 (August to December). Combined with the isolation rate of ST23, this indicates that 2015 was the year with the highest detection of ST23 as well as the year with the highest prevalence of clone transmission. However, we have not discovered any reports of global epidemics around the year 2015, so it is possible that the localised outbreak was only temporary. Clonal dissemination was both vertical and horizontal.35 Previous research has demonstrated that the clb colibactin synthesis locus plays a significant role in the spread of CG23, which promotes bacterial colonisation and metastatic infection.36 This study suggests that the evolutionary spread of ST23 Kp has facilitated a change in the mode of infection, such that the categories of infections have become more diverse. The systematic evolution and transmission events further confirmed the previously hypothesised expansion of ST23 infections from the community to hospitals in clinical practice. Our Sankey diagram also made this flow direction more evident.
Limitation of Study
Several limitations exist in our investigation. First, the current study examined the virulence of the bacteria at the molecular level and did not conduct in vitro-in vivo experiments, such as serum resistance and the Galleria mellonella model, or mouse infection models. Secondly, this was a retrospective study as well. Several clinical examinations and a portion of the strains have been lost. Thirdly, it is difficult to establish statistically significant differences between MDR and non-MDR isolates due to the low number of MDR isolates. We will expand the sample size in the follow-up study and pay attention to the risk factors of poor prognosis.
Conclusion
In conclusion, this single-centre study describes the current status of the most virulent CG from a multidimensional perspective of clinical characteristics, drug resistance, virulence, and evolutionary relationship, and serves as an excellent resource for clinicians and infection control specialists. This study revealed that the ability of ST23 strains to acquire drug resistance has gradually increased, primarily among strains resistant to three generations of cephalosporins, but not among strains resistant to carbapenems. Nevertheless, the infection has spread extensively in clinical practice and is dominated by CG23-1 lineages, with both community and hospital-based expansion. Consequently, the invention of new antivirulence pharmaceuticals and other anti-infection strategies is required. We will continue to monitor these data to observe the evolution of this crucial clinical pathogen.
Data Sharing Statement
The data supporting this study have been deposited in the database (CNGBdb) and logged into the website https://db.cngb.org/search/project/ CNP0001198/.
Author Contributions
WL, QY, and YL conceived and designed the project; ZJ, ZW, and JW participated in the sample collection; YL, ZJ, QY, PL, and BT were responsible for bacterial culture and drug sensitivity test; YL, ZW, ZJ, and AY conducted bioinformatics analysis and statistical analysis on the data. YL wrote the manuscript. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of China (grant number 81672066 to WL).
Disclosure
The author declares that this study was conducted without any commercial or financial relationship that could be interpreted as a potential conflict of interest.
References
1. Russo TA, Marr CM. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev. 2019;32(3). doi:10.1128/cmr.00001-19
2. Choby JE, Howard-Anderson J, Weiss DS. Hypervirulent Klebsiella pneumoniae - clinical and molecular perspectives. J Intern Med. 2020;287(3):283–300. doi:10.1111/joim.13007
3. Dong N, Yang X, Chan EW, Zhang R, Chen S. Klebsiella species: taxonomy, hypervirulence and multidrug resistance. EBioMed. 2022;79:103998. doi:10.1016/j.ebiom.2022.103998
4. Zhang S, Zhang X, Wu Q, et al. Clinical, microbiological, and molecular epidemiological characteristics of Klebsiella pneumoniae-induced pyogenic liver abscess in southeastern China. Antimicrob Resist Infect Control. 2019;8:166. doi:10.1186/s13756-019-0615-2
5. Wyres KL, Nguyen TNT, Lam MMC, et al. Genomic surveillance for hypervirulence and multi-drug resistance in invasive Klebsiella pneumoniae from South and Southeast Asia. Genome Med. 2020;12(1):11. doi:10.1186/s13073-019-0706-y
6. Tian D, Wang M, Zhou Y, Hu D, Ou HY, Jiang X. Genetic diversity and evolution of the virulence plasmids encoding aerobactin and salmochelin in Klebsiella pneumoniae. Virulence. 2021;12(1):1323–1333. doi:10.1080/21505594.2021.1924019
7. Lam MMC, Wyres KL, Judd LM, et al. Tracking key virulence loci encoding aerobactin and salmochelin siderophore synthesis in Klebsiella pneumoniae. Genome Med. 2018;10(1):77. doi:10.1186/s13073-018-0587-5
8. Lam MMC, Wyres KL, Duchêne S, et al. Population genomics of hypervirulent Klebsiella pneumoniae clonal-group 23 reveals early emergence and rapid global dissemination. Nat Commun. 2018;9(1):2703. doi:10.1038/s41467-018-05114-7
9. Lam MMC, Wick RR, Wyres KL, et al. Genetic diversity, mobilisation and spread of the yersiniabactin-encoding mobile element ICEKp in Klebsiella pneumoniae populations. Microb Genom. 2018;4:9. doi:10.1099/mgen.0.000196
10. Vizcaino MI, Crawford JM. The colibactin warhead crosslinks DNA. Nat Chem. 2015;7(5):411–417. doi:10.1038/nchem.2221
11. Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Sus-Ceptibility Testing.
12. Magiorakos AP, Srinivasan A, Carey RB, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18(3):268–281. doi:10.1111/j.1469-0691.2011.03570.x
13. Miller JM, Binnicker MJ, Campbell S, et al. A guide to utilization of the microbiology laboratory for diagnosis of infectious diseases: 2018 update by the Infectious Diseases Society of America and the American Society for Microbiology. Clin Infect Dis. 2018;67(6):e1–e94. doi:10.1093/cid/ciy381
14. Chi X, Meng X, Xiong L, et al. Small wards in the ICU: a favorable measure for controlling the transmission of carbapenem-resistant Klebsiella pneumoniae. Intensive Care Med. 2022;48(11):1573–1581. doi:10.1007/s00134-022-06881-0
15. van Duin D, Paterson DL. Multidrug-resistant bacteria in the community: an update. Infect Dis Clin North Am. 2020;34(4):709–722. doi:10.1016/j.idc.2020.08.002
16. Sun J, Sun W, Tang Y, et al. Clinical characteristics and risk factors for poor prognosis among HIV patients with Talaromyces marneffei bloodstream infection. BMC Infect Dis. 2021;21(1):514. doi:10.1186/s12879-021-06232-2
17. Bortolaia V, Kaas RS, Ruppe E, et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother. 2020;75(12):3491–3500. doi:10.1093/jac/dkaa345
18. Carattoli A, Zankari E, García-Fernández A, et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58(7):3895–3903. doi:10.1128/aac.02412-14
19. Zankari E, Hasman H, Cosentino S, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67(11):2640–2644. doi:10.1093/jac/dks261
20. Lam MM, Wick RR, Wyres KL, et al. Frequent emergence of pathogenic lineages of Klebsiella pneumoniae via mobilisation of yersiniabactin and colibactin. Microbial Genomics. 2018;4:9. doi:10.1099/mgen.0.000196
21. Page AJ, Cummins CA, Hunt M, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31(22):3691–3693. doi:10.1093/bioinformatics/btv421
22. Lemoine F, Correia D, Lefort V, et al. NGPhylogeny.fr: new generation phylogenetic services for non-specialists. Nucleic Acids Res. 2019;47(W1):W260–W265. doi:10.1093/nar/gkz303
23. Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44(W1):W242–5. doi:10.1093/nar/gkw290
24. Siu LK, Yeh KM, Lin JC, Fung CP, Chang FY. Klebsiella pneumoniae liver abscess: a new invasive syndrome. Lancet Infect Dis. 2012;12(11):881–887. doi:10.1016/s1473-3099(12)70205-0
25. Yang PW, Lin HD, Wang LM. Pyogenic liver abscess associated with septic pulmonary embolism. J Chin Med Assoc. 2008;71(9):442–447. doi:10.1016/s1726-4901(08)70146-1
26. Juan CH, Fang SY, Chou CH, Tsai TY, Lin YT. Clinical characteristics of patients with pneumonia caused by Klebsiella pneumoniae in Taiwan and prevalence of antimicrobial-resistant and hypervirulent strains: a retrospective study. Antimicrob Resist Infect Control. 2020;9(1):4. doi:10.1186/s13756-019-0660-x
27. Yan Q, Zhou M, Zou M, Liu WE. Hypervirulent Klebsiella pneumoniae induced ventilator-associated pneumonia in mechanically ventilated patients in China. Eur J Clin Microbiol Infect Dis. 2016;35(3):387–396. doi:10.1007/s10096-015-2551-2
28. Liu C, Guo J. Characteristics of ventilator-associated pneumonia due to hypervirulent Klebsiella pneumoniae genotype in genetic background for the elderly in two tertiary hospitals in China. Antimicrob Resist Infect Control. 2018;7:95. doi:10.1186/s13756-018-0371-8
29. Du P, Liu C, Fan S, Baker S, Guo J. The role of plasmid and resistance gene acquisition in the emergence of ST23 multi-drug resistant, hypervirulent Klebsiella pneumoniae. Microbiol Spectr. 2022;10(2):e0192921. doi:10.1128/spectrum.01929-21
30. He Z, Xu W, Zhao H, et al. Epidemiological characteristics an outbreak of ST11 multidrug-resistant and hypervirulent Klebsiella pneumoniae in Anhui, China. Front Microbiol. 2022;13:996753. doi:10.3389/fmicb.2022.996753
31. Wang S, Ding Q, Zhang Y, et al. Evolution of virulence, fitness, and carbapenem resistance transmission in ST23 hypervirulent Klebsiella pneumoniae with the capsular polysaccharide synthesis gene wcaj inserted via insertion sequence elements. Microbiol Spectr. 2022;10(6):e0240022. doi:10.1128/spectrum.02400-22
32. Biedrzycka M, Izdebski R, Urbanowicz P, et al. MDR carbapenemase-producing Klebsiella pneumoniae of the hypervirulence-associated ST23 clone in Poland, 2009–19. J Antimicrob Chemother. 2022;77(12):3367–3375. doi:10.1093/jac/dkac326
33. Hallal Ferreira Raro O, Nordmann P, Dominguez Pino M, Findlay J, Poirel L. Emergence of carbapenemase-producing hypervirulent Klebsiella pneumoniae in Switzerland. Antimicrob Agents Chemother. 2023;67(3):e0142422. doi:10.1128/aac.01424-22
34. Tian D, Liu X, Chen W, et al. Prevalence of hypervirulent and carbapenem-resistant Klebsiella pneumoniae under divergent evolutionary patterns. Emerg Microbes Infect. 2022;11(1):1936–1949. doi:10.1080/22221751.2022.2103454
35. Marimuthu K, Venkatachalam I, Koh V, et al. Whole genome sequencing reveals hidden transmission of carbapenemase-producing enterobacterales. Nat Commun. 2022;13(1):3052. doi:10.1038/s41467-022-30637-5
36. Lu MC, Chen YT, Chiang MK, et al. Colibactin contributes to the hypervirulence of pks(+) K1 CC23 Klebsiella pneumoniae in mouse meningitis infections. Front Cell Infect Microbiol. 2017;7:103. doi:10.3389/fcimb.2017.00103
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.